
Abstract
Comparative gene identification-58 (CGI-58), also known as α/β-hydrolase domain-containing 5 (ABHD5), is a member of a large family of proteins containing an α/β-hydrolase-fold. CGI-58 is well-known as the co-activator of adipose triglyceride lipase (ATGL), which is a key enzyme initiating cytosolic lipid droplet lipolysis. Mutations in either the human CGI-58 or ATGL gene cause an autosomal recessive neutral lipid storage disease, characterized by the excessive accumulation of triglyceride (TAG)-rich lipid droplets in the cytoplasm of almost all cell types. CGI-58, however, has ATGL-independent functions. Distinct phenotypes associated with CGI-58 deficiency commonly include ichthyosis (scaly dry skin), nonalcoholic steatohepatitis, and hepatic fibrosis. Through regulated interactions with multiple protein families, CGI-58 controls many metabolic and signaling pathways, such as lipid and glucose metabolism, energy balance, insulin signaling, inflammatory responses, and thermogenesis. Recent studies have shown that CGI-58 regulates the pathogenesis of common metabolic diseases in a tissue-specific manner. Future studies are needed to molecularly define ATGL-independent functions of CGI-58, including the newly identified serine protease activity of CGI-58. Elucidation of these versatile functions of CGI-58 may uncover fundamental cellular processes governing lipid and energy homeostasis, which may help develop novel approaches that counter against obesity and its associated metabolic sequelae.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
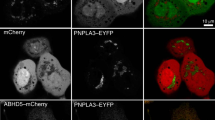

Keywords
13.1 Introduction
The human comparative gene identification-58 (CGI-58) gene was identified through comparative gene identification studies using the Caenorhabditis elegans proteome and human expressed sequence tag (EST) nucleotide database [109]. Human CGI-58 gene is located at the chromosome 3p21.33 locus, spanning about 32kb and producing several splice variants. The full-length human CGI-58 cDNA is transcribed from seven exons and encodes a 349 amino acid protein of ~39 kDa (Fig. 13.1a). CGI-58 is also known as α/β hydrolase domain-containing 5 (ABHD5). The ABHD subfamily belongs to a large protein family defined by an α/β hydrolase fold [146, 258]. The α/β hydrolase fold has a highly conserved catalytic triad containing a nucleophile (serine, cysteine, or aspartic acid), an acidic residue, and histidine that are close in 3D structure, though apart from each other in sequence [116, 258]. The ABHD subfamily has a total of 19 members in humans and 15 members in mice [128, 202], yet the functions of most remain unknown. CGI-58 differs from other members in this subfamily in that the critical serine in the catalytic triad is substituted by asparagine [116].

(a) The amino acid sequence of human CGI-58 protein. The amino acids in red circles highlight those mutated in patients with CDS. Some altered splice donor or acceptor sites are not highlighted. According to the two studies using the mouse CGI-58 protein [16, 74], the amino acids 16–30 in the human CGI-58 protein are likely required for LD anchoring. (b) CGI-58 mutations reported in humans before March 2020. Biallelic mutations in red color are associated with the full phenotypes of CDS, biallelic mutations in blue color are associated with the partial phenotypes (no ichthyosis) of CDS, and those in black color denote monoallelic mutations associated with nonalcoholic fatty liver disease
Mutations in the human CGI-58 gene were identified as the cause of Chanarin-Dorfman syndrome (CDS, OMIM 275630) (Fig. 13.1), an autosomal recessive neutral lipid storage disease (NLSD) with ichthyosis (thickened dry skin) [58, 116]. CDS is characterized by the accumulation of triglyceride (TAG)-rich cytoplasmic lipid droplets (LDs) in most cell types, including leukocytes (Jordans’ anomaly) [96], hepatocytes, myocytes, and cells in the epidermis, dermis, and intestinal mucosa [33, 46, 183, 205]. Patients with CDS often manifest hepatomegaly (hepatic steatosis and steatohepatitis), myopathy, microcephaly, cataracts, hearing loss, ataxia, mild mental retardation, and short stature [33, 46, 90, 183]. Since the initial description of the disease by Dorfman and Chanarin [33, 46], about 130 cases with more than 40 different mutations spanning the entire protein sequence have been reported worldwide [7, 49]. Types of mutations include deletion, insertion, missense, nonsense, and frameshift mutations (Fig. 13.1b) [1, 3, 5, 7, 9, 12, 22,23,24, 49, 52, 54, 89, 92, 116, 130, 150, 151, 169, 174, 181, 187, 192, 208, 219, 224, 243, 255]. While loss-of-function mutations cause CDS (Fig. 13.1), it is currently unknown whether gain-of-function exists for CGI-58 gene.
CGI-58 is ubiquitously expressed in mammals [18, 112, 211]. It is predicted to be cytosolic [116]. Interest in the scientific community regarding the functions of CGI-58 started in the early 2000s when three laboratories simultaneously reported that CGI-58 localizes at cytosolic LDs [121, 211, 244]. This was the time when biomedical scientists started to appreciate the cytosolic LD as an organelle that dynamically regulates energy storage and mobilization, rather than as an inert liposome-like structure that passively stores excess energy. The conceptual innovation placed cytosolic LDs at the center of cellular energy metabolism whose dysregulation is a hallmark of metabolic diseases, such as obesity, insulin resistance, type II diabetes, fatty liver, and cardiovascular disease. It was believed that excessive deposition of cytosolic lipid droplets would cause lipotoxicity, a biochemical mechanism that was widely used to explain impairments of cellular metabolism, cell signaling transduction, and redox imbalance associated with overnutrition-driven metabolic diseases [221]. Mutations in the human CGI-58 gene were known to cause LD deposition in almost all cell types examined, which provided the biomedical research community an excellent opportunity to test how LD accumulation promotes lipotoxicity. Over the past 15 years, we have learned a great deal about the pros and cons of cytosolic LDs by studying the biochemistry, cell biology, and tissue-specific pathophysiology of CGI-58. This chapter summarizes the current knowledge about the role of CGI-58 in LD lipolysis (i.e., hydrolysis of TAGs stored in cytosolic LDs) and discusses how CGI-58-dependent metabolic and signaling pathways regulate the pathogenesis of common metabolic diseases.
13.2 CGI-58 Interacts with Lipolysis-Regulatory Proteins
13.2.1 The PAT (Perilipin, Adipophilin, TIP47) Protein Family
Biochemical and cell biology studies have demonstrated that CGI-58 binds to cytosolic LDs and interacts with perilipin 1 (PLIN1), adipose differentiation-related protein (ADRP, also known as adipophilin or PLIN2), TIP47 (PLIN3), and muscle LD protein (MldP or PLIN5) [18, 63, 121, 161, 211, 244, 245]. These are members of the PAT (perilipin, adipophilin, TIP47) family that also includes S3-12 (or PLIN4) [105, 126, 133, 236]. The PAT family proteins share a highly conserved N-terminal structure. They localize at the surface of intracellular LDs of different lipid compositions and sizes, regulating energy storage and mobilization in response to nutritional fluctuations and various stimuli [126]. Using the two frame shift mutants (Leu-404fs and Val-398fs) that cause partial lipodystrophy in humans, Savage and associates have shown that the C-terminal region of human PLIN1 is essential for binding to CGI-58, and this interaction stabilizes CGI-58 localization on the LDs [63].
13.2.2 The PNPLA (Patatin-Like Phospholipase Domain Containing) Protein Family
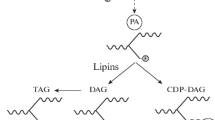
The process that mobilizes the energy (mainly as TAGs) stored in intracellular LDs for utilization is called intracellular lipolysis (Fig. 13.2) [253]. During LD lipolysis, the three fatty acyl chains in a TAG molecule are sequentially cleaved into diacylglycerol (DAG), monoacylglycerol (MAG), and glycerol, releasing a fatty acid molecule at each step. The first enzyme that was discovered to catalyze hydrolysis of cytosolic LD-embedded TAGs is hormone-sensitive lipase (HSL) [86, 111, 177, 223]. The substrate spectrum of HSL appears to be quite broad, including DAGs, TAGs, MAGs, cholesteryl esters, and retinyl esters [40, 113, 234]. Monoacylglycerol lipase (MAGL) was reported, shortly after HSL, as a lipase that specifically hydrolyzes MAGs [98, 223]. Both HSL and MGL belong to the α/β-hydrolase fold family. For years, HSL was thought to be responsible for hydrolyzing TAGs in adipocyte LDs. However, HSL-null mice showed the accumulation of DAGs rather than TAGs in multiple tissues [77, 157, 179, 227], indicating that other enzyme(s) are involved in the TAG hydrolysis. In 2004, three groups independently reported a new lipase possessing abundant TAG hydrolase activity, and the enzyme was named calcium-independent phospholipase A2ζ (iPLA2ζ), desnutrin, or adipose triglyceride lipase (ATGL), respectively [95, 226, 264]. This newly discovered lipase turned out to be the rate-limiting enzyme of cytosolic LD lipolysis (Fig. 13.2), and, thus, the name ATGL became more popular than the other names. ATGL is also known as patatin-like phospholipase domain containing 2 (PNPLA2). The PNPLA protein family consists of a total of nine members, including PNPLA1 through PNPLA9, all of which seem to be implicated in lipid metabolism through their phospholipase or lipase activities, or other functions [104, 144]. Comparative studies of ATGL and CGI-58 in the context of adipose lipolysis have resulted in a major breakthrough regarding the biochemical function of CGI-58. In 2016, Dr. Rudolf Zechner and associates reported that CGI-58 functions as a coactivator of ATGL to promote in vitro TAG hydrolysis [112]. Subsequent studies were consistent with this original finding [70, 71, 161, 228, 233, 250]. Furthermore, CGI-58 was shown to release from perilipin proteins following lipolytic stimulation, which allowed CGI-58 to interact with ATGL and activate TAG hydrolysis [70, 71, 211, 228]. In this scenario, the interaction of CGI-58 and with perilipins functions as a brake of lipolysis (Fig. 13.2), though its efficiency may be cell-type specific due to distinct perilipin compositions and the different abilities of individual perilipins in sequestering CGI-58 in various cell types [161].

Proposed model for CGI-58 regulation of cytosolic lipid droplet lipolysis. Lipolysis regulation differs between basal and stimulated conditions. Under the basal conditions, CGI-58 binds to PLIN1 in adipocytes or PLIN5 in oxidative nonadipocytes, preventing its interaction with ATGL. Thus, the lipolytic activity of ATGL is limited. After stimulation, perilipins are phosphorylated, resulting in the dissociation of CGI-58 from perilipins. CGI-58 then interacts with ATGL and substantially activates ATGL’s TAG hydrolase activity to stimulate lipolysis, producing DAGs and fatty acids (FAs). The DAGs are then hydrolyzed to produce MAGs and FAs by HSL that was phosphorylated and recruited to the LDs during the lipolytic stimulation. Finally, the MAGs are hydrolyzed by MAGL to release the last fatty acyl chain from the glycerol backbone of a TAG molecule
Although CGI-58 activates ATGL’s TAG hydrolase activity [112], mutations in CGI-58 and ATGL cause distinct phenotypes in humans and mice [58, 65, 75, 78, 116, 135, 172, 237]. For example, human CGI-58 mutations cause NLSD with ichthyosis [33, 46, 90, 116, 183], whereas human ATGL mutations cause NSLD without skin defects but with mild myopathy [58]. Global CGI-58 knockout mice die ~16h after birth due to a skin barrier defect [172], yet global ATGL knockout mice are viable [78]. These phenotypic differences associated with mutations of the two genes indicate that CGI-58 must have ATGL-independent functions. Recently, CGI-58 was shown to interact with PNPLA1, another member of the PNPLA protein family, to stimulate PNPLA1-mediated ω-O-acylceramide production in skin [102, 154], providing a potential mechanism for skin barrier defect seen in patients with CDS. CGI-58 was also shown to functionally interact with the wild-type PNPLA3, the fatty liver-promoting PNPLA3(I148M) variant [180], and a lipase dead PNPLA3 mutant [32], suggesting that CGI-58 may coordinate with PNPLA3 and other lipases to regulate LD turnover independently of PNPLA3’s lipase activity. Consistent with this scenario, two laboratories reported that PNPLA3, the fatty liver-causing PNPLA3(I148M) variant, in particular, competes with ATGL (PNPLA2) to bind with CGI-58, reducing TAG hydrolysis in the liver and brown adipocytes [233, 250]. These observations provided a mechanism for how the PNPLA3(I148M) variant promotes fat deposition. However, such observations cannot explain why PNPLA3, including PNPLA3(I148M) but not a lipase dead mutant, retains its ability to reduce LD sizes when co-expressed with CGI-58 in the absence of ATGL [32]. It remains possible that PNPLA3 displays an in vivo lipase or transacylase activity toward specific substrates under some pathophysiological or nutritional conditions. In addition to PNPLA1-3, other members of PNPLA protein family may also interact with CGI-58 to fulfill unique functions under specific pathophysiological and nutritional conditions.
13.2.3 The Fatty Acid-Binding Protein (FABP) Family
Another group of proteins that interact with CGI-58 is the fatty acid binding protein (FABP) family members [85]. It was hypothesized that FABP interacts with CGI-58 to promote ATGL-mediated intracellular lipolysis by serving as an acceptor of free fatty acids released from TAG hydrolysis [85]. This was an important finding because it provided a mechanism for the handling of lipolytic products. Intriguingly, the lipolytic product long-chain acyl CoA was shown to bind CGI-58 and promote CGI-58 interactions with perilipins to suppress lipolysis [188]. This phenomenon seems to be an end-product feedback mechanism that fine-tunes hydrolysis of TAGs stored in intracellular LDs. The interactions of CGI-58 with LD coat proteins, lipases, and lipids suggest that CGI-58 likely play a key role in organizing major components of LD lipolysis into a functional “lipolysome” [85].
13.3 CGI-58 as a Serine Protease
The latest member of the CGI-58 interactome is histone deacetylase 4 (HDAC4). Backs and associates reported that CGI-58 functions in vitro and in vivo as a serine protease that cleaves HDAC4 in the heart in response to catecholamine stimulation, generating an N-terminal polypeptide of HDAC4 (HDAC4-NT) to protect cardiac functions [94]. This study was conceptually paradigm shifting, because it was the first to demonstrate that CGI-58 can function as a serine protease. CGI-58 was previously shown to function as a coactivator of a lipase that promotes lipolysis, and it was never thought to be a protease that promotes proteolysis. Perhaps, CGI-58 is a protein of dual function that promotes both lipolysis and proteolysis. This novel function of CGI-58 raises many new and exciting questions regarding the core function of the protein. For example, does CGI-58 cleave other proteins interacting with it? If yes, is this proteolytic function required for CGI-58 to activate LD lipolysis? Does a lipase require proteolytic cleavage prior to digesting a lipid molecule? Answers to these questions are expected to provide fundamental insights into the molecular and biochemical bases of lipolysis and its potential crosstalk with proteolysis.
13.4 Molecular Basis for CGI-58 Activation of ATGL-Dependent Lipolysis
The cellular, structural, and biochemical bases for CGI-58 and ATGL interaction to promote TAG hydrolysis remain incompletely understood. The N-terminal amino acids 1–30 of mouse CGI-58 were shown to form a lipophilic tryptophan-rich stretch, which is essential for CGI-58 to localize at the LD and activate ATGL in cultured cells [74]. This tryptophan-rich stretch appears to anchor CGI-58 to the LD surface through its three tryptophan residues serving as the left and right anchor arms [16]. A comparative study of mouse ABHD5 (CGI-58) and ABHD4, an ABHD family member that is closely related to ABHD5 but does not activate ATGL, identified R299 and G328 as essential residues for activating ATGL’s TAG hydrolase activity. However, these two amino acids of ABHD5 did not affect ATGL translocation to LDs or ABHD5 binding to PLIN1 [189]. These studies collectively suggest that the LD localization is a prerequisite for a functional CGI-58 to activate ATGL in vivo.
Studies with ATGL mutants associated with NLSD have showed that the mutations result in the expression of either enzymatically inactive proteins localizing to LDs or active TAG hydrolase lacking LD localization [196]. Whereas CGI-58 was identified as a coactivator of ATGL [112], G0/G1 switch gene 2 (G0S2) was subsequently discovered as an inhibitor of ATGL function [246, 247]. It was further demonstrated that G0S2 and CGI-58 do not appear to compete with each other for binding to ATGL in cultured cells transfected with tag-proteins [131]. The 254 N-terminal amino acids of mouse ATGL were reported to be the minimal domain that can be activated by CGI-58 and inhibited by G0S2 [41]. Interestingly, deleting ~220 amino acids from the C-terminus of human ATGL protein increases its interaction and activation by CGI-58 in vitro in the test tube, despite defective LD localization in vivo in cultured cells [196]. This finding indicates that the C-terminal region of ATGL is required for its targeting to LDs and plays a regulatory role in ATGL activation by CGI-58. Considering the newly identified protease function of CGI-58 [94], it would be interesting to test whether CGI-58 activates ATGL by a two-step process. In the first step, CGI-58 may cleave ATGL to release the suppressive role of ATGL’s C-terminal region on its enzymatic activity, which would be consistent with the observation that ATGL protein levels are often increased in the absence of CGI-58 [75, 242, 263]. The second step may involve conformational changes of the two proteins, resulting in tight interactions and correct positioning of “lipolysome” components on the surface of LD for hydrolysis of TAG in vivo.
13.5 CGI-58 Regulation of Autophagy and Lipophagy
The role of CGI-58 as the coactivator of ATGL to promote intracellular lipolysis has been established and reproduced in a series of in vitro and in vivo studies. ATGL is a cytosolic neutral lipase that initiates cytosolic/neutral lipolysis by cleaving a fatty acyl chain from a TAG molecule stored in cytosolic LDs, thus playing a critical role in intracellular lipolysis [95, 226, 257, 264]. Recently, the lipid-specific macroautophagy (lipophagy) was shown to also digest cytosolic LDs by delivering LD-associated fat to lysosomes for degradation by lysosomal acidic lipase (lysosomal/acidic lipolysis) [203]. Autophagy refers to the “self-eating” of a cell in response to starvation or nutrient deprivation for generating energy essential for its survival [155]. It is also a catabolic pathway for recycling of excessive or damaged organelles, such as mitochondria (mitophagy) [217]. In humans, insulin resistance suppresses CGI-58 mRNA expression in liver [99]. The nutritional and hormonal regulations of “neutral” lipolysis and lipophagy (“acidic” lipolysis) are strikingly similar. Both are induced by nutrient deprivation [45], and both are activated by glucagon or inhibited by insulin [45, 57]. It is currently unknown if CGI-58 promotes fat lipolysis by mediating lipophagy in addition to activating ATGL. It was demonstrated that ATGL, a lipase target of CGI-58, promotes autophagy and lipophagy in a sirtuin 1 (SIRT1)-dependent manner and that lipophagy is required for ATGL to promote LD catabolism and associated fatty acid oxidation in hepatocytes [190]. The crosstalk between ATGL-dependent lipolysis and autophagy was also seen in macrophages, though this crosstalk may be indirect or compensatory [66]. Some studies appear to suggest a role of CGI-58 in regulating autophagy and lipophagy. For example, in C2C12 muscle cells, CGI-58 overexpression increases, whereas CGI-58 knockdown decreases, autophagy and mitophagy through regulation of AMPK and mTORC1 signaling pathways [259]. CGI-58 was shown to bind Beclin1, a major regulator of autophagy [159, 163]. Many autophagy components can localize to LDs under some conditions [48, 51, 100, 160, 200, 203], though it is not known whether they interact with CGI-58 or other LD proteins to specifically regulate lipophagy. PLIN2, a major LD coat protein interacting with CGI-58 [244, 245], also binds the heat shock cognate protein of 70 kDa (Hsc70) for degradation via chaperone-mediated autophagy (CMA) [100]. The inhibition of CMA reduces both neutral and acidic lipolysis [100]. Hepatic CMA deficiency, like CGI-58 deletion, induces severe hepatic steatosis with liver damage and inflammation [220]. More studies are needed before the direct role of CGI-58 in the mediation of autophagy and lipophagy can be established.
13.6 Tissue-Specific Roles of CGI-58 in Energy and Lipid Metabolism
13.6.1 Adipose CGI-58 in Thermoregulation and Metabolic Health
CGI-58 is ubiquitously expressed in mammals, with the highest expression in adipose tissue. Adipose tissue is classically divided into white adipose tissue (WAT) and brown adipose tissue (BAT) that have distinct locations and opposite functions in energy balance. In general, WAT stores excess energy as TAGs in the large unilocular LD of white adipocytes, whereas BAT dissipates metabolic energy as heat for adaptive nonshivering thermogenesis in multilocular LD-containing brown adipocytes.
During prolonged fasting or increased energy demand, such as exercise and inflammation, the stored energy in WAT is mobilized via adipose LD lipolysis for utilization by cell types and pathways critical in sustaining life, meeting energetic demand, clearing infectious agents, or resolving inflammation. This process is generally defined as the stimulated adipose lipolysis, because it involves activation of a cell membrane receptor and its downstream signal transduction by neural and humoral factors in response to various stimuli [47, 108, 213, 253]. The classical signal-stimulating adipose lipolysis is the activation of β-adrenergic receptors by catecholamines released from the sympathetic nerves innervating adipose tissue. Binding of a catecholamine to the β receptor activates adenylate cyclase, which is an enzyme that uses ATP as the substrate to produce cAMP [55]. Elevation in cellular cAMP activates protein kinase A (PKA), which then phosphorylates several lipolytic components, such as PLIN1 and HSL, to stimulate lipolysis (Fig. 13.2) [213]. Thus, any stimulus that activates PKA or increases cellular cAMP levels is thus expected to stimulate adipose lipolysis. Phosphorylation of a perilipin, perhaps together with phosphorylation of CGI-58 on S239 [185], causes CGI-58 disassociation from the perilipin for CGI-58 to interact with ATGL (Fig. 13.2) [70, 71, 211, 213, 244, 251]. It was shown that the in vitro TAG hydrolase activity of ATGL can be increased up to 20-fold with CGI-58 interaction [112]. The in vivo significance of CGI-58 as an essential mediator of the stimulated lipolysis was demonstrated in a study showing that adipose-specific inactivation of CGI-58 abolishes the isoproterenol-stimulated increase in plasma levels of free fatty acids in mice [201].
The nonshivering thermogenesis in BAT is mainly mediated by uncoupling protein 1 (UCP-1), which resides in the inner membrane of a mitochondrion, uncoupling chemical energy from ATP synthesis and dissipating the energy as heat [27]. Under some environmental and pathophysiological conditions, such as cold exposure and β-adrenergic receptor activation, a cell type with features of both white and brown adipocytes appears in the classically white fat depots. This type of adipocytes is named brite or beige adipocytes that often express UCP-1 and produce heat [165, 238]. The process that drives the appearance of brite/beige adipocytes in WAT is called WAT browning or beigeing [97]. The origin of beige adipocytes may include mature white adipocyte transdifferentiation and/or de novo adipogenesis, depending on the condition that induces WAT browning [39, 83, 114, 115, 182, 229, 230].
Cytosolic LD lipolysis was thought to be central in nonshivering thermogenesis [27]. Several animal and human studies suggested the essential role of brown fat lipolysis in thermogenesis, though the genetic or pharmacological manipulation of adipose lipolysis employed in the studies inhibited intracellular lipolysis in both BAT and WAT [4, 15, 44, 78, 107]. We created mice deficient in CGI-58 in UCP1-positive brown and beige adipocytes (BAT-KO mice) and mice lacking CGI-58 in all adipocytes (FAT-KO mice), which allowed us to directly test the role of brown adipocyte LD lipolysis in thermoregulation. To our surprise, BAT-KO mice were not cold sensitive even when food was unavailable [201]. The mice became cold sensitive only when the following two conditions were met simultaneously: (1) deletion of CGI-58 in both WAT and BAT and (2) removal of food. Similar phenotypes were observed in mice lacking ATGL in BAT or the total adipose tissue [195]. When CGI-58 or ATGL was deleted in the total adipose tissue in mice, the in vivo lipolysis (fatty acid release from the tissue to the blood circulation) stimulated by isoproterenol, a β-adrenergic receptor agonist, was completely abolished in mice [195, 201]. The results demonstrated the indispensable role of CGI-58 or ATGL in mediating the stimulated lipolysis in the whole animal. These two animal studies also demonstrated a key role of WAT in regulating adaptive nonshivering thermogenesis, likely by providing the heat-producing cells with the metabolic fuels and/or by exposing the temperature sensors in the body to the thermogenically important adipokines or signaling molecules. It is currently unclear how food rescues the cold sensitivity of mice lacking CGI-58 or ATGL in the total adipose tissue [195, 201]. A simple explanation is that food serves as another source of metabolic fuels that may energize the heat-generating cells with glucose, fatty acids, and/or amino acids. However, we observed that only gastric gavage, but not intraperitoneal injections, of glucose can efficiently slow down hypothermia in mice lacking CGI-58 in both WAT and BAT (Wang H et al. unpublished data). This finding strongly supports a critical role of the gastrointestinal track in regulating the diet-induced thermogenesis. The gastrointestinal track is abundantly innervated and has special endocrine cells that secrete various incretins, which are important in local environment sensing and whole-body energy metabolism. Interestingly, secretin, a gut hormone that is derived from the S cells in the duodenum and jejunum of small intestine, was shown to mediate postprandial thermogenesis by activating its receptor in brown adipocytes to stimulate lipolysis and energy expenditure and to subsequently suppress satiation through the brain [119]. However, mice lacking CGI-58 or ATGL in BAT are defective in brown adipocyte lipolysis, yet they are capable of producing heat after a meal, suggesting that either other gastrointestinal factors or non-lipolytic pathways also mediate the postprandial thermogenesis. Nonetheless, it would be interesting to test whether secretin mediates postprandial heat production in mice lacking CGI-58 or ATGL in BAT and, if not, what other gastrointestinal factors are involved.
CGI-58 deletion in UCP1-positive cells in mice increases sympathetic innervation in both BAT and WAT. The animals also exhibit enhanced WAT browning, especially after cold exposure or β3-adrenergic receptor activation [201]. This observation implies that some signals and/or BATokines (factors secreted by BAT) are generated as a result of BAT CGI-58 deficiency. These signals and batokines can somehow be sensed by the central nervous system in the brain, thereby increasing the sympathetic outflow to activate compensatory thermogenic mechanisms. It is currently unknown what these signals and batokines are and whether they work locally or remotely or must be secreted into the blood circulation, which represents an important area for future research in BAT biology. It is important to note that BAT lipolysis deficiency induced by ATGL deletion in UCP1-positive cells does not increase WAT browning as evidenced by unaltered expression levels of UCP-1 protein in the inguinal subcutaneous fat [195], suggesting that deficiency of CGI-58’s ATGL-independent functions in BAT promotes browning in WAT.
Genetic deletion of BAT CGI-58 in mice improves several fat-induced metabolic disorders, such as glucose intolerance, insulin resistance, and hepatic steatosis [201]. This improvement is more profound when CGI-58 is deleted in both BAT and WAT (our unpublished data). ATGL deletion in whole body or adipose tissue also protects mice from fat-induced metabolic abnormalities [4, 87, 194, 239]. These observations indicate that inhibiting adipose lipolysis may improve whole-body glucose handling as a result of failed mobilization of fatty acids for utilization, which would be consistent with the glucose fatty acid cycle (or the Randle cycle) theory [173].
13.6.2 Epidermis CGI-58 and Skin Barrier Function
A major phenotypic distinction of human patients with CGI-58 mutations from those with ATGL mutations is ichthyosis (scaly dry skin) [58, 116]. In mice, whole body ablation of CGI-58, but not ATGL, causes skin barrier defects [78, 172]. Using whole body and cell type-specific transgenic and knockout mouse models, it was shown that CGI-58 promotes the biosynthesis of the skin barrier lipids, ω-O-acylceramides, locally in the keratinocytes of suprabasal epidermal layers, and such function is ATGL independent [73]. It was further shown that CGI-58 interacts directly with PNPLA1 and recruits PNPLA1 to LDs where it functions as the coactivator of PNPLA1 for the biosynthesis of ω-O-acylceramides [102, 154]. Like CGI-58, PNPLA1 mutations in humans also cause ichthyosis [69]. Using biochemical approaches, cell cultures, and tissue-specific PNPLA1 knockout mice, several groups have demonstrated that PNPLA1 has transacylase or acyltransferase activity, which utilizes TAGs as an acyl donor and catalyzes the esterification of ω-hydroxy ceramides with linoleic acid to synthesize ω-O-acylceramides [73, 84, 102, 153]. Collectively, these studies strongly suggest that the defective activation of PNPLA1 is the molecular mechanism underlying CGI-58 mutation-induced ichthyosis in humans.
13.6.3 Muscle CGI-58 in Cardiomyopathy and Insulin Sensitivity
Patients with CDS accumulate neutral lipids in their skeletal muscle [138]. Heart murmurs, muscle weakness, and mild myopathy were reported in some CDS patients [90, 138, 235]. Two laboratories have generated muscle-specific CGI-58 knockout mice using MCK-cre transgenic mice [242, 263]. MCK-cre transgenic mice express cre recombinase in both skeletal and cardiac muscles, thereby deleting a loxP-floxed gene in both tissues [21]. Muscle CGI-58 knockout mice display intramyocellular deposition of neutral lipids in both cardiac and oxidative skeletal muscles [242, 263], implying that muscle fat deposition in human patients with CDS likely results from local CGI-58 deficiency in muscle. Neutral lipid deposition was not detected in the glycolytic skeletal muscle fibers in these animals [242]. The restriction of LD accumulation to the cardiac and oxidative (soleus) muscles highlights an essential role of CGI-58 in fatty acid oxidation in oxidative muscle types, which is consistent with other studies [10, 72].
CGI-58 deficiency in all muscles induces cardiac fibrosis, cardiac remodeling, and heart failure. The similar phenotypes were observed in muscle ATGL knockout mice [79]. In cardiac and oxidative skeletal muscles, CGI-58 interacts with PLIN3 and PLIN5, and this interaction regulates its association with ATGL [132, 167, 228]. These observations collectively suggest that CGI-58 may function through ATGL, promoting intracellular TAG hydrolysis in the muscle fibers. It was shown that cardiac ATGL-dependent TAG hydrolysis sustains mitochondrial functions by activating the PPAR-α pathway through the generation of endogenous ligands for PPAR-α [79]. CGI-58 may facilitate this pathway by activating ATGL in the cardiac muscle. Interestingly, CGI-58 was recently shown to function as a serine protease to protect heart failure by generating an N-terminal polypeptide from histone deacetylase 4 (HDAC4) through proteolysis [94]. The cardiac protective role of the HDAC4’s N-terminal polypeptide generated by CGI-58 was not associated with reduction in cardiac TAG content. Although it is currently unclear whether similar mechanisms operate in other cell types, this study nonetheless uncovered a completely novel function of CGI-58 and emphasized a future direction for CGI-58 research.
Intramyocellular fat deposition in skeletal muscle is often associated with systemic insulin resistance due to accumulation of insulin signaling-suppressing lipids, such as diacylglycerols and ceramides that cause lipotoxicity [186, 222]. Despite intramyocellular accumulation of neutral lipids, mice lacking CGI-58 or ATGL in muscle are not glucose intolerant or insulin resistant [103, 204, 242]. This dissociation of cellular lipid deposition from insulin resistance suggests that how versus how much lipids are accumulated may be more important in driving tissue insulin resistance, which may be due to the differences in the molecular species of lipids deposited. Alternatively, cytosolic LD deposition, if not extremely excessive, may sequester insulin signaling-suppressing metabolites, protecting cells against lipotoxicity. Such scenario would be consistent with an observation that unsaturated fatty acids promote TAG accumulation, yet protect cells against lipotoxicity [120]. In addition, lipid deposition in different skeletal muscle fiber types may lead to different metabolic consequences [118, 123]. Mice overexpressing diacylglycerol acyltransferase 2 (DGAT2) in glycolytic (type II) muscle accumulate TAG in muscle and are insulin resistant [118]. However, mice overexpressing diacylglycerol acyltransferase 1 (DGAT1), another TG synthesis enzyme, in muscle accumulate TAG in the soleus, and these animals are not insulin resistant [122]. Endurance-trained athletes display increased fat content in their skeletal muscle, and they have enhanced insulin sensitivity (“athlete paradox”) [67]. It seems that fat deposition in the glycolytic muscle is more problematic than in the oxidative muscle.
13.6.4 Liver CGI-58 in Nonalcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease in the United States and worldwide [254]. Patients with CDS (CGI-58 mutations) almost always display characteristics of advanced NAFLD, including severe hepatic steatosis, NASH, fibrosis, and cirrhosis [3, 24, 38, 76, 90, 139, 181, 205, 208, 215]. The CDS-causative mutations span the entire human CGI-58 protein sequence (Fig. 13.1). Interestingly, monoallelic mutations in the human CGI-58 gene are also associated with NAFLD (Fig. 13.1b). The prevalence of CGI-58 monoallelic mutations that are associated NAFLD was estimated to be 1 in 1,131 individuals in a normal population [255]. This study highlights an important role of CGI-58 in the pathogenesis of NAFLD in the general population. More importantly it was recently demonstrated that CGI-58 interacts with PNPLA3 [233, 250], a variant (I148M) of which is a major risk factor for fatty liver disease in all populations examined [149, 180, 207, 216]. CGI-58’s association with PNPLA3 interferes with its ATGL interaction, thus inhibiting LD lipolysis [11, 233, 250]. CGI-58 is required for wildtype PNPLA3 and the PNPLA3(148M) variant to localize to hepatic LDs and for the overexpressed PNPLA3(148M) to promote hepatic steatosis [233]. It was shown that PNPLA3 accumulation on LDs, not its catalytic activity, is responsible for PNPLA3(148M)-induced hepatic steatosis [11]. While these studies provided an important mechanism for how CGI-58 coordinates with PNPLA3 and PNPLA2 (ATGL) to control cytosolic LD turnover, more research on the PNPLA3/CGI-58 interaction is needed to address why PNPLA3, including the PNPLA3(I148M) variant but not a lipase dead mutant, can substantially reduce LD size when co-expressed with CGI-58 in the absence of ATGL [32].
Antisense oligonucleotide (ASO)-mediated knockdown of CGI-58 in adult mice induced severe hepatic steatosis, though this study cannot establish a causal relationship between hepatic CGI-58 and fatty liver disease due to knockdown of CGI-58 in multiple tissues, including liver, adipose tissue, and macrophages [19, 28, 127, 129]. Selective inactivation of CGI-58 or ATGL in the liver of mice causes hepatic steatosis [75, 237], implying that fatty liver disease seen in patients with NLSD induced by CGI-58 or ATGL mutations is likely a local effect of hepatic CGI-58 or ATGL deficiency. These studies unequivocally demonstrated an important role of LD lipolysis in controlling lipid homeostasis in the liver. Besides TAGs, other species of lipids, such as DAGs, are also accumulated in mouse livers lacking CGI-58, especially when a high fat diet is used [19, 28, 75]. Although hepatic steatosis is often associated with insulin resistance and DAG accumulation is well-known to suppress insulin signaling [186], liver CGI-58 deficiency-induced hepatic steatosis and DAG accumulation are not associated with insulin resistance in mice [19, 28, 75]. One study demonstrated that this dissociation results from the sequestration of DAGs to LDs and ER, rather than the cell membrane, which prevented PKCε translocation to the plasma membrane to inhibit insulin-receptor kinase activity [28]. The dissociation of hepatic steatosis and insulin resistance is not restricted to the CGI-58 deficiency-induced fatty liver. For instance, hepatic overexpression of DGAT2 or liver-specific deletion of histone deacetylase 3 (HDAC3) in mice induces severe hepatic accumulation of lipids including TAGs, DAGs, and ceramides without causing insulin resistance [141, 212]. In humans, a genetic variant (I148M) of PNPLA3 confers susceptibility to NAFLD in multiple populations without affecting the index of insulin resistance [149, 180, 207, 216]. African-American descendants have significantly less hepatic steatosis despite a relatively high prevalence of obesity and diabetes, while Hispanic-American descendants are the opposite [175, 193]. The variation in correlation between hepatic steatosis and insulin resistance among ethnicities suggests that other factors should also be considered. It should be emphasized that clinical studies of NAFLD only found the association between insulin resistance and hepatic steatosis whereas the relationship between insulin resistance and other liver pathologies, such as NASH and hepatic fibrosis, has yet to be established.
It is currently unknown how liver CGI-58 deficiency induces NASH and hepatic fibrosis in addition to hepatic steatosis. The albumin-cre transgenic mice (Stock #: 003574; The Jackson Laboratory) used for liver-specific inactivation of CGI-58 and ATGL can delete a gene floxed by loxP sites in hepatocytes, biliary epithelial cells (cholangiocytes), and hepatic stellate cells [50, 64, 148, 168, 171, 184, 206, 214]. Each of these cell types has distinct physiological and pathological functions. For instance, injuries of hepatocytes and other liver cells stimulate inflammatory responses, causing NASH [20, 60]. Liver damage and inflammation often trigger ductular reaction (increases in the number of small biliary ductules lined by cholangiocytes) that may contribute to hepatic fibrogenesis to some extent [59, 176, 191]. Hepatic stellate cells increase collagen production after activation by various liver injuries, and this cell type is well accepted to be the major source of hepatic fibrosis [81, 82, 142]. Given that liver ATGL deficiency induced by the same albumin-cre transgenic mouse line does not cause these advanced pathological changes in liver [237], the mechanism underlying liver CGI-58 deficiency-induced NASH and hepatic fibrosis cannot be the inhibition of ATGL-mediated LD lipolysis in hepatocytes, cholangiocytes, or hepatic stellate cells. Consistently, patients with ATGL mutations do not develop NASH and hepatic fibrosis [6, 25, 58, 166, 197]. CGI-58, therefore, must have ATGL-independent functions in the liver. One of such functions may be its interaction with PNPLA3 [233, 250]. Like CGI-58 mutations, the PNPLA3(I148M) variant is also associated with NASH [180]. Another distinct function of CGI-58 is its interactions with almost all perilipins. This interaction may be needed for cellular processes, such as autophagy and lipophagy, besides activation of ATGL. Perilipins are coat proteins of cytosolic LDs. They are required for the biogenesis and turnover of cytosolic LDs. It has been shown that perilipins play an important role in the pathogenesis of hepatic steatosis, NASH, and hepatic fibrosis [29, 30, 34, 35, 61, 88, 91, 145, 162, 231]. Patients with NAFLD accumulate perilipins in the liver [61, 162, 209]. While perilipins may passively accumulate in the steatotic liver due to increased LDs, they may also actively increase to protect cells against lipotoxicity of free lipids. Other CGI-58 functions, such as its newly identified serine protease activity in the heart [94], may also exist in the liver and other tissues. This protease activity of CGI-58, like its lipase coactivator function, may target multiple proteins to regulate a variety of cellular processes important in lipid and energy metabolism.
Liver CGI-58 knockout mice on a regular low-energy chow diet develop a full spectrum of pathologies observed in human patients with advanced NAFLD [75]. The progression of these pathologies can be substantially facilitated by challenging the animals with a typical Western-type diet alone or in combination with fructose in drinking water (our unpublished data). Future studies are needed to discern whether CGI-58 needs to be deleted simultaneously in hepatocytes, cholangiocytes, and stellate cells or in a specific cell type to trigger NASH and fibrosis in liver. Studies are also needed to identify CGI-58’s ATGL-independent mechanisms responsible for fatty liver progression, including testing the known ATGL-independent functions of CGI-58. Detailed comparative studies of liver CGI-58 and ATGL knockout mice may reveal mechanisms important in the etiology of NASH and hepatic fibrosis in general and shed light on novel drug targets against NAFLD progression.
13.6.5 Myeloid CGI-58 in Insulin Resistance, Inflammation, and Atherosclerosis
CGI-58 protein is expressed in mouse and human macrophages [13, 134]. It has been shown that myeloid cell-specific deletion of CGI-58 in mice worsens fat-induced tissue/systemic inflammation, proinflammatory activation of adipose tissue macrophages, glucose intolerance, and insulin resistance [134]. CGI-58-deficient macrophages accumulate cytosolic LDs and show reduced PPAR-γ signaling [134, 248]. Although the underlying mechanism remains unknown, sequestration of free fatty acids in cytosolic LDs may prevent these endogenous PPAR ligands from activating PPAR signaling as seen in ATGL-null cardiomyocytes [79]. As a result of PPARγ signaling suppression, CGI-58-null macrophages show mitochondrial dysfunction and accumulate reactive oxygen species, which activates NLRP3 inflammasome to promote secretion of proinflammatory cytokines [134]. Consistently, overexpression of CGI-58 in macrophages reduces inflammation in vitro and in vivo [241, 248]. The proinflammatory (M1-like) phenotype of CGI-58-null macrophages was also observed in other studies [65, 135]. In contrast, ATGL-deficient macrophages were shown to display the anti-inflammatory M2-like phenotype [2, 65, 110]. These collective observations indicate that CGI-58 also has ATGL-independent functions in myeloid cells, including macrophages.
The anti-inflammatory role of macrophage CGI-58 is expected to protect against atherosclerosis. One study with CGI-58 overexpression in macrophages did show such an atheroprotective role through the promotion of the PPAR/LXR-dependent cholesterol efflux without altering blood cholesterol levels [241]. However, the deletion of CGI-58 in myeloid cells of apoE knockout mice, or simultaneous knockdown of CGI-58 in multiple cell types including hepatocytes, adipocytes, and macrophages in LDLR-KO mice, does not worsen atherosclerosis or alter plasma cholesterol levels [65]. It is difficult to assess atherosclerosis risk in patients with CGI-58 mutations due to the rarity of disease, existence of other abnormalities, and relatively young subjects reported. The role of macrophage CGI-58 in atherogenesis has yet to be clarified. Macrophage CGI-58 deficiency causes foam cell formation [134]. Lipopolysaccharide (LPS) and saturated fatty acids downregulate CGI-58 expression in macrophages [134]. LPS and fatty acids are atherosclerosis risk factors, and many studies have shown that they promote foam cell formation and atherosclerosis [8, 14, 53, 56, 62, 101, 117, 137, 143, 156, 170]. Oxidized (ox)-LDL, a common atherosclerosis risk factor, inhibits CGI-58 expression in THP1 human macrophages (our unpublished data). These findings suggest a potential role of CGI-58 in modulating atherosclerosis risk factor-induced atherogenesis.
13.6.6 Intestine CGI-58 in Fat Absorption and Turnover
A major function of the small intestine is the absorption of nutrients including fats. Fat absorption occurs mainly in duodenum and jejunum. After digestion by pancreatic lipases, in the intestinal lumen, fat (mainly TAGs) becomes free fatty acids and monoacylglycerols (MAGs), which then enter the absorptive enterocytes and travel to the endoplasmic reticulum (ER) for re-esterification into TAGs for packaging into chylomicrons. Intestinal fat absorption is a very efficient process. Chylomicrons are quickly secreted into the lymphatic system heading to the blood circulation. Some of absorbed fat may be temporarily stored in the cytosolic LDs, especially after ingestion of a high fat diet [31, 164, 178, 262]. The TAGs stored in the cytosolic LDs have to be hydrolyzed before they can be assembled into primordial chylomicron particles in the ER lumen. CGI-58 and ATGL are expressed in the enterocytes. Genetic deletion of CGI-58 in these cells in mice induced the accumulation of cytosolic LDs predominantly in the nutrient absorptive segment of small intestine, regardless of dietary compositions and nutritional conditions [106, 240]. These observations demonstrated an important role of intestinal CGI-58 in mobilizing intestinal LDs for local and/or systemic utilization. Consistently, hepatic steatosis is attenuated in the intestine CGI-58 single or CGI-58/ATGL double knockout mice [106, 240]. Using intestine-specific CGI-58 knockout mice fed a synthetic diet containing 40% energy from lard and 0.2% (w/w) cholesterol, our laboratory has shown that intestinal absorption of total fat and long-chain fatty acids is significantly reduced, which is associated with reduced postprandial TAG section into the blood circulation and increased plasma concentrations of free and esterified cholesterol [240]. For reasons currently unknown, another group did not find similar changes in their intestine CGI-58 and ATGL single or double knockout mice fed a diet containing 60% energy from fat [34% (w/w) crude fat] and 1% (w/w) cholesterol. They instead showed a role of intestinal CGI-58 and ATGL in the turnover of lipids derived from the basolateral side of the absorptive enterocytes [106, 152]. More studies are clearly needed to address these controversial findings.
13.7 CGI-58 and Cancer
Cancer cells often accumulate LDs in the cytoplasm [17, 210]. The underlying mechanisms remain elusive. Sequestration of lipids in cytosolic LDs may protect cancer cells from lipotoxic stress [93]. Mutations in CGI-58 cause LD deposition in cells, which led to the first study exploring the role of CGI-58 in colorectal cancer development [158]. It was shown that CGI-58 deficiency promotes the epithelial-mesenchymal transition (EMT) and invasiveness of colorectal cancer cells by increasing aerobic glycolysis (the Warburg Effect) [158]. The increase in aerobic glycolysis in CGI-58-deficient cells may result from limited availability of fatty acids due to defective LD lipolysis. In addition, CGI-58 was shown to promote colorectal tumorigenesis by impairing Beclin1-mediated autophagy [163]. A subsequent study with prostate cancer cells was consistent with the tumor suppressor role of CGI-58 [37]. However, another group using the same prostate cancer cell line found that CGI-58 sustains cancer cell growth by inhibiting cell apoptosis and death [140]. CGI-58 was recently shown to be oncogenic in endometrial cancer [261]. It was reported that CGI-58 in tumor-associated macrophages indirectly promotes colorectal cancer growth by suppressing spermidine synthesis [136]. The same group also reported that CGI-58 suppresses NFκB-dependent metalloproteinase production in macrophages to indirectly inhibit colorectal cancer cell metastasis [199]. Besides regulating tumorigenesis directly and indirectly, CGI-58 was reported to inhibit the sensitivity of colorectal cancer cells to the chemotherapy drug fluorouracil [159]. CGI-58 expression patterns and levels may serve as markers for differentiating benign and malignant tumors in some tissues [36, 158]. DNA methylation and deletion may influence CGI-58 expression in some cancer types, such as cervical cancer [198]. CGI-58 is not the only LD-associated protein that is implicated in cancer development and progression. It was shown that ATGL mediates cancer-associated cachexia [42], correlates with the risk of pancreatic ductal adenocarcinoma [68], and promotes malignancies of breast cancer and hepatocellular carcinoma [43, 124, 125, 232, 249]. It was reported that ATGL deletion is linked to the aggressiveness of A549 lung carcinoma cells [218]. Inhibition of ATGL by the lipolysis suppressor protein G0S2 or a small molecule Atglistatin was found to attenuate the growth of cancer cells [256]. G0S2 was also observed to suppress oncogenic transformation of immortalized mouse embryonic fibroblasts [252]. Interestingly, inhibition of ATGL by hypoxia-inducible gene 2 (HIG2), unlike G0S2, was demonstrated to promote survival of colorectal cancer and renal cell carcinoma cell lines in hypoxia [260]. The role of LD-associated proteins CGI-58, ATGL, G0S2, and HIG2 in tumorigenesis may be cell type-specific, depending on how each cell type handles energy metabolism and signal transduction under different pathophysiological conditions.
13.8 CGI-58 and HCV Infection
A large proportion of patients chronically infected with hepatitis C virus (HCV) manifest LD deposition in the liver in the absence of other steatotic factors [147]. It was shown that the HCV nucleocapsid core, which is the major structural component of HCV virions, localizes at the surface of LDs to inhibit LD turnover in cultured cells and mouse livers [80]. The same group further showed that the HCV core inhibits ATGL-dependent LD lipolysis, but it unexpectedly enhances ATGL interaction with CGI-58 and the recruitment of the ATGL/CGI-58 complex to LDs [26]. Interestingly, an siRNA-based screen identified CGI-58 as a host factor that assists HCV assembly and release without affecting virus entry and replication [225]. They showed that several CDS-causing mutants of CGI-58 fail to localize at the surface of LDs, and those mutants are unable to support HCV production. Moreover, they identified a tribasic motif (KRK233-235) that is required for CGI-58 to promote lipolysis and HCV production, though not essential for CGI-58 localization to LDs. While this study may suggest that it is its lipase coactivator function that mediates HCV assembly and release, it remains unknown whether the newly identified serine protease function of CGI-58 is implicated in HCV production [94].
13.9 Concluding Remarks
Patients with CDS accumulate TAG-rich LDs in all cell types examined. Since the discovery of CGI-58 gene mutations as the cause of CDS in 2001, enormous interest on the function of CGI-58 has been generated in the scientific community of lipid and energy metabolism. It has been well established that CGI-58 is a LD-associated protein that promotes intracellular LD lipolysis by activating ATGL’s TAG hydrolase activity. In addition to ATGL, CGI-58 interacts with many other proteins and regulates LD dynamics and functions in a cell type-specific manner. Such broad protein-protein interactions of CGI-58 have provided important insights into the biochemical basis for its ATGL-independent functions. Future studies are needed to dissect the molecular itineraries of these interactions in regulation of intracellular LD biogenesis and turnover. As a versatile regulator of intracellular LD homeostasis, CGI-58 plays a central role in governing cellular and whole-body energy balance. Genetic deletion of CGI-58 in mice has uncovered distinct effects of LD deposition in different cell types on the pathogenesis of metabolic disease. CGI-58 was recently identified to possess the serine protease activity in the heart. It is unknown if CGI-58 has this protease activity in other tissue. If yes, what are the substrates and functional significance? Is the serine protease activity of CGI-58 coordinate with its lipase coactivator function to activate intracellular lipolysis? Clearly, more studies are needed to answer these exciting new questions.
References
Adant I, Declercq M, Bird M, Bauters M, Boeckx N, Devriendt K, Cassiman D, Witters P (2020) Two cases of non-alcoholic fatty liver disease caused by biallelic ABHD5 mutations. J Hepatol 72(5):1030–1032
Aflaki E, Balenga NA, Luschnig-Schratl P, Wolinski H, Povoden S, Chandak PG, Bogner-Strauss JG, Eder S, Konya V, Kohlwein SD, Heinemann A, Kratky D (2011) Impaired Rho GTPase activation abrogates cell polarization and migration in macrophages with defective lipolysis. Cell Mol Life Sci 68:3933–3947
Aggarwal S, Maras JS, Alam S, Khanna R, Gupta SK, Ahuja A (2012) Novel nonsense mutation of ABHD5 in Dorfman-Chanarin syndrome with unusual findings: a challenge for genotype-phenotype correlation. Eur J Med Genet 55:173–177
Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, Wang Y, Duncan RE, Kang C, Sul HS (2011) Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab 13:739–748
Akiyama M, Sawamura D, Nomura Y, Sugawara M, Shimizu H (2003) Truncation of CGI-58 protein causes malformation of lamellar granules resulting in ichthyosis in Dorfman-Chanarin syndrome. J Invest Dermatol 121:1029–1034
Akiyama M, Sakai K, Ogawa M, McMillan JR, Sawamura D, Shimizu H (2007) Novel duplication mutation in the patatin domain of adipose triglyceride lipase (PNPLA2) in neutral lipid storage disease with severe myopathy. Muscle Nerve 36:856–859
Al-Hage J, Abbas O, Nemer G, Kurban M (2019) Chanarin-Dorfman syndrome: a novel homozygous mutation in the ABHD5 gene. Clin Exp Dermatol 45(2):257–259
Ameziane N, Beillat T, Verpillat P, Chollet-Martin S, Aumont MC, Seknadji P, Lamotte M, Lebret D, Ollivier V, de Prost D (2003) Association of the Toll-like receptor 4 gene Asp299Gly polymorphism with acute coronary events. Arterioscler Thromb Vasc Biol 23:e61–e64
Badeloe S, van Geel M, Nagtzaam I, Rubio-Gozalbo ME, Oei RL, Steijlen PM, van Steensel MA (2008) Chanarin-Dorfman syndrome caused by a novel splice site mutation in ABHD5. Br J Dermatol 158:1378–1380
Badin PM, Loubiere C, Coonen M, Louche K, Tavernier G, Bourlier V, Mairal A, Rustan AC, Smith SR, Langin D, Moro C (2012) Regulation of skeletal muscle lipolysis and oxidative metabolism by the co-lipase CGI-58. J Lipid Res 53:839–848
BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH (2019) Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci U S A 116:9521–9526
Ben Selma Z, Yilmaz S, Schischmanoff PO, Blom A, Ozogul C, Laroche L, Caux F (2007) A novel S115G mutation of CGI-58 in a Turkish patient with Dorfman-Chanarin syndrome. J Invest Dermatol 127:2273–2276
Bergman R, Aviram M, Bitterman-Deutsch O, Oiknine Y, Shemer A, Srebnik A, Brook JG, Friedman-Birnbaum R (1991) Neutral lipid storage disease with ichthyosis: serum apolipoprotein levels and cholesterol metabolism in monocyte-derived macrophages. J Inherit Metab Dis 14:241–246
Bjorkbacka H (2006) Multiple roles of Toll-like receptor signaling in atherosclerosis. Curr Opin Lipidol 17:527–533
Blondin DP, Frisch F, Phoenix S, Guerin B, Turcotte EE, Haman F, Richard D, Carpentier AC (2017) Inhibition of intracellular triglyceride lipolysis suppresses cold-induced brown adipose tissue metabolism and increases shivering in humans. Cell Metab 25:438–447
Boeszoermenyi A, Nagy HM, Arthanari H, Pillip CJ, Lindermuth H, Luna RE, Wagner G, Zechner R, Zangger K, Oberer M (2015) Structure of a CGI-58 motif provides the molecular basis of lipid droplet anchoring. J Biol Chem 290:26361–26372
Bozza PT, Viola JP (2010) Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids 82:243–250
Brown JM, Chung S, Das A, Shelness GS, Rudel LL, Yu L (2007) CGI-58 facilitates the mobilization of cytoplasmic triglyceride for lipoprotein secretion in hepatoma cells. J Lipid Res 48:2295–2305
Brown JM, Betters JL, Lord C, Ma Y, Han X, Yang K, Alger HM, Melchior J, Sawyer J, Shah R, Wilson MD, Liu X, Graham MJ, Lee R, Crooke R, Shulman GI, Xue B, Shi H, Yu L (2010) CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res 51:3306–3315
Browning JD, Horton JD (2004) Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 114:147–152
Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, Kahn CR (1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2:559–569
Bruno C, Bertini E, Di Rocco M, Cassandrini D, Ruffa G, De Toni T, Seri M, Spada M, Li Volti G, D’Amico A, Trucco F, Arca M, Casali C, Angelini C, Dimauro S, Minetti C (2008) Clinical and genetic characterization of Chanarin-Dorfman syndrome. Biochem Biophys Res Commun 369:1125–1128
Cakir M, Bruno C, Cansu A, Cobanoglu U, Erduran E (2010) Liver cirrhosis in an infant with Chanarin-Dorfman syndrome caused by a novel splice-site mutation in ABHD5. Acta Paediatr 99:1592–1594
Cakmak E, Alagozlu H, Yonem O, Ataseven H, Citli S, Ozer H (2012) Steatohepatitis and liver cirrhosis in Chanarin-Dorfman syndrome with a new ABDH5 mutation. Clin Res Hepatol Gastroenterol 36:e34–e37
Campagna F, Nanni L, Quagliarini F, Pennisi E, Michailidis C, Pierelli F, Bruno C, Casali C, DiMauro S, Arca M (2008) Novel mutations in the adipose triglyceride lipase gene causing neutral lipid storage disease with myopathy. Biochem Biophys Res Commun 377:843–846
Camus G, Schweiger M, Herker E, Harris C, Kondratowicz AS, Tsou CL, Farese RV Jr, Herath K, Previs SF, Roddy TP, Pinto S, Zechner R, Ott M (2014) The hepatitis C virus core protein inhibits adipose triglyceride lipase (ATGL)-mediated lipid mobilization and enhances the ATGL interaction with comparative gene identification 58 (CGI-58) and lipid droplets. J Biol Chem 289:35770–35780
Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359
Cantley JL, Yoshimura T, Camporez JP, Zhang D, Jornayvaz FR, Kumashiro N, Guebre-Egziabher F, Jurczak MJ, Kahn M, Guigni BA, Serr J, Hankin J, Murphy RC, Cline GW, Bhanot S, Manchem VP, Brown JM, Samuel VT, Shulman GI (2013) CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci U S A 110:1869–1874
Carr RM, Ahima RS (2016) Pathophysiology of lipid droplet proteins in liver diseases. Exp Cell Res 340:187–192
Carr RM, Patel RT, Rao V, Dhir R, Graham MJ, Crooke RM, Ahima RS (2012) Reduction of TIP47 improves hepatic steatosis and glucose homeostasis in mice. Am J Physiol Regul Integr Comp Physiol 302:R996–R1003
Cartwright IJ, Plonne D, Higgins JA (2000) Intracellular events in the assembly of chylomicrons in rabbit enterocytes. J Lipid Res 41:1728–1739
Chamoun Z, Vacca F, Parton RG, Gruenberg J (2013) PNPLA3/adiponutrin functions in lipid droplet formation. Biol Cell 105:219–233
Chanarin I, Patel A, Slavin G, Wills EJ, Andrews TM, Stewart G (1975) Neutral-lipid storage disease: a new disorder of lipid metabolism. Br Med J 1:553–555
Chang BH, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, Chan L (2006) Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol 26:1063–1076
Chen W, Chang B, Wu X, Li L, Sleeman M, Chan L (2013a) Inactivation of Plin4 downregulates Plin5 and reduces cardiac lipid accumulation in mice. Am J Physiol Endocrinol Metab 304:E770–E779
Chen WS, Chen PL, Li J, Lind AC, Lu D (2013b) Lipid synthesis and processing proteins ABHD5, PGRMC1 and squalene synthase can serve as novel immunohistochemical markers for sebaceous neoplasms and differentiate sebaceous carcinoma from sebaceoma and basal cell carcinoma with clear cell features. J Cutan Pathol 40:631–638
Chen G, Zhou G, Aras S, He Z, Lucas S, Podgorski I, Skar W, Granneman JG, Wang J (2017) Loss of ABHD5 promotes the aggressiveness of prostate cancer cells. Sci Rep 7:13021
Ciesek S, Hadem J, Fischer J, Manns MP, Strassburg CP (2006) A rare cause of nonalcoholic fatty liver disease. Ann Intern Med 145:154–155
Cinti S (2002) Adipocyte differentiation and transdifferentiation: plasticity of the adipose organ. J Endocrinol Invest 25:823–835
Cook KG, Yeaman SJ, Stralfors P, Fredrikson G, Belfrage P (1982) Direct evidence that cholesterol ester hydrolase from adrenal cortex is the same enzyme as hormone-sensitive lipase from adipose tissue. Eur J Biochem 125:245–249
Cornaciu I, Boeszoermenyi A, Lindermuth H, Nagy HM, Cerk IK, Ebner C, Salzburger B, Gruber A, Schweiger M, Zechner R, Lass A, Zimmermann R, Oberer M (2011) The minimal domain of adipose triglyceride lipase (ATGL) ranges until leucine 254 and can be activated and inhibited by CGI-58 and G0S2, respectively. PLoS One 6:e26349
Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M, Zimmermann R, Vesely P, Haemmerle G, Zechner R, Hoefler G (2011) Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 333:233–238
Di Leo L, Vegliante R, Ciccarone F, Salvatori I, Scimeca M, Bonanno E, Sagnotta A, Grazi GL, Aquilano K, Ciriolo MR (2019) Forcing ATGL expression in hepatocarcinoma cells imposes glycolytic rewiring through PPAR-alpha/p300-mediated acetylation of p53. Oncogene 38:1860–1875
Doi K, Ohno T, Kurahashi M, Kuroshima A (1979) Thermoregulatory nonshivering thermogenesis in men, with special reference to lipid metabolism. Jpn J Physiol 29:359–372
Dong H, Czaja MJ (2011) Regulation of lipid droplets by autophagy. Trends Endocrinol Metab 22:234–240
Dorfman ML, Hershko C, Eisenberg S, Sagher F (1974) Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol 110:261–266
Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS (2007) Regulation of lipolysis in adipocytes. Annu Rev Nutr 27:79–101
Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T, Deretic V (2014) Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol 24:609–620
Durdu M, Missaglia S, Moro L, Tavian D (2018) Clinical and genetic characterization of a Chanarin Dorfman Syndrome patient born to diseased parents. BMC Med Genet 19:88
Dutton JR, Chillingworth NL, Eberhard D, Brannon CR, Hornsey MA, Tosh D, Slack JM (2007) Beta cells occur naturally in extrahepatic bile ducts of mice. J Cell Sci 120:239–245
Eid N, Ito Y, Maemura K, Otsuki Y (2013) Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. J Mol Histol 44:311–326
Emre S, Unver N, Evans SE, Yuzbasioglu A, Gurakan F, Gumruk F, Karaduman A (2010) Molecular analysis of Chanarin-Dorfman syndrome (CDS) patients: Identification of novel mutations in the ABHD5 gene. Eur J Med Genet 53:141–144
Epstein SE, Zhu J, Burnett MS, Zhou YF, Vercellotti G, Hajjar D (2000) Infection and atherosclerosis: potential roles of pathogen burden and molecular mimicry. Arterioscler Thromb Vasc Biol 20:1417–1420
Eskiocak AH, Missaglia S, Moro L, Durdu M, Tavian D (2019) A novel mutation of ABHD5 gene in a Chanarin Dorfman patient with unusual dermatological findings. Lipids Health Dis 18:232
Fain JN, Garcija-Sainz JA (1983) Adrenergic regulation of adipocyte metabolism. J Lipid Res 24:945–966
Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, Moser A, Grunfeld C (2012) Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol 92:829–839
Finn PF, Dice JF (2006) Proteolytic and lipolytic responses to starvation. Nutrition 22:830–844
Fischer J, Lefevre C, Morava E, Mussini JM, Laforet P, Negre-Salvayre A, Lathrop M, Salvayre R (2007) The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet 39:28–30
Friedman SL (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134:1655–1669
Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ (2018) Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24:908–922
Fujii H, Ikura Y, Arimoto J, Sugioka K, Iezzoni JC, Park SH, Naruko T, Itabe H, Kawada N, Caldwell SH, Ueda M (2009) Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. J Atheroscler Thromb 16:893–901
Funk JL, Feingold KR, Moser AH, Grunfeld C (1993) Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis 98:67–82
Gandotra S, Lim K, Girousse A, Saudek V, O’Rahilly S, Savage DB (2011) Human frame shift mutations affecting the carboxyl terminus of perilipin increase lipolysis by failing to sequester the adipose triglyceride lipase (ATGL) coactivator AB-hydrolase-containing 5 (ABHD5). J Biol Chem 286:34998–35006
Geisler F, Nagl F, Mazur PK, Lee M, Zimber-Strobl U, Strobl LJ, Radtke F, Schmid RM, Siveke JT (2008) Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology 48:607–616
Goeritzer M, Schlager S, Radovic B, Madreiter CT, Rainer S, Thomas G, Lord CC, Sacks J, Brown AL, Vujic N, Obrowsky S, Sachdev V, Kolb D, Chandak PG, Graier WF, Sattler W, Brown JM, Kratky D (2014) Deletion of CGI-58 or adipose triglyceride lipase differently affects macrophage function and atherosclerosis. J Lipid Res 55:2562–2575
Goeritzer M, Vujic N, Schlager S, Chandak PG, Korbelius M, Gottschalk B, Leopold C, Obrowsky S, Rainer S, Doddapattar P, Aflaki E, Wegscheider M, Sachdev V, Graier WF, Kolb D, Radovic B, Kratky D (2015) Active autophagy but not lipophagy in macrophages with defective lipolysis. Biochim Biophys Acta 1851:1304–1316
Goodpaster BH, He J, Watkins S, Kelley DE (2001) Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 86:5755–5761
Grace SA, Meeks MW, Chen Y, Cornwell M, Ding X, Hou P, Rutgers JK, Crawford SE, Lai JP (2017) Adipose triglyceride lipase (ATGL) expression is associated with adiposity and tumor stromal proliferation in patients with pancreatic ductal adenocarcinoma. Anticancer Res 37:699–703
Grall A, Guaguere E, Planchais S, Grond S, Bourrat E, Hausser I, Hitte C, Le Gallo M, Derbois C, Kim GJ, Lagoutte L, Degorce-Rubiales F, Radner FP, Thomas A, Kury S, Bensignor E, Fontaine J, Pin D, Zimmermann R, Zechner R, Lathrop M, Galibert F, Andre C, Fischer J (2012) PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat Genet 44:140–147
Granneman JG, Moore HP, Granneman RL, Greenberg AS, Obin MS, Zhu Z (2007) Analysis of lipolytic protein trafficking and interactions in adipocytes. J Biol Chem 282:5726–5735
Granneman JG, Moore HP, Krishnamoorthy R, Rathod M (2009a) Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J Biol Chem 284:34538–34544
Granneman JG, Moore HP, Mottillo EP, Zhu Z (2009b) Functional interactions between Mldp (LSDP5) and Abhd5 in the control of intracellular lipid accumulation. J Biol Chem 284:3049–3057
Grond S, Radner FPW, Eichmann TO, Kolb D, Grabner GF, Wolinski H, Gruber R, Hofer P, Heier C, Schauer S, Rulicke T, Hoefler G, Schmuth M, Elias PM, Lass A, Zechner R, Haemmerle G (2017) Skin barrier development depends on CGI-58 protein expression during late-stage keratinocyte differentiation. J Invest Dermatol 137:403–413
Gruber A, Cornaciu I, Lass A, Schweiger M, Poeschl M, Eder C, Kumari M, Schoiswohl G, Wolinski H, Kohlwein SD, Zechner R, Zimmermann R, Oberer M (2010) The N-terminal region of comparative gene identification-58 (CGI-58) is important for lipid droplet binding and activation of adipose triglyceride lipase. J Biol Chem 285:12289–12298
Guo F, Ma Y, Kadegowda AK, Xie P, Liu G, Liu X, Miao H, Ou J, Su X, Zheng Z, Xue B, Shi H, Yu L (2013) Deficiency of liver Comparative Gene Identification-58 causes steatohepatitis and fibrosis in mice. J Lipid Res 54:2109–2120
Gurakan F, Kaymaz F, Kocak N, Ors U, Yuce A, Atakan N (1999) A cause of fatty liver: neutral lipid storage disease with ichthyosis--electron microscopic findings. Dig Dis Sci 44:2214–2217
Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, Sattler W, Magin TM, Wagner EF, Zechner R (2002) Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J Biol Chem 277:4806–4815
Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R (2006) Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312:734–737
Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R (2011) ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med 17:1076–1085
Harris C, Herker E, Farese RV Jr, Ott M (2011) Hepatitis C virus core protein decreases lipid droplet turnover: a mechanism for core-induced steatosis. J Biol Chem 286:42615–42625
Hernandez-Gea V, Friedman SL (2011) Pathogenesis of liver fibrosis. Annu Rev Pathol 6:425–456
Higashi T, Friedman SL, Hoshida Y (2017) Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev 121:27–42
Himms-Hagen J, Melnyk A, Zingaretti MC, Ceresi E, Barbatelli G, Cinti S (2000) Multilocular fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. Am J Physiol Cell Physiol 279:C670–C681
Hirabayashi T, Anjo T, Kaneko A, Senoo Y, Shibata A, Takama H, Yokoyama K, Nishito Y, Ono T, Taya C, Muramatsu K, Fukami K, Munoz-Garcia A, Brash AR, Ikeda K, Arita M, Akiyama M, Murakami M (2017) PNPLA1 has a crucial role in skin barrier function by directing acylceramide biosynthesis. Nat Commun 8:14609
Hofer P, Boeszoermenyi A, Jaeger D, Feiler U, Arthanari H, Mayer N, Zehender F, Rechberger G, Oberer M, Zimmermann R, Lass A, Haemmerle G, Breinbauer R, Zechner R, Preiss-Landl K (2015) Fatty acid-binding proteins interact with comparative gene identification-58 linking lipolysis with lipid ligand shuttling. J Biol Chem 290:18438–18453
Holm C, Kirchgessner TG, Svenson KL, Fredrikson G, Nilsson S, Miller CG, Shively JE, Heinzmann C, Sparkes RS, Mohandas T et al (1988) Hormone-sensitive lipase: sequence, expression, and chromosomal localization to 19 cent-q13.3. Science 241:1503–1506
Hoy AJ, Bruce CR, Turpin SM, Morris AJ, Febbraio MA, Watt MJ (2011) Adipose triglyceride lipase-null mice are resistant to high-fat diet-induced insulin resistance despite reduced energy expenditure and ectopic lipid accumulation. Endocrinology 152:48–58
Hsieh K, Lee YK, Londos C, Raaka BM, Dalen KT, Kimmel AR (2012) Perilipin family members preferentially sequester to either triacylglycerol-specific or cholesteryl-ester-specific intracellular lipid storage droplets. J Cell Sci 125:4067–4076
Huigen MC, van der Graaf M, Morava E, Dassel AC, van Steensel MA, Seyger MM, Wevers RA, Willemsen MA (2015) Cerebral lipid accumulation in Chanarin-Dorfman Syndrome. Mol Genet Metab 114:51–54
Igal RA, Rhoads JM, Coleman RA (1997) Neutral lipid storage disease with fatty liver and cholestasis. J Pediatr Gastroenterol Nutr 25:541–547
Imai Y, Varela GM, Jackson MB, Graham MJ, Crooke RM, Ahima RS (2007) Reduction of hepatosteatosis and lipid levels by an adipose differentiation-related protein antisense oligonucleotide. Gastroenterology 132:1947–1954
Israeli S, Pessach Y, Sarig O, Goldberg I, Sprecher E (2012) Beneficial effect of acitretin in Chanarin-Dorfman syndrome. Clin Exp Dermatol 37:31–33
Jarc E, Eichmann TO, Zimmermann R, Petan T (2018) Lipidomic data on lipid droplet triglyceride remodelling associated with protection of breast cancer cells from lipotoxic stress. Data Brief 18:234–240
Jebessa ZH, Shanmukha Kumar D, Dewenter M, Lehmann LH, Xu C, Schreiter F, Siede D, Gong XM, Worst BC, Federico G, Sauer SW, Fischer T, Wechselberger L, Muller OJ, Sossalla S, Dieterich C, Most P, Grone HJ, Moro C, Oberer M, Haemmerle G, Katus HA, Tyedmers J, Backs J (2019) The lipid droplet-associated protein ABHD5 protects the heart through proteolysis of HDAC4. Nat Metab 1:1157–1167
Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW (2004) Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem 279:48968–48975
Jordans GH (1953) The familial occurrence of fat containing vacuoles in the leukocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva (ERB.). Acta Med Scand 145:419–423
Kajimura S, Spiegelman BM, Seale P (2015) Brown and beige fat: physiological roles beyond heat generation. Cell Metab 22:546–559
Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C (1997) cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J Biol Chem 272:27218–27223
Kato M, Higuchi N, Enjoji M (2008) Reduced hepatic expression of adipose tissue triglyceride lipase and CGI-58 may contribute to the development of non-alcoholic fatty liver disease in patients with insulin resistance. Scand J Gastroenterol 43:1–2
Kaushik S, Cuervo AM (2015) Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 17:759–770
Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Willeit J, Schwartz DA (2002) Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med 347:185–192
Kien B, Grond S, Haemmerle G, Lass A, Eichmann TO, Radner FPW (2018) ABHD5 stimulates PNPLA1-mediated omega-O-acylceramide biosynthesis essential for a functional skin permeability barrier. J Lipid Res 59:2360–2367
Kienesberger PC, Lee D, Pulinilkunnil T, Brenner DS, Cai L, Magnes C, Koefeler HC, Streith IE, Rechberger GN, Haemmerle G, Flier JS, Zechner R, Kim YB, Kershaw EE (2009a) Adipose triglyceride lipase deficiency causes tissue-specific changes in insulin signaling. J Biol Chem 284:30218–30229
Kienesberger PC, Oberer M, Lass A, Zechner R (2009b) Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. J Lipid Res 50(Suppl):S63–S68
Kimmel AR, Brasaemle DL, McAndrews-Hill M, Sztalryd C, Londos C (2010) Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J Lipid Res 51:468–471
Korbelius M, Vujic N, Sachdev V, Obrowsky S, Rainer S, Gottschalk B, Graier WF, Kratky D (2019) ATGL/CGI-58-dependent hydrolysis of a lipid storage pool in murine enterocytes. Cell Rep 28:1923-34 e4
Labbe SM, Caron A, Bakan I, Laplante M, Carpentier AC, Lecomte R, Richard D (2015) In vivo measurement of energy substrate contribution to cold-induced brown adipose tissue thermogenesis. FASEB J 29:2046–2058
Lafontan M, Langin D (2009) Lipolysis and lipid mobilization in human adipose tissue. Prog Lipid Res 48:275–297
Lai CH, Chou CY, Ch’ang LY, Liu CS, Lin W (2000) Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res 10:703–713
Lammers B, Chandak PG, Aflaki E, Van Puijvelde GH, Radovic B, Hildebrand RB, Meurs I, Out R, Kuiper J, Van Berkel TJ, Kolb D, Haemmerle G, Zechner R, Levak-Frank S, Van Eck M, Kratky D (2011) Macrophage adipose triglyceride lipase deficiency attenuates atherosclerotic lesion development in low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol 31:67–73
Langin D, Laurell H, Holst LS, Belfrage P, Holm C (1993) Gene organization and primary structure of human hormone-sensitive lipase: possible significance of a sequence homology with a lipase of Moraxella TA144, an antarctic bacterium. Proc Natl Acad Sci U S A 90:4897–4901
Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R (2006) Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab 3:309–319
Lee FT, Adams JB, Garton AJ, Yeaman SJ (1988) Hormone-sensitive lipase is involved in the hydrolysis of lipoidal derivatives of estrogens and other steroid hormones. Biochim Biophys Acta 963:258–264
Lee YH, Petkova AP, Mottillo EP, Granneman JG (2012) In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell Metab 15:480–491
Lee YH, Petkova AP, Konkar AA, Granneman JG (2015) Cellular origins of cold-induced brown adipocytes in adult mice. FASEB J 29:286–299
Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, Ozguc M, Lathrop M, Prud’homme JF, Fischer J (2001) Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet 69:1002–1012
Lehr HA, Sagban TA, Ihling C, Zahringer U, Hungerer KD, Blumrich M, Reifenberg K, Bhakdi S (2001) Immunopathogenesis of atherosclerosis: endotoxin accelerates atherosclerosis in rabbits on hypercholesterolemic diet. Circulation 104:914–920
Levin MC, Monetti M, Watt MJ, Sajan MP, Stevens RD, Bain JR, Newgard CB, Farese RV Sr, Farese RV Jr (2007) Increased lipid accumulation and insulin resistance in transgenic mice expressing DGAT2 in glycolytic (type II) muscle. Am J Physiol Endocrinol Metab 293:E1772–E1781
Li Y, Schnabl K, Gabler SM, Willershauser M, Reber J, Karlas A, Laurila S, Lahesmaa M, Din U, Bast-Habersbrunner MA, Virtanen KA, Fromme T, Bolze F, O’Farrell LS, Alsina-Fernandez J, Coskun T, Ntziachristos V, Nuutila P, Klingenspor M (2018) Secretin-activated brown fat mediates prandial thermogenesis to induce satiation. Cell 175:1561-74 e12
Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, Schaffer JE (2003) Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 100:3077–3082
Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, Anderson RG (2004) Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem 279:3787–3792
Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ (2009a) DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem 284:36312–36323
Liu L, Shi X, Choi CS, Shulman GI, Klaus K, Nair KS, Schwartz GJ, Zhang Y, Goldberg IJ, Yu YH (2009b) Paradoxical coupling of triglyceride synthesis and fatty acid oxidation in skeletal muscle overexpressing DGAT1. Diabetes 58:2516–2524
Liu X, Liang Y, Song R, Yang G, Han J, Lan Y, Pan S, Zhu M, Liu Y, Wang Y, Meng F, Cui Y, Wang J, Zhang B, Song X, Lu Z, Zheng T, Liu L (2018) Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol Cancer 17:90
Liu M, Yu X, Lin L, Deng J, Wang K, Xia Y, Tang X, Hong H (2019) ATGL promotes the proliferation of hepatocellular carcinoma cells via the p-AKT signaling pathway. J Biochem Mol Toxicol 33:e22391
Londos C, Sztalryd C, Tansey JT, Kimmel AR (2005) Role of PAT proteins in lipid metabolism. Biochimie 87:45–49
Lord CC, Betters JL, Ivanova PT, Milne SB, Myers DS, Madenspacher J, Thomas G, Chung S, Liu M, Davis MA, Lee RG, Crooke RM, Graham MJ, Parks JS, Brasaemle DL, Fessler MB, Brown HA, Brown JM (2012) CGI-58/ABHD5-derived signaling lipids regulate systemic inflammation and insulin action. Diabetes 61:355–363
Lord CC, Thomas G, Brown JM (2013) Mammalian alpha beta hydrolase domain (ABHD) proteins: lipid metabolizing enzymes at the interface of cell signaling and energy metabolism. Biochim Biophys Acta 1831:792–802
Lord CC, Ferguson D, Thomas G, Brown AL, Schugar RC, Burrows A, Gromovsky AD, Betters J, Neumann C, Sacks J, Marshall S, Watts R, Schweiger M, Lee RG, Crooke RM, Graham MJ, Lathia JD, Sakaguchi TF, Lehner R, Haemmerle G, Zechner R, Brown JM (2016) Regulation of hepatic triacylglycerol metabolism by CGI-58 does not require ATGL Co-activation. Cell Rep 16:939–949
Louhichi N, Bahloul E, Marrakchi S, Othman HB, Triki C, Aloulou K, Trabelsi L, Mahfouth N, Ayadi-Mnif Z, Keskes L, Fakhfakh F, Turki H (2019) Thyroid involvement in Chanarin-Dorfman syndrome in adults in the largest series of patients carrying the same founder mutation in ABHD5 gene. Orphanet J Rare Dis 14:112
Lu X, Yang X, Liu J (2010) Differential control of ATGL-mediated lipid droplet degradation by CGI-58 and G0S2. Cell Cycle 9:2719–2725
MacPherson RE, Ramos SV, Vandenboom R, Roy BD, Peters SJ (2013) Skeletal muscle PLIN proteins, ATGL and CGI-58, interactions at rest and following stimulated contraction. Am J Physiol Regul Integr Comp Physiol 304:R644–R650
Martin S, Parton RG (2006) Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol 7:373–378
Miao H, Ou J, Ma Y, Guo F, Yang Z, Wiggins M, Liu C, Song W, Han X, Wang M, Cao Q, Chung BH, Yang D, Liang H, Xue B, Shi H, Gan L, Yu L (2014) Macrophage CGI-58 deficiency activates ROS-inflammasome pathway to promote insulin resistance in mice. Cell Rep 7:223–235
Miao H, Ou J, Zhang X, Chen Y, Xue B, Shi H, Gan L, Yu L, Liang H (2015) Macrophage CGI-58 deficiency promotes IL-1beta transcription by activating the SOCS3-FOXO1 pathway. Clin Sci (Lond) 128:493–506
Miao H, Ou J, Peng Y, Zhang X, Chen Y, Hao L, Xie G, Wang Z, Pang X, Ruan Z, Li J, Yu L, Xue B, Shi H, Shi C, Liang H (2016) Macrophage ABHD5 promotes colorectal cancer growth by suppressing spermidine production by SRM. Nat Commun 7:11716
Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M (2004) Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A 101:10679–10684
Miranda A, DiMauro S, Eastwood A, Hays A, Johnson WG, Olarte M, Whitlock R, Mayeux R, Rowland LP (1979) Lipid storage myopathy, ichthyosis, and steatorrhea. Muscle Nerve 2:1–13
Missaglia S, Valadares ER, Moro L, Faguntes ED, Quintao Roque R, Giardina B, Tavian D (2014) Early onset of Chanarin-Dorfman syndrome with severe liver involvement in a patient with a complex rearrangement of ABHD5 promoter. BMC Med Genet 15:32
Mitra R, Le TT, Gorjala P, Goodman OB Jr (2017) Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer 17:631
Monetti M, Levin MC, Watt MJ, Sajan MP, Marmor S, Hubbard BK, Stevens RD, Bain JR, Newgard CB, Farese RV Sr, Hevener AL, Farese RV Jr (2007) Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 6:69–78
Moreira RK (2007) Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med 131:1728–1734
Mullick AE, Tobias PS, Curtiss LK (2005) Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest 115:3149–3156
Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K (2011) Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog Lipid Res 50:152–192
Najt CP, Senthivinayagam S, Aljazi MB, Fader KA, Olenic SD, Brock JR, Lydic TA, Jones AD, Atshaves BP (2016) Liver-specific loss of Perilipin 2 alleviates diet-induced hepatic steatosis, inflammation, and fibrosis. Am J Physiol Gastrointest Liver Physiol 310:G726–G738
Nardini M, Dijkstra BW (1999) Alpha/beta hydrolase fold enzymes: the family keeps growing. Curr Opin Struct Biol 9:732–737
Negro F (2006) Mechanisms and significance of liver steatosis in hepatitis C virus infection. World J Gastroenterol 12:6756–6765
Newberry EP, Xie Y, Lodeiro C, Solis R, Moritz W, Kennedy S, Barron L, Onufer E, Alpini G, Zhou T, Blaner WS, Chen A, Davidson NO (2019) Hepatocyte and stellate cell deletion of liver fatty acid binding protein reveals distinct roles in fibrogenic injury. FASEB J 33:4610–4625
Nischalke HD, Berger C, Luda C, Berg T, Muller T, Grunhage F, Lammert F, Coenen M, Kramer B, Korner C, Vidovic N, Oldenburg J, Nattermann J, Sauerbruch T, Spengler U (2011) The PNPLA3 rs738409 148M/M genotype is a risk factor for liver cancer in alcoholic cirrhosis but shows no or weak association in hepatitis C cirrhosis. PLoS One 6:e27087
Nur BG, Gencpinar P, Yuzbasioglu A, Emre SD, Mihci E (2015) Chanarin-Dorfman syndrome: genotype-phenotype correlation. Eur J Med Genet 58:238–242
Oberer M, Boeszoermenyi A, Nagy HM, Zechner R (2011) Recent insights into the structure and function of comparative gene identification-58. Curr Opin Lipidol 22:149–158
Obrowsky S, Chandak PG, Patankar JV, Povoden S, Schlager S, Kershaw EE, Bogner-Strauss JG, Hoefler G, Levak-Frank S, Kratky D (2013) Adipose triglyceride lipase is a TG hydrolase of the small intestine and regulates intestinal PPARalpha signaling. J Lipid Res 54:425–435
Ohno Y, Kamiyama N, Nakamichi S, Kihara A (2017) PNPLA1 is a transacylase essential for the generation of the skin barrier lipid omega-O-acylceramide. Nat Commun 8:14610
Ohno Y, Nara A, Nakamichi S, Kihara A (2018) Molecular mechanism of the ichthyosis pathology of Chanarin-Dorfman syndrome: stimulation of PNPLA1-catalyzed omega-O-acylceramide production by ABHD5. J Dermatol Sci 92:245–253
Ohsumi Y (2014) Historical landmarks of autophagy research. Cell Res 24:9–23
Ostos MA, Recalde D, Zakin MM, Scott-Algara D (2002) Implication of natural killer T cells in atherosclerosis development during a LPS-induced chronic inflammation. FEBS Lett 519:23–29
Osuga J, Ishibashi S, Oka T, Yagyu H, Tozawa R, Fujimoto A, Shionoiri F, Yahagi N, Kraemer FB, Tsutsumi O, Yamada N (2000) Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc Natl Acad Sci U S A 97:787–792
Ou J, Miao H, Ma Y, Guo F, Deng J, Wei X, Zhou J, Xie G, Shi H, Xue B, Liang H, Yu L (2014) Loss of abhd5 promotes colorectal tumor development and progression by inducing aerobic glycolysis and epithelial-mesenchymal transition. Cell Rep 9:1798–1811
Ou J, Peng Y, Yang W, Zhang Y, Hao J, Li F, Chen Y, Zhao Y, Xie X, Wu S, Zha L, Luo X, Xie G, Wang L, Sun W, Zhou Q, Li J, Liang H (2019) ABHD5 blunts the sensitivity of colorectal cancer to fluorouracil via promoting autophagic uracil yield. Nat Commun 10:1078
Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL (2011) Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 13:655–667
Patel S, Yang W, Kozusko K, Saudek V, Savage DB (2014) Perilipins 2 and 3 lack a carboxy-terminal domain present in perilipin 1 involved in sequestering ABHD5 and suppressing basal lipolysis. Proc Natl Acad Sci U S A 111:9163–9168
Pawella LM, Hashani M, Eiteneuer E, Renner M, Bartenschlager R, Schirmacher P, Straub BK (2014) Perilipin discerns chronic from acute hepatocellular steatosis. J Hepatol 60:633–642
Peng Y, Miao H, Wu S, Yang W, Zhang Y, Xie G, Xie X, Li J, Shi C, Ye L, Sun W, Wang L, Liang H, Ou J (2016) ABHD5 interacts with BECN1 to regulate autophagy and tumorigenesis of colon cancer independent of PNPLA2. Autophagy 12:2167–2182
Petit V, Arnould L, Martin P, Monnot MC, Pineau T, Besnard P, Niot I (2007) Chronic high-fat diet affects intestinal fat absorption and postprandial triglyceride levels in the mouse. J Lipid Res 48:278–287
Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J (2010) Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem 285:7153–7164
Piva E, Pajola R, Binotto G, Plebani M (2009) Jordans’ anomaly in a new neutral lipid storage disease. Am J Hematol 84:254–255
Pollak NM, Jaeger D, Kolleritsch S, Zimmermann R, Zechner R, Lass A, Haemmerle G (2015) The interplay of protein kinase A and perilipin 5 regulates cardiac lipolysis. J Biol Chem 290:1295–1306
Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA (1999) Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274:305–315
Pujol RM, Gilaberte M, Toll A, Florensa L, Lloreta J, Gonzalez-Ensenat MA, Fischer J, Azon A (2005) Erythrokeratoderma variabilis-like ichthyosis in Chanarin-Dorfman syndrome. Br J Dermatol 153:838–841
Pussinen PJ, Mattila K (2004) Periodontal infections and atherosclerosis: mere associations? Curr Opin Lipidol 15:583–588
Pyzik M, Rath T, Kuo TT, Win S, Baker K, Hubbard JJ, Grenha R, Gandhi A, Kramer TD, Mezo AR, Taylor ZS, McDonnell K, Nienaber V, Andersen JT, Mizoguchi A, Blumberg L, Purohit S, Jones SD, Christianson G, Lencer WI, Sandlie I, Kaplowitz N, Roopenian DC, Blumberg RS (2017) Hepatic FcRn regulates albumin homeostasis and susceptibility to liver injury. Proc Natl Acad Sci U S A 114:E2862–E2E71
Radner FP, Streith IE, Schoiswohl G, Schweiger M, Kumari M, Eichmann TO, Rechberger G, Koefeler HC, Eder S, Schauer S, Theussl HC, Preiss-Landl K, Lass A, Zimmermann R, Hoefler G, Zechner R, Haemmerle G (2010) Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin barrier defect in mice lacking comparative gene identification-58 (CGI-58). J Biol Chem 285:7300–7311
Randle PJ, Garland PB, Hales CN, Newsholme EA (1963) The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1:785–789
Redaelli C, Coleman RA, Moro L, Dacou-Voutetakis C, Elsayed SM, Prati D, Colli A, Mela D, Colombo R, Tavian D (2010) Clinical and genetic characterization of Chanarin-Dorfman syndrome patients: first report of large deletions in the ABHD5 gene. Orphanet J Rare Dis 5:33
Rich NE, Oji S, Mufti AR, Browning JD, Parikh ND, Odewole M, Mayo H, Singal AG (2018) Racial and ethnic disparities in nonalcoholic fatty liver disease prevalence, severity, and outcomes in the United States: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 16:198–210 e2
Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, Bhathal PS, Dixon JB, Weltman MD, Tilg H, Moschen AR, Purdie DM, Demetris AJ, Clouston AD (2007) Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology 133:80–90
Rizack MA (1964) Activation of an epinephrine-sensitive lipolytic activity from adipose tissue by adenosine 3′,5′-Phosphate. J Biol Chem 239:392–395
Robertson MD, Parkes M, Warren BF, Ferguson DJ, Jackson KG, Jewell DP, Frayn KN (2003) Mobilisation of enterocyte fat stores by oral glucose in humans. Gut 52:834–839
Roduit R, Masiello P, Wang SP, Li H, Mitchell GA, Prentki M (2001) A role for hormone-sensitive lipase in glucose-stimulated insulin secretion: a study in hormone-sensitive lipase-deficient mice. Diabetes 50:1970–1975
Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH (2008) Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40:1461–1465
Ronchetti A, Prati D, Pezzotta MG, Tavian D, Colombo R, Callea F, Colli A (2008) Severe steatohepatitis in a patient with a rare neutral lipid storage disorder due to ABHD5 mutation. J Hepatol 49:474–477
Rosenwald M, Perdikari A, Rulicke T, Wolfrum C (2013) Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol 15:659–667
Rozenszajn L, Klajman A, Yaffe D, Efrati P (1966) Jordans’ anomaly in white blood cells. report of case. Blood 28:258–265
Russell JO, Lu WY, Okabe H, Abrams M, Oertel M, Poddar M, Singh S, Forbes SJ, Monga SP (2019) Hepatocyte-specific beta-catenin deletion during severe liver injury provokes cholangiocytes to differentiate into hepatocytes. Hepatology 69:742–759
Sahu-Osen A, Montero-Moran G, Schittmayer M, Fritz K, Dinh A, Chang YF, McMahon D, Boeszoermenyi A, Cornaciu I, Russell D, Oberer M, Carman GM, Birner-Gruenberger R, Brasaemle DL (2015) CGI-58/ABHD5 is phosphorylated on Ser239 by protein kinase A: control of subcellular localization. J Lipid Res 56:109–121
Samuel VT, Shulman GI (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148:852–871
Samuelov L, Fuchs-Telem D, Sarig O, Sprecher E (2011) An exceptional mutational event leading to Chanarin-Dorfman syndrome in a large consanguineous family. Br J Dermatol 164:1390–1392
Sanders MA, Madoux F, Mladenovic L, Zhang H, Ye X, Angrish M, Mottillo EP, Caruso JA, Halvorsen G, Roush WR, Chase P, Hodder P, Granneman JG (2015) Endogenous and synthetic ABHD5 ligands regulate ABHD5-perilipin interactions and lipolysis in fat and muscle. Cell Metab 22:851–860
Sanders MA, Zhang H, Mladenovic L, Tseng YY, Granneman JG (2017) Molecular basis of ABHD5 lipolysis activation. Sci Rep 7:42589
Sathyanarayan A, Mashek MT, Mashek DG (2017) ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep 19:1–9
Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G (2019) Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology 69:420–430
Schleinitz N, Fischer J, Sanchez A, Veit V, Harle JR, Pelissier JF (2005) Two new mutations of the ABHD5 gene in a new adult case of Chanarin Dorfman syndrome: an uncommon lipid storage disease. Arch Dermatol 141:798–800
Schneider AL, Lazo M, Selvin E, Clark JM (2014) Racial differences in nonalcoholic fatty liver disease in the U.S. population. Obesity (Silver Spring) 22:292–299
Schoiswohl G, Stefanovic-Racic M, Menke MN, Wills RC, Surlow BA, Basantani MK, Sitnick MT, Cai L, Yazbeck CF, Stolz DB, Pulinilkunnil T, O’Doherty RM, Kershaw EE (2015) Impact of reduced ATGL-mediated adipocyte lipolysis on obesity-associated insulin resistance and inflammation in male mice. Endocrinology 156:3610–3624
Schreiber R, Diwoky C, Schoiswohl G, Feiler U, Wongsiriroj N, Abdellatif M, Kolb D, Hoeks J, Kershaw EE, Sedej S, Schrauwen P, Haemmerle G, Zechner R (2017) Cold-Induced thermogenesis depends on ATGL-mediated lipolysis in cardiac muscle, but not brown adipose tissue. Cell Metab 26:753-63 e7
Schweiger M, Schoiswohl G, Lass A, Radner FP, Haemmerle G, Malli R, Graier W, Cornaciu I, Oberer M, Salvayre R, Fischer J, Zechner R, Zimmermann R (2008) The C-terminal region of human adipose triglyceride lipase affects enzyme activity and lipid droplet binding. J Biol Chem 283:17211–17220
Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R (2009) Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab 297:E289–E296
Senchenko VN, Kisseljova NP, Ivanova TA, Dmitriev AA, Krasnov GS, Kudryavtseva AV, Panasenko GV, Tsitrin EB, Lerman MI, Kisseljov FL, Kashuba VI, Zabarovsky ER (2013) Novel tumor suppressor candidates on chromosome 3 revealed by NotI-microarrays in cervical cancer. Epigenetics 8:409–420
Shang S, Ji X, Zhang L, Chen J, Li C, Shi R, Xiang W, Kang X, Zhang D, Yang F, Dai R, Chen P, Chen S, Chen Y, Li Y, Miao H (2019) Macrophage ABHD5 suppresses NFkappaB-dependent matrix metalloproteinase expression and cancer metastasis. Cancer Res 79:5513–5526
Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, Arai H, Tanaka K, Kominami E, Uchiyama Y (2009) The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun 382:419–423
Shin H, Ma Y, Chanturiya T, Cao Q, Wang Y, Kadegowda AKG, Jackson R, Rumore D, Xue B, Shi H, Gavrilova O, Yu L (2017) Lipolysis in brown adipocytes is not essential for cold-induced thermogenesis in mice. Cell Metab 26:764-77 e5
Simon GM, Cravatt BF (2006) Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for alpha/beta-hydrolase 4 in this pathway. J Biol Chem 281:26465–26472
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458:1131–1135
Sitnick MT, Basantani MK, Cai L, Schoiswohl G, Yazbeck CF, Distefano G, Ritov V, DeLany JP, Schreiber R, Stolz DB, Gardner NP, Kienesberger PC, Pulinilkunnil T, Zechner R, Goodpaster BH, Coen P, Kershaw EE (2013) Skeletal muscle triacylglycerol hydrolysis does not influence metabolic complications of obesity. Diabetes 62:3350–3361
Slavin G, Wills EJ, Richmond JE, Chanarin I, Andrews T, Stewart G (1975) Morphological features in a neutral lipid storage disease. J Clin Pathol 28:701–710
Sparks EE, Huppert KA, Brown MA, Washington MK, Huppert SS (2010) Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatology 51:1391–1400
Speliotes EK, Butler JL, Palmer CD, Voight BF, Giant Consortium, M. IGen Consortium, Nash CRN, Hirschhorn JN (2010) PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 52:904–912
Srinivasan R, Hadzic N, Fischer J, Knisely AS (2004) Steatohepatitis and unsuspected micronodular cirrhosis in Dorfman-Chanarin syndrome with documented ABHD5 mutation. J Pediatr 144:662–665
Straub BK, Stoeffel P, Heid H, Zimbelmann R, Schirmacher P (2008) Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 47:1936–1946
Straub BK, Herpel E, Singer S, Zimbelmann R, Breuhahn K, Macher-Goeppinger S, Warth A, Lehmann-Koch J, Longerich T, Heid H, Schirmacher P (2010) Lipid droplet-associated PAT-proteins show frequent and differential expression in neoplastic steatogenesis. Mod Pathol 23:480–492
Subramanian V, Rothenberg A, Gomez C, Cohen AW, Garcia A, Bhattacharyya S, Shapiro L, Dolios G, Wang R, Lisanti MP, Brasaemle DL (2004) Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J Biol Chem 279:42062–42071
Sun Z, Miller RA, Patel RT, Chen J, Dhir R, Wang H, Zhang D, Graham MJ, Unterman TG, Shulman GI, Sztalryd C, Bennett MJ, Ahima RS, Birnbaum MJ, Lazar MA (2012) Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med 18:934–942
Sztalryd C, Brasaemle DL (2017) The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim Biophys Acta Mol Cell Biol Lipids 1862:1221–1232
Taguchi K, Hirano I, Itoh T, Tanaka M, Miyajima A, Suzuki A, Motohashi H, Yamamoto M (2014) Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol Cell Biol 34:900–913
Takeda K, Tanaka K, Kumamoto T, Morioka D, Endo I, Togo S, Shimada H (2010) Living donor liver transplantation for Dorfman-Chanarin syndrome with 1 year follow-up: case report. Transplant Proc 42:3858–3861
Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA (2010) Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet 42:21–23
Tolkovsky AM (2009) Mitophagy. Biochim Biophys Acta 1793:1508–1515
Tomin T, Fritz K, Gindlhuber J, Waldherr L, Pucher B, Thallinger GG, Nomura DK, Schittmayer M, Birner-Gruenberger R (2018) Deletion of adipose triglyceride lipase links triacylglycerol accumulation to a more-aggressive phenotype in A549 lung carcinoma cells. J Proteome Res 17:1415–1425
Ujihara M, Nakajima K, Yamamoto M, Teraishi M, Uchida Y, Akiyama M, Shimizu H, Sano S (2010) Epidermal triglyceride levels are correlated with severity of ichthyosis in Dorfman-Chanarin syndrome. J Dermatol Sci 57:102–107
Umemura A, Park EJ, Taniguchi K, Lee JH, Shalapour S, Valasek MA, Aghajan M, Nakagawa H, Seki E, Hall MN, Karin M (2014) Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab 20:133–144
Unger RH (1995) Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 44:863–870
Unger RH (2002) Lipotoxic diseases. Annu Rev Med 53:319–336
Vaughan M, Berger JE, Steinberg D (1964) Hormone-sensitive lipase and monoglyceride lipase activities in adipose tissue. J Biol Chem 239:401–409
Verma SB, Mittal A, Wollina U, Eckstein GH, Gohel K, Giehl K (2017) Chanarin-Dorfman syndrome with rare renal involvement. Br J Dermatol 176:545–548
Vieyres G, Welsch K, Gerold G, Gentzsch J, Kahl S, Vondran FW, Kaderali L, Pietschmann T (2016) ABHD5/CGI-58, the Chanarin-Dorfman syndrome protein, mobilises lipid stores for Hepatitis C virus production. PLoS Pathog 12:e1005568
Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS (2004) Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem 279:47066–47075
Wang SP, Laurin N, Himms-Hagen J, Rudnicki MA, Levy E, Robert MF, Pan L, Oligny L, Mitchell GA (2001) The adipose tissue phenotype of hormone-sensitive lipase deficiency in mice. Obes Res 9:119–128
Wang H, Bell M, Sreenevasan U, Hu H, Liu J, Dalen K, Londos C, Yamaguchi T, Rizzo MA, Coleman R, Gong D, Brasaemle D, Sztalryd C (2011) Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J Biol Chem 286:15707–15715
Wang QA, Tao C, Gupta RK, Scherer PE (2013) Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med 19:1338–1344
Wang W, Kissig M, Rajakumari S, Huang L, Lim HW, Won KJ, Seale P (2014) Ebf2 is a selective marker of brown and beige adipogenic precursor cells. Proc Natl Acad Sci U S A 111:14466–14471
Wang C, Zhao Y, Gao X, Li L, Yuan Y, Liu F, Zhang L, Wu J, Hu P, Zhang X, Gu Y, Xu Y, Wang Z, Li Z, Zhang H, Ye J (2015) Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 61:870–882
Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A, Gilhodes J, Lazar I, Alet N, Laurent V, Le Gonidec S, Biard D, Herve C, Bost F, Ren GS, Bono F, Escourrou G, Prentki M, Nieto L, Valet P, Muller C (2017) Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2:e87489
Wang Y, Kory N, BasuRay S, Cohen JC, Hobbs HH (2019) PNPLA3, CGI-58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology 69:2427–2441
Wei S, Lai K, Patel S, Piantedosi R, Shen H, Colantuoni V, Kraemer FB, Blaner WS (1997) Retinyl ester hydrolysis and retinol efflux from BFC-1beta adipocytes. J Biol Chem 272:14159–14165
Williams ML, Koch TK, O’Donnell JJ, Frost PH, Epstein LB, Grizzard WS, Epstein CJ (1985) Ichthyosis and neutral lipid storage disease. Am J Med Genet 20:711–726
Wolins NE, Brasaemle DL, Bickel PE (2006) A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett 580:5484–5491
Wu JW, Wang SP, Alvarez F, Casavant S, Gauthier N, Abed L, Soni KG, Yang G, Mitchell GA (2011) Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 54:122–132
Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, Huang K, Tu H, van Marken Lichtenbelt WD, Hoeks J, Enerback S, Schrauwen P, Spiegelman BM (2012a) Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150:366–376
Wu JW, Wang SP, Casavant S, Moreau A, Yang GS, Mitchell GA (2012b) Fasting energy homeostasis in mice with adipose deficiency of desnutrin/adipose triglyceride lipase. Endocrinology 153:2198–2207
Xie P, Guo F, Ma Y, Zhu H, Wang F, Xue B, Shi H, Yang J, Yu L (2014a) Intestinal Cgi-58 deficiency reduces postprandial lipid absorption. PLoS One 9:e91652
Xie P, Zeng X, Xiao J, Sun B, Yang D (2014b) Transgenic CGI-58 expression in macrophages alleviates the atherosclerotic lesion development in ApoE knockout mice. Biochim Biophys Acta 1841:1683–1690
Xie P, Kadegowda AK, Ma Y, Guo F, Han X, Wang M, Groban L, Xue B, Shi H, Li H, Yu L (2015) Muscle-specific deletion of comparative gene identification-58 (CGI-58) causes muscle steatosis but improves insulin sensitivity in male mice. Endocrinology 156:1648–1658
Yamaguchi T, Osumi T (2009) Chanarin-Dorfman syndrome: deficiency in CGI-58, a lipid droplet-bound coactivator of lipase. Biochim Biophys Acta 1791:519–523
Yamaguchi T, Omatsu N, Matsushita S, Osumi T (2004) CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J Biol Chem 279:30490–30497
Yamaguchi T, Omatsu N, Morimoto E, Nakashima H, Ueno K, Tanaka T, Satouchi K, Hirose F, Osumi T (2007) CGI-58 facilitates lipolysis on lipid droplets but is not involved in the vesiculation of lipid droplets caused by hormonal stimulation. J Lipid Res 48:1078–1089
Yang X, Lu X, Lombes M, Rha GB, Chi YI, Guerin TM, Smart EJ, Liu J (2010) The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab 11:194–205
Yang X, Zhang X, Heckmann BL, Lu X, Liu J (2011) Relative contribution of adipose triglyceride lipase and hormone-sensitive lipase to tumor necrosis factor-alpha (TNF-alpha)-induced lipolysis in adipocytes. J Biol Chem 286:40477–40485
Yang D, Chen H, Zeng X, Xie P, Wang X, Liu C (2016) Macrophage CGI-58 attenuates inflammatory responsiveness via promotion of PPARgamma signaling. Cell Physiol Biochem 38:696–713
Yang D, Li Y, Xing L, Tan Y, Sun J, Zeng B, Xiang T, Tan J, Ren G, Wang Y (2018) Utilization of adipocyte-derived lipids and enhanced intracellular trafficking of fatty acids contribute to breast cancer progression. Cell Commun Signal 16:32
Yang A, Mottillo EP, Mladenovic-Lucas L, Zhou L, Granneman JG (2019) Dynamic interactions of ABHD5 with PNPLA3 regulate triacylglycerol metabolism in brown adipocytes. Nat Metab 1:560–569
Yen CL, Farese RV Jr (2006) Fat breakdown: a function for CGI-58 (ABHD5) provides a new piece of the puzzle. Cell Metab 3:305–307
Yim CY, Sekula DJ, Hever-Jardine MP, Liu X, Warzecha JM, Tam J, Freemantle SJ, Dmitrovsky E, Spinella MJ (2016) G0S2 suppresses oncogenic transformation by repressing a MYC-regulated transcriptional program. Cancer Res 76:1204–1213
Young SG, Zechner R (2013) Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev 27:459–484
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M (2016) Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64:73–84
Youssefian L, Vahidnezhad H, Saeidian AH, Pajouhanfar S, Sotoudeh S, Mansouri P, Amirkashani D, Zeinali S, Levine MA, Peris K, Colombo R, Uitto J (2019) Inherited non-alcoholic fatty liver disease and dyslipidemia due to monoallelic ABHD5 mutations. J Hepatol 71:366–370
Zagani R, El-Assaad W, Gamache I, Teodoro JG (2015) Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget 6:28282–28295
Zechner R, Strauss JG, Haemmerle G, Lass A, Zimmermann R (2005) Lipolysis: pathway under construction. Curr Opin Lipidol 16:333–340
Zhang L, Godzik A, Skolnick J, Fetrow JS (1998) Functional analysis of the Escherichia coli genome for members of the alpha/beta hydrolase family. Fold Des 3:535–548
Zhang J, Xu D, Nie J, Han R, Zhai Y, Shi Y (2014) Comparative gene identification-58 (CGI-58) promotes autophagy as a putative lysophosphatidylglycerol acyltransferase. J Biol Chem 289(47):33044–33053
Zhang X, Saarinen AM, Hitosugi T, Wang Z, Wang L, Ho TH, Liu J (2017) Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. Elife 6:e31132
Zhou Q, Wang F, Zhou K, Huang K, Zhu Q, Luo X, Yu J, Shi Z (2019) Oncogenic role of ABHD5 in endometrial cancer. Cancer Manag Res 11:2139–2150
Zhu J, Lee B, Buhman KK, Cheng JX (2009) A dynamic, cytoplasmic triacylglycerol pool in enterocytes revealed by ex vivo and in vivo coherent anti-Stokes Raman scattering imaging. J Lipid Res 50:1080–1089
Zierler KA, Jaeger D, Pollak NM, Eder S, Rechberger GN, Radner FP, Woelkart G, Kolb D, Schmidt A, Kumari M, Preiss-Landl K, Pieske B, Mayer B, Zimmermann R, Lass A, Zechner R, Haemmerle G (2013) Functional cardiac lipolysis in mice critically depends on comparative gene identification-58. J Biol Chem 288:9892–9904
Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R (2004) Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306:1383–1386
Acknowledgment
This work was partially supported by the grants DK111052 (to L.Y.) and DK116496 (to L. Y.) from the National Institute of Diabetes and Digestive and Kidney Diseases, the grant 1-18-IBS-346 (to L.Y.) from the American Diabetes Association Research Foundation, and the grant 17GRNT33670590 (to L.Y.) from the American Heart Association.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Yu, L., Li, Y., Grisé, A., Wang, H. (2020). CGI-58: Versatile Regulator of Intracellular Lipid Droplet Homeostasis. In: Jiang, XC. (eds) Lipid Transfer in Lipoprotein Metabolism and Cardiovascular Disease. Advances in Experimental Medicine and Biology, vol 1276. Springer, Singapore. https://doi.org/10.1007/978-981-15-6082-8_13
Download citation
DOI: https://doi.org/10.1007/978-981-15-6082-8_13
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-6081-1
Online ISBN: 978-981-15-6082-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)