
Abstract
The exposure to high glucocorticoids (GC) triggers neuronal atrophy and cognitive deficits, but the exact cellular mechanisms underlying the GC-associated dendritic remodeling and spine loss are still poorly understood. Previous studies have implicated sustained GC elevations in neurodegenerative mechanisms through GC-evoked hyperphosphorylation of the cytoskeletal protein Tau while Tau mislocation has recently been proposed as relevant in Alzheimer’s disease (AD) pathology. In light of the dual cytoplasmic and synaptic role of Tau, this study monitored the impact of prolonged GC treatment on Tau intracellular localization and its phosphorylation status in different cellular compartments. We demonstrate, both by biochemical and ultrastructural analysis, that GC administration led to cytosolic and dendritic Tau accumulation in rat hippocampus, and triggered Tau hyperphosphorylation in epitopes related to its malfunction (Ser396/404) and cytoskeletal pathology (e.g., Thr231 and Ser262). In addition, we show, for the first time, that chronic GC administration also increased Tau levels in synaptic compartment; however, at the synapse, there was an increase in phosphorylation of Ser396/404, but a decrease of Thr231. These GC-triggered Tau changes were paralleled by reduced levels of synaptic scaffolding proteins such as PSD-95 and Shank proteins as well as reduced dendritic branching and spine loss. These in vivo findings add to our limited knowledge about the underlying mechanisms of GC-evoked synaptic atrophy and neuronal disconnection implicating Tau missorting in mechanism(s) of synaptic damage, beyond AD pathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stress, largely through the elevation of circulating glucocorticoids (GCs), impacts on brain structure and function [1–4]. One of the most vulnerable brain areas is the hippocampus, which exhibits remarkable dendritic atrophy and spine loss after GC administration as well as in stress-related pathologies characterized by high GC levels [1, 4]. However, the mechanisms underlying these GC-induced deleterious effects that damage hippocampus structural and functional integrity are still poorly understood.
Microtubule-associated protein Tau is implicated in cytoskeletal dynamics, as it stabilizes the microtubule (MT) network [5]. While being mainly an axonal protein, Tau was recently found in dendrites and synapses where it is suggested to have novel signaling and scaffolding role(s), regulating synaptic structure and function [5–8]. Recent human and animals studies suggest that Tau hyperphosphorylation and its mislocation in synapses may be related to synaptic pathology in Alzheimer’s disease (AD) [9–12]. Our previous work showed that stress and GC trigger Tau hyperphosphorylation in neuronal somata [13, 14], but to date, there is no evidence about the potential impact of dendritic/synaptic Tau on GC-induced dendritic remodeling and spine loss. Thus, in the present study, we used subcellular fractionation-based biochemical analysis and electron microscopy to monitor Tau dynamics and localization in hippocampal neurons of GC-treated rats adding to our understanding of cellular phenomena of GC-driven hippocampal malfunction and dendritic remodeling.
Methods
Animals and Treatment
Three- to four-month-old male Wistar rats (Charles River Laboratories, Spain) were paired housed under standard laboratory conditions (8:00 A.M. to 8:00 P.M.; 22 °C; ad libitum access to food and drink). Half of the animals were receiving daily subcutaneous injections of the synthetic glucocorticoid, dexamethasone (DEX) (300 μg/kg; Sigma D1756; dissolved in sesame oil containing 0.01 % ethanol; sesame oil Sigma S3547) for 14 sequential days, while control animals received daily subcutaneous injections of sesame oil [1]. All experimental procedures were approved by the local ethical committee of University of Minho and national authority for animal experimentation; all experiments were in accordance with the guidelines for the care and handling of laboratory animals, as described in the Directive 2010/63/EU.
Behavioral Test
Spatial reference memory was assessed using the Morris water maze (MWM) test at the end of DEX treatment period. As previously described [1, 13], testing was conducted in a circular black tank (170-cm diameter) filled with opaque water (22 °C) and placed in a dimly lit room with extrinsic clues. The tank was divided into virtual quadrants and had a black, escaping platform (12-cm diameter) placed in one of them. Animals (N = 7–8 per group) were asked to find the escaping platform over four consecutive days (four trials/day; 120-s trial period). Note that there are no differences of swimming speed between control and DEX-treated animals (data not shown). On the 5 day (probe test), the animal had to search (120 sec) for the escaping platform that was absent. Swim paths of each animal were monitored and recorded by a CCD camera, using a video tracking system (Viewpoint).
Neurostructural Analysis
As previously described [15], after animal perfusion with saline, half of each brain (N = 7–8 per group) were immersed in Golgi-Cox solution for 14 days. After transfer to 30 % sucrose solution, vibratome-cut coronal brain sections (200 μm thick) were used. After development, fixation and dehydration, slides were used to perform three-dimensional morphometric analysis. Dendritic arborization and spines were analyzed in dorsal hippocampus (CA1 area). All branches of neuronal dendritic tree (6–8 neurons/animal) were reconstructed at ×600 (oil) magnification using a motorized microscope (Axioplan2, Zeiss) and Neurolucida software (MBF Bioscience). For spine analysis, proximal and distal apical dendritic segments (30 μm) were randomly selected and spines were counted and further classified in immature (thin) and complex/mature (mushroom, wide/thick, and ramified) categories as previously described [15].
For electron microscope analysis, isolated hippocampi were fixed (4 % PFA; 3 days; 4 °C), transferred to 4 %PFA/0.8 % gluteraldehyde in 0.1 M of phosphate buffer (PB; pH 7.4) for 1 h and then, to 0.1 M PB (4 °C). Vibratome-cut axial sections of the dorsal hippocampus (300 μm thick) were collected, and the entire CA1 area was surgically removed. Tissue was then carefully oriented and embedded in Epon resin and ultrathin sections (500 Å), encompassing the superficial-to-deep axis, were cut onto nickel grids. For Tau-immunogold staining, sections were treated with heated citrate buffer (1×; Thermo Scientific, USA) for 30 min followed by 5 % BSA (Sigma, USA). Grids were incubated overnight with the following primary antibodies diluted in 1 % BSA (in PB): Tau-5 (1:30, Abcam), pSer199/202-Tau (1:50 Abcam), pSer262-Tau (1:20, Santa Cruz Biotechnology), pSer396 (1:50; Abcam), followed by appropriate secondary gold antibody (1:15; Abcam). For manual calculation of density of the above phospho-Tau, more than 50 nonoverlapping TEM (30,000×) of counterstained ultrathin sections (60 nm) were used. Grids were observed on a JEOL JEM-1400 transmission electron microscope equipped with a Orious Sc1000 digital camera. Analysis was performed by an experimenter blind to the samples provenience.
Subcellular Fractionation and Western Blot Analysis
For fractionation, we have used a well-established fractionation protocol [8, 16]. As shown in Fig. 1g, dorsal hippocampi (N = 5 per group) were homogenized with 10 volumes of homogenization buffer [sucrose 9 %; 5 mM DTT; 2 mM EDTA; 25 mM Tris pH 7.4; complete protease inhibitor (Roche), and phosphatase inhibitor cocktails II and III (Sigma)] using a Dounce glass homogenizer. Post-nuclear supernatant was subjected to centrifugation (12,500g) and divided into the crude synaptosomal fraction and synaptosome-depleted fraction. The latest fraction was further subjected to ultracentrifugation (176,000g), and separated into light membrane and Golgi fraction (P3) and cytoplasmic fraction (S3) while crude synaptosomal fraction was further lysed hypoosmotically and centrifuged (25,000g) for 20 min in order to obtain the synaptosomal membrane fraction (LP1). S3 (1x) and LP1 (2x) fractions were used for Western blot analysis. Samples were electrophoresed on 10 % acrylamide gels and transferred onto nitrocellulose membranes (Trans-Blot® Turbo™ Blotting System, BioRad). Membranes were blocked in 5 % nonfat milk in TBS-T buffer before incubation with the following antibodies: PSD-95 (1:20000, NeuroMab), GluA2 (1:1000, Abcam), GluN2B (1:1000, Abcam), pTyr1472-GluN2B (1:1000, Cell Signaling) pan-Shank (1:20, NeuroMab), Fyn (1:200, Santa Cruz), detyrosinated-Tubulin (1:1000, Abcam), acetylated-Tubulin (1:500, Abcam), α-Tubulin (1:2000, DSHB), Tau-5 (1:2000, Abcam), p199/202-Tau (1:1000, Abcam), pThr231-Tau (1:1000 Abcam), pSer262-Tau (1:250, Santa Cruz), p396-Tau (1:5000, Abcam), PHF1 (1:1000; kindly provided by Dr. P. Davies, Albert Einstein College, USA) cdk5 (1:2000, Millipore), total GSK3 α/β (1:2000, Invitrogen), phospho-Tyr279/216-GSK3α/β (1:2000, Invitrogen), and actin (1:2000, DSHB). After incubation with the appropriate secondary antibody, antigens were revealed by ECL (Clarity, Bio-Rad). ECL films (GE Healthcare) were used for detection of antigen signal and films were scanned and quantified using TINA 3.0 bioimaging software (Raytest). All values were normalized and expressed as percentages of controls. As previously shown [16], the protocol fractionation efficiency of the hippocampal tissue was confirmed using different synaptic proteins (see above) that were exclusively found in synaptosomal but not in cytosolic fraction (see Fig. 1g).

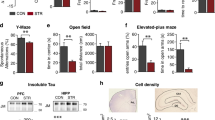
Glucocorticoid-driven biochemical and structural correlates of neuronal atrophy and memory deficits: a–c GC-treated animals exhibited increased latency to escaping platform during the four learning days of Morris water maze test compared to control (saline) animals (a) as well as reduced numbers crossing from the platform area (b) while no difference of percentage of time swam in target quadrant between the two groups was found in probe test (c). d–g Morphometric analysis of Golgi-impregnated hippocampal neurons showed that GC treatment reduced the number of branches in apical but not basal dendrites (d) followed by reduced spine density in both proximal and distal parts of apical dendrites (e, g). Specifically, GC evoked a clear reduction of density of mature/complex spines (f, g (i, iii, iv)) but not immature/thin ones (e, f (ii)). h Schematic representation of the subcellular fractionation protocol followed in this study for separating P1 (nuclear pellet and debri), P2 (crude synaptosomal fraction), P3 (light membranes), S3 (cytosolic fractions), LP1 (synaptosomal membrane fractions), LP2 (synaptic vesicle-enriched fraction), LS2 (soluble synaptosomal fraction)—for details, see Methods. i Western blot analysis of cytosolic and synaptosomal fraction where PSD-related scaffold proteins and receptors are found at synaptosomal, but not at cytosolic, fraction. j, k Representative blots and quantitative WB analysis of scaffold proteins showing that GC significantly decreased PSD-95 and Shank levels in synaptosomal fraction compared to control animals. l, m Levels of stable detyrosinated, but not acetylated, tubulin were reduced in dorsal hippocampus of GC-treated animals. All graphical data is shown as group mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001
Statistical Analysis
All data were evaluated by GraphPad software (version 6, La Jolla, CA, USA) using Student t test while Morris water maze learning curve data were analyzed by repeated-measurements ANOVA. Differences were considered to be significant if p < 0.05. Results are expressed as group means ± SEM.
Results
Behavioral and Neurostructural Correlates of Prolong Exposure to Glucocorticoids
For evaluating the impact of prolong GC treatment on hippocampus-dependent spatial memory, we used the MWM test and we found a significant increase in the time that GC-treated (GC) animals needed to find the escaping platform, confirming a deficit in spatial reference memory (p < 0.05) (Fig. 1a). In addition, we found that animals exposed to GC exhibited lower number of crosses at the platform area supporting further the memory-impairing role of GC (p = 0.028) (Fig. 1b). However, there is no difference between the two groups in the percentage of time that animals swam into target quadrant during probe test (Fig. 1c; p = 0.14), indicating that at the end of the experiment all animals were able to learn the task.
As neuroplastic changes and synaptic loss are robust correlates of impaired cognitive behavior, we next analyzed the entire dendritic tree of hippocampal CA1 pyramidal neurons using unbiased 3D morphometric analysis of Golgi-impregnated pyramidal neurons. We found that GC treatment altered the dendritic arborization in pyramidal neurons by diminishing the number of branches in apical, but not basal, dendrites (p = 0.03 and p = 0.37, respectively; Fig. 1d). Furthermore, spine density was reduced in both proximal and distal segments of apical dendrites (p = 0.017 and p = 0.007, respectively; Fig. 1e, g). Moreover, when we clustered the spine analysis in immature (thin) and mature (complex) categories based on morphological characteristics [15] (see Fig. 1g (i–iv)), we found that the density of mature/complex was reduced by GC treatment (p = 0.038) but no differences were found in immature ones (Fig. 1f). Note that immature (thin) spines are thought to be particularly plastic and linked to learning process while mature spines (e.g., mushroom) are more stable type of spines and believed to be involved in memory formation.
Glucocorticoids Triggers Tau Hyperphosphorylation and Missorting in Hippocampal Synapses
For monitoring the molecular underpinnings of the GC-evoked dendritic remodeling and synaptic loss, we then performed a detailed subcellular fractionation and organelle enrichment protocol which allows us to distinguish between cytosolic (excluding light membrane and Golgi fraction; S3) and synaptosomal fractions (LP1; see Fig. 1h; for more details, see “Methods” sections); as a confirmation of the efficacy of the separation protocol, postsynaptic density proteins (e.g., postsynaptic density protein 95; PSD-95) and synaptic receptors (e.g., GluN2B) were not found in cytoplasmic fraction (Fig. 1i). Next, we compared the synaptic and cytoskeletal proteins whose alterations reflect plastic changes and found that GC treatment reduced total levels of two proteins that play a central role in the assembling of the synaptic molecular components and transmission, PSD-95 and Shank scaffold proteins in synaptosomal fraction (p = 0.008 and p = 0.015, respectively; Fig. 1j, k); of notice, Fyn levels were not altered. In addition, we found a significant reduction of stable, detyrosinated tubulin levels (p < 0.0001), while acetylated tubulin was not different between GC-treated and controls (Fig. 1l, m).
We next monitored the influence of GC on intracellular distribution of Tau proteins and their phosphorylated isoforms. As shown at Fig. 2a, b, our subcellular fractionation-based WB analysis revealed that chronic GC exposure induced a significant increase in total cytosolic Tau levels (p = 0.003), as detected by the pan-Tau antibody Tau5 (that recognizes both phosphorylated and nonphosphorylated protein isoforms), which indicates a cytoplasmic accumulation of Tau. In addition, we observed a significant increase in cytosolic fractions of normalized levels of phosphorylated isoforms, pThr231-Tau (p = 0.023), pSer262-Tau (p = 0.015), and pSer396 (p = 0.03) in the hippocampus of GC-treated rats, but no effect of GC on pSer199/202 (Fig. 2a, b). Next, our analysis focused on the synaptosomal fraction where we found that overall Tau protein levels were also elevated by GC treatment (p = 0.01; Fig. 2c, d) pointing to synaptic accumulation of Tau. Similar to the cytosolic fraction, levels of normalized pSer199/202-Tau were not altered by GC treatment, whereas pSer396-Tau was increased in the synaptosomal fraction (p < 0.001). In contrast to the cytosolic compartment, pThr231-Tau synaptosomal levels were reduced after GC treatment (p < 0.02), while no changes were detected on pSer262-Tau levels. Next, we monitored the protein levels of cyclin-dependent kinase 5 (cdk5) and glycogen synthase kinase 3 (GSK-3), two key kinases that essentially contribute to Tau hyperphosphorylation and malfunction and known to be involved in regulation of synaptic function. We found that GC trigger increased protein levels of cdk5 (p = 0.01) but did not change the total levels of the two isoforms of GSK3 (α and β). However, levels of active-GSK3β (phospho-Tyr216-GSK3β) were elevated by GC (p = 0.001) while this GC effect was not true for active-GSK3α (phospho-Tyr279-GSK3α) (Fig. 2e). In the light of the recently suggested interaction of Tau with synaptic proteins and receptors [7], we also monitored the impact of GC on these proteins finding an clear increase in the levels of phospho-Tyr1472 GluN2R, a phosphorylation type suggestive to reflect activated GluN2R.

GC-triggered Tau missorting at hippocampal synapses. a–d Subcellular distribution of Tau and assessment of its phosphorylation state at different biochemically separated fractions in control and GC-treated hippocampi. GC evoked an accumulation of total Tau protein as detected by increased Tau-5 levels in both cytosolic (a, b) and synaptosomal fractions (c, d). In addition, normalized levels of pThr231-Tau, pSer262-Tau, pSer396-Tau, and p396/404-Tau (PHF-1) were increased in cytosolic fraction of GC-treated hippocampi but no GC effect was detected on pSer199/202-Tau. In contrast, synaptosomal levels of pThr231-Tau were decreased by GC while pSer262-Tau remained unaltered. Similarly to cytosol, p-Ser396-Tau and PHF-1 Tau levels were increased after GC treatment while levels of pSer199/202 were not altered. e–f GC increased protein levels of two key kinases of Tau hyperphosphorylation, cdk5 and active-GSK3β (pTyr216-GSK3β) accompanied by elevated levels of p-Tyr1472-Glu2B receptors. Numeric data shown represent ± SEM values as percentage of controls; *p < 0.05; **p < 0.01; ***p < 0.001
Ultrastructural Evidence of Tau Accumulation and Missorting in Dendrites and Spines of GC-Treated Hippocampus
As Tau intracellular localization and association to MTs and membranes seems to be regulated by its phosphorylation state [9, 11, 17, 18], we next monitor the localization and distribution of Tau, as well as its different phosphorylated forms, using transmission electron microscopy (TEM). As it was recently shown by us and others [8, 19], we found that Tau protein was detected at the rat hippocampal dendrites (Fig. 3 (I, IV)), as well as in synapses (Fig. 3 (II, III)). Moreover, as summarized in Table 1, we found that the density of total Tau was increased in both dendrites and synapses of hippocampal neurons of GC-treated animals (p < 0.0001 for both); these findings confirm our molecular data obtained by WB analysis. In addition, chronic GC treatment resulted in increased density of pSer262-Tau at dendrites (p < 0.001), but no significant alterations in synapses (p = 0.10). Similarly, pSer396-Tau density in dendrites of hippocampus neurons was also significantly increased by GC treatment (p = 0.044), but there was only a trend for an increase at the synaptic level. Note that p199/202-Tau levels were not affected by GC in both dendrites and synapses in line with our WB analysis findings. In addition, we found a characteristic PHF1 immunogold staining in hippocampal neurons of GC-treated animals (Fig. 2g (iv)), indicating the presence of Tau aggregates [20]. In summary, electron microscope analysis of different Tau isoforms (both phosphorylation-dependent and independent ones) suggest that GC trigger Tau accumulation and missorting in dendrites and spines of hippocampal neurons providing further support to the above described biochemical findings.

Ultrastructural detection of different Tau isoforms in hippocampal dendrites and spines. Electron microscope image of Tau-immunogold in dorsal hippocampus showing that Tau protein is detected in dendrites (I) and synapses (II, III) followed by negative controls (V). In addition, note a characteristic form of PHF1-detected Tau immunostaining resembling to aggregates in GC-treated neurons
Discussion
Through its interaction with several cellular partners such as tubulin, F-actin, and Src family kinases, Tau seems to play an important role in mediating alterations in the cytoskeletal structure [21]. Besides its predominant localization at axons, several observations suggest the presence and accumulation of Tau at somatodendritic compartment (e.g., dendritic spines) using cellular or animals models of Tau overexpression [6, 7, 9]. The present study confirms the presence of endogenous Tau in dendrites and synapses of the hippocampus of wild-type, nontransgenic, rats, in line with previous cell culture and in vivo studies [8, 19]. In an extension of our previous findings [13, 14], we now show that GC trigger the accumulation of different isoforms of Tau in both dendritic and synaptic compartments, which is an indicator of neuronal malfunction and pathology. Indeed, accumulation of hyperphosphorylated Tau has been shown to result in destabilization of the dendritic cytoskeleton and compromised intracellular trafficking [22–24]. As we found no GC-evoked changes in mRNA levels of Tau (data not shown), which is in line with previous cellular and animals studies [14, 25], the source of this Tau accumulation could be attributed to GC-induced reduction of Tau turnover [14] probably involving diminished Tau degradation [26]; alternatively, changes in Tau phosphorylation state might disrupt MT-Tau interactions liberating Tau from MTs into the dendritic shafts from where it may diffuse to spines and even to the synaptic compartment [6].
The intracellular distribution of Tau is largely regulated by its phosphorylation status [9, 11, 17, 18]. In agreement with previous studies [13, 14], we found that GC triggers Tau hyperphosphorylation at specific sites (Thr231, Ser262, Ser396, and/or Ser396/404); for example, Tau phosphorylation at the pSer199/202 epitope is not affected by GC suggesting that GC impact of Tau phosphorylation is not global. Moreover, in contrast to pThr231-Tau which plays an important role in early AD pathology, hyperphosphorylation of the 199/202 and 212/214 epitopes of Tau appear at later stages of the disease [27]. Thus, our GC-driven findings may be relevant to the earliest stages of cytoskeletal disturbances where Tau is believed to contribute to synaptic dysfunction and atrophy. This notion is further supported by the fact that GC-triggered dendritic remodeling was limited to reduced dendritic branching and spine loss. It is also pertinent to mention that Tau hyperphosphorylation at epitopes Thr231 and Ser262, both altered by GC, correlates strongly with reduced microtubule binding capacity of Tau and therefore, cytoskeletal disturbances [28, 29]. Consistently, we observed that GC-evoked Tau hyperphosphorylation occurs in tandem with decreased levels of the stable detyrosinated microtubules, suggestive of reduced microtubule stability [30, 31] that may be causally related to the dendritic remodeling that occurs after stress [3, 32].
Interestingly, GC impact on Tau phosphorylation state exhibited a subcellular and distinct pattern. Normalized pThr231-Tau levels were increased in cytosol, but decreased in synaptosomal fraction while p262-Tau levels were increased only in cytosol (not in synaptic fraction detected by WB or at spines as monitored by TEM), suggesting a preferential subcellular localization and/or accumulation of different Tau isoforms in both intracellular compartments after GC treatment. It is suggested that different pools of Tau with different phosphorylation state exist within the neuron exhibiting potentially distinct roles. For example, blockage of Tau phosphorylation at Ser396/404 is necessary for Aβ-driven mislocation of Tau at synapses [6], while Tau phosphorylation at pThr231, but not at other epitopes, regulates its synaptic binding to PSD-95 [19]. Interestingly, we found that the levels of PSD95, a crucial protein for synaptic structure and function, were reduced by the exposure to GCs, consistent with our finding of synaptic loss using Golgi-based neuronal structural analysis. Moreover, the reduction of PSD95 levels was accompanied by GC-driven decrease in levels of pThr231-Tau within synaptosomal fraction, further supporting the previously suggested interrelationship between this phospho-Tau isoform and PSD95 [6]. Note that, with one minor exception (p396-Tau), similar results regarding the differential distribution of phospho-Tau isoforms in different parts of the cell were obtained using two different technical approaches (electron microscopy and WB analysis of different subcellular fractions) (see Fig. 2d and Table 1). Nevertheless, the complementary approaches (subcellular fractionation-based biochemical analysis and in-situ ultrastructural detection) allowed a detailed “mapping” of the presence of Tau in different intracellular compartments. Our future efforts will aim to clarify the role of GC on the intraneuronal dynamics of Tau, with particular focus on individual phosphorylation epitopes as well as on the mechanisms through which GC and/or stress interfere with the Tau sorting and clearance machinery in cytosolic and synaptic compartments.
Recent studies show that Tau mislocation at dendritic spine is a common feature observed in AD [11] as well as other Tauopathies (FTP, FTDP-17) [9]; in parallel, increasing attention and support has been given to the essential involvement of Tau missorting in spine toxicity and synaptic pathology [19, 33]. A previous study demonstrated that local Tau missorting induced by Aβ or glutamate was followed by local spine loss and cytoskeletal disruption [24]; importantly, this Tau mislocation at synapses are shown to depend on Tau hyperphosphorylation [19, 34]. Besides Tau hyperphosphorylation, GCs are known to trigger APP misprocessing and Aβ generation [14, 25, 35] while Aβ is shown to trigger Tau hyperphosphorylation by inducing different kinases such as GSK3β and cdk5 [36]. While further studies are needed to clarify the interplay between Aβ, glutamate, and Tau in stress/GC-triggered neuronal remodeling and spine atrophy, the present study provides evidence that supports the notion that GC can also influence the intracellular trafficking of specific phospho-Tau isoforms and their missorting in synapses. Indeed, as previously suggested by studies in animal models of AD, abnormal Tau hyperphosphorylation and synaptic missorting results in damage to synaptic structure and function, including postsynaptic receptor targeting and function of excitatory glutamate receptor in dendritic spines and leading to dendritic spine loss.[7, 9, 37]. In line with this, we here showed that prolonged exposure to elevated GC levels upregulate active (phosphoTyr1472) GluN2B receptor units and subsequently, reduced levels of synaptic scaffold/anchor proteins, such as PSD-95 and Shank, and loss of spines. These findings provide new insights into the cellular cascades triggered by GC and, in particular, highlight the potentially important role of Tau hyperphosphorylation in neuronal and synaptic malfunction and atrophy underlying prolong exposure to elevated GC levels and related hippocampal pathology.
In summary, the results reported here represent the first description of GC-induced Tau missorting as a mechanism(s) underlying synaptic atrophy and damage, beyond Alzheimer’s disease pathology adding to our knowledge about the poorly understood cellular cascades responsible for stress-related brain pathology.
Abbreviations
- GC:
-
Glucocorticoids
- MT:
-
Microtubules
- AD:
-
Alzheimer’s disease
- MWM:
-
Morris water maze
- PSD-95:
-
Postsynaptic density protein 95
- DEX:
-
Dexamethasone
- GluN2B:
-
NMDA receptor 2B
- GluA2:
-
AMPA receptor 2
- ANOVA:
-
Analysis of variance
- TEM:
-
Transmission electron microscopy
References
Cerqueira JJ, Pego JM, Taipa R, Bessa JM, Almeida OF, Sousa N (2005) Morphological correlates of corticosteroid-induced changes in prefrontal cortex-dependent behaviors. J Neurosci 25(34):7792–7800. doi:10.1523/JNEUROSCI.1598-05.2005
Soares JM, Sampaio A, Ferreira LM, Santos NC, Marques P, Marques F, Palha JA, Cerqueira JJ et al (2013) Stress impact on resting state brain networks. PLoS ONE 8(6):e66500. doi:10.1371/journal.pone.0066500
Sousa N, Almeida OF (2012) Disconnection and reconnection: the morphological basis of (mal)adaptation to stress. Trends Neurosci 35(12):742–751. doi:10.1016/j.tins.2012.08.006
de Kloet ER, Joels M, Holsboer F (2005) Stress and the brain: from adaptation to disease. Nat Rev Neurosci 6(6):463–475. doi:10.1038/nrn1683
Gotz J, Xia D, Leinenga G, Chew YL, Nicholas H (2013) What Renders TAU Toxic. Front Neurol 4:72. doi:10.3389/fneur.2013.00072
Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I, Lante F, Buisson A (2014) Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci 34(17):6084–6097. doi:10.1523/JNEUROSCI.4261-13.2014
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC et al (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142(3):387–397. doi:10.1016/j.cell.2010.06.036
Kimura T, Whitcomb DJ, Jo J, Regan P, Piers T, Heo S, Brown C, Hashikawa T et al (2014) Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc Lond B Biol Sci 369(1633):20130144. doi:10.1098/rstb.2013.0144
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA et al (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68(6):1067–1081. doi:10.1016/j.neuron.2010.11.030
Kimura T, Yamashita S, Fukuda T, Park JM, Murayama M, Mizoroki T, Yoshiike Y, Sahara N et al (2007) Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J 26(24):5143–5152. doi:10.1038/sj.emboj.7601917
Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT (2012) The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol 181(4):1426–1435. doi:10.1016/j.ajpath.2012.06.033
Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, Mandelkow EM (2013) Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J 32(22):2920–2937. doi:10.1038/emboj.2013.207
Sotiropoulos I, Catania C, Pinto LG, Silva R, Pollerberg GE, Takashima A, Sousa N, Almeida OF (2011) Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J Neurosci 31(21):7840–7847. doi:10.1523/JNEUROSCI.0730-11.2011
Sotiropoulos I, Catania C, Riedemann T, Fry JP, Breen KC, Michaelidis TM, Almeida OF (2008) Glucocorticoids trigger Alzheimer disease-like pathobiochemistry in rat neuronal cells expressing human tau. J Neurochem 107(2):385–397. doi:10.1111/j.1471-4159.2008.05613.x
Cerqueira JJ, Taipa R, Uylings HB, Almeida OF, Sousa N (2007) Specific configuration of dendritic degeneration in pyramidal neurons of the medial prefrontal cortex induced by differing corticosteroid regimens. Cereb Cortex 17(9):1998–2006. doi:10.1093/cercor/bhl108
Sahara N, Murayama M, Higuchi M, Suhara T, Takashima A (2014) Biochemical distribution of tau protein in synaptosomal fraction of transgenic mice expressing human P301L tau. Front Neurol 5:26. doi:10.3389/fneur.2014.00026
Merino-Serrais P, Benavides-Piccione R, Blazquez-Llorca L, Kastanauskaite A, Rabano A, Avila J, DeFelipe J (2013) The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer’s disease. Brain 136(Pt 6):1913–1928. doi:10.1093/brain/awt088
Pooler AM, Usardi A, Evans CJ, Philpott KL, Noble W, Hanger DP (2012) Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging 33(2):431. doi:10.1016/j.neurobiolaging.2011.01.005, e427-438
Mondragon-Rodriguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, Boehm J (2012) Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem 287(38):32040–32053. doi:10.1074/jbc.M112.401240
Campbell SN, Zhang C, Monte L, Roe AD, Rice KC, Tache Y, Masliah E, Rissman RA (2015) Increased tau phosphorylation and aggregation in the hippocampus of mice overexpressing corticotropin-releasing factor. J Alzheimers Dis 43(3):967–976. doi:10.3233/JAD-141281
Morris M, Maeda S, Vossel K, Mucke L (2011) The many faces of tau. Neuron 70(3):410–426. doi:10.1016/j.neuron.2011.04.009
Hall GF, Chu B, Lee G, Yao J (2000) Human tau filaments induce microtubule and synapse loss in an in vivo model of neurofibrillary degenerative disease. J Cell Sci 113(Pt 8):1373–1387
Hall GF, Lee VM, Lee G, Yao J (2001) Staging of neurofibrillary degeneration caused by human tau overexpression in a unique cellular model of human tauopathy. Am J Pathol 158(1):235–246, doi:10.1016/S0002-9440(10)63962-4
Zempel H, Thies E, Mandelkow E, Mandelkow EM (2010) Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci 30(36):11938–11950. doi:10.1523/JNEUROSCI.2357-10.2010
Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, Almeida OF (2009) The amyloidogenic potential and behavioral correlates of stress. Mol Psychiatry 14(1):95–105. doi:10.1038/sj.mp.4002101
Sotiropoulos I, Silva J, Kimura T, Rodrigues AJ, Costa P, Almeida OF, Sousa N, Takashima A (2015) Female hippocampus vulnerability to environmental stress, a precipitating factor in tau aggregation pathology. J Alzheimers Dis 43(3):763–774. doi:10.3233/JAD-140693
Luna-Munoz J, Chavez-Macias L, Garcia-Sierra F, Mena R (2007) Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer’s disease. J Alzheimer’s Dis 12(4):365–375
Lauckner J, Frey P, Geula C (2003) Comparative distribution of tau phosphorylated at Ser262 in pre-tangles and tangles. Neurobiol Aging 24(6):767–776
Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K (1998) Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys 357(2):299–309. doi:10.1006/abbi.1998.0813
Cho JH, Johnson GV (2004) Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J Neurochem 88(2):349–358
Yoshiyama Y, Zhang B, Bruce J, Trojanowski JQ, Lee VM (2003) Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau-induced degeneration in astrocytes. J Neurosci 23(33):10662–10671
Sotiropoulos I, Cerqueira JJ, Catania C, Takashima A, Sousa N, Almeida OF (2008) Stress and glucocorticoid footprints in the brain-the path from depression to Alzheimer’s disease. Neurosci Biobehav Rev 32(6):1161–1173. doi:10.1016/j.neubiorev.2008.05.007
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ et al (2007) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316(5825):750–754. doi:10.1126/science.1141736
Miller EC, Teravskis PJ, Dummer BW, Zhao X, Huganir RL, Liao D (2014) Tau phosphorylation and tau mislocalization mediate soluble Abeta oligomer-induced AMPA glutamate receptor signaling deficits. Eur J Neurosci 39(7):1214–1224. doi:10.1111/ejn.12507
Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM (2006) Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci 26(35):9047–9056. doi:10.1523/JNEUROSCI.2797-06.2006
Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K (1993) Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc Natl Acad Sci U S A 90(16):7789–7793
McKinney RA (2010) Excitatory amino acid involvement in dendritic spine formation, maintenance and remodelling. J Physiol 588(Pt 1):107–116. doi:10.1113/jphysiol.2009.178905
Acknowledgments
We would like to thank Rui Fernandes for TEM technical support. IS was supported by the Portuguese Foundation for Science and Technology (FCT). This work was funded by the Portuguese Foundation for Science and Technology (FCT) (grant NMC-113934 to IS and grant SFRH/BPD/80118/2011 to JC), Canon Foundation and project DoIT - Desenvolvimento e Operacionalização da Investigação de Translação (N° do projeto 13853), funded by Fundo Europeu de Desenvolvimento Regional (FEDER) through the Programa Operacional Fatores de Competitividade (POFC).
Conflict of Interest
None of the authors report competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Sara Pinheiro and Joana Silva contributed equally to this work.
Rights and permissions
About this article

Cite this article
Pinheiro, S., Silva, J., Mota, C. et al. Tau Mislocation in Glucocorticoid-Triggered Hippocampal Pathology. Mol Neurobiol 53, 4745–4753 (2016). https://doi.org/10.1007/s12035-015-9356-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9356-2