
Abstract
Autophagy plays an important role in the renewal of cellular components, which function in energy production, metabolism, and clearance of damaged organelles. Both macroautophagy and microautophagy are involved in these processes. Although it was thought that nonselective macroautophagy is responsible for the clearance of damaged or old organelles, recent studies show that the clearance of cellular organelles depends on selective processes. Mitophagy is a process for selective degradation of mitochondria, which is well documented. The selective autophagy for other organelles includes endoplasmic reticulum autophagy (reticulophagy) and peroxisome autophagy (pexophagy). Autophagy is a routine pathway for cells to degrade unused proteins and damaged organelles in cells. Autophagy selectively removes dysfunctional cellular components but not damages the normally functioning organelles, to maintain the homeostasis of cells. In addition to the maintenance of the homeostasis of cells, autophagy clears the damaged organelles in disease or injury conditions to achieve cellular quality control. In some differentiated cells, such as red blood cells, some organelles are removed during the maturation, including mitochondria. The autophagy system can selectively clear the mitochondria and other organelles, which lead to the maturation of red blood cells. Dysfunction of autophagy impairs the clearance of damaged organelles, which results in injury of cells. In the maturation of red blood cells, failure to clear the cellular organelles by autophagy will disturb the normal differentiation of red blood cells, leading to a series of diseases such as anemia.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Mitochondria
In the selective autophagic clearance for cellular organelles, mitophagy is the most studied. Because of the linkage of mitochondrial damage to various neurological diseases, especially in Parkinson’s disease in which the disease-related proteins Parkin and PINK1 are associated with selective autophagy, it blooms the field of mitophagy research.
1.1 Mitochondria
The word mitochondrion comes from a combination of the Greek words that “mitos” means “thread” and the “khondrion”, “granule”. Mitochondria are organelles existing in most eukaryotic cells, including plants, animals, fungi, and others. The protozoan trypanosome has only one large mitochondrion, but most cells have hundreds of mitochondria in cells. The specific number of mitochondria in cells is related to the metabolic level of the cells. The cells that have higher metabolic activity have more mitochondria, even occupying 25% of the cytoplasmic volume. The mitochondria are double-membrane-bound organelles. A mitochondrion contains outer and inner membranes composed of phospholipid bilayers and proteins, with a cavity between the two membranes that is called intermembrane space of mitochondria (ISM). The mitochondria contain all the enzymes required for the tricarboxylic acid cycle. On the inner membrane, there are numerous cristae, which expand the surface area of the inner membrane. There are respiratory chain system and an ATPase complex on the inner membrane, which produces ATP. Mitochondria are placed to provide energy for cell activities. Mitochondria have a variety of shapes. The diameter is generally between 0.15 and 0.5 μm, and varies greatly in length from 1.5 to 3 μm, even as long as 10 μm.
In addition to providing energy to cells, mitochondria are also involved in the process of apoptosis. The Bcl-2 family is one of the most important proteins in the apoptotic system, which is divided into proapoptotic and antiapoptotic families. Once apoptosis is induced, the proapoptotic proteins damage the mitochondria, leading to a release of proteins from mitochondria, such as cytochrome c and Bid, which cause apoptosis. The anti-apoptotic Bcl-2 family functions in maintaining the stability and integrity of the mitochondrial membrane and inhibiting mitochondrial damage and protein release. Therefore, the functional mitochondria with balanced dynamics are extremely important for cellular physiology. The biogenesis of new mitochondria and the degradation of aging mitochondria are dynamically balanced. Autophagy is an important system for the removal of damaged mitochondria (El-Hattab et al. 2018; Tilokani et al. 2018).
Mitochondria are dynamic networks of double-membrane organelles with energy-production sites in the inner membrane of mitochondria (IMM). The mitochondrial electron transport chain transfers electrons to O2 through four protein complexes and two membrane electron carriers. During electron transfer, the mitochondrial respiratory chain complexes I, III, and IV pump protons from the mitochondrial matrix across the IMM to the ISM, which form an electrochemical gradient of H+, the mitochondrial membrane potential (ΔΨm). The ΔΨm of –160 mV (a high proton concentration in the ISM) drives the protons across the F0 subunit of the ATP synthase to produce ATP via the F1 subunit of the ATP synthase. The aerobic respiration coupled to the ATP synthase via an electron transport chain refers to oxidative phosphorylation (OXPHOS). In addition to energy production, mitochondria are the metabolic site for amino acids, fatty acids, and carbon, thus providing sources for protein, lipid, and carbohydrate synthesis. Dynamic changes by mitochondrial fusion and fission form a mitochondrial network for cellular metabolic demands. Mitochondrial fusion is dependent on normal OXPHOS and ΔΨm. Mitochondrial fusion increases the mitochondrial volume and space, further enhancing the function of the electron transport chain. Meanwhile, the fusion of mitochondria protects mitochondria from degradation by mitophagy. In contrast, mitochondrial injury causes depolarization of mitochondria, which increases mitochondrial fission and degradation (El-Hattab et al. 2018; Tilokani et al. 2018).
1.2 Mitophagy
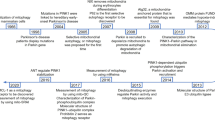
Although it is thought that mitochondria can be eliminated by macroautophagy, it is currently realized that mitochondria undergo a selective degradation by the autophagic system, which is called mitophagy. The earliest report on mitochondrial clearance by autophagy came in 1962. Using electron microscopy, the intact mitochondria and mitochondria in various stages of degradation are engulfed in lysosomes, which are the basis for the conception of autophagy/autophagosome that was proposed in 1963. Although there are other organelles in those autophagosomes, almost every lysosome contains a mitochondrion. At that time, the process for mitochondrial degradation and the fusion process of lysosomes were not understood, but it demonstrated that mitochondria can be degraded by lysosomes. In 2001, Elmore et al. proposed mitochondrial selective autophagy, a new conception of mitophagy. After that, more and more evidence suggest that the clearance of mitochondria in cells is specific and selective. The molecular mechanism for selective degradation of mitochondria has been revealed in both yeast and mammalian cells.
In mitophagy, mitochondrial fission is an important step for their degradation. Fission of mitochondria produces new mitochondria with a smaller size, which facilitates the engulfment of mitochondria by autophagosomes and fusion of autophagosomes to lysosomes. Fission of mitochondria also removes damaged parts from a mitochondrion, which allows normal parts to form a new healthy mitochondrion. It is therefore that the fission of mitochondria plays an important role in the clearance process for mitochondria. In mitophagy, the recognition of damaged mitochondria is critical for their degradation by lysosomes.
2 Mitochondrial Dynamics, Homeostasis, and Mitochondrial Autophagy
Mitochondria are dynamic organelles that undergo motion, fusion, fission, and degradation. The fusion and fission of mitochondria controls mitochondrial shape, size, and number. Mitochondria form a dynamic interconnecting network through fission and fusion, which accommodates the metabolic demands of the cells. Mitochondrial fission produces new mitochondria, allowing them to be transported and redistributed in cells. Mitochondrial fission promotes mitochondria to remove damaged parts that undergo mitophagy. Mitochondrial fusion promotes the exchange of mitochondrial components and enhances mitochondrial function. Mitochondria are the energy-production organelles in cells, which are distributed according to the energy demand in different regions in the cells. Intracellular movement of mitochondria is very important, which allows mitochondria to be transported intracellularly to areas of high metabolic demand. When mitochondria are damaged, they can be selectively engulfed by autophagosomes and fused to lysosomes for degradation, which maintains the healthy mitochondrial population and homeostasis of mitochondria. Mitochondrial dynamic processes, including movement, fusion, division, and autophagy, are regulated by a number of proteins and signals that maintain mitochondrial morphology, distribution, and function.
2.1 Mitochondrial Fission
A major regulator of mitochondrial fission is the dynein-related protein, Dnm1p in yeast, and Drp1 (Dynamin-related protein 1) in mammals. Mitochondrial fission is regulated by mitochondrial fission protein FIS1 and Drp1. Drp1 is a GTPase with a GTPase effector domain. When mitochondrial fission is initiated, the cytosolic distributed Drp1 is recruited to mitochondria, forming a polymer and wrapping around the scission site of the mitochondria. When GTP is bound and hydrolyzed, oligomerized Drp1 changes the conformation to cut mitochondria. FIS1, Mff, and MiD49/51 are anchored to the OMM and bound to Drp1 to recruit Drp1 to the OMM. Mitochondrial fission also requires actin and myosin IIA that provide a mechanical force for the cleavage of mitochondria.
2.2 Mitochondrial Fusion
Mitochondrial fusion is mainly performed by dynamin-related GTPases, the mitochondrial fusion protein MFN1/2 (Mitofusin 1/2), and OPA1 (Optic atrophy 1 protein). The fusion of mitochondria undergoes two processes, fusion of the OMM and fusion of the IMM, which occur simultaneously.
The fusion of the OMM is mediated by MFN1 and MFN2 that are anchored to the OMM via the C-terminal transmembrane domain and that have a conserved catalytic GTP-binding domain at the N-terminus. MFN1 and MFN2 form homo- or heterodimers that mediate fusion of the OMM dependent on GTP hydrolysis. Interactions of MFN1 and MFN2 on the OMM from two mitochondria pull them across each other, promoting fusion of the OMM.
The fusion of the IMM is mediated by OPA1, a dynamin-like GTPase that is anchored to the IMM via the N-terminal transmembrane domain, and that exposes the GTP-binding and GTPase effector domains to the ISM. OPA1 has different forms due to alternative splicing and proteolysis. Alternative splicing of OPA1 produces a long-form OPA1 (L-OPA1) that is cleaved into a short form (S-OPA1) via a proteolytic enzyme on the IMM. The proteolysis of L-OPA1 forms S-OPA1 that promotes the fusion of IMM.
Another protein that affects mitochondrial fusion is the F-box and leucine-rich repeat 4 (FBXL4), a mitochondrial protein localized on the IMM. It forms a quaternary protein complex through its leucine-rich repeat domain that is responsible for protein interactions. FBXL4 acts as a fusion protein or interacts with other mitochondrial fusion proteins to regulate mitochondrial fusion.
2.3 Mitochondrial Homeostasis
The fusion of mitochondria produces tubular or elongated mitochondria that are interconnected to form a dynamic network. Mitochondrial fusion allows material exchange between mitochondria and promotes molecules to diffuse throughout different parts of the mitochondria. Exchange of mitochondrial DNA, proteins, lipids, and metabolites among mitochondria is essential to maintain genetic and biochemical homogeneity within the mitochondrial population. It improves mitochondrial function and avoids accumulation of mitochondrial DNA (mtDNA) mutations during aging. In addition, mitochondrial fusion prevents mitochondria from mitophagy.
Mitochondrial fusion is critical for mtDNA maintenance. The cells require a sufficient amount of mtDNA to transactivate complex subunits that are mtDNA-encoded and function in a tricarboxylic acid cycle for energy production. Defects in mitochondrial fusion lead to an impairment of mtDNA synthesis, which causes mtDNA depletion or mtDNA mutations.
When mitochondria are damaged, mitochondrial fission increases. The damaged part is separated from the mitochondria, which forms a new healthy mitochondrion and a damaged mitochondrion. The damaged mitochondria are cleared by mitophagy, thus maintaining the normal function of mitochondria. Defects in mitochondrial fission or mitophagy lead to an accumulation of damaged mitochondria, which is tightly associated with diseases.
2.4 The Effect of Mitochondrial Fission and Fusion on Autophagy
In mammalian cells, mitochondria have a dynamic property. Fission of mitochondria makes mitochondria smaller, but fusion increases mitochondrial volume and makes mitochondria to form a connecting network. For mitophagy, the smaller mitochondria after fission are easily engulfed by autophagosomes. Decrease of ΔΨm caused by mitochondrial damage is important for mitophagy induction and also affects the process of mitochondrial dynamics. The healthy mitochondria with normal ΔΨm can be fused, but mitochondria with low ΔΨm cannot be fused and are localized to microtubule-associated protein light chain 3 (LC3)-positive autophagosomes, suggesting that damaged mitochondria undergo mitophagy. Since the loss of ΔΨm occurs before mitophagy, a depolarized state of mitochondria prevents mitochondrial fusion and promotes mitophagy.
Mitophagy is extremely important for mitochondrial homeostasis and the clearance of damaged mitochondria. Upon mitochondrial damage, the mitochondrial fission process “cuts off” the damaged parts of the mitochondrion to form two mitochondria, a damaged one and a healthy one. The healthy mitochondrion is fused to other healthy mitochondria by the action of MFN1/2 and OPA1. The damaged mitochondrion after mitochondrial fission has lower respiratory capacity and ΔΨm. Since mitochondrial fusion depends on normal ΔΨm, the damaged mitochondria with low ΔΨm are unable to fuse to other mitochondria. Meanwhile, loss of ΔΨm is an important signal for mitophagy. Decreased ΔΨm induces OPA1 to be hydrolyzed by proteases and MFN1/2 to be degraded by the proteasome. Degradation of these two mitochondrial fusion proteins reduces mitochondrial fusion capability, which in turn promotes mitochondrial fission and mitophagy. A loss of the function of Drp1 in mammalian cells causes a decrease of mitophagy and induces the formation of tubular mitochondria. However, in Drp1 knockout cells, although LC3-II is increased, the colocalization of mitochondria and LC3 labeled autophagosomes is rare. Overexpression of Drp1 promotes mitochondrial fission and induces mitophagy. Therefore, mitochondrial fusion and fission are directly associated with mitophagy.
3 Key Signals and Proteins in Mitophagy
Autophagy-related proteins (Atg) are critical for autophagy, and many Atgs are involved in mitophagy although they may not directly act on mitophagy. Atg is extremely important for the basal autophagy and selective mitophagy in the cells. The functional defects of Atg that affect the autophagy will also influence mitophagy. Recently, mitophagy has been extensively studied. Many proteins and signaling pathways are involved in mitophagy regulation, and more diseases have been found to be associated with mitophagy.
3.1 Mitophagy and Mitochondrial Receptors in Yeast
In 2009, two groups, Ohsumi and Klionsky, independently identified that Atg32 specifically participates in mitochondrial selective autophagy. In addition, Atg33 that was identified by Klionsky group is also a factor that selectively affects mitophagy. Thus, Atg32 is the earliest identified mitophagy receptor on the mitochondrial membrane.
During growth, yeast cells are converted from anaerobic to aerobic respiration when they are cultured in media containing lactose, ethanol or glycerol. Mitochondrial oxidative stress and damage occur during aerobic respiration. The aerobic respiration in yeast induces the expression of Atg32 that is recruited to the OMM. Atg32 is an Atg protein unique in yeast, for which no homolog has been identified in animals. Atg32 is required for mitophagy. Atg32 is a 59 kDa transmembrane protein on the OMM. The N-terminus of Atg32 faces to the cytosol and the C-terminus is located in the ISM. Inhibition of Atg32 expression reduces mitophagy, while overexpression enhances mitophagy. The 43 kDa N-terminus of Atg32 that faces to the cytosol carries two conserved motifs that bind Atg8 and Atg11, thus greatly influencing mitophagy. The binding motif of Atg32 bound to Atg8 is W/YXXI/L/V that is called AIM (Atg8-family interacting motif), which can bind to free form of Atg8 (a homolog of mammal LC3) or Atg8 coupled to PE (phosphatidylethanolamine) (Fig. 19.1). Interestingly, the AIM of Atg32 is also presented in Atg19 and the mammalian p62 protein, in which this functional motif binds to Atg8 and LC3. Mutation in AIM significantly decreases the binding of Atg32 to Atg8, which decreases mitophagy. In addition to the effects of Atg8 on Atg32-mediated mitophagy, interactions between Atg11 and Atg32 also greatly influence Atg32-mediated mitophagy. Atg32 contains two serines (114 and 119), and mutation of the serine site greatly reduces mitophagy. In yeast, the damaged mitochondria are accumulated in Atg11 and Atg32 deletion mutants after exposure to ROS. Thus, Atg32 is an important autophagy receptor identified in yeast mitochondria, which binds to Atg8 coupled PE on autophagophores to transfer mitochondria to autophagosomes. It is now well accepted that Atg32 is a mitochondrial receptor for mitophagy (Fig. 19.1).

In yeast, the mitochondrial outer membrane protein Atg32 binds to Atg8 through its AIM (Atg8-family interacting motif) that can bind to free form of Atg8 or Atg8 coupled to PE (phosphatidylethanolamine), which anchors mitochondria to phagophore. In mammals, NIX or FUNDC1 has an LC3-interacting region (LIR) that binds to LC3 that is coupled to PE that is bound to the phagophore a membrane. Binding of LIR on NIX to LC3 drives mitochondria to be engulfed by autophagosomes, thus inducing mitophagy
Similar to Atg32, Atg33, approximately 20 kD, is a protein resident on the OMM. In yeast, the Atg33 deletion mutant strain has lower mitophagy than wild-type stain after starvation. Mitophagy cannot be induced after logarithmic growth phase in Atg33 mutants, suggesting a role of Atg33 in mediating aging mitochondria for mitophagy, rather than mediating mitophagy in all phases by Atg32.
Among the genes in the regulation of mitophagy in yeast, Atg32 has been recognized as a selective autophagy receptor for mitophagy. Atg genes are necessary for autophagy as well as mitophagy. No autophagy or mitophagy can be induced in yeast strains even with single-gene deletion of ΔAtg1/ΔAtg6/ΔAtg8/ΔAtg12. The mitochondrial protein levels are significantly increased in Atg deletion mutants of yeast strains, suggesting that the core ATG functions in autophagy as well in the selective mitophagy. In addition, autophagy inactivation caused by Atg deficiency limits mitochondria-dependent cell growth, resulting in a significant increase in the number of cells at G1 phase. The mutants show decreases in ΔΨm and mitochondrial electron transport chain activity and increases in ROS levels and mtDNA mutations. Thus, mitophagy plays an important role in the maintenance of mitochondrial normal function.
3.2 Membrane Receptors on the OMM in Mammals
Atg32 is an autophagy receptor on the OMM, which mediates mitophagy through its binding to Atg8 in yeast. In mammalian cells, LC3 is a homolog of Atg8 and has a similar function as yeast Atg8. On the OMM of mammalian cells, there are two proteins that are functionally similar to yeast Atg32 and are considered to be receptors for mitophagy (Kanki et al. 2009; Okamoto et al. 2009). One is the OMM protein NIX identified by Dikic group (Novak et al. 2010), and another is FUNDC1 that is identified by Chinese scientist Chen’s Group (Liu et al. 2012).
NIX, also known as BNIP3L, is homologous to BNIP and was first identified as a protein that binds to Bcl-2 and that is involved in the regulation of apoptosis. NIX is a protein localized to the OMM, partially distributed in the endoplasmic reticulum. The involvement of NIX in mitophagy was first noticed in the regulation of maturation of reticulocytes. It was found that NIX knockout animals show residual mitochondria in reticulocytes, leading to failure of maturation for red blood cells, suggesting that NIX is involved in mitophagy. NIX has a LC3-interacting region (LIR) that binds to LC3. Binding of LIR on NIX to LC3 drives mitochondria to be engulfed by autophagosomes, thus inducing mitophagy, which is similar to AIM of yeast Atg32, binding to Atg8 in yeast for mitophagy induction (Fig. 19.1). NIX is therefore considered to be a mammalian mitophagy receptor. However, in NIX knockout animals, some reticulocytes can undergo maturation and develop to red blood cells, suggesting that some other factors may be also involved in reticulocyte maturation.
FUNDC1 is an OMM protein with its N-terminus facing to the cytosol and carrying a typical LIR motif, the YXXL amino acid sequence, in which tyrosine (Y) 18 and leucine (L) 21 are required for FUNDC1 bound to LC3. Mutations of Y18 and L21 or deletion of LIR in FUNDC1 lead to FUNDC1 failure to binding to LC3, thus failing to induce mitophagy. Interestingly, FUNDC1 seems to be involved in the induction of mitochondrial fission before mitophagy. Overexpression of FUNDC1 induces massive mitochondrial fission prior to autophagy, whereas knockdown of FUNDC1 promotes mitochondrial fusion. In addition, FUNDC1, in the absence of LIR, is still able to induce mitochondrial fission although it is completely unable to induce mitophagy. Thus, FUNDC1 is a membrane receptor that not only mediates mitophagy but also plays an important role in mitochondrial fission.
4 Ubiquitin-Dependent Mitophagy
Mitochondrial receptor-mediated mitophagy occurs through a direct binding of LC3 to the mitochondrial receptor on the OMM, thereby mediating mitophagy. However, in most cases, mitophagy is initiated by a series of signals, followed by ubiquitination of mitochondria on which the OMM proteins are polyubiquitinated. The autophagy receptors such as p62, NBR1, and OPTN recognize the polyubiquitin chains coupled to mitochondria. Meanwhile, the autophagy receptors also bind to LC3, thus driving polyubiquitinated mitochondria to be engulfed by the autophagosomes. In mammalian cells, loss of ΔΨm is required for mitophagy induction. In addition, mitochondrial dynamics, mitochondrial fission and fusion, is extremely important for mitophagy. Recently, the functions of Parkinson’s disease-related proteins PINK1 and Parkin in mitochondrial ubiquitination and mitophagy have been well studied.
4.1 Parkin and PINK1
Parkin is a cytosolic E3 ligase that was first identified to be linked to familial Parkinson’s disease by Shimizu group in 1998. Point or deletion mutations in PARK2 that encode Parkin cause autosomal recessive familial Parkinson’s disease. Since Parkin is a cytosolic E3 ligase, early studies focus on the identification of Parkin’s cytosolic ubiquitination substrate. Subsequently, using the Drosophila model, Pallank group found that Parkin plays an important role in the maintenance of mitochondrial morphology and function (Greene et al. 2003). Parkin-deficient Drosophila exhibits mitochondrial morphological abnormalities and degeneration of dopaminergic neurons and muscle tissue.
PINK1 was first identified as a PTEN-inducible kinase that is widely expressed in various tissues and organs, which was considered to be a tumor-associated factor at the beginning of its discovery. Soon, PINK1 was identified as a protein associated with Parkinson’s disease. Mutations of PINK1 encoding gene PARK6 cause autosomal recessive Parkinson’s disease. PINK1 is a protein of 581 amino acids with a mitochondrial targeting signal, a transmembrane helix and a serine/threonine kinase domain at the N-terminus. In Drosophila, PINK1 deletion mutants show phenotypes similar to Parkin mutants. Overexpression of Parkin in PINK1-deficient Drosophila can partially rescue phenotypes, while overexpression of PINK1 in Parkin-deficient Drosophila cannot rescue phenotypes, suggesting that Parkin acts downstream of PINK1 (Clark et al. 2006; Park et al. 2006).
Under normal conditions, PINK1 is transported into mitochondria through the recognition of PINK1 N-terminal mitochondrial targeting sequence by the mitochondrial complexes, the translocase of the outer membrane (TOM), and the translocase of the inner membrane (TIM). The N-terminal mitochondrial targeting signal of PINK1 is cleaved by mitochondrial processing peptidase in the matrix and rhomboid family protease presenilin-associated rhomboid-like protein (PARL) on the IMM, resulting in a cleavage at N-terminal Ala104. The N-terminal cleaved PINK1 returns to the cytosol and is degraded by the proteasome. In 2008, Youle group first showed that Parkin is accumulated on mitochondria after mitochondrial uncoupler CCCP (carbonyl cyanide m-chlorophenyl hydrazone) treatment (Narendra et al. 2008). Due to the uncoupling of mitochondria by CCCP, an increase of proton permeability eliminates ΔΨm of the IMM. Loss of ΔΨm induced by CCCP allows PINK1 to cross only the OMM but not the IMM, so that its N-terminal mitochondrial targeting signal cannot be processed by MPP in the matrix or the PARL on the IMM, resulting in an enrichment of the full-length PINK1 on the OMM. Enrichment of PINK1 on the OMM promotes Parkin translocation from cytosol to mitochondria and induces mitophagy (Narendra et al. 2009, 2010). Since Parkin is an E3 ligase that ubiquitinates mitochondrial proteins after its recruitment onto mitochondria, PINK1-induced Parkin recruitment onto mitochondria produces the polyubiquitin chains that are recognized by autophagy receptors (also see the section “Protein modification and autophagy activation”), thus the mitochondrial ubiquitination by PINK1/Parkin pathway is an important process for mitophagy (Fig. 19.2).

In most cases, mitophagy is mediated by autophagy receptors such as p62, NBR1, and OPTN that can interact with polyubiquitin chains as well as LC3. The accumulation of PINK1 on the outer membrane of mitochondria (OMM) recruits Parkin to mitochondria, leading to ubiquitination of substrates by Parkin. Thus, the autophagy receptors bind to ubiquitinated mitochondria (the ubiquitinated proteins on the OMM) and LC3, driving ubiquitinated mitochondria to be engulfed by the autophagosomes
PINK1 is autophosphorylated on the OMM, and its autophosphorylation is important for PINK1 activation and enrichment on the OMM (Aerts et al. 2015). Enrichment of PINK1 on the OMM recruits Parkin to mitochondria and phosphorylates Parkin at serine (S) 65 site on Parkin ubiquitin-like domain (Ubl) (Ordureau et al. 2015). Phosphorylation of Parkin at S65 alters Parkin conformation, which removes Parkin’s self-inhibition and increases its E3 ligase activity, allowing Parkin to catalyze its substrate ubiquitination. PINK1 also phosphorylates the S65 site on ubiquitin. Phosphorylation of ubiquitin on S65 activates ubiquitin and leads to resistance to deubiquitinases, thus promoting mitochondrial ubiquitination (Wauer et al. 2015). In addition, activated ubiquitin binds to Parkin to further activate Parkin, promoting Parkin to ubiquitinate substrates. Therefore, phosphorylation of Parkin and ubiquitin by PINK1 plays an important role in PINK1/Parkin-mediated mitophagy.
4.2 Autophagy Receptor and Mitophagy
One of the important features of the autophagy receptor proteins is that they have ubiquitin interacting motif, which allows them to bind to ubiquitinated substrates. Meanwhile, they also have a LIR binding to LC3, so that the bound ubiquitinated substrates can be delivered to autophagosomes. Youle group found that NDP52 and OPTN are repaired for mitophagy using rescue approaches in the cells where all five autophagy receptors, p62, NBR1, NDP52, OPTN, and TAX1BP, are knocked out (Vargas et al. 2019). PINK1 kinase activity is required for autophagy receptor enrichment on the OMM. Enrichment of PINK1 and Parkin on the OMM and interaction between autophagic receptors and ubiquitin activate TBK1. TBK1 activation increases mitochondrial recruitment of phosphorylated OPTN, NDP52, and p62. In addition, phosphorylation of OPTN by TBK1 at serines 473 and 513 increases the affinity of OPTN to ubiquitin chains as well as phosphorylated ubiquitin chains. Furthermore, phosphorylation of OPTN by TBK1 induces retention of OPTN on mitochondria, which further recruits ubiquitin and autophagy receptors onto mitochondria.
In mitophagy, phosphorylated ubiquitin is functionally similar to an autophagy receptor that induces Parkin and autophagy receptors onto the OMM. On mitochondria, Parkin is activated by PINK1 and phosphorylated ubiquitin, which promotes TBK1 to activate OPTN and NDP52. The positive feedback pathway further functions in mitophagy through phosphorylation of ubiquitin and activation of PINK1/Parkin.
5 Physiological Functions of Mitophagy
Mitophagy plays an important role in the formation and maturation of lens, red blood cells, and sperm as mitochondria are degraded for the maturation and development of these organs and tissues. These organs serve as ideal models for exploring the mechanism of mitophagy as the clearance of mitochondria is required for the development. In ATG5−/− mice, no mitochondrial clearance defects occur in the lens and red blood cells. In ATG7−/− mice, the clearance of mitochondria in reticulocytes occurs normally. Thus, the induction of mitophagy seems unrelated to ATG5 or ATG7. However, more reticulocytes with mitochondria appear in ULK1−/− mice. The clearance of mitochondria in reticulocytes is delayed in ULK1−/− mice. Upon CCCP treatment, the efficiency of the clearance of mitochondria in reticulocytes from ULK1−/− cells is similar to wild-type cells in vitro although the efficiency is lower in ULK1−/− cells than wild-type cells without CCCP. It seems that NIX is more related to mitophagy than ULK1 or ATG5 and ATG7. NIX−/− reticulocytes are immature and susceptible to apoptotic stimulation, leading to anemia, suggesting that NIX-mediated mitophagy is closely related to the pathology in blood cells.
6 Mitochondrial Autophagy and Cell Quality Control
Mitochondrial damage is closely related to apoptosis. During a mitochondrial injury, many mitochondrial proteins are released from damaged mitochondria to cytosol, leading to apoptosis. As mitophagy is the only way to clear the damaged mitochondria, mitophagy has an important role in protecting cells from mitochondrial-associated apoptosis. Under normal conditions, mitochondria pump protons across the IMM into the ISM, thus forming ΔΨm, which can block release of mitochondrial components to cytosol. Inhibition of the respiratory chain increases mitochondrial oxidative metabolites and causes a decrease in ΔΨm, resulting in an increase of mitochondrial membrane permeability. The superoxides produced by mitochondrial respiratory chain further damage the mitochondria that allow cytochrome c and other apoptotic factors releasing to the cytosol. Cytochrome c activates caspase-9 that further activates caspase-3, leading to apoptosis.
Mitophagy is an important cell quality control system for the clearance of damaged mitochondria. Injury of mitochondria induces mitochondrial fission so that the damaged mitochondria will be recognized and undergo mitophagy. Mitochondrial damage can be induced by increased ROS production during cellular stress, disease, and aging. Mitophagy effectively removes damaged mitochondria to avoid the release of mitochondrial proteins, which protect cells from apoptosis.
The selective autophagy in mitochondria interests scientists much in recent years, because mitochondria play crucial roles in the maintenance of physiological functions in cells, as well as in the pathogenesis of many diseases. Studies on selective mitophagy can expand our knowledge to understanding the functions of mitochondria as well as their roles in diseases.
Abbreviations
- AIM:
-
Atg8-family interacting motif
- Atg:
-
Autophagy-related protein
- CCCP:
-
Carbonyl cyanide m-chlorophenyl hydrazone
- Drp1:
-
Dynamin-related protein 1
- Fis1:
-
Mitochondrial fission 1 protein
- LC3:
-
Microtubule-associated protein light chain 3
- LIR:
-
LC3-interacting region
- MFN1/2:
-
Mitofusin1/2
- mtDNA:
-
Mitochondrial DNA
- OPA1:
-
Optic atrophy 1
- PE:
-
Phosphatidylethanolamine
References
Aerts L, Craessaerts K, De Strooper B et al (2015) PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem 290:2798–2811
Clark IE, Dodson MW, Jiang C et al (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–1166
El-Hattab AW, Suleiman J, Almannai M et al (2018) Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol Genet Metab 125:315–321
Greene JC, Whitworth AJ, Kuo I et al (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100:4078–4083
Kanki T, Wang K, Cao Y et al (2009) Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17:98–109
Liu L, Feng D, Chen G et al (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14:177–185
Narendra D, Tanaka A, Suen DF et al (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
Narendra D, Tanaka A, Suen DF et al (2009) Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 5:706–708
Narendra DP, Jin SM, Tanaka A et al (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298
Novak I, Kirkin V, McEwan DG et al (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11:45–51
Okamoto K, Kondo-Okamoto N, Ohsumi Y (2009) Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17:87–97
Ordureau A, Heo JM, Duda DM et al (2015) Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci USA 112:6637–6642
Park J, Lee SB, Lee S et al (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441:1157–1161
Tilokani L, Nagashima S, Paupe V et al (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem 62:341–360
Vargas JNS, Wang C, Bunker E et al (2019) Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Molecular cell
Wauer T, Simicek M, Schubert A et al (2015) Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524:370–374
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Science Press and Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Wang, R., Wang, G. (2019). Autophagy in Mitochondrial Quality Control. In: Qin, ZH. (eds) Autophagy: Biology and Diseases. Advances in Experimental Medicine and Biology, vol 1206. Springer, Singapore. https://doi.org/10.1007/978-981-15-0602-4_19
Download citation
DOI: https://doi.org/10.1007/978-981-15-0602-4_19
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-0601-7
Online ISBN: 978-981-15-0602-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)