
Abstract
It has been observed globally that primary groundwater arsenic (As) is derived from modern and ancient magmatic arcs at continental convergent margins of some of the most prominent orogenic systems worldwide (e.g., Andes, Himalaya). It ends up in arc-derived sediments in the adjacent foreland basin through rapid and continuous erosion of exhuming arc sequences, where groundwater could be contaminated through secondary As in the aquifer matrix, mostly allochthonous arc-derived sediments. Apart from the origin and development of magmatic arc, geotectonic control for other primary As sources is also evident in the form of (i) global distribution of geothermal and volcanic centers, where As-enriched hydrothermal and magmatic fluids contaminate surface and groundwater, (ii) ore (especially metal sulfide) deposits through natural leaching that releases As to surrounding environments, which is accelerated by metal mining and processing activities, and (iii) coal deposits and hydrocarbon reservoirs, together with the exploitation processes of both these fuel resources, are other geogenic sources of As contamination that is accelerated by the extraction and refining processes and subsequent usage. Whereas the aforementioned solid and liquid phase As inputs to groundwater are attributed to the lithology and geothermal and volcanic fluids, respectively, As speciation can also be associated with the basin's structural settings driven by geotectonics. Basin morphology, prolongation of faults, especially division of the underlying basement into blocks can produce discrete compartmentalized hydrodynamic environments, resulting in spatial variations in hydrochemical composition, including As speciation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Arsenic
- Environment
- Contamination
- Groundwater
- Geotectonics
- Speciation
- Arc magmatism
- Geothermal
- Volcanism
- Metallogeny
- Orogenesis
- Foreland basin
- Basement structure
1 Introduction
Although arsenic (As) content of crustal rocks varies widely, for most known and quite extensive instances of groundwater As contamination, its sources are geogenic (Smedley and Kinniburgh 2002; Bhattacharya et al. 2002, 2017; Nriagu et al. 2007; Coomar et al. 2019). Arsenic is the most ubiquitous toxic element present in all sorts of geological environments that gets transported by all kinds of media. Tracing all its origin and pathways for its human exposure has been a challenging task. However, this challenge has been met through multidisciplinary global collaborative and team efforts by deciphering As sources and pathways. Arsenic can get disseminated in air and carried by wind (e.g., volcanic ash in Argentina and Chile, Bhattacharya et al. 2006a, b, Quintanilla et al. 2009; Bundschuh et al. 2004, 2010, 2012; Nicolli et al. 2012; Morales-Simfors et al. 2020; Aullón Alcaine et al. 2020; Mariño et al. 2020), dissolved and transported by water (e.g., groundwater in Argentina, Bangladesh, Bolivia, Chile, India, Peru, Tanzania, Bhattacharya et al. 1997, 2001; Biswas et al. 2014a, b, c; Chakraborti et al. 2015; Bhowmick et al. 2018; Tapia et al. 2019a, b, Chakraborty et al. 2015, 2020; Mukherjee et al. 2021; Quino Lima et al. 2020, Quino Lima 2021a, b, Ijumulana et al. 2020), or stay in solid form in As-bearing minerals (e.g., mineralized zones, coupled with the residual products of mining and metallurgical processes, e.g., Latin America and the Caribbean, Quintanilla et al. 2009; Ramos et al. 2012, 2014; Bundschuh et al. 2012, 2020).
The ubiquitous nature of As in the environment can be understood simply by considering that hypothetical leaching of its average crustal concentration (1.8 mg/kg, Henke 2009) into groundwater is sufficient to cause toxic levels of As concentration (0.01 mg/L, WHO 2017). Moreover, surface sediments and soils in various parts of the world contain higher and more variable concentrations (ranging from 2 to 20 mg/kg on average) than the average crustal concentration of 1.8 mg/kg (Taylor and McLennan 1985; Wedepohl 1995; Mandal and Suzuki 2002; Bhattacharya et al. 2002; Henke 2009; Mukherjee et al. 2009a, b, and references therein). The average and maximum As concentration in 3,024 marine sediment samples from coastal Japan was determined to be 14 and ~580 mg/kg, respectively, of which As concentration in 65% samples was <10 mg/kg, while in 87.5% it was <20 mg/kg (Masuda 2018). On the other hand, Ohta et al. (2010) attributed the sediments with high As concentrations in Japan’s inland areas to coal and sulfide mines. Consequently, even a trace amount of As in solid-phase aquifer matrices is sufficient for groundwater under conducive environments (Korte and Fernando 1991; Stüben et al. 2003; Sracek et al. 2004; Guilliot and Charlet 2007; Henke 2009). All these imply that any lithological unit in the crust can be considered a potential As source (Matschullat 2000; Bhattacharya et al. 2002, 2007; Nordstrom 2002; Smedley and Kinniburgh 2002; Saunders et al. 2005). Nonetheless, considering “average value” being a theoretical concept and also, as a matter of fact, not all aquifers around the world are As-contaminated, as only certain segments of the lithosphere are enriched in As. Delineation and segmentation of such enriched portions are the basis for discussing the role of geotectonics in As contamination of aquifers in this chapter, based on some insightful works and representative examples from across the world (e.g., Mukherjee et al. 2007, 2008, 2009a, b; Mukherjee et al. 2014, 2019; Barringer and Reilly 2013; Giménez-Forcada and Smedley 2014; Masuda 2018; Morales-Simfors et al. 2020; Bundschuh et al. 2020).
2 Some Considerations and Constraints
Arsenic, a metalloid, being siderophile and chalcophile element, behaves as a metal and combines with sulfur; however, it shows volatile behavior in molten rock, similar to the light rare earth elements (Sims et al. 1990; Bhattacharya et al. 2002; Nriagu et al. 2007). The partition coefficient of As between sulfide and silicate melts varies as a function of oxygen fugacity, \({\mathrm{f}}_{{\mathrm{O}}_{2}}\) (Li and Audétat 2012, 2015). Accordingly, at a specific temperature and FeO content, As fraction in silicate and monosulfide melts increases with \({\mathrm{f}}_{{\mathrm{O}}_{2}}\) (Li and Audétat 2012, 2015). Although silicate and carbonate mineral phases do not host As as a major element, i.e., it does not appear in their formula (Majzlan et al. 2014), at low temperature, high \({\mathrm{f}}_{{\mathrm{O}}_{2}}\) and low sulfur fugacity \({\mathrm{f}}_{{\mathrm{S}}_{2}}\), silicates and carbonates can incorporate As, e.g., filatovite K(Al,Zn)2(As,Si)2O8), a variety of feldspar first reported at Tolbachik volcano, Kamchatka, Russia (Vergasova et al. 2004). Charnock et al. (2007) found As incorporated into garnet in the overgrowth of a hydrothermal vein. Pascua et al. (2005) reported As-enriched smectite in hydrothermal precipitates at a geothermal plant in Akita, Japan. Moreover, Ryan et al. (2011) found fractured aquifers in ultramafic rocks contaminated with As derived from serpentine and magnesite through chemical weathering.
Arsenic sources that can potentially contaminate the aquifers, i.e., leading the As concentration to toxic levels (0.01 mg/kg, WHO 2017), may develop through complex interactions between tectonic, geochemical, and biological processes (e.g., Saunders et al. 2005; Zheng 2006; Nordstrom 2009; Barringer and Reilly 2013; Mukherjee et al. 2014, 2019; Masoodi and Rahimzadeh 2018; Masuda 2018; Polya et al. 2019). Although there is no controversy about As mobilization from the aquifer matrix leading to As enrichment of groundwater and there are plenty of works on it (e.g., Smedley and Kinniburgh 2002; Saunders et al. 2005 and references therein), the primary source of As is often controversial and studied by few researchers in detail (e.g., Mukherjee et al. 2011; Raychowdhury et al. 2014). Bundschuh et al. (2021) have identified different sources and pathways for As exposure for Latin America, namely, (i) volcanism and geothermalism, taking into account (a) volcanic rocks, fluids (e.g., gases), and ash, including large-scale transport of the latter through different mechanisms (b) geothermal fluids and their exploitation, (ii) natural lixiviation and accelerated mobilization from (mostly sulfidic) metal ore deposits by mining and related activities, (iii) coal deposits and their exploitation, (iv) hydrocarbon reservoirs and co-produced water during exploitation, (v) solute and sediment transport through rivers to the sea, (vi) atmospheric As (dust and aerosol), and (vii) As exposure through geophagy and involuntary ingestion. Of these, authors identified the two most important and well-recognized sources and mechanisms for As release into environments affecting the Latin American population: (i) volcanism and geothermalism, and (ii) the strongly accelerated release from geogenic sources by mining and related activities. This is in line with the findings of Smedley and Kinniburgh (2002) that typical environments containing highly As-contaminated groundwater include (i) low temperature (including mining and non-mining) and (ii) high temperature (hydrothermal) settings.
Since As-contaminated groundwater is often distal from the (primary) As source(s), it has been argued continuously that understanding As mobilization is more critical than deciphering the (primary) source (e.g., Nordstrom 2002; Smedley and Kinniburgh 2002; Mukherjee and Fryar 2008; Scanlon et al. 2009; Ravenscroft et al. 2009). Consequently, many published peer-reviewed works deal with the hydrogeochemical processes controlling As mobilization processes in the contaminated areas, which hugely outnumber the studies on primary or ultimate As source. Saunders et al. (2005) emphasized the need for the latter, putting forward a case for a better understanding of the ultimate As source, considering that there is a consensus about mobilization processes leading to As enrichment within the aquifers (Saunders et al. 2005). van Geen (2011), on the other hand, put this view in another term. He emphasized the importance of understanding the etiological aspects of As enrichment in groundwater and the mobilization in aquifers and its control through in-situ reactive mobilization within the aquifer or transported advectively along flow paths from upstream enriched sources, i.e., ex situ, to areas with low As initially. van Geen (2011) proposed that mobilization processes, viz. water chemistry, with possible influence of local geology, lithology in particular, control the spatial distribution of As in aquifers. The duration of such hydrochemical processes leading to As enrichment of groundwater could be from few decades to thousands of years (van Geen 2011). Several workers have recognized the importance of ex situ sources, in addition to the in situ mobilization processes (viz. leaching) for As enrichment of groundwater, e.g., Smedley and Kinniburgh (2002), Nordstrom (2002), Mukherjee et al. (2009a, b), Ravenscroft et al. (2009). Accordingly, high As in young sedimentary aquifers should not be attributed only to local geology, as the time required for in situ hydrochemical processes might be more than the time elapsed since the deposition of those sediments and diagenesis, instead to larger, regional-scale geodynamic processes (Bhattacharya et al. 2004; Scanlon et al. 2009; Mukherjee et al. 2014, 2019). In this regard, although Ravenscroft et al. (2009) did highlight the proximity of young fold mountain belts to many As-enriched areas, they did not elaborate further on the relationship between the two. Saunders et al. (2005), on the other hand, proposed that As-contaminated groundwater is a product of interactions between tectonic, geochemical, and biological processes. Zheng (2006) and Nordstrom (2009) suggested a link between As enrichment and crustal evolution. Moreover, regional-scale geochemical processes lead to chemical weathering of a variety of As-bearing rocks, as noted by analyses of surficial material (viz. rocks, sediments, etc.) in different parts of the continental crust (e.g., Ahmed et al. 2004; Grosz et al. 2004; Saunders et al. 2005; Mukherjee et al. 2014, 2019). For example, As source in the contaminated aquifers of the major fluvial systems of the Indian subcontinent (e.g., Ganges–Brahmaputra, Indus, etc.) have been traced to the Himalayas, which acts as the sediment provenance, as well as the headwater for the rivers draining the basin (Stanger 2005; Hattori and Guillot 2003; Guilliot and Charlet 2007; Fendorf et al. 2010; Mukherjee et al. 2014, 2019; Chakraborty et al. 2015, 2020).
Arc magmatism and related hydrothermal activity, together with high rates of erosion due to rapid uplift of the arc zones and transport of the eroded sediments to the downstream basins, contribute to As contamination of the aquifers through in situ leaching of As-bearing minerals present in arc derived sediments (Mukherjee et al. 2019). However, geotectonics play a significant role in arc magmatism and erosion rates of the arc zone lithologies (Hecht and Oguchi 2017; Masuda 2018), which implies the tectonic control on geodynamic processes responsible for the geologic distribution and dissemination of As.
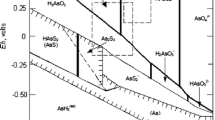
Another vital consideration is As speciation, since not all its species are equally toxic to humans, animals, and plants. Moreover, its toxicity varies widely with its oxidation state. WHO (1981, 2001) has documented that inorganic species are generally more toxic than organic ones, and arsenite [As(III)] is 60 times more toxic than arsenate [As(V)], which is 70 times more toxic than methylated species, monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA), which are consequently considered to be moderately toxic, whereas arsenobetaine (AsB) and arsenocholine (AsC) are considered to be nontoxic (Kumaresan and Riyazuddin 2001). Therefore, for an accurate environmental and human health risk assessment, identification and quantification of the As species are essential, especially the more toxic ones, rather than simply reporting total As concentrations, which has been the norm for many years. In this regard, Eh–pH diagrams (also known as Pourbaix diagrams, Pourbaix 1974) are conventionally used to represent and describe the speciation of As and identify significant geochemical processes (Welch and Stollenwerk 2002; Nicolli et al. 2012). Figure 1 shows the Eh–pH diagram of aqueous As species in water at 25 °C.

Eh–pH diagram illustrating the stability fields of the oxidized and reduced species of As in water at 25 °C. The blue rectangle represents the general pH and redox condition of groundwater systems. Modified after Herath et al. (2016)
Arsenic may exist with oxidation states of −3, 0, + 3, and +5; however, the oxidation states −3 and 0 (native As) are rare in nature (Panagiotaras et al. 2012). Consequently, As chemistry primarily focuses on As(V) or As(III), depending on redox potential and pH of groundwater (Sracek et al. 2004). Both As(III) and As(V) may form protonated oxyanions in aqueous solutions (Panagiotaras et al. 2012). However, the extent of protonation depends mainly on the pH of the medium. Under oxidizing conditions, As(V) hydrolyzes to four possible species, depending on the pH range encountered in surface and groundwater systems. In oxygenated water systems, arsenious acid (H3AsO4) is the predominant species only at extremely low pH (<2), whereas, As exists in the form of H2AsO4− and HAsO42− in a pH range of 2–11. In reducing environments and at low pH, arsenious acid converts into H2AsO3−, and at pH > 12, it occurs in the form of HAsO32− (Welch and Stollenwerk 2002; Panagiotaras et al. 2012; Gong et al. 2006). In the pH range of 4–10, As(V) species is negatively charged in water, whereas the predominant As(III) species is neutral because of redox potential and the presence of complexing ions such as sulfur, iron, and calcium and microbial activity (Katsoyiannis and Zouboulis 2015). However, complete dissociation of As(V) is rare, because very few water systems reach a pH > 11.5 (Brookins 2012). In reducing environments, completely protonated arsenite species predominate over a wide range of pH without being ionized; hence, arsenite cannot adsorb As strongly as arsenate species, thus making the removal of the As species more difficult during remediation of As-contaminated water (Maity et al. 2011a). These authors found high As concentration in the geothermal fluids in southern Taiwan to be associated with low redox (Eh) conditions (Maity et al. 2011b).
Sulfide and arsenide minerals have low solubility in water and are insoluble in anoxic environments, e.g., the solubility coefficient (log K) of realgar (AsS) and orpiment (As2S2) is −83.13 and −180.43, respectively, under standard temperature and pressure (STP) conditions, however, these minerals readily oxidize and then dissolve in oxic waters at STP (Boyle and Jonasson 1973). During chemical weathering of ore deposits and subsequent soil formation, As-bearing mineral phases change as follows: arsenopyrite and löllinite in the ore body become scorodite, pharmacosiderite, and arsenosiderite in the weathered rock, and finally goethite in the soil, latter As-bearing phases being more susceptible to getting dissolved and may contaminate surface and groundwater (Majzlan et al. 2014). Under neutral to alkaline conditions, arsenate ion substitutes for sulfate ion in the alteration minerals (Fernández-Martínez et al. 2008; Raychowdhury et al. 2014). Arsenite and arsenate minerals (e.g., scorodite) are the most likely sources of As contamination for their high solubility in aqueous solution, except under strongly acidic conditions (Masuda 2018). Due to secondary enrichment and their higher solubility, weathering products of the primary As-bearing rocks and minerals are more critical (and direct) sources of As contamination (Masuda 2018). For example, Savage et al. (2000) found that As concentration in soils, sediments, and weathered crust (>2000 mg/kg) from the fluvial basins in the Mother Lode District, California, have higher than nearby source rocks (<600 mg/kg).
In contaminated Cenozoic aquifers, As is primarily present in iron oxyhydroxides/oxides (usually goethite) through chemical weathering of iron-bearing minerals (Masuda 2018). Some studies examining iron oxyhydroxides/oxide precipitation and As incorporation into the aquifer sediments are as follows. According to Berg et al. (2007), in the Red River delta, the principal mechanism of As contamination of groundwater is the reduction of As-bearing iron oxyhydroxide minerals in the delta sediments, which are likely chemical weathering products transported from an upstream source. Other mechanisms include in situ decomposition of As-bearing pyrite with seasonal changes of the groundwater level (e.g., Munshuganj, Bangladesh, Pollizzotto et al. 2005) and in situ chemical weathering of chlorite (e.g., Sonragaon, Bangladesh, Masuda et al. 2012, 2013). Both the Ganges–Brahmaputra delta examples imply that the source of As contamination in the aquifers is in situ dissolution of As from primary minerals rather than the dissolution of or desorption from As-bearing iron oxyhydroxides. Such dissolutions could be accelerated further through enhanced inductive recharge of surface water if there is overexploitation of the aquifers (Masuda 2018).
3 Geotectonic Arc Distribution and Recycling
The As concentration of Earth’s primitive mantle is estimated to be 0.05 mg/kg (McDonough and Sun 1995). Hattori et al. (2002) found As highly concentrated in microscale sulfide minerals in mantle xenoliths from Lesotho, moreover, relatively higher As concentration in lherzolite (>200 mg/kg As) than harzburgite (<14 mg/kg As). Besides, comparative analyses of mantle xenoliths of Ichinomegata, Japan by Hattori et al. (2002) showed that As is depleted in wedge mantle, where As concentrations of <50 mg/kg were found in microscale sulfide minerals.
Noll et al. (1996) showed enriching arc magmas in As (~10 mg/kg) by hydrothermal fluids derived from dehydrating subducting slab during metamorphism and confirmed the As release from subducting slab through laboratory experiments as well as analyses of the field samples. Through a similar experiment, You et al. (1996) demonstrated As enrichment of a hydrothermal solution at temperatures ~300 °C by leaching the sediments from the Nankai Trough (where the Philippine Sea plate is subducting beneath the Eurasian plate) with a synthetic NaCl–CaCl2 solution. With the example of the relatively cool subduction zone associated with Catalina Schist on Santa Catalina Island of the Channel Islands, California, Bebout et al. (1999) proposed that devolatilization of As in fore-arc is low, so As is subducted at depth, resulting in As-enriched arc lavas in such subduction zones. On the other hand, Hattori et al. (2005) attributed the formation of As-bearing serpentine to alteration of the fore-arc mantle in the Himalayas, wherein As was derived from hydrothermal fluids releasing from the subducting slab, as observed in the aquifers of Ladakh Himalayas Indus-Tsangpo Suture Zone (Lone et al. 2020). This was corroborated by the documentation of an 85% (in some cases as much as 95%) decrease in the As concentration of arc lavas from fore-arc to back-arc along the convergent margins (Noll et al. 1996). Fujiwara et al. (2011) reported As release from deep continental and island-arc crust during the low-temperature and high-pressure metamorphism (forming pelitic schists) in the rocks from the Sambagawa metamorphic belt, Japan. These authors also reported a decrease in As concentration from ~10 mg/kg to 0 (below detection limit) with the increasing temperature (from 300 to 450 °C). In striking contrast, Breuer and Pichler (2013) reported that As concentration in submarine hydrothermal fluids from the East Pacific Rise (up to 80.5 μg/L) and the Mid-Atlantic Ridge (up to 24 μg/L) one degree lesser than those from the back-arc basin (1386 μg/L) and island-arc (5850 μg/L). Sigufusson et al. (2011) found that As in hydrothermal solutions in basalts of Iceland are complexed with sulfur, i.e., As(OH)S22−, AsS3H2−, AsS33−, As(SH)4−. The chemical species formed by covalent bonds between As and sulfur (S) precipitate from the hydrothermal solution as arsenide and sulfide minerals; consequently, many sulfide minerals, viz. pyrite, contain As (Masuda 2018). With decreasing temperature and increasing \({\mathrm{f}}_{{\mathrm{O}}_{2}}\) of the hydrothermal fluids, As–S complexes are decomposed and oxidized to arsenites, commonly found near the hot-spring water, where these fluids discharge on the surface (Masuda 2018).
High As concentration in hydrothermal systems than its average concentration in groundwater is reported globally (Maity et al. 2017). Arsenic-enriched hydrothermal discharges impact downstream surface and groundwater mixing, including related hydro(bio)geochemical processes (Maity et al. 2017). These processes are further influenced by respective changes in pressure, temperature, redox, and pH conditions (Mukherjee and Fryar 2008; Shimada et al. 2004). Such impacts of geothermal water on freshwater resources (surface and groundwater) lead to significant degradation, making affected resources unsuitable for drinking or irrigation without treatment (Maity et al. 2017). Hydrothermal discharges, even with similar geological/tectonic settings, may have very different As concentrations (Bundschuh and Maity 2015; López et al. 2012; Maity et al. 2011a, b; Maity et al. 2016, 2017). For example, significantly higher As concentrations (up to 75 mg/L) have been recorded for the hydrothermal discharges at the western margin of the Pacific Plate, e.g., El Tatio in northern Chile, Copper River and Yellowstone National Park in the USA, Los Humeros in Mexico (Ellis and Mahon 1967; Ball et al. 1998; Motyka et al. 1998; Gonzalez-Partida et al. 2001; Romero et al. 2003), as compared to those (up to 6.2 mg/L) at its eastern margin, e.g., Wairakei, Waiotapu, Ohaaki and Broadlands in New Zealand, Mt Apo in the Philippines (Ritchie 1961; Ellis and Mahon 1977; Webster 1990, 1999; Guo et al. 2007). A better understanding of As cycling in hydrothermal discharges will help better understand hydrothermal fluids’ role in the geochemical evolution of global hydrological systems and the related environmental risk (Bundschuh et al. 2013; Maity et al. 2017) (Fig. 2).

Redistribution of As during endogenic and exogenic geologic processes at the convergent plate boundary. Numbers in parenthesis indicate average As concentrations in the representative geologic bodies (references within this section). Purple and blue colored letters indicate the releasing and fixing mechanism, respectively, of As with source to sink redistribution cycle. Modified after Masuda (2018)
4 Geotectonic Distribution and Disposition of As-Contaminated Aquifers
Despite intrinsic heterogeneity of As concentrations (primarily due to local geology) in more than 70 countries on six continents, worldwide distribution of naturally As-contaminated aquifers show their existence within foreland basins adjacent to the orogenic belts on the global tectonic map (Fig. 3; Smedley and Kinniburgh 2002; Mukherjee et al. 2014, 2019; Ravenscroft et al. 2009).

Global distribution of major As contaminated aquifers (groundwater resources data from BGR & UNESCO under World-wide Hydrogeological Mapping and Assessment Programme (WHYMAP) available at http://www.whymap.org, Margat 2008), orogenic belts (based on the global tectonic map available on the website of the USGS Earthquake Hazard Program, http://earthquake.usgs.gov), in addition to naturally As-enriched aquifers and geothermal fluids (based on Smedley and Kinniburgh 2002; Nriagu et al. 2007; Mukherjee et al. 2007, 2008, 2009a, b, 2011, 2012, 2014, 2019; Ravenscroft et al. 2009; Morales-Simfors et al. 2020). Modified after Mukherjee et al. (2019)
A large-scale source-sink relationship between orogenesis and As concentration in groundwater has motivated workers like Mukherjee et al. (2007, 2008, 2009a, b, 2011, 2012, 2014, 2019, 2021) to identify universal primary As source-aggradation mechanism(s) for contaminated aquifers across the globe, instead of explaining individual cases of As contamination. With three examples, these authors corroborated that the primary As source in groundwater is ex situ. Also, As in affected foreland basin aquifers is derived through a series of geodynamic processes, viz. tectonic cycles, sedimentation, and regional-scale water flow. This As mobilizes with the availability of conducive surface geochemical and biogeochemical environments (Mukherjee et al. 2014, 2019). In this context, it is essential to consider modern and ancient orogenic belts and corresponding foreland basins. Present-day examples include (i) the Alpine–Himalayan orogen that stretches from Western Europe (the Alps), through the Indian subcontinent, to eastern China (the Himalayas), (ii) the Cordilleran orogens in western North America, (iii) the Andean orogen of western South America, and the (iv) Lachlan orogen of southeastern Australia, etc. Ancient equivalents are the Appalachian of the eastern USA and the Caledonian orogen of northern Europe and East Greenland (Mukherjee et al. 2014, 2019).
The aforementioned orogenic belts have associated foreland basins, which are extensive depressions developed during the mountain building process through lithospheric flexure (DeCelles and Giles 1996). These foreland basins act as sinks for the sediments eroded from the rapidly exhuming adjacent orogens and typically have thick sedimentary successions, thinning away from the orogenic belts. Examples of such basins include the Indus–Ganges–Brahmaputra, Northern Tarim, Southern Junggar basins of Asia, the Northern Alpine, Po, Ebro, Guadalquivir, Aquitaine basins of Europe, the Appalachian and Western Canada Sedimentary basins of North America. Based on their structures, DeCelles and Giles (1996) classified foreland basins as (i) peripheral, where the basins occur on the subducted plate, i.e., the outer arc of the orogen, e.g., the Indus–Ganges–Brahmaputra basin and (ii) retroarc, where the basins occur on the overriding plate during the convergence and are situated behind the magmatic arcs, e.g., the basins of the Andean or Western Cordilleran orogens.
Arsenic, being an incompatible element, is mainly concentrated in differentiated magmas; and as a result, volcanic rocks of intermediate (andesitic) to felsic (rhyolitic) composition have relatively higher As concentrations when compared to mafic (basaltic) and ultramafic rocks (Mukherjee et al. 2019), as shown by Welch et al. (1988, 2000) through a dataset of lava samples obtained from the western USA. This association of As with felsic rocks has been corroborated by Peters et al. (1999), Peters and Blum (2003), and Peters (2008), who attributed the As content in groundwater of Maine and New Hampshire, USA, to pegmatites (up to 60 mg/kg As), granites (46 mg/kg As) and granofels (39 mg/kg As), and also by Brown and Chute (2002), who attributed groundwater As to the weathering of pegmatite veins in Connecticut, USA. Moreover, in California, USA, As concentrations up to 48 mg/L have been reported in groundwater associated with acid volcanics, viz. tuffs and rhyolites, containing As-bearing sulfide minerals as the primary source of As (Welch et al. 1988, and references therein). On the other hand, it must be emphasized that although As content of mafic rocks is known to be relatively low, as pointed out by Welch et al. (1988, 2000), up to 327 μg/L of As in groundwater has been attributed to fractured ultramafic rocks in Vermont, USA, by Ryan et al. (2011).
5 Arsenic Sources and Associated Geotectonic Settings
5.1 Metal (mostly Sulfidic) Ore Deposits
Ore deposits are one of the significant and widely recognized sources of As contamination, e.g., As released from marcasite, present in 5 Ma old hydrothermal ore deposits in New Zealand, causes groundwater contamination with >100 μg/L As (Craw et al. 2000). Although the distribution of geogenic As contamination around localized ore bodies is limited and defined mainly through geotectonic setting prevalent at the time of their formation, anthropogenic materials associated with ore mining and processing, viz. tailings and sludge, expand the area of contamination into the hydrosphere, thus affecting surface and groundwater, and pedosphere, i,e., soil horizons and near-surface ecosystems (Masuda 2018). Moreover, chemical weathering of the excavated ore bodies, exposed to exogenic forces of weathering and erosion, produce As-contaminated sediments, which might be the potential sources of As contamination of the environment and consequent human As exposure (Masuda 2018). Mining and metallurgical activities cause As contamination to spread to air, sediments, and streams, which might remain contaminated long after the cessation of these activities. For example, long after mining was ceased at Toroku mine, Miyazaki (Kyushu, Japan), 146 people were found to be suffering from arsenicosis due to inhalation of refining soot and drinking contaminated water at this site in 1990, where arsenite was mined from the 1820s to 1962 (Shimada 2009). On the other hand, in the absence of such activities, the contamination is restricted to a limited area (locality), close to the exposure or contact of ore bodies and alteration zones to water (surface or groundwater) or air.
Metal mining (mainly gold, copper, silver) in Latin America has been the key for As mobilization from the geologic materials and aggravating contamination of groundwater resources (McClintock et al. 2012; Tapia and Audry 2013; Tapia et al. 2012, 2018, 2019a, b, c, 2021). A summary table of the worldwide occurrence of As in groundwater by Nordstrom (2002) lists As bearing ores and other geologic sources of As, which include 24 As-bearing minerals that commonly occur in hydrothermal veins, ore deposits, and rocks and dominate the environmental sources and conditions responsible for As mobilization to groundwater. According to the Committee on Medical and Biologic Effects of Environmental Pollutants of the National Research Council (NRC 1977), high concentrations of As are found in sulfide deposits, in native form or as alloys (4 minerals), arsenides (27 minerals), sulfides (13 minerals), sulfosalts (sulfides of As with lead, copper, silver and other metals, 65 minerals), and their oxidation products (2 oxides 11 arsenites 116 arsenates, 7 silicates). Altogether, As has been shown to occur in 568 different mineral species (Nordstrom 2002).
Most primary As minerals correspond to metal sulfides, which present As in solid solution with other metals, of which arsenopyrite is the most common (Ehrlich and Newman 2009). In contrast, secondary minerals tend to be less common and are related to arsenates and oxides (Ehrlich and Newman 2009). WHO (2001) provided a list of these secondary minerals, which Mandal and Suzuki (2002) also tabulated. Moreover, arsenosulfides of almost any metal cation can be found (NRC 1977). Arsenic, which is both chalcophile and siderophile, is found associated with pyrite (FeS2) or iron hydroxides (FeOOH) and oxides (Fe2O3 and Fe3O4) in crustal rocks (Nordstrom 2002). Although the maximum As content of pyrite is ~5%, its typical range is 0.02–0.5% (NRC 1977). Under surface conditions, readily oxidizing As-bearing sulfides and sulfosalts transform to As trioxide and arsenate stages, and subsequently, secondary minerals tend to be arsenates and oxides (NRC 1977). Although igneous rocks show a relatively uniform distribution of As among the major constituent minerals, their sulfide mineral constituents show slight As enrichment (NRC 1977).
Through weathering, As present in sulfides ores are converted into a highly mobile [As(V)], which can enter the As biogeochemical cycle directly in either solid-state as dust particles or through dissolution in the rain, surface, and groundwater (Maity et al. 2011a). Consequently, soil overlying sulfide ore deposits commonly contain several hundred ppm of As, up to 8000 ppm (NRC 1977). In this way, As may be present in unweathered sulfide minerals or in an inorganic anion state. Inorganic arsenate is usually bound to iron and aluminum but could form bonds with any other cation present, viz. calcium, magnesium, lead, zinc (NRC 1977). The composition of the recharging water, water-aquifer matrix interactions (e.g., cation exchange, mineral dissolution), availability of reactive organic matter, and groundwater residence time within the aquifer primarily control groundwater chemistry (e.g., Edmunds et al. 1981; Stallard and Edmond 1983; Chapelle 1993; Drever 1997; Faure 1998; Mukherjee et al. 2009, 2012). Accordingly, elevated concentrations of chalcophile and siderophile As in contaminated aquifers may result from the dissolution of primary or secondary sulfidic minerals (viz. pyrite), or desorption from secondary concentrations in hydrous metal oxides (Nordstrom 2002), which is common in the aquifer matrix in many large unconsolidated sedimentary provinces (Mukherjee et al. 2014, 2019).
A simple analysis of the basic types of sulfide ore deposits based on associated geotectonic regimes reveals a systematic pattern. For example, calc-alkaline magmatism at convergent plate boundaries gives rise to (i) Kuroko-type (massive sulfide) deposits in submarine volcanic environments and (ii) Cordilleran-type (postmagmatic) ore deposits in epizonal plutonic settings (Sawkins 1990). Porphyry copper deposits, an important subtype of Cordilleran-type deposits, show a spatial relationship to present or ancient convergent plate boundary regimes (Sawkins 1990). Copper-nickel deposits occurring in ultramafic extrusive or penecontemporaneous intrusive rocks are also related to plate convergence (Sawkins 1990). Sulfide ore deposits generated at divergent plate boundaries (spreading-center systems) are rare as a function of their submarine habitat and are unknown at transform plate boundaries (Sawkins 1990).
On the other hand, stratiform copper deposits and most of the Mississippi Valley-type lead–zinc deposits are formed in continental intraplate environments. In contrast, magmatic deposits in layered mafic complexes also form in intraplate environments, but with spreading-center activity limited in time and space (Masuda 2018). Moreover, gold deposits in greenstone-type volcanic arc sequences can be attributed to a two-stage concentration process at inferred (ancient) convergent plate boundaries (Masuda 2018). Arsenic, being an integral part of all these deposits, implies that primary As distribution is tectonically controlled, transported by secondary processes in solution through leaching or solid-state through physical erosion to the sedimentary basins (see Sect. 5.2).
It is important to emphasize that As contamination, even in a mining district, can sometimes be associated with other lithologies instead of the ore deposits themselves. A study from Osaka Prefecture in western Honshu (Japan) pointed out that although many sulfide mines are present in the Hokusetsu Mountains, they are not essentially sources of As contamination as much as the arsenide disseminated in hornfels and shales (Masuda 2018). On the eastern edge of the Cretaceous–Neogene magmatic bodies in the area, Even et al. (2017) reported high As content in hornfels near the granitic complex that effectively led to As contamination of surface and groundwater. Similarly, As leaching and oxidation in disseminated Miocene sediment-hosted hydrothermal gold (Au) ore deposits led to As contamination of shallow groundwater in Sulawesi, Indonesia (Iskandar et al. 2012). In both cases, the As contamination mechanism was oxidation and decomposition reactions of oxic water infiltrating through a fault system with diffusively distributed sulfides and arsenides (Masuda 2018). Section 6 deals with the basement structure's role in controlling the hydrochemical environment, thus As speciation.
In the mining area of Wassa West district in Ghana, Bhattacharya et al. (2012) found As (besides manganese and iron) contamination in some wells potentially affected by acid mine drainage (AMD) likely to be controlled by the adsorption processes, evident by supersaturation of groundwater with respect to several mineral phases including iron-hydroxides/oxides in the hydrogeochemical models.
5.2 Arc Magmas
Reported concentrations of As in various lithologies worldwide suggest a close association with magmatic arcs at convergent continental margins (Fig. 2) and collision belts that inevitably contain arc-derived components (Mukherjee et al. 2014, 2019). Accordingly, high As content in arc magmas can be derived from (a) the subducting oceanic crust with its sediment cover, (b) the mantle component of the subducting slab, (c) the mantle wedge component overlying the slab, (d) the overlying continental crust through which the arc magma ascends to the surface (Hattori and Gulliot 2003; Hattori et al. 2005; Gulliot and Charlet 2007). Ocean-floor basalts, comprising the oceanic crust are formed by partial melting of the depleted mantle at the mid-oceanic ridge, are not enriched in As (<0.01–1 mg/kg, Deschamp et al. 2013). This implies that As in the oceanic crust is derived from the sediments derived from the continents (terrigenous detritus).
The As concentration in the continental crust is at least two orders of magnitude higher than in the depleted mantle, the source region for mid-oceanic ridge basalts (Matschullat 2000). It is worth mentioning that the arc magma of some volcanoes in the southern part of the Japan-Kurile volcanic chain has low As content, comparable to normal depleted mantle-derived magmas. In contrast, several volcanoes in the northern part of the same region, e.g., Esan (Japan) and Onekotan (Kuriles), which are underlain by thick arc basement, have very high As concentration in their magmas (Noll et al. 1996; Henke 2009). Similarly, medium to high As concentration have been reported from the arc extrusives at continent to ocean convergence-related volcanics of Cordilleran orogenic belts of British Columbia (Boyle et al. 1998), Alaska (Crock et al. 1999), Mexico (Armienta et al. 2001), Chile (Noll et al. 1996) and Perú (Noble et al. 1998). This tells that the As content is almost always high in the settings where the arc magma ascends through an overlying continental crust, implying that interactions of volatile-enriched arc magma with the continental crust is the most important source of primary As enrichment compared to other tectonic settings. This further suggests that (i) in any tectonic regime, the continental crust forms the ultimate source of groundwater As, (ii) As in continental crust is mobilized hydrothermally by young magmatic hydrous fluids of metasomatic origin, and (iii) As mobilization is enhanced through prolonged interaction between continental crust and hot hydrous fluids or magmas.
Arsenic mobilization from the sediment cover of the subducting plate to the fluid is most effective in continental domains with elevated geothermal gradients, such as near active, e.g., Andes (e.g., Raychowdhury et al. 2014) or ancient magmatic activity centers, e.g., Indus-Tsangpo Suture Zone lithologies of the Himalayas (e.g., Hattori et al. 2005; with up to As 257 mg/kg, Lone et al. 2020). Meteoric water in such domains percolate to depth, becomes heated, and forms hydrothermal systems, e.g., geothermal wells in Costa Rica (As ~25,900 µg/L, Hammarlund et al. 2009) that can efficiently remove As and other mobile elements from the crust. The limits of such As mobilization by thermal fluids are controlled and/or governed by the extent of water–rock interaction, which in turn is constrained by the maximum penetration depth of water can (Gleeson and Ingebritsen 2016). Arsenic can be mobilized from deeper parts of the continental crust only if it comes in contact with hot hydrous fluids and gets dissolved. This is evident at active continental margins (mostly hot orogens, Mukherjee et al. 2014, 2019), where hot hydrous fluids mobilize As derived from the subducting slab (Tatsumi 1989). These fluids may or may not be As enriched at the time of slab dehydration. The As content of these fluids depends on the proportions of continent-derived sediments (terrigenous detritus) and hydrated oceanic crust associated with the subducted slab (Mukherjee et al. 2014, 2019). The fluids, which may also be associated with slab-derived melts, subsequently metasomatize the depleted continental lithospheric wedge, lower the mantle solidus and initiate the production of hydrous arc magmas (Mukherjee et al. 2014, 2019). Fractionation of such magmas leads to the generation of high-Al basalts/gabbros, andesites/diorites and dacites/granodiorites (Sisson and Grove 1993) in active or ancient magmatic arcs, which are known to have high to very high (up to 1500 mg/kg) solid-phase As (Henke 2009). The chemistry of the arc magmas may thus be influenced by sedimentary input from the continent into the trench (Plank and Langmuir 1993). At active continental margins, the ascending magma invariably interacts with the (thick) continental crust as it rises, unlike at an ocean-ocean setting; thus, continental arc volcanics tend to have the highest primary As enrichment (Mukherjee et al. 2014, 2019).
As in the Andes, Arc volcanism leads to the deposition of As-rich volcanic ash and other continent-derived sediments (Fig. 2) that can be carried to downstream basins. Arsenic enriched rhyolitic glass, volcanic ash, and associated silicates in sediments and metamorphic deposits, with As up to 10 mg/kg (Raychowdhury et al. 2014), undergo hydrolytic dissolution and produce an influx of major (e.g., Na+, K+, Si4+, and HCO3−) and minor (e.g., oxyanions of As, V, and Mo) solutes to groundwater, which leads to precipitation of clays (Mukherjee et al. 2007, 2008; Mukherjee and Fryar 2008; Raychowdhury et al. 2014), under favorable climatic conditions. Himalayan arc-derived lithologies also comprise significant proportions of mafic and intermediate rocks (predominantly andesites), enriched in As compared to their non-arc equivalents, owing to interaction with subduction-zone fluids (Hattori et al. 2005; Guillot and Charlet 2007).
Chemical weathering and leaching of more soluble and labile, solid-phase As from the metastable, high-temperature arc-derived mafic minerals (e.g., olivine, pyroxenes, serpentine) in these rocks (As ~19 mg/kg, Verma et al. 2016) facilitates the release of this As (up to 4100 µg/L, Mukherjee et al. 2009a, b) into the hydrological system. Some As-carrying (up to 25,900 µg/L (Hammarlund et al. 2009) hydrothermal fluids may also discharge on the surface as hot springs and fumaroles (For more details, see Sect. 5.4 Geothermal and volcanic fluids). Subsequently, with orogeny initiation at active continental margins, the As-bearing arc volcanics coalesce with sediments and rocks of the exhuming orogen. With continuing convergence, slices of serpentinized and, therefore, As-enriched mantle rock (As ~275 mg/kg, Hattori et al. 2005) are also incorporated into the orogen through thrusting (Hattori and Guillot 2003). Sediments derived from the rapidly exhuming orogenic belt by weathering of the As-bearing arc volcanics and obducted, serpentinized mantle rocks are subsequently deposited in adjacent foreland and even back-arc basins (Verma et al. 2016). During protracted convergence over geologic time, multiple generations of foreland basins can form and coalesce with the main orogenic belt, e.g., Siwaliks in the Himalayas (As ~10 mg/kg, Guillot et al. 2015).
Rivers or glaciers cutting through the orogenic belt erode the foreland sediments and exhumed arc derived magmatic rocks (Guillot and Charlet 2007; Verma et al. 2016), and deposit weathered As-rich sediments in the most recent foreland basins, e.g., Terai, Nepal (As up to 27 mg/kg, Guillot et al. 2015). Proximity to these orogenic As sources enhances the possibility of increased chemical weathering and leaching of solid-phase primary As, from meta-stable, high-temperature arc-derived rocks, which constitute primary As source (Verma et al. 2016). With increasing distance from these As sources, weathering and transport lead primary As to remain chemically sorbed mainly in the residual state, more recalcitrant or more strongly bonded, more crystalline or mineralogically mature phases, viz. low-temperature/late-phase silicates (Onishi and Sandell 1955), which are less prone to chemical weathering (Henke 2009). Conversely, with distance, the released As gets physisorbed in more labile secondary solid phases, which act as the immediate (and secondary) source of groundwater As enrichment in adjoining foreland basin aquifers by redox-dominated reactions (Mukherjee et al. 2014, 2019). This liberated As can co-precipitate with/or get adsorbed onto secondary Fe(III) (oxyhydr)oxides (Smedley and Kinniburgh 2002) or can be re-sequestered by re-sorption or co-precipitation with authigenic Fe(II) sulfides under sulfate-reducing conditions, i.e., aquifer sediment sink (Mukherjee and Fryar 2008; Mukherjee et al. 2008).
To integrate the knowledge of orogen-sourced sediments and groundwater chemistry of ancient (e.g., Himalayan system) and modern (e.g., Andean system) orogens, Mukherjee et al. (2014, 2019) correlated the tectonic settings and groundwater As-enrichment data from 63 major aquifers across the world, to show that a distinctly high proportion of As-affected aquifers are located in convergent (~90%), particularly magmatic arc settings. In stark contrast, only 5% of the basins in other tectonic settings contain groundwater As. However, rift-related basin aquifers are an exception to this generalization (Mukherjee et al. 2014, 2019), although all such rift basins receive magmatic arc detritus, commonly andesite (Peters 2008). Similarly, As-contaminated aquifers in the Red River and the Mekong basins have a mixed convergent/non-convergent setting, with sediment sources from ancient Qamdo-Simao arc rocks (Stanger 2005). Dissolution of volcanic glasses in ash layers and leaching of loess-type deposits in the Chaco-Pampean plain of Argentina have led to groundwater As concentrations from <10 to 5300 μg/L (Nicolli et al. 2012). These examples corroborate that magmatic arcs are the predominant source of primary As in groundwater across the globe.
5.3 Allochthonous As-bearing Sediments
The third primary source, preferably a secondary source (see the previous section), of contamination comprises the Cenozoic (mostly Holocene) aquifer sediments. It is worth highlighting that the total As concentration in contaminated groundwater is not as high when compared to the average concentration in soils and sediments, for example, despite total As concentration in Holocene bulk aquifer sediments in Bangladesh ranges from 5 to 16 mg/kg, groundwater has only little over 1 mg/L As (Seddique et al. 2011). This is because groundwater As contamination depends mostly on the chemical form of the As bearing solid phase (mineral species) and changes in the aquatic environment leading to the release of As from aquifer sediments to water (Seddique et al. 2011). The mechanisms for As release from Cenozoic, especially Holocene, aquifer sediments have been described in this section.
As mentioned in the previous section, As-contaminated aquifers are often hosted within extensive clastic sedimentary basins associated with young mountains (Ravenscroft et al. 2009) within or around foreland basins at both active and ancient convergent continental margins (Mukherjee et al. 2014, 2019, 2021), e.g., Indus–Ganges–Brahmaputra basin, the Mekong and Red River deltas in southeast Asia (Alpine–Himalayan Orogenic System), the North American foreland of the western USA (American and Pacific Cordillera) and Canada (Laramide-Nevadan-Sonoma Orogenic System), the Appalachian foreland in New England and adjacent areas of the eastern USA (Appalachian-Ouachita Orogenic System), parts of Argentina, Bolivia, Chile and Peru in the Andean forelands of South America (Andean-Pampean-Toco Orogenic System). Arsenic found in such aquifers can be attributed to the solid phase concentration of As-bearing clasts in their provenance located in major magmatic arcs and orogenic belts at convergent boundaries (Mukherjee et al. 2014, 2019). Thus, As sourced from magmatic arcs may be referred to as primary for both Andean (Ramos Ramos et al. 2012; Raychowdhury et al. 2014; Ormachea Muñoz et al. 2015) and Himalayan (Hattori and Guillot 2003; Hattori et al. 2005; Guillot and Charlet 2007; Mukherjee et al. 2014, 2019; Verma et al. 2016) type orogenic systems, at continental convergent margins across the globe, which after exogenic processes of erosion and transport, ends up in the sediments as a (secondary) source of As, in the aquifers hosted in adjoining foreland basins. Tectonic setting interpretations based on major, trace, and rare earth elements, in conjunction with field measurements of primary sedimentary structures, can help in discriminating provenance and tectonic setting (including ancient, e.g., Proterozoic sedimentary basin in Rajasthan, India, Banerjee and Bhattacharya 1994) of the As-bearing sediments.
5.4 Geothermal and Volcanic Fluids
Arsenic contamination of the aquifers—clastic or fractured—might not involve leaching of As from allochthonous clastics or any other host formation lithology. Instead, it might be from As rich geothermal and volcanic fluids that interact with groundwater (e.g., Latin America, Morales-Simfors et al. 2020; Western Iran, Keshavarzi et al. 2011), which are the sources as well as pathways of As. In a recent study covering 15 countries in Latin America (Morales-Simfors et al. 2020), 423 sites were characterized with As originating from geothermal sources, mostly related to present volcanic activity (0.001 < As < 73 mg/L, mean: 36.5 mg/L) and the transboundary Guarani Aquifer System (0.001 < As < 0.114 mg/L, mean: 0.06 mg/L).
High As concentrations in geothermal fluids indicate that As is a volatile magmatic component, which is highly soluble in water., e.g., hot springs in Yellowstone National Park, USA have up to 10 mg/L As (Stauffer and Thompson 1984; Webster and Nordstrom 2003), which is released to groundwater and surface water from geothermal fluids within and near the park (Ball et al. 1998, 2002; Nimick et al. 1998). Geothermal fluids in Dominica, the Lesser Antilles, have high As concentrations (>50 μg/L, McCarthy et al. 2005). Moreover, As from hot springs can be mobilized to surface waters and also enter riverbed sediments, e.g., arsenite released from hot springs along the Owens River (USA) gets oxidized to arsenate by microbial activity, and subsequently precipitates and settles to the bottom, thus becoming part of the riverbed sediments with high As content (up to 60 mg/kg, Nimick et al. 1998). It has been observed that geothermal well fluids occasionally contain higher As concentrations (up to 50 mg/L) than associated surface discharges through host springs (Webster and Nordstrom 2003 and references therein). Arsenic content in the outflow (drainage water) from Waikato Power Station, New Zealand was 145 t/year till 2000; however, it has been lowered to ~70 t/year (Webster-Brown et al. 2000 and references therein). This discharge has led to elevated As concentrations (61–1790 mg/kg) in the Waikato River sediments (Webster-Brown et al. 2000).
Geothermal areas occur along plate boundaries, e.g., the Pacific Ring of Fire, in tectonic rift areas, e.g., East Africa, Central India and at seafloor spreading centers, e.g., Iceland, and at hot spots, where mantle-derived plumes ascend, e.g., Hawaii and Yellowstone National Park, USA. Arsenic is one of the incompatible elements, together with antimony (Sb), boron (B), fluoride (F), lithium (Li), mercury (Hg), selenium (Se), and thallium (Tl), and hydrogen sulfide, in high-temperature geothermal settings (Webster and Nordstrom 2003). Moreover, As concentrations are high mainly in thermal waters that leach continental rocks, whereas thermal waters in basaltic rocks, e.g., in Iceland, contain lesser amounts of As. Stauffer and Thompson (1984) demonstrated that As in thermal fluids at Yellowstone National Park in Wyoming, USA, it is derived mainly from leaching host rocks rather than magmas. Arsenic in thermal water is present as [As(III)] in arsenious acid (H3AsO3). In contrast, in low-sulfide fluids, the [As(III)] in the arsenious acid is oxidized to [As(V)]. The ascending fluid interacts with cold oxygenated groundwater or comes in contact with the atmosphere. On the other hand, in high sulfide solutions, As could be present as thioarsenate complexes (Webster and Nordstrom 2003; Planer-Friedrich et al. 2007).
Since the 1950s, As contamination has been attributed to hydrothermal activity in many geothermal areas of Japan, as reflected by some old works, e.g., Beppu, Kyushu (Kawakami et al. 1956), Tamagawa, Akita, Northeast Japan (Minami et al. 1958), Hakone, Yugawara, Kanagawa (Awaya et al. 2002), and Toyohira, Hokkaido (Jin et al. 2012). In Kusatsu-Shirane (Gunma), thermal water discharges with up to 7 mg/L of As concentration was reported flowing into the Yukawa River for a decade until the 1990s (Kikawada et al. 2006, 2009). Up to 3.4 mg/L As was reported in thermal water at Kakkonda Geothermal Plant, Akita, Japan (Okada et al. 2002). Acidic (pH 1.2) thermal water with about 2600 μg/L As was reported by Noguchi and Nakagawa (1969) to precipitate out as As sulfides and lead (Pb) As sulfides in surface-water sediments containing ~5–56 wt.% of As. Up to 4.8 mg/L As in thermal waters from geothermal fields in New Zealand are attributed to the leaching of variable composition host rocks. This was considered the mechanism for As enrichment of thermal waters from various geothermal areas in New Zealand.
In Latin America, volcanic and geothermal activity along the convergent plate boundary discharges As-rich waters and gases on the surface in the form of hot springs and fumaroles (Raychowdhury et al. 2014; Mukherjee et al. 2014, 2019, 2021; Coomar et al. 2019). The As concentration of ~50,000 μ/L has been reported by López et al. (2012) in Na-Cl type thermal waters at El Tatio geothermal field in Chile (Landrum et al. 2009). Up to 73,600 μg/L of As concentration has been reported in thermal waters ascending through a wide range of lithologies—sandstones and shales, lava flows and pyroclastics, and metamorphosed carbonate rocks, basalts, and hornblende andesites—in Mexican geothermal fields (López et al. 2012). In the coastal volcanic zones of Central and South America, where the predominant lithological composition is andesitic or rhyolitic, As concentration in thermal waters varies widely, and although As concentrations of several thousand μg/L are frequently reported, none of them is as high as the highest reported As concentration in Mexican geothermal fields (i.e., 73.6 mg/L, López et al. 2012). Reported As concentrations, apparently is contributed by oxidation of sulfide deposits, are as high as 4600 mg/L (?) in the Bolivian Altiplano, where hot springs and fumaroles discharge water and gases in Poopó Lake (López et al. 2012).
In Europe, As concentrations >50 μg/L in groundwater are reported from the Massif Central's geothermal fields in France and in Greece (Brunt et al. 2004; Karydakis et al. 2005). Arsenic concentration in groundwater in Iceland often exceeds 10 μg/L (Arnorssón 2003), although basaltic lava typically contains less As than silicic lavas (Onishi and Sandell 1955; Baur and Onishi 1969; Ure and Berrow 1982). Geothermal systems in northern and northeastern Spain are characterized by high As concentrations and As-rich mineral deposits around surface geothermal manifestations (Navarro et al. 2011), e.g., hot springs with 50–80 μg/L As in the Caldes de Malavella geothermal field in northern Spain contributes a substantial amount of As (from <1 to 200 μg/L) to groundwater (Piqué et al. 2010). The As concentrations in groundwater (including brines) range from 1.6 to 6900 μg/L in the Phlegraean geothermal fields in southern Italy (Aiuppa et al. 2006). Quaternary volcanic rocks with hydrothermal activity on Ischia (a volcanic island offshore from Naples, Italy, in the Tyrrhenian Sea) contribute As to groundwater at concentrations ranging up to 3800 μg/L (Aiuppa, et al. 2006; Daniele 2004).
In the Middle East, reported As concentrations in thermal waters are as high as 3500 μg/L near Mt. Sabalan, a stratovolcano in northwestern Iran (Haeri et al. 2011). In western Anatolia, Turkey, natural leaching, aided by pumping and discharge of waste geothermal fluids from an active geothermal system, has mobilized As from metamorphic, igneous, and sedimentary rocks to groundwater in a shallow alluvial aquifer, where highest As concentrations in groundwater and geothermal fluids were 561 and 594 μg/L, respectively (Gunduz et al. 2010). Baba et al. (2021) reported that geothermal fluids circulating through volcano-sedimentary units in the Anatolian plate caused a more effective alteration, increasing As concentrations in geothermal systems. Moreover, they determined that high As concentrations within the convective-non-magmatic extensional geothermal play types, viz. Western Anatolian Extensional System and the North Anatolian Fault. Bundschuh et al. (2013) found the mixing of geothermal fluids primarily responsible for contamination of freshwater resources, making them unsuitable for drinking or irrigation, based on hydrogeochemical investigations of geothermal fluids from deep wells and hot springs in western Anatolia, Turkey. Baba and Sözbilir (2012) deciphered that the enhanced dilation ~ E–W-strikes of the graben faults in west Turkey, accommodating the deep circulation of hydrothermal fluids of meteoric origin, provide a suitable environment for the presence of high As levels in geothermal fluids.
Groundwater in early Proterozoic silicic volcanics and granites of the Chhattisgarh Basin (India) contains >10 μg/L of As, emplaced there by hydrothermal fluids (Acharyya 2005).
Geothermal systems are controlled by global tectonic and local structural settings, which ultimately define heat and fluid flow regimes. See the chapter Tectonic and Structural Control on Geothermal Systems in the same book for more details. This association of geothermal systems with geotectonic settings advocates for geotectonic control in distributing and disseminating As.
5.5 Coal and Hydrocarbon Resources
Together with both of these fuel resources’ exploitation processes, coal deposits and hydrocarbon reservoirs are other geogenic sources of As contamination that is accelerated by the extraction and refining processes and subsequent usage. Geotectonics also controls their formation in ancient and modern sedimentary basins. The average world concentration of As in coal is 9.0 ± 0.8 and 7.4 ± 1.4 mg/kg for bituminous coals and lignites, while in their ash, it is 50 ± 5 and 49 ± 8 mg/kg, respectively (Yudovich and Ketris 2005).
Petroleum reservoir waters from sedimentary basins can contain As as a result of the evaporation of seawater that concentrates As in oil reservoirs (Birkle et al. 2010). For example, Birkle et al. (2010) reported As concentration up to 2000 µg/L at depths of 2900–6100 m below sea level in oil fields of southeastern Mexico. Moreover, formation water (fresh or saline water trapped in geological formations for thousands to millions of years) forms the most significant part of the volume of co-produced water in hydrocarbon production (Neff et al. 2011). Stromgren et al. (1995) found that the formation water has particularities of the geological formation and is polluted by metals, inorganic compounds, and minerals, including As. Also, their concentrations can differ considerably from seawater.
6 Structural Controls on As Concentration and Distribution in Aquifers
Apart from the aforementioned solid and liquid phase inputs of As, attributed to the lithology and geothermal and volcanic fluids, respectively, there is yet another essential aspect of As enrichment of aquifers, which has to do with the structural settings of the aquifers in a basin, e.g., Duero Basin, Spain (Giménez-Forcada and Smedley 2014), wherein basin morphology, prolongation of faults, especially division of the underlying basement into blocks, produces discrete hydrodynamic environments, resulting in spatial variations in hydrochemical composition, including As speciation (Fig. 4, Giménez-Forcada and Smedley 2014).

Groundwater (springs and boreholes) Arsenic distribution, along with the prominent structural fractures mapped by Simón Gómez (1996), in Duero Basin. Deep boreholes B1 and B2 with the highest As concentrations are represented in the cross-section A–B. Blue contours indicate the piezometric surface. Symbol size reflects arsenic concentration. Modified after Giménez-Forcada and Smedley (2014)
Although pH and redox conditions play essential roles in controlling the As occurrence, distribution, and mobility in groundwater, the basement structure plays a significant control, e.g., faults behave as effective conduits for groundwater flow and influence the recharge of the basin (Giménez-Forcada and Smedley 2014). The groundwater flow could be controlled by the faults (or fault zones), which act as preferential flow conduits and zones in tectonic depressions (Vilanova et al. 2008), particularly those faults oriented parallel to the general flow path in the basin (Giménez-Forcada and Smedley 2014). Moreover, horst and graben structures can generate discrete hydrogeological cells (Giménez-Forcada and Smedley 2014).
Groundwater chemistry varies laterally and with depth, depending upon climatic and hydrological factors, viz. evaporation (Giménez-Forcada and Smedley 2014). For example, in the shallow sedimentary aquifer (<40 m), evaporation can increase the concentrations of dissolved elements, including trace elements like As (e.g., Fujii and Swain 1995; Welch and Lico 1998; Smedley and Kinniburgh 2002). For shallow wells (<40 m) in Duero Basin, Spain, oxidizing conditions and evaporation control groundwater composition. At 40–160 m depth, reducing conditions affect the concentrations of As and other redox-sensitive species, viz. SO4, NO3, Fe, and Mn (Giménez-Forcada and Smedley 2014). The low As concentration at depth is consistent with As substitution into sulfide minerals under reducing conditions (Giménez-Forcada and Smedley 2014).
However, in deeper boreholes within the sedimentary aquifer (200–450 m) in Duero Basin, the dominant waters are Na-HCO3 type with alkaline pH (>9) and high As (up to 241 μg/L, borehole B2), which implies the presence of an oxidizing environment (Fig. 4, Giménez-Forcada and Smedley 2014). These authors explained it in terms of the contribution of oxidizing flows following the main direction of faults in Duero Basin. These deep flow paths (coming directly from the basement and also through the discontinuities within the sedimentary basin) can also transport dissolved As from other As-enriched zones under oxic and alkaline groundwater conditions (Giménez-Forcada and Smedley 2014).
The basement structures consisting of an alternation of horsts and grabens, as in the case of Duero Basin, which is associated with late or post-Variscan tectonism (Ubanell 1985; Ares Yañez et al. 1995; Gómez Ortiz and Babín Vich 1996; Simón Gómez 1996; Herrero 1999; Giménez-Forcada and Smedley 2014), have their manifestation on the surface as large lineaments or areas with a high density of smaller lineaments (Fig. 4) and define subsequent sedimentation and lithological discontinuities (Colmenero et al. 2001; Armenteros et al. 2002; Alonso Gavilán et al. 2004), which in turn control the hydrogeological and hydrochemical features in the basin (Alberruche Del Campo 2010).
The major fault structures thus control the occurrence of groundwater As, as in Duero Basin (Giménez-Forcada and Smedley 2014), where groundwater extracted from the deep boreholes along the main fault lines, taking advantage of the direction of preferential recharge flows, are oxidizing, alkaline, Na-HCO3 type with relatively high concentrations of As, up to 241 μg/L As, albeit with limited data, appear to have high percentages of [As(V)], along with U, V, and F (Giménez-Forcada and Smedley 2014).
Another structural control could be the hydraulic gradient, the lower one providing more residence time for groundwater (Bhattacharya et al. 2002; Nriagu et al. 2007). Smedley and Kinniburgh (2002) pointed out that whether released As remains at problematic levels in groundwater depends not only on whether there are biogeochemical reactions that retard the transport of As but also upon the hydrologic and hydrogeologic properties of the aquifer, such as flow velocity and dispersion. If the kinetics of As release is slow, and groundwater residence time is short, then As concentrations may not increase to the point where groundwater would be considered contaminated. Conversely, with rapid As mobilizing reactions and prolonged residence, As can accumulate in groundwater such that concentrations become hazardous, e.g., Bangladesh (Zheng et al. 2004; Bhattacharya et al. 2009). Eventually, if the biogeochemical conditions that lead to As release and mobilization continue to be present (within a geologic timeframe), the source could become exhausted.
Bhattacharya et al. (2012) highlighted the importance of groundwater divides, which leads to many local flows, thus forming different groundwater systems controlling As and other toxic element concentrations in Wassa West district's mining area in Ghana. The aquifers in this hilly area are not affected severely by the leaching of minerals because of short groundwater residence time and intense flushing because of the high hydraulic gradient. Thus, groundwater flow systems’ local character prevents AMD’s significant impact on groundwater systems on a regional scale.
7 Conclusions
Based on the average crustal concentration and presence in different geological materials, any aquifer is potentially susceptible to contamination. Instead, there is heterogeneity in the level of As contamination of groundwater across the globe, albeit with a pattern defined mainly by geotectonics. For example, major occurrences of contaminated aquifers within sedimentary basins located in the vicinity of active or ancient magmatic arcs at continental convergent margins are controlled by geodynamic processes operating at a regional-scale over millions of years. They are not mere manifestations of heterogeneous As-mobilizing (bio) geochemical conditions.
As enrichment of aquifer matrix in the forelands basins and subsequently the residing groundwater therein is also related to exhumation and erosion of arc-related lithologies that supply As-enriched sediments to these basins. Although rift basins seem to be apparent exceptions to the aforementioned conclusion of associating As contamination to convergent boundaries; however, those basins also derive sediments, at least substantially if not alone, from nearby magmatic arcs. So this observation regarding As contamination of aquifers in the rift basin does not contradict the previous conclusion. Arsenic contamination is widespread globally; however, contaminated areas tend to concentrate in and around active tectonic zones (modern/active and ancient), manifested along with previously mentioned arc magmatism, through volcanics center, geothermal fields, mineralized zones (especially metal sulfides), etc.
Among geogenic sources of As contamination, the most common occurrences are metal sulfides and sorbed species on Fe hydroxides, although As occurs within some silicate and carbonate rocks as well. Although geotectonics control the primary geologic sources of As through regional scale geodynamic processes, the basement structure of the basin, also driven by (paleo)geotectonics, may control As speciation locally through the development of compartmentalized hydrodynamic environments, resulting in spatial variations in hydrochemical composition, including As speciation.
References
Acharyya SK, Shah BA, Ashyiya ID et al (2005) Arsenic contamination in groundwater from parts of Ambagarh-Chowki block, Chhattisgarh, India: source and release mechanism. Environ Geol 49:148–158. https://doi.org/10.1007/s00254-005-0074-3
Ahmed KM, Bhattacharya P, Hasan MA et al (2004) Arsenic enrichment in groundwater of the alluvial aquifers in Bangladesh: an overview. Appl Geochem 19:181–200. https://doi.org/10.1016/j.apgeochem.2003.09.006
Aiuppa A, Avino R, Caliro S et al (2006) Mineral control of arsenic content in thermal waters from volcanic-hosted hydrothermal systems: insights from the island of Ischia and Phlegrean fields (Campanian Volcanic Province, Italy). Chem Geol 229:313–330. https://doi.org/10.1016/j.chemgeo.2005.11.004
Alberruche Del Campo ME (2010) Atlas del medio natural y de los recursos hídricos de la provincia de Ávila. Instituto Geológico y Minero de España-Diputación de Ávila, Madrid
Alonso Gavilán G, Armenteros I, Carballeira J et al (2004) Cuenca del Duero. In: Vera JA (ed) Geología de España. SGE-IGME, Madrid, pp 550–556
Ares Yañez M, Gutiérrez Alonso G, Díez Balda MA et al (1995) La prolongación del Despegue de Salamanca (segunda fase de deformación varisca) en el Horst de Mirueña (Zona Centro Ibérica). Rev Soc Geol Esp 8:175–191
Armenteros I, Corrochano A, Alonso Gavilán G et al (2002) Duero basin (northern Spain). In: Gibbons W, Moreno T (eds) The geology of Spain. Geological Society, London, pp 304–315
Armienta MA, Villaseñor G, Rodríguez R et al (2001) The role of arsenic-bearing rocks in groundwater pollution at Zimapan Valle, Mexico. Environ Geol 40(4–5):571–581. https://doi.org/10.1007/s002540000220
Arnorssón S (2003) Arsenic in surface- and up to 90°C ground waters in a basalt area, N-Iceland: processes controlling its mobility. Appl Geochem 18:1297–1312. https://doi.org/10.1016/S0883-2927(03)00052-0
Aullón Alcaine A, Schulz C, Bundschuh J et al (2020) Hydrogeochemical controls on the mobility of arsenic, fluoride and other geogenic co-contaminants in the shallow aquifers of northeastern La Pampa Province in Argentina. Sci Total Environ 715:136671. https://doi.org/10.1016/j.scitotenv.2020.136671
Awaya T, Oyama M, Ishizaka N et al (2002) The amount of arsenic loads of river waters and hot springs in the Hakone-Yugawara area. Ann Rep Kanagawa Onsen Chigaku Kenkyujo 33:49–70. Japanese
Baba A, Sözbilir, H (2012) Source of arsenic based on geological and hydrogeochemical properties of geothermal systems in Western Turkey. Chem Geol 334:364–377. https://doi.org/10.1016/j.chemgeo.2012.06.006
Baba A, Uzelli T, Sözbilir H (2021) Distribution of geothermal arsenic in relation to geothermal play types: a global review and case study from the Anatolian Plate (Turkey). J Hazard Mat 414:125510. https://doi.org/10.1016/j.jhazmat.2021.125510
Ball JW, Nordstrom DK, Jenne EA et al (1998) Chemical analyses of hot springs, pools, geysers, and surface waters of Yellowstone National Park, Wyoming and vicinity 1972–1975. U.S. Geological Survey Open-File Report 98–182, U.S. Geological Survey, Reston, VA. https://doi.org/10.3133/ofr98182
Ball JW, McCleskey RB, Nordstrom, DK et al (2002) Water chemistry data for selected springs, geysers, and streams in Yellowstone National Park, Wyoming 1999–2000. U.S. Geological Survey Open-File Report 2002–382, U.S. Geological Survey, Reston, VA. https://doi.org/10.3133/ofr02382
Banerjee DM, Bhattacharya P (1994) Petrology and geochemistry of greywackes from the Aravalli Supergroup, Rajasthan, India and the tectonic evolution of a Proterozoic sedimentary basin. Precambrian Res 67(1–2):1–35. https://doi.org/10.1016/0301-9268(94)90003-5
Barringer JL, Reilly PA (2013) Arsenic in groundwater: a summary of sources and the biogeochemical and hydrogeologic factors affecting Arsenic occurrence and mobility. In: Bradley PM (ed) Current perspectives in contaminant hydrology and water resources sustainability. IntechOpen, Rijeka, Croatia. https://doi.org/10.5772/55354
Baur WH, Onishi BMH (1969) Arsenic. In: Wedepohl KH (ed), Handbook of geochemistry. Springer-Verlag, Berlin, pp 33-A-1-33-0-5
Bebout GE, Ryan JG, Leeman WP et al (1999) Fractionation of trace elements by subduction-zone metamorphism – effect of convergent-margin thermal evolution. Earth Planet Sci Lett 171:63–81. https://doi.org/10.1016/S0012-821X(99)00135-1
Berg M, Stengel C, Trang PTK et al (2007) Magnitude of arsenic pollution in the Mekong and Red River deltas – Cambodia and Vietnam. Sci Total Environ 372:413–425. https://doi.org/10.1016/j.scitotenv.2006.09.010
Bhattacharya P, Chatterjee D, Jacks G (1997) Occurrence of Arsenic-contaminated groundwater in alluvial aquifers from delta plains, Eastern India: options for safe drinking water supply. Int J Water Resour Dev 13(1):79–92. https://doi.org/10.1080/07900629749944
Bhattacharya P, Jacks G, Sracek A, et al (2001) Geochemistry of the holocene alluvial sediments in the Bengal delta plains: implication on arsenic contamination in the groundwater. In: Jacks G, Bhattacharya P, Khan AA (eds) Proceedings of the KTH - Dhaka University Seminar on Groundwater Arsenic Contamination in the Bengal Delta Plains of Bangladesh, Department of Geology, Dhaka, Bangladesh, 7–8 February, 1999, KTH Special Publication TRITA-AMI Report 3084, Stockholm, Sweden. Available at: https://www.divaportal.org/smash/get/diva2:500376/FULLTEXT01.pdf
Bhattacharya P, Frisbie SH, Smith E et al (2002) Arsenic in the environment: a global perspective. In: Sarkar B (ed) Handbook of heavy metals in the environment. Marcell Dekker, New York, pp 147–215
Bhattacharya P, Welch AH, Ahmed KM et al (2004) Arsenic in groundwater of sedimentary aquifers. Appl Geochem 19(2):163–167. https://doi.org/10.1016/j.apgeochem.2003.09.004
Bhattacharya P, Welch AH, Stollenwerk KG et al (2007) Arsenic in the environment: biology and chemistry. Sci Total Environ 379:109–120. https://doi.org/10.1016/j.scitotenv.2007.02.037
Bhattacharya P, Hasan MA, Sracek O et al (2009) Groundwater chemistry and arsenic mobilization in the Holocene flood plains in south-central Bangladesh. Environ Geochem Health 31:23–43. https://doi.org/10.1007/s10653-008-9230-5
Bhattacharya P, Sracek O, Eldvall B et al (2012) Hydrogeochemical study on the contamination of water resources in a part of Tarkwa mining area, Western Ghana. J African Earth Sci 66–67:72–84. https://doi.org/10.1016/j.jafrearsci.2012.03.005
Bhattacharya P, Classon M, Bundschuh J et al (2006b) Distribution and mobility of arsenic in the Río Dulce alluvial aquifers in Santiago del Estero Province, Argentina. Sci Total Environ 358:97–120. https://doi.org/10.1016/j.scitotenv.2005.04.048
Bhattacharya P, Ahmed KM, Hasan MA et al (2006a) Mobility of arsenic in groundwater in a part of Brahmanbaria district, NE Bangladesh. In: Naidu R, Smith E, Owens G, Bhattacharya P, Nadebaum P (eds) Managing Arsenic in the environment: from soil to human health. CSIRO Publishing, Melbourne, Australia, pp 95–115
Bhattacharya, P, Polya DA, Jovanovic D (eds) (2017) Best practice guide for the control of arsenic in drinking water. International Water Association, London. https://doi.org/10.2166/9781780404929
Bhowmick S, Pramanik S, Singh P et al (2018) Arsenic in groundwater of West Bengal, India: a review of human health risks and assessment of possible intervention options. Sci Total Environ 612:148–169. https://doi.org/10.1016/j.scitotenv.2017.08.216
Birkle P, Bundschuh J, Sracek O (2010) Mechanisms of arsenic enrichment in geothermal and petroleum reservoirs fluids in Mexico. Water Res 44(19):5605–5617. https://doi.org/10.1016/j.watres.2010.05.046
Biswas A, Gustafsson JP, Neidhardt H, et al (2014a) Role of competing ions in the mobilization of arsenic in groundwater of Bengal Basin: insight from surface complexation modeling. Water Res 55:30–39. https://doi.org/10.1016/j.watres.2014.02.002
Biswas A, Bhattacharya P, Mukherjee A, et al (2014b) Shallow hydrostratigraphy in an arsenic affected region of Bengal Basin: implication for targeting safe aquifers for drinking water supply. Sci Total Environ 485–486: 12–22
Biswas A, Neidhardt H, Kundu AK, et al (2014c) Spatial, vertical and temporal variation of arsenic in shallow aquifers of the Bengal Basin: controlling geochemical processes. Chem Geol 387:157–169. https://doi.org/10.1016/j.chemgeo.2014.08.022
Biswas A, Nath B, Bhattacharya P, et al (2012) Hydrogeochemical contrast between brown and grey sand aquifers in shallow depth of Bengal Basin: consequences for sustainable drinking water supply. Sci Total Environ 431, 402–412. https://doi.org/10.1016/j.scitotenv.2012.05.031
Biswas A, Majumder S, Neidhardt H, et al (2011) Groundwater chemistry and redox processes: depth dependent arsenic release mechanism. Appl Geochem 26:516–525. https://doi.org/10.1016/j.apgeochem.2011.01.010
Boyle RW, Jonasson IR (1973) The geochemistry of arsenic and its use as an indicator element in geochemical prospecting. J Geochem Explor 2:251–296. https://doi.org/10.1016/0375-6742(73)90003-4
Boyle DR, Turner RJW, Hall GEM (1998) Anomalous arsenic concentrations in groundwaters of an island community, Bowen Island, British Columbia. Environ Geochem Health 20:199–212. https://doi.org/10.1023/A:1006597311909
Breuer C, Pichler T (2013) Arsenic in marine hydrothermal fluids. Chem Geol 348:2–14. https://doi.org/10.1016/j.chemgeo.2012.10.044
Brookins DG (2012) Eh-pH diagrams for geochemistry. Springer, Berlin Heidelberg
Brown, CJ, Chute SK (2002) Arsenic in bedrock wells in Connecticut (Abstract). Arsenic in New England: a multidisciplinary scientific conference, national institute of environmental health sciences, superfund basic research program, Manchester, New Hampshire, 2002
Brunt R, Vasak L, Griffioen J (2004) Arsenic in groundwater: probability of occurrence of excessive concentration on a global scale. International Groundwater Resources Assessment Centre: Report no. SP 2004-1. https://www.un-igrac.org/resource/arsenic-groundwater-probability-occurrence-excessive-concentration-global-scale. Accessed 31 January 2021
Bundschuh J, Maity JP (2015) Geothermal arsenic: occurrence, mobility and environmental implications. Renew Sustain Energy Rev 42:1214–1222. https://doi.org/10.1016/j.rser.2014.10.092
Bundschuh J, Bhattacharya P, Hoinkis J et al (2010) Groundwater arsenic: from genesis to sustainable remediation. Water Res 44(19):5511. https://doi.org/10.1016/j.watres.2010.10.028
Bundschuh J, Litter MI, Parvez F et al (2012) One century of arsenic exposure in Latin America: a review of history and occurrence from 14 countries. Sci Total Environ 429:2–35. https://doi.org/10.1016/j.scitotenv.2011.06.024
Bundschuh J, Maity JP, Nath B et al (2013) Naturally occurring arsenic in terrestrial geothermal systems of western Anatolia, Turkey: potential role in contamination of freshwater resources. J Hazard Mat 262:951–959. https://doi.org/10.1016/j.jhazmat.2013.01.039
Bundschuh J, Armienta MA, Morales-Simfors N et al (2020) Arsenic in Latin America: new findings on source, mobilization and mobility in human environments in 20 countries based on decadal research 2010–2020. Crit Rev Env Sci Tec. https://doi.org/10.1080/10643389.2020.1770527
Bundschuh J, Schneider J, Alam MA et al (2021) Seven potential sources of arsenic pollution in Latin America and their environmental and health impacts. Sci Total Environ. https://doi.org/10.1016/j.scitotenv.2021.146274
Bundschuh J, Farias B, Martin R et al (2004) Groundwater arsenic in the Chaco-Pampean Plain, Argentina: case study from Robles County, Santiago del Estero Province. Appl Geochem 19(2):231–243. https://doi.org/10.1016/j.apgeochem.2003.09.009
Chakraborti D, Rahman MM, Mukherjee A et al (2015) Groundwater arsenic contamination in Bangladesh—21 Years of research. J Trace Elem Med Biol 31:237–248. https://doi.org/10.1016/j.jtemb.2015.01.003
Chakraborty M, Mukherjee A, Ahmed KM (2015) A review of groundwater arsenic in the Bengal Basin, Bangladesh and India: from source to sink. Curr Pollution Rep 1:220–247. https://doi.org/10.1007/s40726-015-0022-0
Chakraborty M, Sarkar S, Mukherjee A et al (2020) Modeling regional-scale groundwater arsenic hazard in the transboundary Ganges River Delta, India and Bangladesh: infusing physically-based model with machine learning. Sci Total Environ 748:141107. https://doi.org/10.1016/j.scitotenv.2020.141107
Chapelle FH (1993) Ground-water microbiology and geochemistry. John Wiley and Sons, New York
Charnock JM, Polya DA, Gault AG et al (2007) Direct EXAFS evidence for incorporation of As5+ in the tetrahedral site of natural andraditic garnet. Am Min 92:1856–1861. https://doi.org/10.2138/am.2007.2541
Colmenero JR, Rodríguez JM, Gómez JJ et al (2001) Estratigrafía del subsuelo y evolución sedimentaria del sector sur de la cuenca terciaria del Duero. Geotemas 3:129–132
Coomar P, Mukherjee A, Bhattacharya P et al (2019) Contrasting controls on hydrogeochemistry of arsenic-enriched groundwater in the homologous tectonic settings of Andean and Himalayan basin aquifers, Latin America and South Asia. Sci Total Environ 689:1370–1387. https://doi.org/10.1016/j.scitotenv.2019.05.444
Craw D, Chappell D, Reay A (2000) Environmental mercury and arsenic sources in fossil hydrothermal systems, Northland, New Zealand. Environ Geol 38:875–887. https://doi.org/10.1007/s002549900068
Crock JG, Gough LP, Wanty RB et al (1999) Regional geochemical results from the analyses of rock, water, soil, stream sediment, and vegetation samples — Fortymile River Watershed, East-Central, Alaska 1998 Sampling. U.S. Department of the Interior, U.S. Geological Survey, Open-File Report 00-511. https://doi.org/10.3133/ofr00511
Daniele L (2004) Distribution of arsenic and other minor trace elements in the groundwater of Ischia Island (southern Italy). Environ Geol 46:96–103. https://doi.org/10.1007/s00254-004-1018-z
DeCelles PG, Giles KA (1996) Foreland basin systems. Basin Res 8(2):105–123. https://doi.org/10.1046/j.1365-2117.1996.01491.x
Deschamp F, Godard M, Guillot S et al (2013) Geochemistry of subduction zone serpentinites: a review. Lithos 178:96–127. https://doi.org/10.1016/j.lithos.2013.05.019
Drever JI (1997) The geochemistry of natural waters, 3rd edn. Prentice-Hall, Upper Saddle River, NJ
Edmunds, WM., Walton, NRG, Howard MP J et al (1981). Geochemical estimation of aquifer recharge. British Geological Survey, Wallingford, Oxfordshire, Report WD/OS/80/17
Ehrlich HL, Newman DK (eds) (2009) Geomicrobiology. CRC Press, Boca Raton, FL
Ellis AJ, Mahon WAJ (1967) Natural hydrothermal systems and experimental hot water/rock interactions (Part II). Geochim Cosmochim Acta 31(4):519–538. https://doi.org/10.1016/0016-7037(67)90032-4
Ellis AJ, Mahon WAJ (1977) Chemistry and geothermal systems. Academic Press, New York
Even E, Masuda H, Shibata T et al (2017) Geochemical distribution and fate of arsenic in water and sediments of rivers from the Hokusetsu area, Japan. J Hydrol Reg Stud 9:34–47. https://doi.org/10.1016/j.ejrh.2016.09.008
Faure G (1998) Principles and applications of geochemistry, 2nd edn. Prentice Hall, Upper Saddle River, NJ
Fendorf S, Holly HA, van Geen A (2010) Spatial and temporal variations of groundwater arsenic in South and Southeast Asia. Science 328:1123–1127. https://doi.org/10.1126/science.1172974
Fernández-Martínez A, Cuello GJ, Johnson MR et al (2008) Arsenate incorporation in gypsum probed by neutron, x-ray scattering and density functional theory modeling. J Phys Chem A 112:5159–5166. https://doi.org/10.1021/jp076067r
Fujii R, Swain WC (1995) Areal distribution of trace elements, salinity, major ions in shallow ground water, Tulare basin, Southern San Joaquin Valley, California. U.S. Geological Survey Water Resources Investigations Report 95-4048, Sacramento, California. https://doi.org/10.3133/wri954048
Fujiwara S, Yamamoto K, Mimura K (2011) Dissolution processes of elements from subducting sediments into fluids: evidence from the chemical composition of the Sanbagawa pelitic schists. Geochem J 45:221–234. https://doi.org/10.2343/geochemj.1.0117
Giménez-Forcada E, Smedley PL (2014) Geological factors controlling occurrence and distribution of arsenic in groundwaters from the southern margin of the Duero Basin, Spain. Environ Geochem Health 36(6):1029–1047. https://doi.org/10.1007/s10653-014-9599-2
Gleeson T, Ingebritsen SE (eds) (2016) Crustal permeability. Wiley-Blackwell, Chichester
Gómez Ortiz D, Babín Vich RB (1996) La tectónica alpina en el sector centro-oriental del borde norte del Sistema Central, Provincia de Segovia, España. Geogaceta 19:19–22
Gong ZL, Lu XF, Watt C et al (2006) Speciation analysis of arsenic in groundwater from Inner Mongolia with an emphasis on acid-leachable particulate arsenic. Anal Chim Acta 555:181–187. https://doi.org/10.1016/j.aca.2005.08.062
Gonzalez-Partida E, Hinojosa ET, Verma MP (2001) Interraccion agua geothermica manantiales en el campo geotermico de Los Humeros, México. Ing Hidraul Mex XVI:185–194
Grosz AE, Grossman JN, Garrett R et al (2004) A preliminary geochemical map for arsenic in surficial materials of Canada and the United States. Appl Geochem 19:257–260. https://doi.org/10.1016/j.apgeochem.2003.09.012
Guilliot S, Charlet L (2007) Bengal arsenic, an archive of Himalaya orogeny and paleohydrology. J Environ Sci Health A 42:1785–1794. https://doi.org/10.1080/10934520701566702
Guillot S, Garcon M, Weinman B et al (2015) Origin of arsenic in Late Pleistocene to Holocene sediments in the Nawalparasi district (Terai, Nepal). Environ Earth Sci 74:2571–2593. https://doi.org/10.1007/s12665-015-4277-y
Gunduz O, Simsek C, Hasozbek A (2010) Arsenic pollution in the groundwater of Simav Plain, Turkey: its impact on water quality and human health. Water Air Soil Pollut 205:43–62. https://doi.org/10.1007/s11270-009-0055-3
Guo H-M, Yang S-Z, Li X-H et al (2007) Groundwater geochemistry and its implications for arsenic mobilization in shallow aquifers of the Hetao basin, Inner Mongolia. Sci Total Environ 393(1):131–144. https://doi.org/10.1016/j.scitotenv.2007.12.025
Haeri A, Strelbitskaya S, Porkhial S et al (2011) Distribution of arsenic in geothermal fluids from Sabalan geothermal field, N-W Iran. In: Proceedings 36th workshop on geothermal reservoir engineering, Stanford University, Stanford, California, USA, 31 January–2 February 2011, SGP-TR-191. https://pangea.stanford.edu/ERE/pdf/IGAstandard/SGW/2011/haeri2.pdf. Accessed 31 January 2021
Hammarlund L, Pionens J, Bhattacharya P et al (2009) Study of geothermal fluid-groundwater interaction and evolution in thermal fields of Costa Rica. Geological Society of America, Abstracts with Programs 41(7):219
Hattori KH, Guillot S (2003) Volcanic fronts form as a consequence of serpentinite dehydration in the forearc mantle wedge. Geology 31(6):525–528. https://doi.org/10.1130/0091-7613(2003)031%3c0525:VFFAAC%3e2.0.CO,2
Hattori KH, Arai S, Clarke DB (2002) Selenium, tellurium, arsenic and antimony contents of primary mantle sulfides. Can Mineral 40:637–650. https://doi.org/10.2113/gscanmin.40.2.637
Hattori K, Takahashi Y, Guillot S et al (2005) Occurrence of arsenic (V) in forearc mantle serpentinites based on X-ray absorption spectroscopy study. Geochim Cosmochim Acta 69(23):5585–5596. https://doi.org/10.1016/j.gca.2005.07.009
Hecht H, Oguchi T (2017) Global evaluation of erosion rates in relation to tectonics. Prog Earth Planet Sci 4:40. https://doi.org/10.1186/s40645-017-0156-3
Henke KR (2009) Arsenic in natural environments. In: Henke KR (ed) Arsenic—environmental chemistry, health threats and waste treatment. John Wiley & Sons, Chichester, pp 69–236
Herath I, Vithanage M, Bundschuh J et al (2016) Natural arsenic in global groundwaters: distribution and geochemical triggers for mobilization. Curr Pollut Rep 2:68–89. https://doi.org/10.1007/s40726-016-0028-2
Herrero M (1999) El papel explicativo de las rocas filonianas en la evolución morfoestructural de áreas de zócalo cristalino: La Sierra de Ávila. Cuaternario Geomorfol 13:51–60
Ijumulana J, Ligate F, Bhattacharya P et al (2020) Spatial analysis and GIS mapping of regional hotspots and potential health risk of fluoride concentrations in groundwater of northern Tanzania. Sci Total Environ 735:139584. https://doi.org/10.1007/s40726-016-0028-2
Iskandar I, Koike K, Sendjaja P (2012) Identifying groundwater arsenic contamination mechanisms in relation to arsenic concentrations in water and host rocks. Environ Earth Sci 65:2015–2026. https://doi.org/10.1007/s12665-011-1182-x
Jin X, She Q, Ang X et al (2012) Removal of boron and arsenic by forward osmosis membrane: influence of membrane orientation and organic fouling. J Membr Sci 389:182–187. http://dx.doi.org/10.1016/j.memsci.2011.10.028
Karydakis G, Arvanitis A, Andritsos N et al (2005). Low enthalpy geothermal fields in the Strymon Basin (Northern Greece). In: Proceedings world geothermal congress 2005, Antalya, 24–29 Turkey April 2005, Paper Number: 0258. https://pangea.stanford.edu/ERE/pdf/IGAstandard/EGC/2007/258.pdf. Accessed 31 January 2021
Katsoyiannis IA, Mitrakas M, Zouboulis AI (2015) Arsenic occurrence in Europe: emphasis in Greece and description of the applied full-scale treatment plants. Desalin Water Treat 54(8):2100–2107. https://doi.org/10.1080/19443994.2014.933630
Kawakami H, Nozaki H, Koga A (1956) Chemical study on Beppu Hoto spring (II) – trace elements of Peppu hot spring (II) distribution of arsenic. Nihon Ishigaku Zasshi 77:1785–1789. Japanese
Keshavarzi B, Moore F, Mosaferi M et al (2011) The source of natural arsenic contamination in groundwater, west of Iran. Water Qual Expo Health 3:135–147. https://doi.org/10.1007/s12403-011-0051-x
Kikawada K, Kawai S, Oi T (2006) Long term changes in the concentration of dissolved arsenic and its present supply in the Kusatsu hot springs Gunma Japan. Chikyukagaku (Geochemistry) 40:125–136. Japanese with English abstract
Kikawada K, Kyomen K, Oi T (2009) Behavior of arsenic in Yukawa River of the Kusatsu hot spring resource area, Gunma prefecture, Japan. J Hot Spring Sci 59:81–87. Japanese with English abstract
Korte NE, Quintus F (1991) A review of arsenic (III) in groundwater. Crit Rev Environ Sci Technol 21(1):1–39. https://doi.org/10.1080/10643389109388408
Kumaresan M, Riyazuddin P (2001) Overview of speciation chemistry of arsenic. Curr Sci 80(7):837–846. https://www.jstor.org/stable/24105734. Accessed 31 January 2021
Landrum JT, Bennet PC, Engel AS et al (2009) Partitioning geochemistry of arsenic and antimony, El Tatio Geyser Field, Chile. Appl Geochem 24:664–676. https://doi.org/10.1016/j.apgeochem.2008.12.024
Li Y, Audétat A (2012) Partitioning of V, Mn Co, Ni, Cu, Zn, As, Mo, Ag, Sn, Sb, W, Au, Pb, and Bi between sulfide phases and hydrous basanite melt at upper mantle conditions. Earth Planet Sci Lett 355–356:327–340. https://doi.org/10.1016/j.epsl.2012.08.008
Li Y, Audétat A (2015) Effects of temperature, silicate melt composition, and oxygen fugacity on the partitioning of V, Mn Co, Ni, Cu, Zn, As, Mo, Ag, Sn, Sb, W, Au, Pb and Bi between sulfide phases and silicate melts. Geochim Cosmochim Acta 162:25–45. https://doi.org/10.1016/j.gca.2015.04.036
Lone SA, Jeelani G, Mukherjee A et al (2020). Geogenic groundwater arsenic in high altitude bedrock aquifers of upper Indus river basin (UIRB), Ladakh. Appl Geochem 113:104497. https://doi.org/10.1016/j.apgeochem.2019.104497
López DL, Bundschuh J, Birkle P et al (2012) Arsenic in volcanic geothermal fluids of Latin America. Sci Total Environ 429:57–75. https://doi.org/10.1016/j.scitotenv.2011.08.043
Maity JP, Chen C-Y, Bundschuh J et al (2017) Hydrogeochemical reconnaissance of arsenic cycling and possible environmental risk in hydrothermal systems of Taiwan. Groundwater Sustainable Dev 5:1–13. https://doi.org/10.1016/j.gsd.2017.03.001
Maity JP, Kar S, Liu J-H et al (2011a) The potential for reductive mobilization of arsenic [As(V) to As(III)] by OSBH2 (Pseudomonas stutzeri) and OSBH5 (Bacillus cereus) in an oil-contaminated site. J Environ Sci Health 46:1239–1246. https://doi.org/10.1080/10934529.2011.598802
Maity JP, Liu C-C, Nath B et al (2011b) Biogeochemical characteristics of Kuan-Tzu-Ling, Chung-Lun and Bao-Lai hot springs in southern Taiwan. J Environ Sci Health, Part A 46(11):1–11. https://doi.org/10.1080/10934529.2011.598788
Maity JP, Chen C-Y, Bundschuh J et al (2016) Investigation of arsenic contamination from geothermal water in different geological settings of Taiwan: hydrogeochemical and microbial signatures. In: Bhattacharya P, Vahter M, Jarsjö, J et al (eds) Arsenic research and global sustainability as 2016. Interdisciplinary book series: “Arsenic in the Environment—Proceedings”. Series Editors: Bundschuh J, Bhattacharya P, CRC Press/Taylor and Francis, London, pp 84–85
Majzlan J, Drahota P, Filippi M (2014) Parageneses and crystal chemistry of arsenic minerals. Rev Mineral Geochem 79:17–184. https://doi.org/10.2138/rmg.2014.79.2
Mandal BK, Suzuki KT (2002) Arsenic round the world: a review. Talanta 58:201–235. https://doi.org/10.1016/S0039-9140(02)00268-0
Margat J (2008) Les eaux souterraines dans le monde [Groundwater around the world]. BRGM, Orleans and UNESCO, Paris
Mariño EE, Teijó Ávila G et al (2020) The occurrence of arsenic and other trace elements in groundwaters of the southwestern Chaco-Pampean plain, Argentina. J S Am Earth Sci 100:102547. https://doi.org/10.1016/j.jsames.2020.102547
Masoodi M, Rahimzadeh M (2018) Tectonic controls on groundwater geochemistry in Hormozgan Province, Southern Iran. Arab J Geosci 11:141. https://doi.org/10.1007/s12517-018-3478-6
Masuda H (2018) Arsenic cycling in the Earth’s crust and hydrosphere: interaction between naturally occurring arsenic and human activities. Prog Earth Planet Sci 5:68. https://doi.org/10.1186/s40645-018-0224-3
Masuda H, Shinoda K, Okudaira T et al (2012) Chlorite – source of arsenic groundwater pollution in the Holocene aquifer of Bangladesh. Geochem J 46:381–391. https://doi.org/10.2343/geochemj.2.0208
Masuda H, Okabayashi K, Maeda S et al (2013) Sequential chemical extraction of arsenic and related elements from the Holocene sediments of Sonargaon, Bangladesh, in relation to formation of arsenic-contaminated groundwater. Geochem J 47:651–666. https://doi.org/10.2343/geochemj.2.0269
Matschullat J (2000) Arsenic in the geosphere – a review. Sci Total Environ 249(1–3):297–312. https://doi.org/10.1016/S0048-9697(99)00524-0
McCarthy KT, Pichler T, Price RE (2005) Geochemistry of Champagne Hot Springs shallow hydrothermal vent field and associated sediments, Dominica, Less Antilles. Chem Geol 224:55–68. https://doi.org/10.1016/j.chemgeo.2005.07.014
McClintock TR, Chen Y, Bundschuh J et al (2012) Arsenic exposure in Latin America: Biomarkers, risk assessments and related health effects. Sci Total Environ 429:76–91. https://doi.org/10.1016/j.scitotenv.2011.08.051
McDonough WF, Sun S-s (1995) The composition of the Earth. Chem Geol 12:223–253. https://doi.org/10.1016/0009-2541(94)00140-4
Minami H, Sato G, Watanuki K (1958). Concentrations of arsenic and lead of Tamagawa hot spring waters, Akita prefecture. Nippon Kagaku Zasshi 79:860–865 Japanese
Morales-Simfors N, Bundschuh J, Herath I et al (2020) Arsenic in Latin America: a critical overview on the geochemistry of arsenic originating from geothermal features and volcanic emissions for solving its environmental consequences. Sci Total Environ 716:135564. https://doi.org/10.1016/j.scitotenv.2019.135564
Motyka RJ, Poreda RJ, Jeffrey AWA (1998) Geochemistry, isotopic composition, and origin of fluids emanating from mud volcanoes in the Copper River basin, Alaska. Geochim Cosmochim Acta 53(1):3302–3309. https://doi.org/10.1016/0016-7037(89)90270-6
Mukherjee A, Fryar AE (2008) Deeper groundwater chemistry and geochemical modeling of the arsenic affected western Bengal basin, West Bengal, India. Appl Geochem 23(4):863–892. https://doi.org/10.1016/j.apgeochem.2007.07.011
Mukherjee A, Fryar AE, Howell PD (2007) Regional hydrostratigraphy and groundwater flow modeling in the arsenic affected areas of the western Bengal Basin, West Bengal, India. Hydrogeol J 15(7):1397–1418. https://doi.org/10.1007/s10040-007-0208-7
Mukherjee A, von Brömssen M, Scanlon BR et al (2008) Hydrogeochemical comparison and effects of overlapping redox zones on groundwater arsenic near the Western (Bhagirathi sub-basin, India) and Eastern (Meghna sub-basin, Bangladesh) margins of the Bengal Basin. J Contam Hydrol 99:31–48. https://doi.org/10.1016/j.jconhyd.2007.10.005
Mukherjee A, Fryar AE, Scanlona BR et al (2011) Elevated arsenic in deeper groundwater of the western Bengal basin, India: extent controls from regional to local scale. Appl Geochem 26:600–613. https://doi.org/10.1016/j.apgeochem.2011.01.017
Mukherjee A, Scanlon BR, Fryar AE et al (2012) Solute chemistry and arsenic fate in aquifers between the Himalayan foothills and Indian craton (including central Gangetic plain): influence of geology and geomorphology. Geochim Cosmochim Acta 90:283–302. https://doi.org/10.1016/j.gca.2012.05.015
Mukherjee A, Verma S, Gupta S et al (2014) Influence of tectonics, sedimentation and aqueous flow cycles on the origin of global groundwater arsenic: paradigms from three continents. J Hydrol 518:284–299. https://doi.org/10.1016/j.jhydrol.2013.10.044
Mukherjee A, Gupta S, Coomar P et al (2019) Plate tectonics influence on geogenic arsenic cycling: From primary sources to global groundwater enrichment. Geochim Cosmochim Acta 683:793–807. https://doi.org/10.1016/j.scitotenv.2019.04.255
Mukherjee A, Fryar AE, O'Shea BM (2009a). Major occurrences of elevated arsenic in groundwater and other natural waters. In: Henke KR (ed) Arsenic—Environmental Chemistry, Health Threats and Waste Treatment. John Wiley & Sons, Chichester, pp 303–350
Mukherjee A, Fryar AE, Thomas WA (2009b). Geologic, geomorphic and hydrologic framework and evolution of the Bengal basin, India and Bangladesh. J Asian Earth Sci 34(3):227–244. https://doi.org/10.1016/j.jseaes.2008.05.011
Mukherjee A, Sarkar S, Chakraborty M et al (2021) Occurrence, predictors and hazards of elevated groundwater arsenic across India through field observations and regional-scale AI-based modeling. Sci Total Environ 759:143511. https://doi.org/10.1016/j.scitotenv.2020.143511
Navarro A, Font X, Viladevall M (2011) Geochemistry and groundwater contamination in the La Selva geothermal system (Girona, Northeast Spain). Geothermics 40:275–285. https://doi.org/10.1016/j.geothermics.2011.07.005
Neff J, Lee K, Deblois EM (2011) Produced water: overview of composition, fates and effects. In: Lee K, Neff J (eds) Produced water. Springer, New York
Nicolli HB, Bundschuh J, Blanco MdC et al (2012) Arsenic and associated trace-elements in groundwater from the Chaco-Pampean plain, Argentina: results from 100 years of research. Sci Total Environ 429:36–56. https://doi.org/10.1016/j.scitotenv.2012.04.048
Nimick DA, Moore JN, Dalby CE, Savka MW (1998) The fate of geothermal arsenic in the Madison and Missouri Rivers, Montana and Wyoming. Water Resour Res 34:3051–3067. https://doi.org/10.1029/98WR01704
Noble DC, Ressel MW, Connors KA (1998) Magmatic As, Sb, Cs and other volatile elements in glassy silicic rocks. Geological Society of America, Abstracts with Programs 30(7):377
Noguchi K, Nakagawa R (1969) Arsenic and arsenic-lead sulfides in sediments from Tamagawa hot springs, Akita Prefecture. Proc Jpn Acad 45:45–50. https://doi.org/10.2183/pjab1945.45.45
Noll PDJr, Newsom HE, Leeman WP et al (1996) The role of hydrothermal fluids in the production of subduction zone magmas: evidence from siderophile and chalcophile trace elements and boron. Geochim Cosmochim Acta 60, 587–611. https://doi.org/10.1016/0016-7037(95)00405-X
Nordstrom DK (2002) Worldwide occurrences of arsenic in ground water. Science 296:2143–2145. https://doi.org/10.1126/science.1072375
Nordstrom DK (2009) Natural arsenic enrichment: effects of diagenetic-tectonic hydrothermal cycle. Geological Society of America, Abstracts with Program 41(7):217
NRC (1977) 3. Distribution of arsenic in the environment. In: Arsenic: medical and biologic effects of environmental pollutants. National Research Council (NRC) Committee on medical and biological effects of environmental pollutants. National Academies Press, Washington, DC. https://www.ncbi.nlm.nih.gov/books/NBK231016/. Accessed 31 January 2021
Nriagu JO, Bhattacharya P, Mukherjee AB et al (2007) Arsenic in soil and groundwater: an overview. In: Bhattacharya P, Mukherjee AB, Bundschuh, J et al (eds) Trace metals and other contaminants in the environment, vol 9. Elsevier, Amsterdam, pp 3–60. https://doi.org/10.1016/S1875-1121(06)09001-8
Ohta A, Imai N, Terashima S et al (2010) Factors controlling regional spatial distribution of 53 elements in coastal sea sediments in northern Japan: comparison of geochemical data derived from stream and marine sediments. Appl Geochem 25:357–376. https://doi.org/10.1016/j.apgeochem.2009.12.003
Okada H, Tada T, Chiba et al (2002) Decontamination of geothermal water – removal of arsenic. Low Temp Eng 37:331–337. Japanese with English abstract
Onishi H, Sandell EB (1955) Geochemistry of arsenic. Geochim Cosmochim Acta 7:1–33. https://doi.org/10.1016/0016-7037(55)90042-9
Ormachea Muñoz M, Bhattacharya P, Sracek O, Ramos Ramos O, Quintanilla Agurre J, Bundschuh J, Maity JP (2015) Arsenic and other trace elements in thermal springs and in cold waters from drinking water wells on the Bolivian Altiplano. J S Am Earth Sci 60:10–20. https://doi.org/10.1016/j.jsames.2015.02.006
Panagiotaras D, Papoulis D, Panagopoulos G et al (2012) Arsenic geochemistry in groundwater system. In: Panagiotaras D (ed) Earth’s system processes. IntechOpen, Rijeka, Croatia. https://doi.org/10.5772/39384
Pascua C, Charnock J, Polya DA et al (2005) Arsenic-bearing smectite from the geothermal environment. Mineral Mag 69:897–906. https://doi.org/10.1180/0026461056950297
Peters SC (2008) Arsenic in groundwaters in the Northern Appalachian Mountain belt: a review of patterns and processes. J Contam Hydrol 99:8–21. https://doi.org/10.1016/j.jconhyd.2008.04.001
Peters SC, Blum JD (2003) The source and transport of arsenic in a bedrock aquifer, New Hampshire, USA. Appl Geochem 18:1773–1787. https://doi.org/10.1016/S0883-2927(03)00109-4
Peters SC, Blum JD, Klaue B, Karagas MR (1999) Arsenic occurrence in New Hampshire drinking water. Environ Sci Technol 33:1328–1333. https://doi.org/10.1021/es980999e
Piqué A, Grandia F, Canals A (2010) Processes releasing arsenic to groundwater in the Caldes de Malavella geothermal area, NE Spain. Water Res 44:5618–5630. https://doi.org/10.1016/j.watres.2010.07.012
Planer-Friedrich B, London J, McCleskey RB et al (2007) Thioarsenates in geothermal waters of Yellowstone National Park: determination, preservation, and geochemical importance. Environ Sci Technol 41:5245–5251. https://doi.org/10.1021/es070273v
Plank T, Langmuir CH (1993) Tracing trace-elements from sediment input to volcanic output at subduction zones. Nature 362(6422):739–743
Pollizzotto ML, Harvey CF, Sutton S et al (2005) Processes conductive to the release and transport of arsenic into aquifers of Bangladesh. Proc Natl Acad Sci USA 102:18819–18823. https://doi.org/10.1073/pnas.0509539103
Polya DA, Sparrenbom C, Datta S, Guo H (2019) Groundwater arsenic biogeochemistry – key questions and use of tracers to understand arsenic-prone groundwater systems. Geosci Front 10(5):1635–1641. https://doi.org/10.1016/j.gsf.2019.05.004
Pourbaix M (1974) Atlas of electrochemical equilibria in aqueous solutions 2nd edn. National Association of Corrosion Engineers, Houston, TX
Quino Lima I, Ramos Ramos OE, Ormachea Muñoz M et al (2020) Spatial dependency of arsenic, antimony, boron and other trace elements in the shallow groundwater systems of the Lower Katari Basin, Bolivian Altiplano. Sci Total Environ 719:137505. https://doi.org/10.1016/j.scitotenv.2020.137505
Quino Lima, I., Ramos Ramos, O.E., Ormachea Muñoz, M et al (2021b) Geochemical mechanisms of natural arsenic mobility in the hydrogeologic system of Lower Katari Basin, Bolivian Altiplano. J Hydrol 594:125778. https://doi.org/10.1016/j.jhydrol.2020.125778
Quino Lima I, Ormachea Muñoz M, Ramos Ramos OE et al (2021a) Hydrogeochemical contrasts in the shallow aquifer systems of the Lower Katari Basin and Southern Poopó Basin, Bolivian Altiplano. J S Am Earth Sci 105:102914. https://doi.org/10.1016/j.jsames.2020.102914
Quintanilla J, Ramos OE, Ormachea M et al (2009) Arsenic contamination, speciation and environmental consequences in the Bolivian Plateau. In: Bundschuh J, Armienta MA, Birkle P et al (eds) Natural arsenic in groundwaters of Latin America. Taylor & Francis, Boca Raton, NJ, pp 91–99
Ramos Ramos OE, Rötting T, French M et al (2014) Geochemical processes controlling mobilization of arsenic and trace elements in shallow aquifers and surface waters in the Antequera and Poopó mining regions, Bolivian Altiplano. J Hydrol 518:421–433. https://doi.org/10.1016/j.jhydrol.2014.08.019
Ramos Ramos OE, Cáceres LF, Ormachea Muñoz MR et al (2012) Sources and behavior of arsenic and trace elements in groundwater and surface water in the Poopó Lake Basin, Bolivian Altiplano. Environ Earth Sci 66(3):793–807. https://doi.org/10.1007/s12665-011-1288-1
Ravenscroft P, Brammer H, Richards KS (2009) Arsenic pollution: a global synthesis. Wiley-Blackwell Publication, Chichester
Raychowdhury N, Mukherjee A, Bhattacharya P et al (2014) Provenance and fate of arsenic and other solutes in the Chaco-Pampean Plain of the Andean foreland, Argentina: from perspectives of hydrogeochemical modeling and regional tectonic setup. J Hydrol 518:300–316. https://doi.org/10.1016/j.jhydrol.2013.07.003
Ritchie JA (1961) Arsenic and antimony in some New Zealand thermal waters. NZ J Sci 4:218–229
Romero L, Alonso H, Campano P et al (2003) Arsenic enrichment in waters and sediments of the Rio Loa (Second region, Chile). Appl Geochem 18(9):1399–1416. https://doi.org/10.1016/S0883-2927(03)00059-3
Ryan PC, Kim J, Wall AJ et al (2011) Ultramafic-derived arsenic in a fractured rock aquifer. Appl Geochem 26:444–457. https://doi.org/10.1016/j.apgeochem.2011.01.004
Saunders JA, Lee MK, Mohammad S (2005) Natural arsenic contamination of Holocene alluvial aquifers by linked tectonic, weathering, and microbial processes. Geochem Geophys Geosyst 6(4):Q04006. https://doi.org/10.1029/2004GC000803
Savage KS, Tingle TN, O’Day PA et al (2000) Arsenic speciation in pyrite and secondary weathering phases, Mother Lode Gold District, Tuolumne County, California. Appl Geochem 15:1219–1244. https://doi.org/10.1016/S0883-2927(99)00115-8
Sawkins FJ (1990) Metal deposits in relation to plate tectonics, 2nd edn. Minerals and Rocks Series 17, Springer-Verlag, New York
Scanlon BR, Nicot JP, Reedy R et al (2009) Elevated naturally occurring arsenic in a semiarid oxidizing system, Southern High Plains aquifer, Texas, USA. Appl Geochem 24:2061–2071. https://doi.org/10.1016/j.apgeochem.2009.08.004
Seddique AA, Masuda H, Mitamura M et al (2011) Mineralogy and geochemistry of shallow sediments of Sonargaon, Bangladesh and implications for arsenic dynamics: focusing on the role of organic matter. App Geochem 2:587–599. https://doi.org/10.1016/j.apgeochem.2011.01.016
Shimada Y, Stute M, van Geen A et al (2004) Redox control of arsenic mobilization in Bangladesh groundwater. Appl Geochem 19(2):201–214. https://doi.org/10.1016/j.apgeochem.2003.09.007
Shimada N (2009) The essence of problems on groundwater and soil pollutions caused by naturally occurring heavy metals and harmful elements: arsenic. Oyo Technical Report 29, pp 31–59. Japanese with English abstract
Sigufusson B, Gislason SR, Meharg AA (2011) A field and reactive transport model study of arsenic in a basaltic rock aquifer. Appl Geochem 26:553–564. https://doi.org/10.1016/j.apgeochem.2011.01.013
Simón Gómez JL (1996) Estudio estructural de la Comarca de La Moraña (provincia de Ávila). Universidad de Zaragoza, Zaragoza, Spain, Departamento de Geología
Sims KWW, Newsom HE, Gladney ES (1990) Chemical fractionation during formation of the Earth’s core and continental crust: clues from As, Sb, W, and Mo. In: Newson HE, Jones JH (eds) Origin of the earth. Oxford University Press, New York, pp 291–317
Sisson TW, Grove TL (1993) Experimental investigations of the role of H2O in calc-alkaline differentiation and subduction zone magmatism. Contrib Mineral Petrol 113:143–166. https://doi.org/10.1007/BF00283225
Smedley PL, Kinniburgh DG (2002) A review of the source, behavior and distribution of arsenic in natural waters. Appl Geochem 17(5):517–568. https://doi.org/10.1016/S0883-2927(02)00018-5
Sracek O, Bhattacharya P, Jacks G et al (2004) Behavior of arsenic and geochemical modeling of arsenic contamination. Appl Geochem 19(2):169–180. https://doi.org/10.1016/j.apgeochem.2003.09.005
Stallard RF, Edmond JM (1983) Geochemistry of the Amazon: 2. The influence of geology and weathering environment on the dissolved load. J Geophys Res Oceans 88:9671–9688. https://doi.org/10.1029/JC088iC14p09671
Stanger G (2005) A palaeo-hydrogeological model for arsenic contamination in southern and south-east Asia. Environ Geochem Health 27:359–367. https://doi.org/10.1007/s10653-005-7102-9
Stauffer RE, Thompson JM (1984) Arsenic and antimony in geothermal waters of Yellow Stone National Park, Wyoming, USA. Geochim Cosmochim Acta 48:2547–2561. https://doi.org/10.1016/0016-7037(84)90305-3
Stromgren T, Sørstrøm SE, Schou L et al (1995) Acute toxic effects of produced water in relation to chemical composition and dispersion. Mar Environ Res 40:147–169. https://doi.org/10.1016/0141-1136(94)00143-D
Stüben D, Berner Z, Chandrasekharam D et al (2003) Arsenic enrichment in groundwater of West Bengal, India: geochemical evidence for mobilization of As under reducing conditions. Appl Geochem 18:1417–1434. https://doi.org/10.1016/S0883-2927(03)00060-X
Tapia J, Davenport J, Townley B et al (2018) Sources, enrichment, and redistribution of As, Cd, Cu, Li, Mo, and Sb in the Northern Atacama Region, Chile: implications for arid watersheds affected by mining. J Geochem Explor 185:33–51. https://doi.org/10.1016/j.gexplo.2017.10.021
Tapia JS, Audry (2013) Control of early diagenesis processes on trace metal (Cu, Zn, Cd, Pb and U) and metalloid (As, Sb) behaviors in mining- and smelting-impacted lacustrine environments of the Bolivian Altiplano. Appl Geochem 31:60–78. https://doi.org/10.1016/j.apgeochem.2012.12.006
Tapia J, Audry S, Townley B et al (2012) Geochemical background, baseline and origin of contaminants from sediments in the mining-impacted Altiplano and Eastern Cordillera of Oruro, Bolivia. Geochem-Explor Eenv A 12(1):3–20. https://doi.org/10.1144/1467-7873/10-RA-049
Tapia J, Audry S, van Beek P (2019a) Natural and anthropogenic controls on particulate metal(loid) deposition in Bolivian highland sediments, Lake Uru Uru (Bolivia). Holocene 30(3):428–440. 10.1177%2F0959683619887425
Tapia J, Murray J, Ormachea M et al (2019b) Origin, distribution, and geochemistry of arsenic in the Altiplano-Puna plateau of Argentina, Bolivia, Chile, and Perú. Sci Total Environ 678, 309–325. https://doi.org/10.1016/j.scitotenv.2019.04.084
Tapia J, Rodríguez MP, Castillo P et al (2019c) Arsenic and copper in Chile and the development of environmental standards. In: Alaniz AJ (ed) Chile: environmental history, perspectives and challenges. Nova Publishers, Hauppauge, NY
Tapia J, Schneider B, Inostroza M et al (2021) Naturally elevated arsenic in the Altiplano-Puna, Chile and the link to recent (Mio-Pliocene to Quaternary) volcanic activity, high crustal thicknesses, and geological structures. J S Am Earth Sci 105:102905. https://doi.org/10.1016/j.jsames.2020.102905
Tatsumi Y (1989) Migration of fluid phases and genesis of basalt magmas in subduction zones. J Geophys Res Solid Earth 94(B4):4697–4707. https://doi.org/10.1029/JB094iB04p04697
Taylor SR, McLennan SM (1985) The continental crust: its composition and evolution. Blackwell, Oxford
Ubanell AG (1985) Características principales de la fracturación tardihercínica en un segmento del Sistema Central Español. Cuad Geog 7:591–605
Ure A, Berrow M (1982) Chapter 3. The elemental constituents of soils. In: Bowen HJM (ed) Environmental chemistry, vol 2. Royal Society of Chemistry, London, pp 94–204
van Geen A (2011) International drilling to recover aquifer sands (IDRAs) and arsenic contaminated groundwater in Asia. Sci Drill 12:49–52. https://doi.org/10.2204/iodp.sd.12.06.20112011
Vergasova LP, Krivovichev SV, Britvin SN et al (2004) Filatovite, K [(Al, Zn)2(As, Si)2O8], a new mineral species from the Tolbachik volcano, Kamchatka peninsula, Russia. Eur J Mineral 16:533–536. https://doi.org/10.1127/0935-1221/2004/0016-0533
Verma S, Mukherjee A, Mahanta C et al (2016) Influence of geology on groundwater–sediment interactions in arsenic enriched tectono-morphic aquifers of the Himalayan Brahmaputra river basin. J Hydrol 540:176–195. https://doi.org/10.1016/j.jhydrol.2016.05.041
Vilanova E, Mas-Pla J, Menció A (2008) Determinación de sistemas de flujo regionales y locales en las depresiones tectónicas del Baix Empordà y La Selva (NE de España) en base a datos hidroquímicos e isotópicos. Bol Geol Min 119(1):51–62
Webster JG (1990) The solubility of As2S3 and speciation of As in dilute and sulphide bearing fluids at 25 and 90°C. Geochim Cosmochim Acta 54:1009–1017. https://doi.org/10.1016/0016-7037(90)90434-M
Webster JG, Nordstrom K (2003) Chapter 4. Geothermal arsenic the source, transport and fate of arsenic in geothermal systems. In: Welch AH, Stollenwerk, KG (eds) Arsenic in groundwater. Kluwer Academic Publishers, Boston, MA
Webster, JG (1999) The source of arsenic (and other elements) in the Marbel – Matingo river catchment, Mindanao, Philippines. Geothermics 28(1):95–111. https://doi.org/10.1016/S0375-6505(98)00046-7
Webster-Brown JG, Lane V (2005) The environmental fate of geothermal arsenic in a major river system, New Zealand. In: Proceedings world geothermal congress 2005, Antalya, Turkey, 24–29 April 2005, Paper Number: 0263. https://www.geothermal-energy.org/pdf/IGAstandard/WGC/2005/0263.pdf. Accessed 31 January 2021
Webster-Brown JG, Lane V, Webster KS (2000) Arsenic in the Waikato River – an update. In: Proceedings of the 22nd New Zealand Geothermal Workshop, pp 63–68
Wedepohl KH (1995) The composition of the continental-crust. Geochim Cosmochim Acta 59:1217–1232. https://doi.org/10.1016/0016-7037(95)00038-2
Welch AH, Lico MS (1998) Factors controlling As and U in shallow ground water, southern Carson Desert, Nevada. Appl Geochem 13:521–539. https://doi.org/10.1016/S0883-2927(97)00083-8
Welch AH, Stollenwerk KG (2002) Arsenic in ground water: geochemistry and occurrence. Springer Science & Business Media, New York
Welch AH, Lico MS, Hughes JL (1988) Arsenic in groundwater of the western United States. Ground Water 26:333–347. https://doi.org/10.1111/j.1745-6584.1988.tb00397.x
Welch AH, Westjohn DB, Helsel DR et al (2000) Arsenic in groundwater of the United States: occurrence and geochemistry. Ground Water 38:589–604. https://doi.org/10.1111/j.1745-6584.2000.tb00251.x
WHO (1981) Arsenic: environmental health criteria 18. World Health Organization, Geneva, Switzerland
WHO (2001) Arsenic and arsenic compounds (Environmental Health Criteria 224), 2nd edn. World Health Organization, Geneva, Switzerland
WHO (2017) Guidelines for drinking-water quality, 4th edn, incorporating the 1st addendum. World Health Organization, Geneva, Switzerland
Yudovich Y, Ketris MP (2005) Arsenic in coal: a review. Int J Coal Geol 61:141–196. https://doi.org/10.1016/j.coal.2004.09.003
You C-F, Castillo PR, Gieskes JM et al (1996) Trace element behavior in hydrothermal experiments: implications for fluid processes at shallow depths in subduction zones. Earth Planet Sci Lett 140:41–52
Zheng Y (2006) The heterogeneity of arsenic in the crust: a linkage to occurrence in groundwater. Geological Society of America, Abstracts with Programs 38(7):179
Zheng Y, Stute M, van Geen A, et al (2004) Redox control of arsenic mobilization in Bangladesh groundwater. Appl Geochem 19(2):201–214. https://doi.org/10.1016/j.apgeochem.2003.09.007
Acknowledgements
The authors would like to thank the two anonymous reviewers for their careful reading of the original manuscript and its revised versions. Their insightful comments and suggestions helped us tremendously in improving the quality of our work. Efficient editorial handling is much appreciated. Mohammad Ayaz Alam would like to thank the Universidad de Atacama for providing the necessary facilities and support to carry out this work, also Adolfo Muñoz for his help with the two figures. Prosun Bhattacharya gratefully acknowledges the financial support on arsenic research from the Swedish International Development Cooperation Agency (Sida) Contributions 75000553, 700 (Bolivia), 75000854 (Bangladesh), and 51170072 (Tanzania).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Alam, M.A., Mukherjee, A., Bhattacharya, P. (2021). A Critical Evaluation of the Role of Geotectonics in Groundwater Arsenic Contamination. In: Shandilya, A.K., Singh, V.K., Bhatt, S.C., Dubey, C.S. (eds) Geological and Geo-Environmental Processes on Earth. Springer Natural Hazards. Springer, Singapore. https://doi.org/10.1007/978-981-16-4122-0_14
Download citation
DOI: https://doi.org/10.1007/978-981-16-4122-0_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-4121-3
Online ISBN: 978-981-16-4122-0
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)