
Abstract
In 1945, K. R. Porter et al. observed mouse embryonic fibroblasts (MEFs) and found that the cytoplasmic part of the cell had an unreported reticular structure, so it was named endoplasmic reticulum (ER). The major functions of the endoplasmic reticulum are: synthesis of intracellular proteins and the modification and processing of proteins. It is an important organelle in eukaryotic cells. It is a three-dimensional network structure in which complex and closed intracellular tubular intimal systems are intertwined. When cells are subjected to various strong stimulating factors such as nutrient deficiencies, Ca2+ metabolic imbalance, toxin stimulation, and sustained oxidative stress stimulation, the cell homeostasis will be broken. In order to survive, a series of cell self-protection event will be initiated including the endoplasmic reticulum stress (ERS). The UPR can further promote the expression of the proteins which can help the misfolded and unfolded proteins restore to its normal structure through the activation of PERK, IRE1, and ATF6. However, the co-working of UPR and the ubiquitin-proteasome system still cannot make the endoplasmic reticulum restoring to its normal state, when the stimuli persist or are too strong. The damaged endoplasmic reticulum can be partially engulfed by the autophagic vesicles for degradation when the ERS persists. The degraded endoplasmic reticulum fragments can be reassembled into a new endoplasmic reticulum to restore the normal state of it. Hence, it seems that the autophagy has become the last mean to restore the homeostasis of endoplasmic reticulum.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Endoplasmic reticulum stress
- Autophagy
- Unfolded protein response
- The endoplasmic reticulum-associated degradation
1 The Structure and Function of Endoplasmic Reticulum
The major functions of the endoplasmic reticulum are: synthesis of intracellular proteins and the modification and processing of proteins. It is an important organelle in eukaryotic cells. It is a three-dimensional network structure in which complex and closed intracellular tubular intimal systems are intertwined. The endoplasmic reticulum is a highly dynamic intracellular organelle, mainly in terms of quantity, type, and morphology. Interestingly, when the same cell undergoes different cell life activities or is in different physiological states, the structure and function of the endoplasmic reticulum will not remain the same. In 1945, K. R. Porter et al. observed mouse embryonic fibroblasts (MEFs) and found that the cytoplasmic part of the cell had an unreported reticular structure, so it was named endoplasmic reticulum (ER). Although it was found in later studies that the endoplasmic reticulum is mainly composed of one small vesicle and also exists in other parts of the cell, it is still customary to extend the name of endoplasmic reticulum.
According to the difference in structure and function, the endoplasmic reticulum can be divided into rough endoplasmic reticulum (rER) and smooth endoplasmic reticulum (sER). There is a large amount of ribosome attachment on the rough endoplasmic reticulum, which is mainly responsible for protein synthesis, while there is no ribosome on the smooth endoplasmic reticulum or only a small amount of ribosome attachment, which is mainly responsible for lipid synthesis. The endoplasmic reticulum is not only a substance synthesis plant in cells but also capable of glycosylation, acylation, hydroxylation of proteins, and the promotion of disulfide bond formation between proteins in the endoplasmic reticulum. Glycosylation is divided into N-linked glycosylation and O-linked glycosylation, the former mainly occurs in the endoplasmic reticulum, and the latter mainly occurs in the Golgi apparatus. GRP78 (glucose-regulated protein 78), also known as Bip (heavy-chain binding protein), is a molecular chaperone localized on the endoplasmic reticulum membrane by promoting the degradation and refolding of the unfolded proteins and incorrectly folded proteins accumulated in the endoplasmic reticulum restoring the homeostasis of endoplasmic reticulum. In addition, the sER is involved in the biotransformation process of hepatocytes, converting fat-soluble poisons into water-soluble and eliminating it from the body. In some cells of the body, the sER is highly specialized, such as in the cardiomyocytes and skeletal muscle cells called sarcoplasmic reticulum, in which a large amount of Ca2+ is stored to regulate the muscle contraction (Ellgaard and Helenius 2003; Yan and Lennarz 2005).
2 The Endoplasmic Reticulum Associated Degradation
The homeostasis of endoplasmic reticulum is essential for the survival of the cells. To achieve this condition, the integrity of protein folding and maturation in the endoplasmic reticulum must be ensured. However, there is no eternal endoplasmic reticulum homeostasis which will eventually be broken by some specific physiological or pathological factors, causing a large amount of misfolded proteins accumulated in the endoplasmic reticulum. The endoplasmic reticulum has two ways to cope with the misfolded proteins accumulated in the ER lumen: one is the unfolded protein response and other is the endoplasmic reticulum associated degradation (ERAD) which mediates the transportation of misfolded proteins back to the cytosol and the degradation in the ubiquitin-proteasome system (Olzmann et al. 2013).
ERAD, a multistep process comprising recognition, transportation, and ubiquitination of ER misfolded proteins for cytosolic proteasomal degradation, was discovered by Ardythe McCracken and Jeffrey Brodsky at University of Pittsburgh in 1996. The misfolded proteins and unfolded proteins can be transport back to the cytosol by the protein sel-1 homolog 1 (SEL1L) for the ubiquitin-associated proteasomal degradation through a putative ER membrane channel HRD1 (an E3-ubiquitin ligase aka SYVN1), which is termed as the SEL1L–HRD1 ERAD. Ablation of SEL1L or HRD1 causes the embryonic lethal of mice in days 11–14, while the adult mice ablated the SEL1L or HRD1 die young within about 3 weeks, and mice knocked out of the SEL1L or HRD1 in adipocytes show lipodystrophy and postprandial hyperlipidemia. Deletion of SEL1L in mice AVP neurons cause polyuria and polydipsia, whereas in POMC neurons cause hyperphagia and obesity (Francisco et al. 2010; Kim et al. 2018).
3 The Endoplasmic Reticulum Stress
When cells are subjected to various strong stimulating factors (nutrient deficiencies, Ca2+ metabolic imbalance, toxin stimulation, sustained oxidative stress stimulation), cell homeostasis will be broken, and in order to survive, a series of cell self-protection event will be initiated, including the endoplasmic reticulum stress (ERS). ERS will initiate three endoplasmic reticulum-related reactions, including unfolded protein response (UPR), endoplasmic reticulum overload response (EOR), and sterol regulation reaction to help clear the unfolded and incorrectly folded proteins accumulated in the endoplasmic reticulum to restore the homeostasis of ER. On the one hand, the ERS-induced UPR can promote the refolding and degradation of proteins by the molecular chaperone GRP78/Bip; on the other hand, it can also reduce the production of protein, finally restoring the homeostasis of the endoplasmic reticulum. Recently, it has been reported in the literature that ERS can restore the normal function of the endoplasmic reticulum or even restore the normal function of the whole cell through autophagy. However, autophagy is a ‘double-edged’ sword for the cells. On the one hand, it can protect cells. For example, when cells are starved, cells can decompose intracellular substances through autophagy to provide cells with the necessary materials for survival and promote cell survival. On the other hand, when the stimulating factors inside and outside the cell persist, the level of autophagy rises, which in turn aggravates cell damage and eventually causes cell death. This cell death caused by excessive autophagy is called type II programmed cell death (Kapoor and Sanyal 2009).
3.1 The Endoplasmic Reticulum Stress Activate UPR
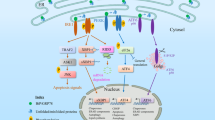
The unfolded protein response (UPR) is a protective mechanism initiated by cells in order to restore the homeostasis of the endoplasmic reticulum when ERS occurs. The UPR can activate some transmembrane proteins located on the endoplasmic reticulum and then initiate a series of intracellular downstream signaling to promote expression of proteins, such as the chaperone GRP78/Bip or to reduce protein synthesis in the endoplasmic reticulum, and eventually restoring the normal state of endoplasmic reticulum. ERS can cause autophagy in cells by inducing the UPR, making the destroyed endoplasmic reticulum engulfed by the autophagic vesicles and restoring the homeostasis of endoplasmic reticulum. Three major branches of UPR are mediated by some transmembrane proteins located on the endoplasmic reticulum, including protein kinase R-like ER kinase (PERK), inositol requiring kinase 1 (IRE1), and activate transcription factor 6 (ATF6). PERK, IRE1, and ATF6 bind to GRP78/Bip when ERS does not happen, leaving it in an inactive state. The abnormally accumulated misfolded and unfolded proteins in the endoplasmic reticulum can compete with the PERK, IRE1, and ATF6 for the binding site of GRP78/Bip, activating the PERK, IRE1, and ATF6 through this competition, when ERS happens. Thus PERK, IRE1, and ATF6 are activated, which initiates the downstream signal transduction, affects the expression of certain genes in the nucleus, increases the synthesis of chaperones, increases the degradation of abnormally accumulated proteins, and finally reduces the synthesis of proteins, leading to the homeostasis of endoplasmic reticulum (Hetz 2012; Liu et al. 2016).
3.1.1 PERK
The N-terminal domain of PERK is bound with GRP78/Bip, so it cannot be activated, when the ERS does not happen. The dimerization and the cross-phosphorylation happen in PERK, causing it no longer bind to GRP78/Bip and leading to the activation of PERK, when ERS occurs. The eukaryotic initiation factor 2α (eIF2α) is the downstream signal factor of PERK. PERK can phosphorylate the eIF2α at Ser-51, and the phosphorylated eIF2α can alleviate the pressure of protein accumulation by inhibiting the level of protein synthesis in the endoplasmic reticulum, thereby achieving the goal of restoring the normal function of endoplasmic reticulum.
3.1.2 IRE1
The endoplasmic reticulum side domain of IRE1 has serine/threonine kinase activity and its cytoplasmic side domain also has RNA endonuclease activity. The abnormally accumulated misfolded and unfolded proteins, which can compete with the GRP78/Bip for binding to IRE1, is the cause for the release of GRP78/Bip from IRE1 when ERS happens and for the cross-phosphorylation and dimerization of IRE1, thus finally leading to the activation of its RNA endonuclease activity. The activated IRE1 can cleave the mRNA of certain proteins. For example, IRE1 can cleave the precursor of the X-box binding protein-1 (XBP-1) mRNA, which can bind to the promoter region of the endoplasmic reticulum stress element (ERSE) upregulating the expression of proteins involved in UPR, increasing the expression of proteins that help misfolded and unfolded proteins restore their normal morphology, and reducing the protein synthesis level in the endoplasmic reticulum restoring the homeostasis of it.
3.1.3 ATF6
ATF6 is a transcription factor localized on the endoplasmic reticulum membrane. When ERS happens, ATF is separated from GRP78/Bip, while unlike the PERK and IRE1, ATF needs to be translocated to the Golgi with the help of coat protein complex II (COPII) vesicles, which can then be cleaved by the Site-1 protease (S1P) and Site-2 protease (S2P) in order to finish the activation of it. The ATF6 can directly promote the expression of proteins involved in the UPR without the activation of downstream signal transduction; for instance, the molecular chaperone GRP78/Bip can also directly promote the expression of the XBP-1, which is the transcription factor of ERSE.
4 The Endoplasmic Reticulum Stress and Autophagy
The abnormal accumulation of misfolded and unfolded proteins in the endoplasmic reticulum can cause the ERS, when a cell is subjected to strong intracellular or extracellular stress. The UPR can further promote the expression of the proteins which can help the misfolded and unfolded proteins restore to its normal structure through the activation of PERK, IRE1, and ATF6. It has been reported that the ubiquitin-proteasome system is also involved in the degradation of misfolded and unfolded proteins to further help the UPR restore the endoplasmic reticulum to its normal state, when the ERS-activated UPR is still insufficient to eliminate the misfolded and unfolded proteins accumulated in the endoplasmic reticulum. However, the co-working of UPR and the ubiquitin-proteasome system still cannot make the endoplasmic reticulum restore to its normal state, when the stimuli persist or are too strong. Hence, it seems that the autophagy has become the last mean to restore the homeostasis of endoplasmic reticulum. The damaged endoplasmic reticulum can be partially engulfed by the autophagic vesicles for degradation when the ERS persists. The degraded endoplasmic reticulum fragments can be reassembled into a new endoplasmic reticulum to restore to the normal state.
4.1 The Endoplasmic Reticulum Stress Activates Autophagy Through UPR
The PERK, IRE1, and ATF6 are the three different signal transduction pathways of UPR, which can not only promote the expression of proteins helping the misfolded and unfolded protein restore to its normal structure but can also further alleviate the ERS through the activation of cell autophagy, eventually restoring the homeostasis of the endoplasmic reticulum. It is noted that the three signal pathways induced by the ERS are all involved in the activation of cell autophagy but depends on the different cell type and cell living environment. The ERS-induced autophagy has a pro-survival effect of cell; hence it may be an important reason for the bad treatment effect of anti-tumor drugs. Indeed, it has been reported that inhibiting the autophagy of tumor cells will increase the c-Myc-dependent cell apoptosis through the pharmacological inhibitor and RNA interfering technology, but the c-Myc-dependent effect of promoting the cell apoptosis will be dramatically decreased when inhibiting the ERS-induced cell autophagy (Fujita et al. 2007; Yorimitsu et al. 2006).
PERK can promote the expression of autophagy-related gene Atg12 and LC3 through the phosphorylation of eIFα. Kouroku et al. (2007) found that gene knockout of PERK can inhibit the cell autophagy activated by the ERS-induced PERK-eIF2α cell signal transduction pathway, dramatically decreasing the numbers of autophagosome in cytoplasm in mouse embryonic cancer cells and embryonic fibroblasts (MEFs), and the above results indicate that the PERK-eIF2α cell signal transduction pathway is required for the ERS-induced cell autophagy. In addition, it is reported that the eIF2α can not only be phosphorylated by the PERK but also by the PKR (double-stranded RNA-activated protein kinase), GCN2 kinase (general control nonderepressible 2 GCN2), and (heme-regulated inhibitor HRI), which can activate the cell autophagy through the phosphorylation of eIF2α in the case of viral infection, nutrient depletion, and heme depletion. Moreover, it was found that the phosphorylated eIF2α, also involved in the amino acid deficiency, mimicked the formation of cell autophagy, although specific mechanism for it still remains unknown, at least indicating that the phosphorylated eIF2α plays a critical role in the ERS-induced cell autophagy (Liu et al. 2010; Talloczy et al. 2002).
The expression of transcriptional factor C/EBP-β will increase when ERS happens, which then further controls the expression of the death-associated protein kinase 1 (DAPK1). DAPK1 can phosphorylate Beclin-1 and free it from the negative regulator of autophagy Bcl-2, finally leading to the formation of cell autophagy. Interestingly, some researchers think that the real cell autophagy signal transduction pathway induced by ERS is IRE1 rather than PERK and ATF6. They found that the IRE1 is required for the formation and aggregation of the autophagosomes rather than PERK and ATF6 in IRE1 or ATF6-deficient mouse MEFs and PERK-deficient mouse embryonic cells through the treatment of tunicamycin and thapsigargin.
In addition, they also found that the IRE1 can form a complex with tumor necrosis factor receptor-associated factor-2 (TRAF-2), which can phosphorylate the apoptosis signal-regulating kinase 1 (ASK1), and then the phosphorylated ASK1 further phosphorylates the JNK, which can promote the activation of the Bcl-2, eventually leading to cell autophagy. In their further experiments they found that the thapsigargin can inhibit the formation of autophagosomes in TRAF-2 KO MEFs through the inhibition of LC3. The same result is obtained by using the inhibitor of JNK, and more importantly the ERS still exists. Hence, the ERS-induced cell autophagy is mediated by the IRE1–TRAF2–JNK pathway (Kouroku et al. 2007; Liu et al. 2010; Talloczy et al. 2002; Ogata et al. 2006).
4.2 The Endoplasmic Reticulum Stress Activates Autophagy Through Releasing of the Ca2+ into the Cytoplasm
The ERS can not only activate the cell autophagy through the UPR but also promote the release of a lot of Ca2+ from the endoplasmic reticulum into the cytoplasm, which can also induce the cell autophagy. Both DAPK1 and Calpain are Ca2+-related protease and are located in the cytoplasm. It has been reported that the DAPK1 and Calpain can be activated by the cytoplasmic Ca2+ upregulating the level of cell autophagy, but the specific mechanism remains unclear. It is reported that many agonists of autophagy still cannot induce the formation of autophagosome in Calpain KO MEFs and human osteosarcoma cells, causing there is no obvious change in the degradation of the misfolded proteins in the endoplasmic reticulum. It is noted that the cell autophagy cannot be activated by the use of rapamycin (inhibitor of mTOR) in the Calpain KO cells, although a lot of degradation of LC3-I and LC3-II emerged in the cytosol (Gozuacik et al. 2008; Hyrskyluoto et al. 2012; Madden et al. 2007).
IP3 (inositol-1,4,5-triphosphate) is an endogenous ligand of IP3R (inositol-1,4,5-triphosphate receptor) which can promote the releasing of Ca2+ from the endoplasmic reticulum into the cytosol. It is reported that the inhibition of IP3R with its siRNA can increase the activity of autophagy rather than decrease it. Indeed, the calmodulin-dependent kinase kinase-β (CaMKK-β) can activate the AMPK (AMP-activated protein kinase), and then the activated AMPK can inhibit the mammalian target of rapamycin (mTOR), causing the inhibition of cell autophagy when ERS induces the release of Ca2+ from the endoplasmic reticulum to the cytosol (Bhutia et al. 2011; Demarchi et al. 2006; Ganley et al. 2011). These above results indicate that the activation of cell autophagy, which is caused by the activation of IP3R, primarily relies on the AMPK–mTOR pathway. However, there are also some reports that the production of IP3 can be promoted by the intracellular ATP, which then promotes the cell autophagy. The ATP-dependent cell autophagy is mediated by the Ca2+–AMPK–mTOR pathway, and this reaction is slow and may take up to 2 days. Interestingly, the cell autophagy can be produced within 2 hours of using the pharmacological inhibitors of IP3R, the activity of mTOR was not inhibited, and there was no significant change in the cytoplasmic Ca2+ content. The above data indicate that the activation or inhibition of IP3R caused by ERS can lead to the occurrence of autophagy, which is likely to be caused by IP3R through different signal transduction pathways, but how ERS can cause autophagy through IP3R and the mechanism remain unclear, hence further research is needed.
4.3 The Endoplasmic Reticulum Stress Inhibits the Bcl-2 Causing the Autophagy of Endoplasmic Reticulum
ERS-induced autophagy is not significantly different from traditional autophagy and is also mediated by various autophagic factors. Beclin-1, also known as BECN1, and also called ATG6 in yeast, plays a key role in the formation of autophagosomes during autophagy. From a certain point of view, the content of Beclin-1 in the cell determines the degree of autophagy, and Beclin-1 is also a marker protein for autophagy formation. B-cell lymphoma-2 (Bcl-2) is mainly found in the endoplasmic reticulum, nuclear membrane, mitochondria, and its function is mainly to inhibit the cell apoptosis. It is reported in the literature that Bcl-2 has both promoting and inhibiting effects in the development of autophagy, but the specific mechanism is still unclear, and it may be associated with different subcellular localization and post-transcriptional modifications of Bcl-2. ERS initiates UPR, and UPR further inhibits Bcl-2 and ultimately causes autophagy by activating CHOP. For the inhibition of autophagy after activation of Bcl-2, it seems that there are two views in the academic world, the first one is Bcl-2 that can inhibit the interaction of Beclin-1, PI3 K and P150, and inhibit the formation of autophagy by blocking the formation of autophagosome membrane, and the second one is Bcl-2 that can inhibit Ca2+-dependent autophagy by inhibiting the release of Ca2+ induced by IP3R. However, it has also been reported in the literature that Bcl-2 can also inhibit autophagy caused by the endoplasmic reticulum IP3R inhibitor Xestospogin B, and autophagy induced in this way does not depend on Ca2+. Therefore, whether Bcl-2-induced inhibition of autophagy requires Ca2+ involvement or other mechanisms involved requires further research to elucidate (Cheng and Yang 2011; Kandala and Srivastava 2012; Marquez and Xu 2012; Pattingre et al. 2005) (Fig. 8.1).

Illustration of the endoplasmic reticulum stress induced cell autophagy
5 Conclusion
The relationship between ERS and autophagy has only become a research hotspot in the research field in recent years. How does ERS cause autophagy in cells? There are still many unresolved issues. When cells are stimulated by various strong stimulating factors inside or outside the cell, the ERS occurs. ERS activates UPR to eliminate misfolded and unfolded proteins accumulated in the endoplasmic reticulum, and the ubiquitin-proteasome system is also involved in the degradation of abnormal accumulation proteins in the endoplasmic reticulum. It is worth noting that UPR and ubiquitin-proteasome cannot remove the misfolded and unfolded proteins from the endoplasmic reticulum in time when the stimulating factors persist. ERS will stimulate the production of autophagy to reduce the degree of swelling of the endoplasmic reticulum, relieving the pressure of protein accumulation in the endoplasmic reticulum, finally restoring the normal state of the endoplasmic reticulum and keeping the cells alive. However, when ERS causes excessive autophagy, it will lead to cell death. The 3-methyladenine (3-MA) was used to inhibit autophagy in MEFs following Atg5 KO and was found to reduce the ERS-induced cell death. The above indicates that if we want to treat diseases caused by abnormal accumulation of proteins in the endoplasmic reticulum or cytoplasm by enhancing autophagy, more in-depth studies are needed to clarify whether autophagy is a pro-survival effect or a death-promoting effect on cells.
References
Bhutia SK, Das SK, Azab B, Dash R, Su ZZ, Lee SG, Dent P, Curiel DT, Sarkar D, Fisher PB (2011) Autophagy switches to apoptosis in prostate cancer cells infected with melanoma differentiation associated gene-7/interleukin-24 (mda-7/IL-24). Autophagy 7:1076–1077
Cheng Y, Yang JM (2011) Survival and death of endoplasmic-reticulum-stressed cells: role of autophagy. World J Biol Chem 2:226–231
Demarchi F, Bertoli C, Copetti T, Tanida I, Brancolini C, Eskelinen EL, Schneider C (2006) Calpain is required for macroautophagy in mammalian cells. J Cell Biol 175:595–605
Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4:181–191
Francisco AB, Singh R, Li S, Vani AK, Yang L, Munroe RJ, Diaferia G, Cardano M, Biunno I, Qi L, Schimenti JC, Long Q (2010) Deficiency of suppressor enhancer Lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J Biol Chem 285:13694–13703
Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T (2007) Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum Mol Genet 16:618–629
Ganley IG, Wong PM, Gammoh N, Jiang X (2011) Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell 42:731–743
Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, Mizushima N, Yoshimori T, Kimchi A (2008) DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ 15:1875–1886
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13:89–102
Hyrskyluoto A, Reijonen S, Kivinen J, Lindholm D, Korhonen L (2012) GADD34 mediates cytoprotective autophagy in mutant huntingtin expressing cells via the mTOR pathway. Exp Cell Res 318:33–42
Kandala PK, Srivastava SK (2012) Regulation of macroautophagy in ovarian cancer cells in vitro and in vivo by controlling glucose regulatory protein 78 and AMPK. Oncotarget 3:435–449
Kapoor A, Sanyal AJ (2009) Endoplasmic reticulum stress and the unfolded protein response. Clin Liver Dis 13:581–590
Kim GH, Shi G, Somlo DR, Haataja L, Song S, Long Q, Nillni EA, Low MJ, Arvan P, Myers MG, Qi L (2018) Hypothalamic ER-associated degradation regulates POMC maturation, feeding, and age-associated obesity. J Clin Invest 128:1125–1140
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T (2007) ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 14:230–239
Liu MQ, Chen Z, Chen LX (2016) Endoplasmic reticulum stress: a novel mechanism and therapeutic target for cardiovascular diseases. Acta Pharmacol Sin 37:425–443
Liu Y, Laszlo C, Liu Y, Liu W, Chen X, Evans SC, Wu S (2010) Regulation of G(1) arrest and apoptosis in hypoxia by PERK and GCN2-mediated eIF2alpha phosphorylation. Neoplasia 12:61–68
Madden DT, Egger L, Bredesen DE (2007) A calpain-like protease inhibits autophagic cell death. Autophagy 3:519–522
Marquez RT, Xu L (2012) Bcl-2: Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res 2:214–221
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26:9220–9231
Olzmann JA, Kopito RR, Christianson JC (2013) The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol 5
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939
Talloczy Z, Jiang W, Virgin HWT, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B (2002) Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 99:190–195
Yan A, Lennarz WJ (2005) Unraveling the mechanism of protein N-glycosylation. J Biol Chem 280:3121–3124
Yorimitsu T, Nair U, Yang Z, Klionsky DJ (2006) Endoplasmic reticulum stress triggers autophagy. J Biol Chem 281:30299–30304
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Science Press and Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Qi, Z., Chen, L. (2019). Endoplasmic Reticulum Stress and Autophagy. In: Qin, ZH. (eds) Autophagy: Biology and Diseases. Advances in Experimental Medicine and Biology, vol 1206. Springer, Singapore. https://doi.org/10.1007/978-981-15-0602-4_8
Download citation
DOI: https://doi.org/10.1007/978-981-15-0602-4_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-0601-7
Online ISBN: 978-981-15-0602-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)