
Abstract
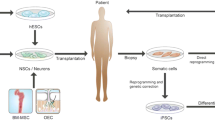
Parkinson’s disease (PD) is one of the most common neurodegenerative diseases caused by specific degeneration and loss of dopamine neurons in substantia nigra of the midbrain. PD is clinically characterized by motor dysfunctions and non-motor symptoms. Even though the dopamine replacement can improve the motor symptoms of PD, it cannot stop the neural degeneration and disease progression. Electrical deep brain stimulation (DBS) to the specific brain areas can improve the symptoms, but it eventually loses the effectiveness. Stem cell transplantation provides an exciting potential for the treatment of PD. Current available cell sources include neural stem cells (NSCs) from fetal brain tissues, human embryonic stem cells (hESCs) isolated from blastocyst, and induced pluripotent stem cells (iPSCs) reprogrammed from the somatic cells such as the fibroblasts and blood cells. Here, we summarize the research advance in experimental and clinical studies to transplant these cells into animal models and clinical patients, and specifically highlight the studies to use hESCs /iPSCs-derived dopaminergic precursor cells and dopamine neurons for the treatment of PD, at last propose future challenges for developing clinical-grade dopaminergic cells for treating the PD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Parkinson’s disease (PD) is caused by eventual neurodegeneration of dopamine neurons in the substantia nigra of middle brain. It is clinically characterized by bradykinesia, tremor, rigidity, and later postural instability of motor symptoms and related non-motor symptoms. Currently there is no treatment to stop the disease progression of PD. However, over the past 30 years studies indicated that PD is mainly caused by synuclein-mediated protein aggregation (lewy body), leading to cell death of dopamine neurons (DA neurons). Further studies have discovered that other neurons such as the cholinergic neurons or astrocytes may also be affected to induce non-motor symptoms such as depression, constipation, pain, and sleeping dysfunctions in the early development of PD. The most effective treatment is to use levodopa/carbidopa, dopamine agonists (both ergot and non-ergot types) to replace dopamine. Neverthless, the levodopa replacement can only relieve the symptoms of PD without affecting the progression of disease. Furthermore, the levodopa replacement can cause the side effect of involuntary muscle movements called dyskinesias. Another useful treatment for PD is called deep brain stimulation (DBS) which is to surgically implant the electrodes to the subthalamic nucleus to improve motor symptoms of PD by the unknown electrophysiological mechanisms. Most possibly the DBS is to increase the dopamine release of the undegenerated DA neurons, but DBS treatment has been found to lose effectiveness over time. Because of the specific cell death of dopamine neurons in PD, the neural stem cell transplantation has been considered the potential treatment and extensively explored for more than two decades (Berg et al. 2014; Olanow et al. 2001; Postuma et al. 2015).
In the 1980s and 1990s, early studies ever got enthusiastic results by transplanting fetal brain-derived neural stem cells (fNSCs) into the striatums of PD patients. Afterward, the expanded large-scale double-blind studies were tried to use fNSCs for PD patients in different clinical trials. However these studies did not convince the early findings, promoting research to explore other stem cell sources for PD. The bone marrow–derived mesenchymal stem cells (BM-MSCs) or umbilical cord–derived mesenchymal stem cells (UC-MSCs) were also transplanted for the treatment of PD in animal models and got some therapeutic effects. After the successful isolation and culturing of human embryonic stem cells (hESCs) from in vitro fertilized blastocysts, researchers have developed efficient differentiation methods to largely produce dopaminergic progenitors or dopamine neurons for transplantation therapy of PD and other neurodegenerative diseases. To overcome the ethical issues and immune rejection of fNSCs and hESCs, induced pluripotent stem cells (iPSCs) reprogrammed from patients’ somatic fibroblasts or blood cells have been recently explored to derive suitable cells for the treatment of PD, even for all other diseases (Han et al. 2015a, b; Lindvall and Björklund 2011; Takahashi et al. 2007).
2 Etiology and Genetic Study
Generally, PD can be divided into familial and sporadic cases. Most of PD cases are sporadic (80–90%) whereas the inherited familial cases only account for 10–20%, which are usually caused by genetic mutations in associated PD genes (Chen et al. 2014; Han et al. 2016; Lesage and Brice 2009). The twin studies also suggested that genetic mutations may not be a major factor causing typical PD, especially for the late-onset patients (Chung et al. 2013; Tanner et al. 1999). However, the discovery of the mutated genes in familial PD has largely contributed to uncover the molecular mechanism and therapeutic targets for PD.
Since the α-synuclein (SNCA) gene was first reported as the PD gene in 1997 (Polymeropoulos et al. 1997), more and more genetic linkage and association studies have identified more than 30 genes or susceptible loci related to familial and sporadic PD (Bandres-Ciga et al. 2020). Different mutations in the SNCA and LRRK2 genes of autosomal dominant PD genes have been extensively investigated to explore how the mutant proteins of SNCA and LRRK2 cause the cell death of the neurons and dopamine neurons in the brain. Soon afterward, the PARK2 (Parkin), PINK1, PARK7, PLA2G6, and ATP13A2 of autosomal recessive genes were identified and shown to mainly contribute to the pathogenesis of PD through the mitochondrial-lysosome pathways. In the meanwhile several susceptible genes or loci of Tau, Nurr1, and GBA were also reported to be associated with PD (Han et al. 2016; Yu et al. 2015). Recently more new PD loci (ACMSD, STK39, MCCC1/LAMP3, SYT11, and CCDC62/HIP1R) were identified through the genome-wide association study (Deng et al. 2018; Nalls et al. 2011).
The most important progress of the molecular pathological studies is to recognize the aggregation of alpha-synuclein to be the key factor for PD cases. A lot of studies have identified different point mutations (A53T, A30P, E46K) of SNCA and its genomic rearrangements including the duplication and triplications in different families with PD. It is now understood that the specific pathological lewy bodies in brains of PD patients are mainly composed of the misfolding and aggregation of α-synuclein, ubiquintin, and other proteins in the dopamine neurons (Surmeier 2018). In addition, another autosomal dominant PD gene of LRRK2 was found to modify the alpha-synuclein and combine to contribute to the pathology of PD. The mutations in LRRK2 occur in each exon and exon-intron boundaries of the LRRK2 gene. The most common pathological LRRK2 mutations are R1441C, Y1699C, G2019A, and I2020T. It was reported that the G2019S and R1441C mutations of LRRK2 account for 1–3% of familial PD cases and sporadic PD cases. These mutations were shown to increase the kinase activity (Gain of function) of LRRK2 protein to play a toxic role in PD (Grimes et al. 2007; Martin et al. 2014). The parkin gene is the second one to be identified in autosome recessive families of PD (PARK2). Most of the parkin mutations are exonic deletions but missense, nonsense mutations, and genomic rearrangements were also found in PD families (Sliter et al. 2018). Molecular studies have found that the parkin has the enzymatic activity domain of ubiquitin and the RING-like structures with some ubiquitin-ligase activity.
Other susceptible genes are also reported to be associated with PD. We had ever screened 202 familial and sporadic PD patients for NURR1 (NR4A2) mutations and identified a novel missense mutation in exon 3 of the NURR1 gene in one sporadic PD individual. This point mutation produced a truncated NURR1 protein which is unable to bind the promoter region of the tyrosine hydroxylase (TH) (Grimes et al. 2006). Pathological mutations in GBA were first reported in patients with lysosome diseases such as GD (gaucher’s disease). Later GBA mutations were also reported to confer more risk to the development of PD. We have recently performed a case control study in a Chinese cohort with PD and a Chinese control cohort by sequencing all the 12 exons of the GBA gene and found the PD patients have significantly higher frequency of mutations in the GBA gene. Totally we found 9 reported and 3 novel GBA mutations in 184 Chinese patients. These novel mutations are 5-bp deletion (c.334_338delCAGAA), L264I and L314V and the nine known GBA mutations are R163Q, F213I, E326K, S364S, F347L, V375L, L444P, RecNciI, and Q497R. Importantly we identified the novel 5-bp deletion (CAGAA) which produces a non-functional GBA protein of 142 amino acids, which loses major enzymatic function domains of the full GBA protein (Yu et al. 2015). The mechanism for mutations in GBA to cause the neural death of dopaminergic neurons is discussed in the following Sect. 3.2.
3 Animal Models and Molecular Mechanisms
3.1 Animal Model of PD
The cell degeneration of DA neurons is usually accompanied by lewy bodies formed by insoluble aggregates of alpha-synuclein, ubiquitin, and other misfolded proteins, and aggregated lewy bodies are toxic to neural stem cells, neurons, and glial cells (Dawson et al. 2010; Surmeier 2018). To explore the pathogenesis and effective treatment of PD, animal models from different species have been generated to recapitulate the phenotypes of PD. However, each of the current PD models has its limitations and no animal models can completely recapitulate clinical and pathologic characteristics of PD. Currently, three kinds of PD animal models can be available, the neurotoxin-induced model, the transgenic model, and the newly synuclein protein–induced model.
The commonly used drug-induced animal models of PD include mouse, rat, and non-human primate models. The classic PD model is 6-OHDA (6-hydroxydopamine)-lesioned acute PD rat model which can have the motor behavior deficits, but pathologically the 6-OHDA-induced rats do not have pathological lewy body aggregation in dopamine neurons of the brain. 6-OHDA is an analogue of dopamine which can be directly injected to substantia nigra of the rat brains to destroy the DA neurons to induce the hemi-Parkinsonism (Chao Chen et al. 2016). MPTP (1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine) is a synthetic heroin analogue, which can also induce the injury to DA neurons in mice and non-human primates, but not in rats, probably because the rats resist the metabolites of MPTP. MPTP-induced primate monkey models of PD are able to reproduce most, although not all, of the clinical and pathological hallmarks of PD and have been widely used to understand disease pathophysiology and develop potential therapeutics including cell transplantation therapy (Fox and Brotchie 2010; Kikuchi et al. 2017). The transgenic PD models were also developed by overexpressing or knock-out the PD-related genes such as LRRK2, SNCA, DJ1, and Parkin. The alpha-synuclein transgenic mice have been developed by overexpressing the wild-type and mutant SNCA (WT, A30P, A53T), but these synuclein-based animal models rarely have the phenotypic characteristics of PD as these models do not have progressive loss of DA neurons in brains of the mice (Chesselet et al. 2008).
Since the transgenic animal models have limitations to recapitulate the specific loss of dopamine neurons, the synuclein protein–based models were created. One study used rAAV vectors to express wild-type or mutant α-synuclein in the dopaminergic neurons in the substantia nigra of midbrains. The motor defects were seen in this model, which include the apomorphine-induced rotation turns. Other AAV serotypes such as rAAV2/1, rAAV2/5, rAAV2/6, rAAV2/7, and rAAV2/8 have also been used to express alpha-synuclein in animal models to increase transduction efficiency and expression level of α-synuclein in the brain of rats or mice (Koprich et al. 2010; Lundblad et al. 2012). The chronic degenerated PD model is to inject the α-synuclein-formed fibrils to the substantia nigra or other brain areas of the animals to induce the degeneration of the dopamine neurons. This PD model has the formation of protein-aggregated inclusions which can grow and propagate throughout the neurons of the brain (Volpicelli-Daley et al. 2011). The advantage of α-synuclein fibrils-induced models and rAAV-α-synuclein models are formation of pathological cytoplasmic inclusion and have the slowly progressive loss of dopamine neurons, which are more similar to the pathological process of human PD (Paumier et al. 2015).
3.2 Molecular Pathogenesis of PD
The pathogenesis of PD is attributed to the environmental, genetic and aging factors. In the development of PD, the dopamine neurons seem to be more vulnerable to toxic proteins and misfolded protein aggregates. Under the pathological conditions, the cytoplasmic soluble α-synucleins are misfolded and eventually form oligomers to form fibrils and insoluble protein aggregates, which eventually induce the cell death of dopamine neurons through different molecular pathways such as the ubiqintination-mediated protein degradation and endoplasmic reticulum-Golgi localization of the regulating factors such as RAB1a for vesicular transport in midbrain dopamine neurons. The genetic mutations in PD genes can lead to an increase in the production of missense proteins or affect the functions of the proteins such as the activation of the LRRK2 kinase or decrease the Parkin ubiqintinase activity. In addition, the protein trafficking and degradation can also be affected and convergently lead to increased membrane permeability and degeneration of dopamine neurons in PD (Fares et al. 2014; Gonzalez-Horta 2015). Mutations in β-glucocerebrosidase (GBA) were reported to decrease the activity of glucocerebrosidase and increase the production of the glucosylceramides to affect lysosomal function. Recent studies revealed that GBA are associated with misfolded α-synucleins to cause neuronal death in PD, highlighting GBA as a new therapeutic target for PD (Blanz and Saftig 2016; Yu et al. 2015).
In the past years the research on pathogenesis of PD has made progress on the abnormalities of mitochondria, lysosomal-proteasome, autophagy, and oxidative stress pathways through in vitro and in vivo studies. Autosomal recessive gene mutations in PINK1, parkin, DJ-1have uncovered the importance of mitochondrial dysfunction in PD. Several studies suggested that a crosstalk between lysosomes and mitochondria in the pathogenesis of PD. Autosomal dominant mutant LRRK2 interact with alpha-synuclein to induce abnormal protein aggregations which have been shown to localize to mitochondria and lysosomes, further supporting the mitochondrial and endo-lysosomal dysfunctions play key roles in PD (Plotegher and Duchen 2017; Zhang et al. 2018a). The molecular pathological mechanism in the development of PD is diagramed as in Fig. 3.1 (Martin et al. 2011; Zhang et al. 2018a).

The molecular pathways leading to the dysfunctions of the mitochondria, lysosomal-proteasome and autophagy in development of PD. Autosomal dominant Mutations of SNCA and LRRK2 in sporadic PD lead to protein aggregation and fibrillization to form oligomers and fibrils which disrupt the functions of autophages, lysosomes and mitochondria, which gradually induce neural cell apoptosis in PD. In addition, autosomal recessive loss of function mutations in Parkin, PINK1 and DJ1 impair the functions of Ubiquintin 3 ligase, Kinase and Chaperone peroxidase activities, which lead to accumulation of some unidentified pathogenic substrates and neurodegeneration in PD
4 Cell Transplantation Therapy in PD
Cell transplantation for PD has been explored for more than 30 years, but no reliable cell line or cell sources are available for the patients until now. However, different stem cell lines have been studied for the treatment of PD in animal models and some clinical patients. Here we discuss each of the stem cell lines for the treatment of PD as outlined in Chaps. 1 and 2.
4.1 Mesenchymal Stem Cells
Mesenchymal stem cells (MSCs) can be isolated from most of the organs and tissues including bone marrow, muscle, skin, dental pulp, peripheral blood, and umbilical cords. These MSCs have been shown to have multipotent differentiation into mesodermal, endothelial, and ectodermal cell lineage including neuronal cells (Hass et al. 2011; Zhang et al. 2016). Mesenchymal stem cells from bone marrow (BM-MSC) and umbilical cords (UC-MSC) have been used in tissue engineering and neural repair because they are easy to be isolated from the adults or newborns and can proliferate for more than 30 passages in vitro without losing their multipotent differentiations. The transplantation of BM-MSCs and UC-MSCs into the 6-OHDA-induced PD rat models was shown to protect damaged DA neurons and differentiate to neural stem cells and mature DA neurons, indicating their therapeutic efficacy for PD. In vitro studies BM-MSCs were able to be induced to neuronal cells expressing markers of neurons (TUJ1) and DA neurons (TH). After transplantation most of the BM-MSC-derived cells survived in striatum, expressed TH of DA neuronal marker, and restored the motor defects of 6-OHDA-induced rats and MPTP-induced mouse models (Blandini et al. 2010; Han et al. 2018; Park et al. 2008a). As LMX1a is a key transcriptional factor to regulate dopaminergic neuron differentiation, over-expressing LMX1a in BM-MSCs was able to improve BM-MSCs toward dopaminergic differentiation fate, with high expression levels of tyrosine hydroxylase (TH) (Barzilay et al. 2009). Our lab showed that human hUC-MSCs were efficiently induced to convert to neurons (73.1 ± 2.9%) and dopamine neurons (36.3 ± 1.8%) by combinations of noggin, CHIR99021, SHH, FGF8, TGFβ, GDNF, and BDNF. The transplantation of the hUC-MSCs with the growth factors into the 6-OHDA-lesioned rat model of PD was shown to improve motor dysfunctions of these rats from weeks 4 to 16 post-grafting. The efficacy and usefulness of the growth factors in combination with hUC-MSC transplantation in 6-OHDA-lesioned rats provided a promising cell-based treatment strategy for the PD (Han et al. 2018).
4.2 Fetal Brain-Derived Neural Stem Cells and Dopamine Neurons
Fetal brain-derived neural stem cells (NSCs) have been widely studied for their expansion, and differentiation, and transplantation for the treatment of neurological diseases such as PD, AD, ALS, and spinal cord injury (SCI) (Courtois et al. 2010; Kallur et al. 2006). These fNSCs can be isolated from various regions of human fetal brains at 11–13 weeks of gestation and cultured as neurospheres for long-term expansion. The differentiation abilities fNSCs derived from the different brain regions seem to be regionally specific and temporally different from each other. NSCs have a strong proliferative ability and can differentiate into specific neurons, astrocytes, and oligodendrocytes for the treatment of different neurological diseases. The midbrain-derived NSCs are rich in mesencephalic DA neurons suitable for the transplantation treatment of PD (Kim et al. 2006).
After transplantation into the 6-OHDA-lesioned rats, human fetal NSCs were shown to improve the motor defects of these PD rats and the transplanted cells were found to survive in host brains (Monni et al. 2014; Studer et al. 1998). Our lab has recently shown that human fetal NSCs transplanted into the brain of the 6-OHDA-lesioned rats survive and differentiate to dopaminergic neurons in vivo (Wang et al. 2015). Moreover, in PD rats with depleted DA levels, engrafted NSCs tended to be sensitive to microenvironment to differentiate preferentially to DA neurons in the middle brain of the rats (Eriksson et al. 1998; Taupin and Gage 2002). In order to increase the survival of the transplanted cells, some studies modified NSCs to overexpress neurotrophic factors. Some homeodomain proteins of Lmx1a and Msx1 were found to increase the induction of DA neurons. NSCs overexpressing the neural-specific transcription factor ASCL1 were found to increase neurogenesis and to produce larger neurons with more neurites (Kim et al. 2009). Because of the limited availability of the human fetal midbrain tissues, one study developed a method to increase expansion of fetal NSCs and generate more than threefold cells by culturing neurospheres on non-adherent tissue culture plates in neural basal medium containing MEM/F12, HEPES buffer, glutamine, Albumax, and N2 supplement with Shh, FGF8, BDNF, and bFGF. These expanded neurospheres of fetal NSCs can be frozen down for cell banking and retain their ability to efficiently differentiate to the dopamine neurons for the transplantation treatment of PD (Ribeiro et al. 2013).
4.3 Human Embryonic Stem Cells (hESCs)–Derived Neural Stem Cells and Dopamine Neurons
Because of the limited sources of fetal NSCs, human embryonic stem cells (hESCs) have also been explored to derive dopaminergic precursors or dopamine neurons for the cell sources of PD. hESCs are pluripotent stem cells and can potentially differentiate to any type of tissue cells such as blood cells, cardiac cells, and neurons. At first hESCs can only propagated and cultured on cell feeder layer of mouse embryonic fibroblasts (MEFs) (Thomson et al. 1998). After more than 20 years of research, different protocols have developed to culture the hESCs in feeder-free and xeno-free medium and induce hESCs to dopaminergic precursors or dopamine neurons by specific patterning molecules to regulate midbrain development. After 3–4 weeks of induction, hESC colonies differentiated to tyrosine hydroxylase (TH)–positive dopamine neurons. The transplantation of hESCs-derived neural precursor cells to the 6-OHDA-lesioned rats was shown to alleviate the motor dysfunctions and the grafted cells are able to differentiate to DA neurons in vivo and integrated into the rat striatums (Nakagawa et al. 2014; Zeng et al. 2004; Ma et al. 2011).
To produce specific neurons for PD therapy, other protocols were also developed for generating specifically midbrain-like DA neurons. The treatment of hESC with neural-patterning growth factors SHH and FGF8a resulted in midbrain projection DA neurons with large cell bodies to coexpress DA markers of TH and engrailed 1 (En1) (Yan et al. 2005). These in vitro generated DA neurons were electrophysiologically active and released DA in an activity-dependent manner. After transplantation, these hESC-derived dopamine neurons were shown to integrate in the host brains and were able to improve the motor deficiency of PD rats. To increase the efficiency of DA neuron differentiation, hESCs were induced by adding Noggin and SB431542 to inhibit SMAD signaling. The addition of Noggin and SB431542 induced more than 80% of hESCs converted to neural fate under adherent culture conditions, but the human DA neuron specification is still limited (Chambers et al. 2009). To get more conversion of DA neurons, the same group further developed a midbrain floor-plate-based protocol for generating DA neurons from hESCs by adding activators of sonic hedgehog (SHH) and canonical WNT signaling in differentiation medium. As a result, midbrain floor plate precursors were derived from hESCs by 11 days and by day 25 midbrain DA neurons expressing TH, LMX1A, and FOXA2 were obtained. Importantly these engraftable DA neurons were able to grow for several months in vitro and restored the motor behavior deficits in transplanted Parkinsonian monkeys and 6-OHDA-lesioned rats (Kriks et al. 2011). Another group induced DA neuron conversion of hESCs by expressing LMX1A to obtain more than 60% ventral mesencephalic DA neurons of all neurons derived from LMX1A-modified hESC (Sanchez-Danes et al. 2012). After being transplanted to 6-OHDA-induced PD rats, these hESC-DA neurons integrated into host rats to form the neural connections with the neurons of host brains and improve the motor deficits of PD rats as similar as the transplanted fetal brain DA neurons. This study provided further preclinical basis of hESC-derived dopamine neurons for the treatment of PD patients (Grealish et al. 2014). In the meanwhile mouse embryonic stem cell–derived neurons and dopamine neurons were also shown to differentiate to TH-positive dopamine neurons and played neuroprotective effects on PD (Liu et al. 2013).
4.4 Induced Pluripotent Stem Cells (iPSCs)–Derived Neural Stem Cells or Dopamine Neurons
As NSCs and dopamine neurons from fetal brain tissues and hESCs have ethical and immune rejection problems, in 2006, the successful generation of iPSCs provided great cells for autologous cell-based treatment of diseases by somatic reprogramming technology to introduce Oct3/4, Sox2, c-Myc, and Klf4 into mouse somatic fibroblasts (Takahashi and Yamanaka 2006). One year afterward, patient-specific iPSCs with PD or other diseases were also generated by expressing OCT4, SOX2, c-MYC, KLF4 or OCT4, SOX2, NANOG, and LIN28 in human fibroblasts (Park et al. 2008b; Yu et al. 2007). iPSCs have the same pluripotency with hESCs, overcome the shortages of ESCs, and can be used for exploring the molecular mechanisms, drug screening, and cell replacement therapy of neurodegenerative diseases such as PD, AD, MS, and ALS (Egawa et al. 2012; Han 2012; Isobe et al. 2015; Li et al. 2011).
To explore the therapeutic effects of iPSCs-derived cells, Wernig et al. reported that iPSCs-derived midbrain-like dopamine neurons, resulting in motor behavioral improvements in rat PD models (Wernig et al. 2008). Because the possibility of integration of viral vectors and transgenes in the genome of iPSCs from patients may induce malignant transformation or affect their differentiation potential, PD patient-iPSCs free of transgenes were derived using Cre-recombinase to excise the reprogramming factors or iPSCs were generated using the non-integrating episomal plasmids to express the genes of OCT4,SOX2, KLF4, NANOG, and tumor-suppressing gene p53 in fibroblasts or blood cells (Kadari et al. 2014; Okita et al. 2011).
After transplantation, the virus-free iPS cells–derived DA neurons were able to survive and improve motor defects of the 6-OHDA-lesioned rats (Hargus et al. 2010). Recently our lab generated iPS cells by retrovirus-mediated expression of OCT4, SOX2, c-MYC, and KLF4 from skin fibroblasts of PD patients and control individuals. We found that the iPSCs carrying the transgenes can also be differentiated to the NSCs and be differentiated to neurons and DA neurons in vitro and in vivo. The grafted iPS cells–derived dopamine neurons integrated to the host brains and significantly alleviated the rotational asymmetry of PD rats (Han et al. 2015b).
As we noticed in transplanted animal models or PD patients, only a small percentage of cells survived whereas a majority of the transplanted are rejected or cannot survive for more than 6 months. This is mainly caused by the immune rejections or no physical integration of the grafted cells to the host neurons to form the functional neural circuit. In order to overcome this issue, we have used the growth cocktail to increase the survival of transplanted cells and improve the host brain microenvironment for the synaptic connections to get the long-term therapeutic effects in PD (Han et al. 2015b; Zhang et al. 2018b) (Fig. 3.2).

Transplanted human iPSC-derived neural stem cells differentiated to the dopamine neurons and integrated to 6-OHDA-induced rat model with PD. Upper panel: human iPSC-derived dopamine neurons were co-stained with HNuc and TH. Lower panel: Transplanted human iPSC-derived dopamine neurons formed synaptic connection with host brain cells in vivo, which is co-stained with TH and synaptophysin
To efficiently derive the patient-specific iPSCs, much work has been done to increase the neural and dopaminergic neuronal differentiation of iPSCs. Studer lab reported that using two inhibitors of SMAD signaling, Noggin and SB431542, is able to produce complete neural conversion of >80% of hESC and iPS cells under adherent culture conditions. Hereafter this group modified the induction method to obtain midbrain DA neurons by 25 days and transplanted the human ESC-derived DA neurons to the monkey model with PD. These grafted DA neurons survived for a longer time and were able to completely restore the amphetamine-induced rotation behavior and improvements in tests of forelimb use, indicating promise for the development of iPSC-based therapies in Parkinson’s disease (Kriks et al. 2011). Other labs used fibroblast growth factor 8 (FGF8) to promote dopaminergic differentiation and sonic hedgehog and GSK3β inhibitor of CHIR99021 to induce the midbrain floor plate (FP) progenitors to establish a robust system for the generation of midbrain dopamine (DA) neurons from human and rhesus monkey embryonic stem cells and induced pluripotent stem cells (PSCs) (Xi et al. 2012). Our lab has also derived the neural stem cells from iPSCs, transplanted the neural stem cells to the 6-OHDA-induced rat PD model, and found the grafted iPSC-NSCs differentiated to the neurons and dopamine neurons in vivo to improve the motor defects of the rats (Wang et al. 2015). The detailed protocol for rapid and efficient conversation of iPSCs to the dopamine neurons is described as in Fig. 3.3.

Derivation of dopamine neuron precursors and dopamine neurons from hESCs/iPSCs. The time schedule for 4–6-week dopaminergic neural differentiation includes 4 stages of neural induction (first 2 weeks), neural stem cell (Third week), neuron precursor (Fourth week) and dopamine neuron (5–6 week). hESCs/iPSCs were cultured in hESC medium with SB + Noggin in the first 5 days and after that were cultured in DMEM/F12 + N2 medium with SHH, GDNF, TGF-β, cAMP, BDNF, ascorbic acid (AA), FGF8b at different time points. (Our lab protocol modified from Kriks et al. (2011) and Ma et al. (2011))
To produce enough and safe clinical-grade midbrain DA neurons from iPS cells, Isacson et al. have developed a protocol to sort ventral mesencephalic DA neurons from both human ESCs and iPS cells by antibodies of NCAM (+)/CD29 (low) to get rid of the undifferentiated iPS cells. Molecular analysis showed that the sorted neurons were positive for TH, FOXA2, and EN1 and had elevated expression levels of dopamine neuron markers of FOXA2, GIRK2, PITX3, LMX1A, TH, and NURR1. In-vivo studies showed that 16 weeks after transplantation the sorted iPSC-DA neurons were able to form neural connections with the rat brains with TH+/hNCAM+ staining in the host striatum and alleviate motor defects of 6-OHDA-lesioned rats. Their results provided experimental support for the clinical use of iPSC-derived DA neuron in PD (Sundberg et al. 2013). For the clinical purpose of iPSC-derived DA neurons, a transgene-free, xeno-free and scalable differentiation protocol is needed. A suspension culture system was created for the neural differentiation of hESCs and human iPSCs. To decrease the effects of transgenes on pluripotency of iPSCs, several labs developed protocols to use two or three factors to generate iPSCs. iPSCs can be reprogrammed from mouse adult and embryonic fibroblasts with the single factor of OCT4 in combination of small molecules of CHIR 99021,VPA, TGF-β inhibitor (616452) (Li et al. 2011). A recent study also indicated that the derivation of naive iPSCs from rhesus monkey fibroblasts can be obtained with only small molecules, omitting the OCT4, which provided a valuable cell source for further use in disease modeling and pre-clinical study (Fang et al. 2014).
To determine if the human iPS cell–derived DA neurons can survive and play therapeutic effects in a non-human primate model of PD, a Japanese group transplanted the CORIN-sorted iPSC-dopaminergic progenitor cells to MPTP-induced PD monkeys and found that the grafted cells further differentiated to the DA neurons which extended their neurites into the host striatum and improved the spontaneous movement of the monkeys. These transplanted cell did not produce any tumor cells in the monkey brains for more than 2 years, indicating the safety of human iPS cell–derived dopaminergic progenitor cells to be used for clinical patients (Kikuchi et al. 2017).
To overcome the possibility of tumoric cells restraining in the iPS-dervied DA neurons, another approach is to generate direct dopaminergic precursor cells/dopamine neurons from somatic fibroblasts. A lot of studies have reported the generation of trans-differentiated DA neurons by directly reprogramming the fibroblasts with different combinations of transcription factors such as Mash1 (Ascl1), Nurr1 (Nr4a2), Ngn2, Sox2, Lmx1a, and Pitx3 (Caiazzo et al. 2011; Kim et al. 2014). Since most of these studies used the lentiviral expression of dopaminergic genes to convert fibroblasts to DA-like neurons and have the risk to produce genome instability, recently Mou et al. used silica nanoparticles (MSNs) as a non-viral delivery system to express the key dopaminergic genes of Ascl1, Brn2, and Myt1l to convert mouse fibroblasts (MFs) into functional dopaminergic neuron-like cells. These DA-like neurons were validated to express the DA neuron-specific markers and have electrophysiological properties dopaminergic neurons (Chang et al. 2018). The problems to use the directly converted neurons is that the percentage of directly converted DA neurons from fibroblasts is still low and their purification should be obtained by sorting or other selection methods.
5 Clinical Trials in Using Stem Cells for the Treatment of PD
Following the beneficial therapeutic results of stem cell transplantation on animal models, the first clinical trials for PD patients were carried out in the late 1980s using fetal brain neural stem cells (fNSCs). After that more clinical studies have been done and significant effects were found by examining behavioral and histological improvement of the PD patients with transplanted fetal tissues (Freed et al. 2001; Lindvall et al. 1994). In a patient transplanted with fNSCs, clinical symptoms were shown to be gradually improved and L-dopa treatment could be withdrawn after 6 years of transplantation. After 10 years of grafting, the PET scan showed that patient still had dopamine release from grafted dopaminergic neurons (Piccini et al. 1999). Interestingly a study by Freed et al. compared the therapeutic effects of fNSC transplantation in patients who are younger than 60 years old with the patients who are older than 60 years and showed that more significant improvement of movement symptoms was seen in younger PD patients than the older patients (Freed et al. 2001). This suggested that the host brain microenvironment might play an important role in the treatment efficiency of grafted neural stem cells (Hagell and Brundin 2001). Although some variable improvements were found from the clinical trials, the therapeutic effects were confirmed by clinical examinations and imaging evaluations (Barker et al. 2013; Lindvall and Bjorklund 2004). Some patients have improved so well that L-DOPA could be withdrawn for several years (Olanow et al. 2003; Piccini et al. 1999). However, some cell-transplanted patients developed side effects such as dyskinesia. It was shown that transplanted cells containing serotonin neurons were easier to induce this side effect, suggesting that dyskinesia may be avoided by using purified dopaminergic neurons (Freed et al. 2001; Lindvall 2015; Olanow et al. 2003; Politis et al. 2010).
To know how long transplanted cells survive in PD patients, some clinical studies analyzed the brain slices of post-mortems 16 years after cells were grafted and showed that transplanted fNSCs were able to survive without pathology (Mendez et al. 2008). The transplanted fetal cells in brains of patients were found to stain with DA neuron marker, TH, indicating the transplanted DA neurons survived (Fig. 3.4a) (Mendez et al. 2008). However other two studies reported that alpha-synuclein-positive lewy bodies were also found in the transplanted cells in brains of PD patients, suggesting the pathological process of the host patients affecting the grafted cells. The lewy bodies were stained with alpha-synuclein and ubiquintin, indicating that the important role of synuclein in aggregated proteins of patients. (Kordower et al. 2008; Li et al. 2008). As other allogeneic transplantations, there is also a risk of graft rejection which may affect the efficacy of the transplanted cells (Michel-Monigadon et al. 2010) (Barker et al. 2013). Some of the clinical trials with fetal brain-derived NSCs or dopamine neurons to treat PD are summarized in Table 3.1.

Lewy bodies were formed in grafted cells transplanted to patients with Parkinson’s disease. (a) Some of the grafted fetal neural stem cells differentiated to the dopamine neurons stained with TH (Red) and serotoninergic neurons stained with TrypOH (Green). (b) TH/Girk2-stained dopamine neurons were preferentially located in the peripheral areas of the grafted cells. (c) TH/Calbindin-stained dopamine neurons were preferentially located in central areas of the grafted cells. (d–f) Double immunolabeling shows colocalization of TH (green, d) and a-synuclein (red, e) in a graft (arrowheads). One dopaminergic neuron does not contain detectable synuclein (arrows, d, f). Modified from Mendez et al. (2008) and Li et al. (2008)
In order to develop uniformed standards to evaluate the therapeutic effects of the transplanted fNSCs, a multicenter and collaborative study of European Union (TRANSEURO) was formed in 2010 to design the guidelines for clinical trials of PD patients with stem cell–based therapy. These screening standards include selection of patients, the time course of their disease (disease duration 2–10 years), and age range of 30–68 years old at the time of inclusion, being responsive to levodopa therapy. These guidelines also contain preparation of cells and location of cells to be transplanted; immunosuppression application after transplantation and time course of follow-up; minimum numbers of patients to be enrolled and clinical assessment standards such as the MRI and PET-CT. The new clinical trial for more than 100 patients suffered with PD has completed in this study, and results are in the analysis (Evans et al. 2012; Moore et al. 2014). A recent clinical study indicated that a patient with transplanted fetal dopamine neurons have significant recovery in striatal dopaminergic function. Pathological examinations showed a graft-derived dopaminergic reinnervation of the putamen can be survived for 24 years with no evidence of immune response in spite of some severe host brain pathology (Li et al. 2016).
Overall the fetal brain-derived dopaminergic neurons can survive for as long as 16–24 years in patients’ brains and improved the functions in some patients, although the outcomes of other clinical studies are variable. Another problem is the availability of the fetal brain tissues which limited the wide use of fNSCs. To overcome this problem of fNSCs, an international corporation was set up for Gfore in 2014 to start the world’s first clinical studies of an iPS cell–derived dopaminergic neuron therapy for PD in Europe, USA, and Japan. Some results of the trials are in the progress. At the same time outside of the Gforce, other clinical trials using iPS cell-DA neurons for the treatment of PD including the Chinese clinical trial for an HLA-matched HESC-derived dopaminergic neuron transplantation therapy for PD (NCT03119636, http://clinicaltrials.gov). Since August 2018, a Japanese group led by Takahashi has started the first clinical trial to use the iPSC-derived dopaminergic precursor cells and the incoming results will soon benefit for the PD patients (Takahashi 2019). The results of clinical trials with HESC- or iPSC-derived DA neurons for treatment of PD will be available soon.
6 Conclusion: Translation to the Clinic
A large number of studies showed that transplantation of fNSCs/hESCs/ iPSCs and their derivatives into animal models has a solid therapeutic effect for PD. One of the major advantages of iPSCs over BM-MSCs, fetal NSCs, and hESCs is that iPSCs can be generated from the autologous cells of the individuals being treated. Once the autologous skin or blood cells are reprogrammed into iPSCs, the iPSCs can be induced to a specific neural lineage such as dopaminergic precursor cells or mature DA neurons (Garitaonandia et al. 2018; Kiskinis and Eggan 2010). Thus, autologous or HLA-matched iPSCs seem to have advantages over other cell sources. Until now, a ton of work has been done to improve the generation, differentiation, and potential clinical applications of iPSCs, especially with great efforts made to bring these therapeutic cells to meet GMP (Good Manufacturing Practice) standards in order to apply the iPSC-derived dopaminergic cells for the treatment of PD and other neurodegenerative diseases. There are several challenges which need to be overcome for clinical uses of iPSCs.
The first challenge is to ensure the efficacy of iPSC-derived dopaminergic neurons after transplantation to the patients. The iPSC-derived dopaminergic neurons have to play similar therapeutic efficacy as human fetal dopaminergic neurons. Since animal research results may not be directly translated to the patients, more clinical trials are needed to define the GMP-graded cells, cell dosages, the injection routes and locations as well as the standard methods to evaluate the neural circuit formation, DA release, and the therapeutic effects of transplanted cells on patients. The second challenge is to ensure the safety of transplanted cells by reducing the tumor formation and the side effects of grafted cells. It is required that the residues of undifferentiated hESCs/iPSCs should be less than 1% to avoid the teratoma formation after transplantation to patients. The patient-derived iPSCs may harbor some mutations in the genes of SNCA, LRRK2, Parkin, Dj-1, GBA, or other loci associated with PD (Lister et al. 2011; Yu et al. 2015). These concerns are related to donor source of iPSCs and the reprogramming approaches which can produce new mutations in iPSCs. To maintain the genomic stability of iPS cells, non-integrating vectors should be used to express the reprogramming genes or combine with small molecules to generate clinical applicable iPSCs (Hou et al. 2013). On the other hand, several gene editing approaches have been developed including the zinc finger nucleases (ZEN), transcription activator-like effector nucleases (TALEN), and CRISPR/Cas9 (Jiaxin Xie et al. 2018; Wang et al. 2016). iPSCs with LRRK2 G2019S mutation were able to be corrected and the LRRK2 mutation correction rescued the phenotypes of differentiated neurons (Reinhardt et al. 2013). Soldner et al. reported that the iPSCs with SNCA mutation (A53T) was repaired using zinc finger nuclease (ZFN)–mediated nuclease approach and genetic repair of the A53T mutation in the patient-derived iPSCs did not affect their differentiation ability to dopaminergic neurons. A recent study combined the Cas9/CRISPR and piggyBac technologies to generate genome-edited footprint-free human iPSCs at the rate of 10–20%, which will largely increase clinical application in editing the iPSCs derived from PD patients and other patients with genetic mutations (Safari et al. 2019; Wang et al. 2017).
The third challenge is the efficiency of generating iPSCs and the purity of iPSC-derived cells. Since the lower efficiency of fully reprogrammed iPS cells through the non-integrated method is affecting the genome editing and their application, some studies tried to resolve his potential problem by the addition of VPA and other chemicals to increase the generation efficiency of iPSCs (Wang et al. 2011). A recent study summarized the generation of non-integration and feeder-free iPSCs by sendai viral vectors to express OCT4, SOX2, KLF4, and c-MYC in peripheral blood mononuclear cells (PBMCs) and reported the reprogramming efficiency is approximately 0.01% (Ye and Wang 2018). For increasing the purity of iPSC-derived dopaminergic precursor cells/dopamine neurons, several efficient differentiation methods were described in the previous sections. Recent studies purified the human iPS cell–derived dopaminergic neural precursor cells by cell surface-specific marker CORIN to sort the CORIN+ cells. The CORIN+ cells accounted for more than 30% of totally differentiated cells. Importantly these sorted CORIN+ cells expressed the dopaminergic precursor markers of NURR1 and FOXA2. After transplantation to the rat and monkey PD models, these CORIN+ cells differentiated to the dopamine neurons in vivo and improved the locomotive defects of these animal models without tumor formation, ruling out the concerns on tumorigenesis of iPS cell-differentiated cells (Kikuchi et al. 2017; Samata et al. 2016).
The fourth challenge is to screen for the most suitable PD patients to be transplanted with reprogrammed iPS cell–derived dopaminergic neurons in the clinical trials. To achieve the best efficacy and safety, patients have to be in a relatively earlier disease progression stage and have a chance of therapeutic benefit when the dopaminergic neuron loss is mainly affecting the caudate-putamen of midbrain and is not dispused in the forebrains of the patients (Barker et al. 2017).
References
S. Bandres-Ciga, M. Diez-Fairen, J.J. Kim, A.B. Singleton, Genetics of Parkinson's disease: an introspection of its journey towards precision medicine. Neurobiol. Dis. 137, 104782 (2020)
R.A. Barker, J. Barrett, S.L. Mason, A. Bjorklund, Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson’s disease. Lancet Neurol. 12, 84–91 (2013)
R.A. Barker, M. Parmar, L. Studer, J. Takahashi, Human trials of stem cell-derived dopamine neurons for Parkinson’s disease: dawn of a new era. Cell Stem Cell 21, 569–573 (2017)
R. Barzilay, T. Ben-Zur, S. Bulvik, E. Melamed, D. Offen, Lentiviral delivery of LMX1a enhances dopaminergic phenotype in differentiated human bone marrow mesenchymal stem cells. Stem Cells Dev. 18, 591–601 (2009)
D. Berg, R.B. Postuma, B. Bloem, P. Chan, B. Dubois, T. Gasser, C.G. Goetz, G.M. Halliday, J. Hardy, A.E. Lang, et al., Time to redefine PD? Introductory statement of the MDS task force on the definition of Parkinson”s disease. Mov. Disord. 29, 454–462 (2014)
F. Blandini, L. Cova, M.T. Armentero, E. Zennaro, G. Levandis, P. Bossolasco, C. Calzarossa, M. Mellone, B. Giuseppe, G.L. Deliliers, et al., Transplantation of undifferentiated human mesenchymal stem cells protects against 6-hydroxydopamine neurotoxicity in the rat. Cell Transplant. 19, 203–217 (2010)
J. Blanz, P. Saftig, Parkinson's disease: acid-glucocerebrosidase activity and alpha-synuclein clearance. J. Neurochem 139, 198 (2016)
M. Caiazzo, M.T. Dell'Anno, E. Dvoretskova, D. Lazarevic, S. Taverna, D. Leo, T.D. Sotnikova, A. Menegon, P. Roncaglia, G. Colciago, et al., Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227 (2011)
S.M. Chambers, C.A. Fasano, E.P. Papapetrou, M. Tomishima, M. Sadelain, L. Studer, Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280 (2009)
J.H. Chang, P.H. Tsai, K.Y. Wang, Y.T. Wei, S.H. Chiou, C.Y. Mou, Generation of functional dopaminergic neurons from reprogramming fibroblasts by nonviral-based mesoporous silica nanoparticles. Sci. Rep. 8, 11 (2018)
J.D. Chao Chen, A. Shen, W. Wang, H. Song, Y. Liu, X. Lu, X. Wang, Z. You, Z. Han, F. Han, Transplantation of human umbilical cord blood-derived mononuclear cells induces recovery of motor dysfunction in a rat model of Parkinson's disease. J. Neurorestoratol. 2016(4), 23–33 (2016)
M.L. Chen, C.H. Lin, M.J. Lee, R.M. Wu, BST1 rs11724635 interacts with environmental factors to increase the risk of Parkinson’s disease in a Taiwanese population. Parkinsonism Relat. Disord. 20, 280–283 (2014)
M.F. Chesselet, S. Fleming, F. Mortazavi, B. Meurers, Strengths and limitations of genetic mouse models of Parkinson’s disease. Parkinsonism Relat. Disord. 14(Suppl 2), S84–S87 (2008)
S.J. Chung, S.M. Armasu, K.J. Anderson, J.M. Biernacka, T.G. Lesnick, D.N. Rider, J.M. Cunningham, J.E. Ahlskog, R. Frigerio, D.M. Maraganore, Genetic susceptibility loci, environmental exposures, and Parkinson’s disease: a case-control study of gene-environment interactions. Parkinsonism Relat. Disord. 19, 595–599 (2013)
E.T. Courtois, C.G. Castillo, E.G. Seiz, M. Ramos, C. Bueno, I. Liste, A. Martinez-Serrano, In vitro and in vivo enhanced generation of human A9 dopamine neurons from neural stem cells by Bcl-XL. J. Biol. Chem. 285, 9881–9897 (2010)
T.M. Dawson, H.S. Ko, V.L. Dawson, Genetic animal models of Parkinson's disease. Neuron 66, 646–661 (2010)
H. Deng, P. Wang, J. Jankovic, The genetics of Parkinson disease. Ageing Res. Rev. 42, 72–85 (2018)
N. Egawa, S. Kitaoka, K. Tsukita, M. Naitoh, K. Takahashi, T. Yamamoto, F. Adachi, T. Kondo, K. Okita, I. Asaka, et al., Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4, 145ra104 (2012)
P.S. Eriksson, E. Perfilieva, T. Bjork-Eriksson, A.M. Alborn, C. Nordborg, D.A. Peterson, F.H. Gage, Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317 (1998)
J.R. Evans, S.L. Mason, R.A. Barker, Current status of clinical trials of neural transplantation in Parkinson's disease. Prog. Brain Res. 200, 169–198 (2012)
R. Fang, K. Liu, Y. Zhao, H. Li, D. Zhu, Y. Du, C. Xiang, X. Li, H. Liu, Z. Miao, et al., Generation of naive induced pluripotent stem cells from rhesus monkey fibroblasts. Cell Stem Cell 15, 488–496 (2014)
M.B. Fares, N. Ait-Bouziad, I. Dikiy, M.K. Mbefo, A. Jovicic, A. Kiely, J.L. Holton, S.J. Lee, A.D. Gitler, D. Eliezer, et al., The novel Parkinson's disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 23, 4491–4509 (2014)
S.H. Fox, J.M. Brotchie, The MPTP-lesioned non-human primate models of Parkinson's disease. Past, present, and future. Prog. Brain Res. 184, 133–157 (2010)
C.R. Freed, P.E. Greene, R.E. Breeze, W.Y. Tsai, W. DuMouchel, R. Kao, S. Dillon, H. Winfield, S. Culver, J.Q. Trojanowski, et al., Transplantation of embryonic dopamine neurons for severe Parkinson's disease. N. Engl. J. Med. 344, 710–719 (2001)
I. Garitaonandia, R. Gonzalez, G. Sherman, A. Semechkin, A. Evans, R. Kern, Novel approach to stem cell therapy in Parkinson's disease. Stem Cells Dev. 27, 951–957 (2018)
A. Gonzalez-Horta, The interaction of alpha-synuclein with membranes and its implication in Parkinson's disease: a literature review. Nat. Prod. Commun. 10, 1775–1778 (2015)
S. Grealish, E. Diguet, A. Kirkeby, B. Mattsson, A. Heuer, Y. Bramoulle, N. Van Camp, A.L. Perrier, P. Hantraye, A. Bjorklund, et al., Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson's disease. Cell Stem Cell 15, 653–665 (2014)
D.A. Grimes, F. Han, M. Panisset, L. Racacho, F. Xiao, R. Zou, K. Westaff, D.E. Bulman, Translated mutation in the Nurr1 gene as a cause for Parkinson's disease. Mov. Disord. 21, 906–909 (2006)
D.A. Grimes, L. Racacho, F. Han, M. Panisset, D.E. Bulman, LRRK2 screening in a Canadian Parkinson's disease cohort. Can. J. Neurol. Sci. 34, 336–338 (2007)
P. Hagell, P. Brundin, Cell survival and clinical outcome following intrastriatal transplantation in Parkinson disease. J. Neuropathol. Exp. Neurol. 60, 741–752 (2001)
P. Hagell, A. Schrag, P. Piccini, M. Jahanshahi, R. Brown, S. Rehncrona, H. Widner, P. Brundin, J.C. Rothwell, P. Odin, et al., Sequential bilateral transplantation in Parkinson's disease: effects of the second graft. Brain 122(Pt 6), 1121–1132 (1999)
F. Han, The applications of the induced pluripotent stem cells in studying the neurodegenerative diseases. Chin. J. Cell Biol. 34, 13 (2012)
F. Han, D. Baremberg, J. Gao, J. Duan, X. Lu, N. Zhang, Q. Chen, Development of Stem Cell-Based Therapy for Parkinson’s Disease. Transl. Neurodegener. 4, 16 (2015a)
F. Han, W. Wang, B. Chen, C. Chen, S. Li, X. Lu, J. Duan, Y. Zhang, Y.A. Zhang, W. Guo, et al., Human induced pluripotent stem cell-derived neurons improve motor asymmetry in a 6-hydroxydopamine-induced rat model of Parkinson's disease. Cytotherapy 17, 665–679 (2015b)
F. Han, D.A. Grimes, F. Li, T. Wang, Z. Yu, N. Song, S. Wu, L. Racacho, D.E. Bulman, Mutations in the glucocerebrosidase gene are common in patients with Parkinson's disease from Eastern Canada. Int. J. Neurosci. 126, 415–421 (2016)
F. Han, C. Chen, W. Wang, H. Song, S. Li, J. Duan, X. Lu, S. Wu, N. Zhang, Q. Chen, Human umbilical cord-derived mesenchymal stromal cells ameliorated motor defects of 6-OHDA-induced rat model of Parkinson’s disease. Oncotarget (2018). https://doi.org/10.18632/oncotarget.24103
G. Hargus, O. Cooper, M. Deleidi, A. Levy, K. Lee, E. Marlow, A. Yow, F. Soldner, D. Hockemeyer, P.J. Hallett, et al., Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. U. S. A. 107, 15921–15926 (2010)
R. Hass, C. Kasper, S. Bohm, R. Jacobs, Different populations and sources of human mesenchymal stem cells (MSC): a comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal 9, 12 (2011)
P. Hou, Y. Li, X. Zhang, C. Liu, J. Guan, H. Li, T. Zhao, J. Ye, W. Yang, K. Liu, et al., Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 341, 651–654 (2013)
K. Isobe, Z. Cheng, N. Nishio, T. Suganya, Y. Tanaka, S. Ito, Reprint of “iPSCs, aging and age-related diseases”. New Biotechnol. 32, 169–179 (2015)
X.D. Jiaxin Xie, F. Yu, N. Cao, X. Zhang, F. Fang, S. Zhang, Y. Feng, Early intradural microsurgery improves neurological recovery of acute spinal cord injury: a study of 87 cases. J. Neurorestoratol. 6, 152–157 (2018)
A. Kadari, M. Lu, M. Li, T. Sekaran, R.P. Thummer, N. Guyette, V. Chu, F. Edenhofer, Excision of viral reprogramming cassettes by Cre protein transduction enables rapid, robust and efficient derivation of transgene-free human induced pluripotent stem cells. Stem Cell Res Ther 5, 47 (2014)
T. Kallur, V. Darsalia, O. Lindvall, Z. Kokaia, Human fetal cortical and striatal neural stem cells generate region-specific neurons in vitro and differentiate extensively to neurons after intrastriatal transplantation in neonatal rats. J. Neurosci. Res. 84, 1630–1644 (2006)
Z. Kefalopoulou, M. Politis, P. Piccini, N. Mencacci, K. Bhatia, M. Jahanshahi, H. Widner, S. Rehncrona, P. Brundin, A. Bjorklund, et al., Long-term clinical outcome of fetal cell transplantation for Parkinson disease: Two case reports. JAMA Neurol. 71, 83–87 (2014)
T. Kikuchi, A. Morizane, D. Doi, H. Magotani, H. Onoe, T. Hayashi, H. Mizuma, S. Takara, R. Takahashi, H. Inoue, et al., Human iPS cell-derived dopaminergic neurons function in a primate Parkinson's disease model. Nature 548, 592–596 (2017)
H.T. Kim, I.S. Kim, I.S. Lee, J.P. Lee, E.Y. Snyder, K.I. Park, Human neurospheres derived from the fetal central nervous system are regionally and temporally specified but are not committed. Exp. Neurol. 199, 222–235 (2006)
H.J. Kim, E. McMillan, F. Han, C.N. Svendsen, Regionally specified human neural progenitor cells derived from the mesencephalon and forebrain undergo increased neurogenesis following overexpression of ASCL1. Stem Cells 27, 390–398 (2009)
H.S. Kim, J. Kim, Y. Jo, D. Jeon, Y.S. Cho, Direct lineage reprogramming of mouse fibroblasts to functional midbrain dopaminergic neuronal progenitors. Stem Cell Res. 12, 60–68 (2014)
E. Kiskinis, K. Eggan, Progress toward the clinical application of patient-specific pluripotent stem cells. J. Clin. Invest. 120, 51–59 (2010)
J.B. Koprich, T.H. Johnston, M.G. Reyes, X. Sun, J.M. Brotchie, Expression of human A53T alpha-synuclein in the rat substantia nigra using a novel AAV1/2 vector produces a rapidly evolving pathology with protein aggregation, dystrophic neurite architecture and nigrostriatal degeneration with potential to model the pathology of Parkinson's disease. Mol. Neurodegener. 5, 43 (2010)
J.H. Kordower, Y. Chu, R.A. Hauser, T.B. Freeman, C.W. Olanow, Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med. 14, 504–506 (2008)
S. Kriks, J.W. Shim, J. Piao, Y.M. Ganat, D.R. Wakeman, Z. Xie, L. Carrillo-Reid, G. Auyeung, C. Antonacci, A. Buch, et al., Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 480, 547–551 (2011)
S. Lesage, A. Brice, Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 18, R48–R59 (2009)
J.Y. Li, E. Englund, J.L. Holton, D. Soulet, P. Hagell, A.J. Lees, T. Lashley, N.P. Quinn, S. Rehncrona, A. Bjorklund, et al., Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503 (2008)
Y. Li, Q. Zhang, X. Yin, W. Yang, Y. Du, P. Hou, J. Ge, C. Liu, W. Zhang, X. Zhang, et al., Generation of iPSCs from mouse fibroblasts with a single gene, Oct4, and small molecules. Cell Res. 21, 196–204 (2011)
W. Li, E. Englund, H. Widner, B. Mattsson, D. van Westen, J. Lätt, S. Rehncrona, P. Brundin, A. Björklund, O. Lindvall, J.-Y. Li, Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc. Natl. Acad. Sci. USA 113(23), 6544–6549 (2016)
O. Lindvall, Treatment of Parkinson's disease using cell transplantation. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 370, 20140370 (2015)
O. Lindvall, A. Bjorklund, Cell therapy in Parkinson's disease. NeuroRx 1, 382–393 (2004)
O. Lindvall, A. Björklund, Cell therapeutics in Parkinson’s disease. Neurotherapeutics 8, 539–548 (2011)
O. Lindvall, P. Brundin, H. Widner, S. Rehncrona, B. Gustavii, R. Frackowiak, K.L. Leenders, G. Sawle, J.C. Rothwell, C.D. Marsden, et al., Grafts of fetal dopamine neurons survive and improve motor function in Parkinson's disease. Science 247, 574–577 (1990)
O. Lindvall, G. Sawle, H. Widner, J.C. Rothwell, A. Bjorklund, D. Brooks, P. Brundin, R. Frackowiak, C.D. Marsden, P. Odin, et al., Evidence for long-term survival and function of dopaminergic grafts in progressive Parkinson's disease. Ann. Neurol. 35, 172–180 (1994)
R. Lister, M. Pelizzola, Y.S. Kida, R.D. Hawkins, J.R. Nery, G. Hon, J. Antosiewicz-Bourget, R. O'Malley, R. Castanon, S. Klugman, et al., Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471, 68–73 (2011)
T.W. Liu, Z.G. Ma, Y. Zhou, J.X. Xie, Transplantation of mouse CGR8 embryonic stem cells producing GDNF and TH protects against 6-hydroxydopamine neurotoxicity in the rat. Int. J. Biochem. Cell Biol. 45, 1265–1273 (2013)
M. Lundblad, M. Decressac, B. Mattsson, A. Bjorklund, Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. U. S. A. 109, 3213–3219 (2012)
Y. Ma, C. Tang, T. Chaly, P. Greene, R. Breeze, S. Fahn, C. Freed, V. Dhawan, D. Eidelberg, Dopamine cell implantation in Parkinson's disease: long-term clinical and (18)F-FDOPA PET outcomes. J. Nucl. Med. 51, 7–15 (2010)
L. Ma, Y. Liu, S.C. Zhang, Directed differentiation of dopamine neurons from human pluripotent stem cells. Methods Mol. Biol. 767, 411–418 (2011)
I. Martin, V.L. Dawson, T.M. Dawson, Recent advances in the genetics of Parkinson's disease. Annu. Rev. Genomics Hum. Genet. 12, 301–325 (2011)
I. Martin, J.W. Kim, V.L. Dawson, T.M. Dawson, LRRK2 pathobiology in Parkinson's disease. J. Neurochem. 131, 554–565 (2014)
I. Mendez, A. Viñuela, A. Astradsson, K. Mukhida, P. Hallett, H. Robertson, T. Tierney, R. Holness, A. Dagher, J.Q. Trojanowski, Dopamine neurons implanted into people with Parkinson’s disease survive without pathology for 14 years. Nat. Med. 14, 507–509 (2008)
D. Michel-Monigadon, V. Nerriere-Daguin, X. Leveque, M. Plat, E. Venturi, P. Brachet, P. Naveilhan, I. Neveu, Minocycline promotes long-term survival of neuronal transplant in the brain by inhibiting late microglial activation and T-cell recruitment. Transplantation 89, 816–823 (2010)
E. Monni, C. Cusulin, M. Cavallaro, O. Lindvall, Z. Kokaia, Human fetal striatum-derived neural stem (NS) cells differentiate to mature neurons in vitro and in vivo. Curr. Stem Cell Res. Ther. 9, 338–346 (2014)
S.F. Moore, N.V. Guzman, S.L. Mason, C.H. Williams-Gray, R.A. Barker, Which patients with Parkinson's disease participate in clinical trials? One centre's experiences with a new cell based therapy trial (TRANSEURO). J. Parkinsons Dis. 4, 671–676 (2014)
M. Nakagawa, Y. Taniguchi, S. Senda, N. Takizawa, T. Ichisaka, K. Asano, A. Morizane, D. Doi, J. Takahashi, M. Nishizawa, et al., A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci. Rep. 4, 3594 (2014)
M.A. Nalls, V. Plagnol, D.G. Hernandez, M. Sharma, U.M. Sheerin, M. Saad, J. Simon-Sanchez, C. Schulte, S. Lesage, S. Sveinbjornsdottir, et al., Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet 377, 641–649 (2011)
K. Okita, Y. Matsumura, Y. Sato, A. Okada, A. Morizane, S. Okamoto, H. Hong, M. Nakagawa, K. Tanabe, K. Tezuka, et al., A more efficient method to generate integration-free human iPS cells. Nat. Methods 8, 409–412 (2011)
C.W. Olanow, R.L. Watts, W.C. Koller, An algorithm (decision tree) for the management of Parkinson's disease (2001): treatment guidelines. Neurology 56, S1–S88 (2001)
C.W. Olanow, C.G. Goetz, J.H. Kordower, A.J. Stoessl, V. Sossi, M.F. Brin, K.M. Shannon, G.M. Nauert, D.P. Perl, J. Godbold, et al., A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson's disease. Ann. Neurol. 54, 403–414 (2003)
H.J. Park, P.H. Lee, O.Y. Bang, G. Lee, Y.H. Ahn, Mesenchymal stem cells therapy exerts neuroprotection in a progressive animal model of Parkinson's disease. J. Neurochem. 107, 141–151 (2008a)
I.H. Park, R. Zhao, J.A. West, A. Yabuuchi, H. Huo, T.A. Ince, P.H. Lerou, M.W. Lensch, G.Q. Daley, Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146 (2008b)
K.L. Paumier, K.C. Luk, F.P. Manfredsson, N.M. Kanaan, J.W. Lipton, T.J. Collier, K. Steece-Collier, C.J. Kemp, S. Celano, E. Schulz, et al., Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 82, 185–199 (2015)
P. Piccini, D.J. Brooks, A. Bjorklund, R.N. Gunn, P.M. Grasby, O. Rimoldi, P. Brundin, P. Hagell, S. Rehncrona, H. Widner, et al., Dopamine release from nigral transplants visualized in vivo in a Parkinson's patient. Nat. Neurosci. 2, 1137–1140 (1999)
N. Plotegher, M.R. Duchen, Crosstalk between lysosomes and mitochondria in Parkinson's disease. Front. Cell Dev. Biol. 5, 110 (2017)
O. Pogarell, W. Koch, F.J. Gildehaus, A. Kupsch, O. Lindvall, W.H. Oertel, K. Tatsch, Long-term assessment of striatal dopamine transporters in Parkinsonian patients with intrastriatal embryonic mesencephalic grafts. Eur. J. Nucl. Med. Mol. Imaging 33, 407–411 (2006)
M. Politis, K. Wu, C. Loane, N.P. Quinn, D.J. Brooks, S. Rehncrona, A. Bjorklund, O. Lindvall, P. Piccini, Serotonergic neurons mediate dyskinesia side effects in Parkinson's patients with neural transplants. Sci. Transl. Med. 2, 38ra46 (2010)
M. Politis, K. Wu, C. Loane, N.P. Quinn, D.J. Brooks, W.H. Oertel, A. Bjorklund, O. Lindvall, P. Piccini, Serotonin neuron loss and nonmotor symptoms continue in Parkinson's patients treated with dopamine grafts. Sci. Transl. Med. 4, 128ra141 (2012)
M.H. Polymeropoulos, C. Lavedan, E. Leroy, S.E. Ide, A. Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer, et al., Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 (1997)
R.B. Postuma, D. Berg, M. Stern, W. Poewe, C.W. Olanow, W. Oertel, J. Obeso, K. Marek, I. Litvan, A.E. Lang, et al., MDS clinical diagnostic criteria for Parkinson's disease. Mov. Disord. 30, 1591–1601 (2015)
P. Reinhardt, B. Schmid, L.F. Burbulla, D.C. Schondorf, L. Wagner, M. Glatza, S. Hoing, G. Hargus, S.A. Heck, A. Dhingra, et al., Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12, 354–367 (2013)
D. Ribeiro, R. Laguna Goya, G. Ravindran, R. Vuono, C.L. Parish, C. Foldi, T. Piroth, S. Yang, M. Parmar, G. Nikkhah, et al., Efficient expansion and dopaminergic differentiation of human fetal ventral midbrain neural stem cells by midbrain morphogens. Neurobiol. Dis. 49, 118–127 (2013)
F. Safari, G. Hatam, A.B. Behbahani, V. Rezaei, M. Barekati-Mowahed, P. Petramfar, F. Khademi, CRISPR system: a high-throughput toolbox for research and treatment of Parkinson's disease. Cell. Mol. Neurobiol 40, 477 (2019)
B. Samata, D. Doi, K. Nishimura, T. Kikuchi, A. Watanabe, Y. Sakamoto, J. Kakuta, Y. Ono, J. Takahashi, Purification of functional human ES and iPSC-derived midbrain dopaminergic progenitors using LRTM1. Nat. Commun. 7, 13097 (2016)
A. Sanchez-Danes, A. Consiglio, Y. Richaud, I. Rodriguez-Piza, B. Dehay, M. Edel, J. Bove, M. Memo, M. Vila, A. Raya, et al., Efficient generation of A9 midbrain dopaminergic neurons by lentiviral delivery of LMX1A in human embryonic stem cells and induced pluripotent stem cells. Hum. Gene Ther. 23, 56–69 (2012)
D.A. Sliter, J. Martinez, L. Hao, X. Chen, N. Sun, T.D. Fischer, J.L. Burman, Y. Li, Z. Zhang, D.P. Narendra, et al., Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018)
F. Soldner, J. Laganiere, A.W. Cheng, D. Hockemeyer, Q. Gao, R. Alagappan, V. Khurana, L.I. Golbe, R.H. Myers, S. Lindquist, et al., Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146, 318–331 (2011)
L. Studer, V. Tabar, R.D. McKay, Transplantation of expanded mesencephalic precursors leads to recovery in parkinsonian rats. Nat. Neurosci. 1, 290–295 (1998)
M. Sundberg, H. Bogetofte, T. Lawson, J. Jansson, G. Smith, A. Astradsson, M. Moore, T. Osborn, O. Cooper, R. Spealman, Improved cell therapy protocols for Parkinson's disease based on differentiation efficiency and safety of hESC-, hiPSC-, and non-human primate iPSC-derived dopaminergic neurons. Stem Cells 31, 1548–1562 (2013)
D.J. Surmeier, Determinants of dopaminergic neuron loss in Parkinson's disease. FEBS J. 285, 3657–3668 (2018)
J. Takahashi, Preparing for first human trial of induced pluripotent stem cell-derived cells for Parkinson's disease: an interview with Jun Takahashi. Regen. Med. 14, 93–95 (2019)
K. Takahashi, S. Yamanaka, Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006)
K. Takahashi, K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda, S. Yamanaka, Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007)
C.M. Tanner, R. Ottman, S.M. Goldman, J. Ellenberg, P. Chan, R. Mayeux, J.W. Langston, Parkinson disease in twins: an etiologic study. JAMA 281, 341–346 (1999)
P. Taupin, F.H. Gage, Adult neurogenesis and neural stem cells of the central nervous system in mammals. J. Neurosci. Res. 69, 745–749 (2002)
J.A. Thomson, J. Itskovitz-Eldor, S.S. Shapiro, M.A. Waknitz, J.J. Swiergiel, V.S. Marshall, J.M. Jones, Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 (1998)
L.A. Volpicelli-Daley, K.C. Luk, T.P. Patel, S.A. Tanik, D.M. Riddle, A. Stieber, D.F. Meaney, J.Q. Trojanowski, V.M. Lee, Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 (2011)
Q. Wang, X. Xu, J. Li, J. Liu, H. Gu, R. Zhang, J. Chen, Y. Kuang, J. Fei, C. Jiang, et al., Lithium, an anti-psychotic drug, greatly enhances the generation of induced pluripotent stem cells. Cell Res. 21, 1424–1435 (2011)
W. Wang, H. Song, A. Shen, C. Chen, Y. Liu, Y. Dong, F. Han, Differentiated cells derived from fetal neural stem cells improve motor deficits in a rat model of Parkinson's disease. Transl. Neurosci. 1, 75–85 (2015)
H. Wang, M. La Russa, L.S. Qi, CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 85, 227–264 (2016)
G. Wang, L. Yang, D. Grishin, X. Rios, L.Y. Ye, Y. Hu, K. Li, D. Zhang, G.M. Church, W.T. Pu, Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat. Protoc. 12, 88–103 (2017)
G.K. Wenning, P. Odin, P. Morrish, S. Rehncrona, H. Widner, P. Brundin, J.C. Rothwell, R. Brown, B. Gustavii, P. Hagell, et al., Short- and long-term survival and function of unilateral intrastriatal dopaminergic grafts in Parkinson's disease. Ann. Neurol. 42, 95–107 (1997)
M. Wernig, J.-P. Zhao, J. Pruszak, E. Hedlund, D. Fu, F. Soldner, V. Broccoli, M. Constantine-Paton, O. Isacson, R. Jaenisch, Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson's disease. Proc. Natl. Acad. Sci. 105, 5856–5861 (2008)
J. Xi, Y. Liu, H. Liu, H. Chen, M.E. Emborg, S.C. Zhang, Specification of midbrain dopamine neurons from primate pluripotent stem cells. Stem Cells 30, 1655–1663 (2012)
Y. Yan, D. Yang, E.D. Zarnowska, Z. Du, B. Werbel, C. Valliere, R.A. Pearce, J.A. Thomson, S.C. Zhang, Directed differentiation of dopaminergic neuronal subtypes from human embryonic stem cells. Stem Cells 23, 781–790 (2005)
H. Ye, Q. Wang, Efficient generation of non-integration and feeder-free induced pluripotent stem cells from human peripheral blood cells by sendai virus. Cell. Physiol. Biochem. 50, 1318–1331 (2018)
J. Yu, M.A. Vodyanik, K. Smuga-Otto, J. Antosiewicz-Bourget, J.L. Frane, S. Tian, J. Nie, G.A. Jonsdottir, V. Ruotti, R. Stewart, et al., Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007)
Z. Yu, T. Wang, J. Xu, W. Wang, G. Wang, C. Chen, L. Zheng, L. Pan, D. Gong, X. Li, et al., Mutations in the glucocerebrosidase gene are responsible for Chinese patients with Parkinson's disease. J. Hum. Genet. 60, 85–90 (2015)
X. Zeng, J. Cai, J. Chen, Y. Luo, Z.B. You, E. Fotter, Y. Wang, B. Harvey, T. Miura, C. Backman, et al., Dopaminergic differentiation of human embryonic stem cells. Stem Cells 22, 925–940 (2004)
N. Zhang, B. Chen, W. Wang, C. Chen, J. Kang, S.Q. Deng, B. Zhang, S. Liu, F. Han, Isolation, characterization and multi-lineage differentiation of stem cells from human exfoliated deciduous teeth. Mol. Med. Rep. 14, 95–102 (2016)
J. Zhang, M.L. Culp, J.G. Craver, V. Darley-Usmar, Mitochondrial function and autophagy: integrating proteotoxic, redox, and metabolic stress in Parkinson's disease. J. Neurochem. 144, 691–709 (2018a)
N. Zhang, X. Lu, S. Wu, X. Li, J. Duan, C. Chen, W. Wang, H. Song, J. Tong, S. Li, Y. Liu, X. Kang, X. Wang, F. Han, Intrastriatal transplantation of stem cells from human exfoliated deciduous teeth reduces motor defects in Parkinsonian rats. Cytotherapy. 20(5), 670–686 (2018b)
Acknowledgment
This work was supported by National Natural Science Foundation of China (NSFC 81571241), Department of Science and Technology of Shandong Province, China (2017GSF18104), Science and Technology Innovation Committee of Shenzhen Municipality, China (JCYJ201803051642562) and Basic Research Fund from Natural Science Foundation of Shandong Province, China (ZR2019ZD39). We also thank Wei Wang, Jing Duan, Xianjie Lu, Hao Song, Nan Zhang, and Yanming Liu in our lab for technically editing the references and preparing figures of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Han, F., Hu, B. (2020). Stem Cell Therapy for Parkinson’s Disease. In: Han, F., Lu, P.(. (eds) Stem Cell-based Therapy for Neurodegenerative Diseases. Advances in Experimental Medicine and Biology, vol 1266. Springer, Singapore. https://doi.org/10.1007/978-981-15-4370-8_3
Download citation
DOI: https://doi.org/10.1007/978-981-15-4370-8_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-4369-2
Online ISBN: 978-981-15-4370-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)