
Abstract
Chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis are regarded as a diseases of accelerated lung ageing and show all of the hallmarks of ageing, including telomere shortening, cellular senescence, activation of PI3 kinase-mTOR signaling, impaired autophagy, mitochondrial dysfunction, stem cell exhaustion, epigenetic changes, abnormal microRNA profiles, immunosenescence and a low grade chronic inflammation due to senescence-associated secretory phenotype (SASP). Many of these ageing mechanisms are driven by exogenous and endogenous oxidative stress. There is also a reduction in anti-ageing molecules, such as sirtuins and Klotho, which further accelerate the ageing process. Understanding these molecular mechanisms has identified several novel therapeutic targets and several drugs and dietary interventions are now in development to treat chronic lung disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Accelerated ageing plays a key role in several pulmonary diseases, particularly chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF), but also may play a role in asthma, bronchiectasis, pulmonary hypertension and lung infections. Although cystic fibrosis (CF) is a disease of childhood, with advances in management patients may survive into middle age so that ageing processes may interact with the disease. In this chapter most focus is on COPD as this is a prevalent disease and most research on lung ageing has been related to this disease.
Lung Ageing
In normal individuals lung functions, such as forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) and increased functional residual capacity (resting lung volume) decline slowly with age after a peak of around 25 years in men (Janssens et al. 1999). There is a change in the flow volume loop that suggests that the loss of lung volumes is largely due to narrowing of peripheral airways. This loss of lung function over time may be associated with loss of lung elasticity and enlargement of alveolar spaces (which has been termed senile emphysema), resulting in a reduction in gas transfer, although this is not associated with alveolar wall destruction as in COPD. None of these changes in lung function with age are sufficient to cause symptoms or impaired oxygenation and do not lead to respiratory symptoms. The aged lung appears to be more susceptible to damage by environmental stresses, such as cigarette smoking and more susceptiblity to infection.
COPD
COPD is a global epidemic, with rising prevalence as populations live longer because of reduced mortality from infectious and cardiovascular diseases. COPD is the third commonest cause of death in developed countries and the fifth ranked cause of disability, but is increasing to a greater extent in developing countries (Lozano et al. 2012). COPD is present in approximately 10% of people over 45 years and in developed countries it as common in women as in men, reflecting the prevalence of smoking in the population (Barnes et al. 2015). The major risk factor for COPD world-wide is cigarette smoking, but in developing countries exposure to biomass smoke and household air pollution, especially in rural areas is also common (Sood et al. 2018). COPD is also linked to low socioeconomic status, which is likely to be explained by a combination of factors, such as smoking, air pollution, poor nutrition and damp housing (Burney et al. 2014).
COPD is characterized by largely irreversible and progressive airway obstruction as a result of fibrosis and obstruction of small airways (chronic obstructive bronchiolitis) and destruction of the lung parenchyma (emphysema) (Hogg and Timens 2009). This results physiologically in air trapping, hyperinflated lungs and dynamic hyperinflation that leads to shortness of breath on exertion, the major symptom of COPD patients. This results in accelerated decline in lung function over time, although only half of the patients diagnosed with COPD show rapid decline, the remainder have a normal decline starting from low peak lung volumes as a result of poor lung growth during childhood (Lange et al. 2015). About 20% of smokers develop COPD but the reasons for this susceptibility have not been fully determined. Although several genes have been implicated in susceptibility, but do not account for most of this susceptibility, other factors such as epigenetic modifications may be important for inherited and environmental influences.
The underlying mechanisms of COPD are poorly understood but it is associated with chronic inflammation that is largely corticosteroid-resistant (Barnes 2016). This inflammation is similar to that found in normal smokers, but appears to be amplified. A significant mechanism of amplification involves a reduction in the nuclear enzyme histone deacetylase-2 (HDAC2), which plays an important role in switching off activated inflammatory genes (Barnes 2009). Many cells and mediators are involved in COPD (Barnes 2014) and increased oxidative stress from inhaled smoke, or endogenously from activated lung inflammatory cells, is a key driving mechanism and results in a reduction in HDAC2 (Kirkham and Barnes 2013).
The global increase in COPD is likely to be related to the ageing population, as this disease predominantly affects the elderly, with peak prevalence around 65 years. Airway obstruction in COPD is slowly progressive and represents an acceleration of the normal decline in lung function with age, so this has suggested that COPD, and especially emphysema, involves acceleration of the normal lung ageing process (Ito and Barnes 2009; Mercado et al. 2015). There is increasing evidence that in COPD there are all of the hallmarks of accelerated ageing, compared to smokers with normal lung function and non-smokers, and this is discussed below (Table 3.1). Age is the most important risk factor for several chronic diseases, including COPD, and drives morbidity and mortality (Kennedy et al. 2014).
Idiopathic Pulmonary Ibrosis
IPF involves slowly progressive fibrosis of the lung parenchyma that is not associated with any causative mechanisms (Sgalla et al. 2018). It is usually diagnosed in people over the age of 70 and has a poor prognosis. The fibrotic process is thought to be due to sustained alveolar microinjury and aberrant repair in genetically predisposed individuals. There is increasing evidence that IPF is due to accelerated ageing and accumulation of senescent cells (Schafer et al. 2017). Removal of senescent cells abrogates the development of fibrosis in animal models of IPF.
Some patients with IPF also have emphysema and this overlap syndrome called combined pulmonary fibrosis and emphysema (CPFE) is increasingly recognized (Cottin and Cordier 2012). It is related to cigarette smoking, which is a defined risk factor for IPF as well as COPD. CPFE is characterized by accelerated ageing and cellular senescence (Chilosi et al. 2013).
Hallmarks of Ageing in Lung Disease
Chronic lung diseases are characterized by increased oxidative stress, which may be a key mechanisms in driving accelerated ageing and cellular senescence in COPD and IPF (Kirkham and Barnes 2013; Fois et al. 2018). Excessive reactive oxygen species (ROS) induce the accumulation of molecular damage, which shortens lifespan and an optimal level of ROS is needed for healthy ageing (Hekimi et al. 2011). Oxidative stress that is greater than that associated with normal ageing, may induce age-related degeneration that favors chronic inflammation.
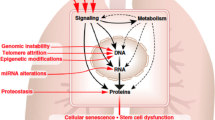
All of the classical hallmarks of ageing are seen in COPD and IPF, including telomere shortening, genomic instability, epigenetic alterations, loss of proteostasis, mitochondrial dysfunction, deregulated nutrient-sensing and stem cell exhaustion (Table 3.1) (Mercado et al. 2015; Meiners et al. 2015; Sgalla et al. 2018). There is an accumulation of senescent cells, which characteristically have irreversible cell cycle arrest, but are metabolically active and display what is termed a “senescence-associated secretory phenotype” (SASP) or “inflammaging”, with secretion of proinflammatory cytokines, chemokines and matrix metalloproteinases (MMP) (Salama et al. 2014). These proinflammatory mediators may induce further senescence in the cell itself (autocrine) and surrounding cells (paracrine), thus amplifying and spreading cellular senescence (Fig. 3.1). Senescent cells accumulate with age in tissues and the SASP proinflammatory state may be an important driving mechanism in age-related lung diseases.

Senescence-associated secretory phenotype (SASP). Senescent cells that express p16 and p21 are metabolically active, with activation of nuclear factor-kappa B (NF-κB) and mitogen- activated protein kinase p38, resulting in the increased expression of multiple inflammatory proteins, including cytokines, chemokines, proteases and growth factors, all of which are increased in chronic lung diseases. This inflammatory response has paracrine effects inducing senescence in adjacent cells and senescent cells also produce reactive oxygen species that further drive the senescence process
Telomere Shortening in Lung Disease
Telomere shortening is greater in COPD and IPF (Birch et al. 2017). Telomere shortening is found in circulating leukocytes from patients with COPD to a greater extent than in smokers with normal lung function and appears to be related to a greater risk of lung cancer (Lee et al. 2012; Houben et al. 2009; Savale et al. 2009; Rutten et al. 2016). Short telomeres increase the risks of developing emphysema amongst smokers (Houben et al. 2009). In a large observational study short telomeres in circulating leukocytes have been linked to reduced lung function, although the independent effect of COPD, after correction for age and smoking, is small. Shorter telomeres have also been described in alveolar epithelial and endothelial cells from patients with emphysema (Tsuji et al. 2006), although this may also be found in smokers with normal lung function (Tomita et al. 2010). Telomere-associated DNA damage is found in small airway epithelial cells from COPD patients compared to age-matched controls (Birch et al. 2015). A family with a genetic defect in telomerase has been linked to early onset emphysema (Alder et al. 2011). Approximately 1% of patients (mainly female) with severe emphysema have mutations of telomere reverse transcriptase (TERT) resulting in short telomeres (Stanley et al. 2015). Telomerase null mice with short telomeres showed increased susceptibility to develop emphysema and increased cellular senescence after chronic cigarette exposure (Alder et al. 2011). Knock-out of telomerase in mice results in replicative senescence of alveolar cells and a low grade lung inflammation, with increased interleukin(IL)-1, IL-6, KC (murine IL-8) and CCL2, indicating how telomere shortening can result in COPD-like lung disease (Chen et al. 2015). Telomerase knockout mice also show increased emphysema after cigarette smoke exposure (Birch et al. 2015). Genetic determinants of telomere length may increase susceptibility to COPD but also to risk of developing comorbidities, such as cardiovascular and metabolic diseases.
The mechanisms leading to telomere shortening in association with chronic diseases such as COPD are not yet understood. Increased oxidative stress impairs telomerase activity and telomerase DNA is particularly susceptible to DNA damage (Passos et al. 2007). Telomere shortening in turn results in activation of p21, resulting in cellular senescence and the release of proinflammatory mediators, such as IL-6 and CXCL8 (IL-8). Cultured pulmonary endothelial cells from COPD patients have reduced telomerase activity, which is associated with shorter telomeres with increased cellular senescence and a SASP response compared to age-matched non-smoking control subjects (Amsellem et al. 2011).
Telomere shortening is also an important feature in IPF. Telomerase defiance mice develop pulmonary fibrosis (Povedano et al. 2015). IPF patients have shorter telomeres in circulating leukocytes and in alveolar type 2 cells (Alder et al. 2008) and there is an increase in telomere DNA damage foci (Schafer et al. 2017). Mutation in telomerase genes is commonly found in patients with IPF, particularly if there is a family history (Armanios 2012). Shortened telomeres and telomere damage have also been described in airway epithelial cells from patients with bronchiectasis (Birch et al. 2016). Shorter telomeres have also been described in circulating leukocytes of patients with life-long chronic persistent asthma, possibly as a result of chronic inflammation and oxidative stress (Belsky et al. 2014).
Cellular Senescence
Diseases of accelerated ageing are characterized by the accumulation of senescent cells in tissues, which cells enter a state of irreversible cell cycle arrest (Fig. 3.2) (Munoz-Espin and Serrano 2014; van Deursen 2014). Mammalian cells have a limited number of divisions and cells enter cellular senescence and subsequently death by apoptosis once DNA damage can no longer be repaired effectively. The accumulation of senescent cells results in age-related loss of function and removal of senescent cells prolongs lifespan of mice and delays the development of organ failure and development of cancers (Baker et al. 2011; Baker et al. 2016a). Cellular senescence results from the activation of the tumor suppression pathways p53 and p21CIP1/WAF1 and the p16INK4a/retinoblastoma protein pathways, which are activated by the DNA damage response (DDR) in response to critical telomere shortening and telomeric and non-telomeric DNA damage. Deletion of cells that express p16INK4a increases the lifespan of normal mice (by up to 30%) and delays several age-related diseases and the onset of cancer (Baker et al. 2011, 2016a). In COPD and IPF there is an accumulation of senescent cells (Tsuji et al. 2006; Rutten et al. 2016; Schafer et al. 2017).

Cellular senescence pathways in lung disease. Repeated cell division leads to progressive telomere shortening (replicative senescence) or DNA (particularly telomeres) is damaged by reactive oxygen species (ROS), activating DNA damage pathways, including the DNA repair kinase ataxia-telangectasia mutated kinase (ATM) which phosphorylates histone 2AX (H2AX), leading to activation of p53 and the activation of p21CIP1/WAF1 which results in cell cycle arrest and senescence. Oxidative stress and other stresses acvtivate p16INK4a , which is a cyclin inhibitor and phosphorylates retinoblastoma protein (pRb) which also activates the cyclin dependent kinase inhibitor p21. Senescent cells secrete cytokines and chemokines – the senescence-activated secretory phenotype (SASP), which amplifies and spreads cellular senescence
Cellular senescence may be enhanced by extraneous stressful stimuli, such as oxidative stress and ultraviolet radiation in the case of skin. Cigarette smoke exposure in mice increases the number of p16 expressing cells in the lung, indicating that smoking can accelerate lung ageing (Sorrentino et al. 2014). Unlike apoptotic cells, senescent cells remain metabolically active and therefore may influence other cells through the SASP response (Salama et al. 2014; Correia-Melo et al. 2014)., The SASP response is triggered by p21, which activates p38 mitogen-activated protein (MAP) kinase and Janus-activated kinases (JAK). This results in the activation of the proinflammatory transcription factor nuclear factor-κB (NF-κB), resulting in secretion of cytokines, such as IL-1β, IL-6, TNF-α, growth factors, such as transforming growth factor (TGF)-β and chemokines, such as CXCL1, CXCL8 and CCL2, and elastolytic matrix metalloproteinases (MMP), such as MMP-2 and MMP-9, all of which are increased in diseases of accelerated ageing including COPD. Plasminogen activator inhibitor-1 (PAI-1) is another characteristic SASP protein and is increased in the sputum of COPD patients (To et al. 2013). This makes it likely that the chronic inflammation seen in COPD is a result of cellular senescence. CXCL8 acts via the chemokine receptor CXCR2, which induces cellular senescence and DNA damage, whereas blocking CXCR2 reduces both replicative and stress-induced senescence (Acosta et al. 2008). The SASP response itself mediates cellular senescence thus amplifying and spreading senescence. Activation of the p16INK4a pathway also activates NADPH oxidases, resulting in increased oxidative stress, which further activates NF-κB (Takahashi et al. 2006). JAK inhibitors inhibit the SASP response and thereby reduce frailty in ageing mice (Xu et al. 2015). SASP is also inhibited by rapamycin through inhibition of MK2, which is downstream of p38 MAPK (Herranz et al. 2015). The SASP also includes activation of the NRLP3 inflammasome and secretion of IL-1β (Acosta et al. 2013).
There is an accumulating of senescent cells in murine lungs with age, which is associated with emphysematous changes and deposition of collagen (Calhoun et al. 2016). Elimination of senescent cells (positive for p19ARF) appears to abrogate the decline in lung elasticity in ageing mice (Hashimoto et al. 2016). Cellular senescence is found in emphysematous lungs with enhanced expression of p21CIP1/WAF1, p16INK4a and senescence-associated β-galactosidase (SA-βGal) activity (Chilosi et al. 2013; Amsellem et al. 2011). Lung macrophages from COPD patients also express the senescence marker p21CIP1/WAF1 (Tomita et al. 2002). Furthermore, in COPD there is an increase expression of SASP components, including IL-1, IL-6, TNF-α, CXCL8, CCL2, TBG-β, MMP2 and MMP9 (Barnes 2004) (Fig. 3.1). Similarly lung fibroblasts and alveolar epithelial cells from IPF patients also show cellular senescence, with increased expression of SA-βGal, p16, p21, p53 and the SASP response, that may important in the fibrotic process (Alvarez et al. 2017; Schafer et al. 2017; Minagawa et al. 2011).
Increased number of senescent airway epithelial cells that express p16 are also seen in cystic fibrosis (Fischer et al. 2013) and in bronchiectasis (Birch et al. 2016).
Senolytic Therapy
Removal of senolytic cells expressing p16INK4a using a caspase-activated system extends the lifespan of mice and delays the development of organ failure (Baker et al. 2016a). Using this approach pulmonary fibrosis induced by bleomycin and its associated inflammatory response is prevented (Schafer et al. 2017). Senolytics are drugs that mimic this effect by inducing apoptosis in senescent cells and have little or no effect on proliferating cells (Kirkland et al. 2017). Several senolytic drugs have been identified by screening and include the naturally occurring polyphonic quercitin and the cytotoxic agent navitoclax (ABT263) which inhibits the Bcl2 family of anti-apoptotic proteins (Zhu et al. 2016). A combination of quercitin and navitoclax is effective in protecting against bleomycin-induced pulmonary fibrosis in mice (Schafer et al. 2017) and navitoclax also reduces the senescence associated with lung irradiation by selectively eliminating senescent type 2 pneumocytes (Pan et al. 2017). A senolytic combination of quercitin and dasitinib also reduced the numbers of senescent type 2 pneumocytes in bleomycin-induced fibrosis in mice, with reduced p16 and SASP proteins (Lehmann et al. 2017). Another approach is to target FOXO4, which is important in maintaining senescent cell viability through binding to p53. A cell penetrant D-retro-inverso peptide FOXO4-DRI interferes with this interaction so that cells enter apoptosis and in vivo it counteracts chemotherapy-induced senescence in normal and a strain of fast ageing mice (Baar et al. 2017). Several biotechnology and pharmaceutical companies are now searching for safe and effective senolytic therapies.
PI3K-mTOR Signaling
Signaling though the phosphoinositide-3-kinase (PI3K)-AKT-mammalian target of rapamycin (mTOR) pathway plays a key role in inducing cellular senescence and ageing and inhibiting this pathway extends the lifespan of many species, from yeast to mammals (Johnson et al. 2013). Rapamycin Inhibits mTOR and increases longevity in mice (Harrison et al. 2009). This may partly be due to a reduction in SASP, which reduces the paracrine spread of senescence (Laberge et al. 2015; Herranz et al. 2015). mTOR comprises two complexes, mTORC1 and mTORC2, with mTORC1 playing the most well defined role on growth and inhibited by rapamycin through its binding to the FK506-binding protein FKB12. mTORC1 is activated by growth factors, stress (including oxidative stress) and caloric nutrients, whereas mTORC2 is activated by growth factors. Inhibition of mTORC1 by rapamycin may results in up-regulation of mTORC2, which may reduce its efficacy so that dual inhibitors of mTORC1 and 2 might be more effective in chronic therapy. mTORC1 activation by oxidative and other cellular stresses and by nutrients results in increased protein synthesis through ribosomal S6 kinases with secretion of growth factors, such as TGF-β and vascular-endothelial growth factor (VEGF).
The mTOR pathway has multiple effects, including inhibition of FOXO transcription factors that are linked to longevity. There is evidence for PI3K activation in the lungs and cells of COPD patients as shown by increased expression of the downstream kinase phosphorylated AKT, which in turn activates mTOR (To et al. 2010) (Fig. 3.3). The mTORC1 complex is activated in COPD peripheral lung and peripheral blood mononuclear cells (PBMC), as shown by increased phosphorylated S6 kinase and is effectively inhibited by rapamycin (Mitani et al. 2016).

Reduced sirtuins and senescence. Sirtuin-1 is reduced by reactive oxygen species (ROS) through the activation of phosphoinositide-3-kinase (PI3K), mammalian target of rapamycin (mTOR) and microRNA-34a (MiR-34a). This accelerates ageing through increased acetylation of several proteins, including Ku70 that is important in double-stranded DNA repair, peroxisome proliferator gamma coactivator-1 (PGC-1), resulting in mitochondrial dysfunction, nuclear factor-kappa B (NF-κB), which orchestrates the senescence-associated secretory phenotype (SASP) and matrix metalloproteinase-9 (MMP-9), forkhead transcription factor-3a (FOXO3a) which reduces antioxidants and further increases oxidative stress. Sirtuin-6 is also reduced by oxidative stress through the same pathway, including miR-34a and this leads to activation of NF-κB, reduced β-catenin leading to reduced vascular-endothelial growth factor (VEGF), reduced telomere length and reduced Nrf2, further increasing oxidative stress. The coordinated reduction of these sirtuins leads to cellular senescence, accelerated aging and emphysema
Not surprisingly, there are important endogenous inhibitory mechanisms to limit the activation of this pathway. PI3K is inhibited endogenously by the membrane tyrosine phosphatases phosphatase and tensin homologue (PTEN) and SH2-containing inositol-5'-phosphatase-1 (SHIP-1). Both have oxidation-susceptible cysteine residues in the active site that is readily modified by oxidative stress, and thus excessive oxidative stress reduces their catalytic activity (Worby and Dixon 2014). PTEN polymorphisms have been shown to be a genetic risk factor for COPD (Hosgood et al. 2009). PTEN activity and expression are markedly reduced in COPD lungs and this is likely to be due to oxidative stress and leads to activation of PI3K (Yanagisawa et al. 2017a).
mTOR is inhibited endogenously by 5’-adenosine monophosphate activating kinase (AMPK) which is activated by low ATP levels in the cell. AMPK is activated by the biguanide metformin, which therefore inhibits mTOR signaling and has been shown to extend the lifespan of several species, including mammals (Anisimov et al. 2008). AMPK is also activated by caloric restriction, also extends lifespan (Colman et al. 2014). This suggests that activation of the mTOR pathway may play an important role in COPD and inhibition of this pathway offers a future therapeutic opportunity (Lamming et al. 2013).
Mitochondrial Dysfunction
Mitochondrial dysfunction is an important feature of ageing and mitochondria are an important intracellular source of ROS (Correia-Melo and Passos 2015). There is a gradual accumulation of mutations in mitochondrial DNA during ageing, with a reduced resistance to damage by oxidative stress (Zheng et al. 2012). Mitochondrial function is regulated by the transcription factors peroxisome proliferator-activated receptor-gamma co-activator PGC-1α and PGC-1β, which are inhibited by telomere shortening and by mTOR activation so that with senescence there is a reduction in mitochondrial membrane potential, fragmentation and reduced mitochondria numbers. In turn mitochondria derived ROS may maintain DNA damage, thus driving further senescence through feedback loops. Damaged mitochondria are removed by autophagy (mitophagy) which is impaired in senescent cells (Garcia-Prat et al. 2016). Mitochondria play an important role in senescence and in driving the SASP response in lung disease (Birch et al. 2017).
COPD is associated with increased mitochondrial ROS production, decreased intracellular anti-oxidants and reduced numbers of mitochondria (Kirkham and Barnes 2013; Sureshbabu and Bhandari 2013). COPD cells show increased mitochondrial ROS generation (Wiegman et al. 2015). Mitochondrial dysfunction is also found in skeletal muscle of COPD patients with muscle weakness, with increased mitochondrial ROS production and decreased numbers (Meyer et al. 2013). Prohibitins (PHB), which are localized to the inner membrane of mitochondria, play a role in mitochondrial biogenesis and maintaining normal function (Artal-Sanz and Tavernarakis 2009). However, PHB1 expression is reduced in epithelial cells of smokers and to a greater extent in COPD patients, suggesting a mechanism for mitochondrial dysfunction in COPD patients (Soulitzis et al. 2012). Mitochondrial fragmentation with increased expression of mitochondrial fission/fusion markers, oxidative phosphorylation (OXPHOS) proteins (Complex II, III and V) are found in epithelial cells from COPD patients to a greater extent than cells from normal smokers, along with SASP expression with increased IL-1β, IL-6, CXCL8 secretion (Hoffmann et al. 2013). The mitochondrial stress markers Parkin and PTEN-induced protein kinase-1 (PINK1) are also increased in COPD patients (Hoffmann et al. 2013; Mizumura et al. 2014). Knockdown of the PINK1 protects mice against mitochondrial oxidative stress induced by cigarette smoke (Mizumura et al. 2014). Damaged mitochondria are normally efficiently removed by mitophagy through highly regulated pathways, including PTEN, which may be reduced by oxidative stress and PINK1. Failure of mitophagy leads to generation of intracellular ROS and eventually cell death. PGC-1α, which is a key regulator of mitochondrial biogenesis and generation of mitochondrial ROS is increased in epithelial cells of mild COPD patients but reduced with increasing COPD severity (Li et al. 2010).
Mitochondrial dysfunction is also evident in IPF and type 2 pneumocytes show the accumulation of abnormal and dysfunctional mitochondria associated with reduced PINK1 and increased expression of profibrotic mediators (Bueno et al. 2015). PINK1 gene knockout in mice is linked to increased susceptibility to lung fibrosis.
Defective Autophagy
Autophagy is a highly regulated process that removes degraded proteins, damaged organelles (including mitochondria, as discussed above) and foreign organisms (such as bacteria), in order to maintain normal cellular function. Defective autophagy is a key characteristic of ageing cells and age-related diseases, including COPD (Mizumura et al. 2012; Mizushima et al. 2008). Autophagy removes damaged proteins, organelles and pathogens via lysosomal degradation. However, ageing cells accumulate damaged and misfolded proteins through a decline in autophagy, leading eventually to cellular senescence. Although autophagy plays a protective role in response to exogenous stress, prolonged and excessive autophagy has been associated with cell death when cytosol and organelles are destroyed irreparably. Inhibition of autophagy increases susceptibility to oxidative damage and apoptosis, whereas activation of autophagy leads to inhibition of apoptosis (Murrow and Debnath 2013). Cigarette smoke and oxidative stress activate autophagy, indicating that autophagy may be a protective response designed to remove damaged proteins and organelles. Alveolar macrophages from smokers show defective autophagy and this could contribute to accumulation of aggregates, abnormal mitochondrial function and defective clearance of bacteria (Monick et al. 2010). Patients and mice with emphysema show increased autophagy markers in lung tissue, with increased activation of autophagic proteins, such as light chain-3 (LC3-II), and autophagy may be contributory to apoptosis and alveolar cell destruction (Chen et al. 2010). Other studies have demonstrated increased autophagic vacuoles (autophagosomes) in COPD. However, it is possible that completion of autophagy may not occur (defective autophagic flux). Macrophages from smokers show defective autophagic flux, resulting in accumulation of the substrate of autophagy, p62, and misfolded proteins due to dysfunctional lysosomal digestion of the autophagosomal load caused by a reduction in the lysosomal protein LAMP2 (Monick et al. 2010). Inhibition of autophagy after cigarette smoke exposure leads to accumulation of p62 and ubiquitinated proteins in airway epithelial cells, resulting in increased cellular senescence and SASP with secretion of CXCL8, thereby mimicking the changes seen in COPD cells (Fujii et al. 2012). Loss of autophagy might account for reduced mitophagy and also contribute to defective phagocytosis of bacteria in COPD (Donnelly and Barnes 2012). Autophagy may be impaired through the activation of PI3K-mTOR signaling, resulting in inhibition of unc-51 like autophagy-activating kinase-1 (ULK1) complex that normally activates autophagy, thereby linking defective autophagy to the accelerated ageing mechanisms (Dunlop and Tee 2014). Defective autophagy and mitochondrial function play a key role in IPF and the generation of profibrotic mediators, such as TGF-β (Mora et al. 2017).
Stem Cell Exhaustion
The clearance of senescent cells means that progenitor cells are needed to maintain cell numbers, but this may become less efficient or may exhaust the regenerative capacity of stem cells. Stem cell exhaustion and depletion are characteristic feature of ageing (Lopez-Otin et al. 2013).
Senescence of mesenchymal stem cells (fibroblast, endothelial cells) may be a mechanism of emphysema and the failure to repair lung injury. Damaged alveolar cells may be replaced by progenitor cells, particularly alveolar type 2 (AT2) cells (Kubo 2012). However, once the alveolar architecture is destroyed by elastases, such as MMP9, progenitors cannot rebuild the appropriate functional lung structure. In stem cells, ROS force cells out of quiescence and into a more proliferative state by activating PI3K/AKT signaling and further promoting the production ROS, thus repressing FOXO-mediated stress response and autophagy. AT2 cells are progenitors for type I alveolar cells and show evidence of senescence in patients with emphysema (Tsuji et al. 2006).
Circulating endothelial progenitor cells (EPC) and specifically blood outgrowth endothelial cells are made in the bone marrow and are important in maintaining endothelial integrity (Fadini et al. 2012). EPC from smokers and COPD patients show cellular senescence and increased DNA double-strand breaks compared to cells from non-smokers and this is correlated with reduced expression of sirtuin-1 (Paschalaki et al. 2013). These senescent stem cells are poorly effective in repairing endothelial damage and provide a link between COPD and vascular ageing that leads to ischaemic heart disease.
In diseases of accelerated ageing, stem cells, including AT2 cells and EPCs, show features of cellular senescence, oxidative stress-induced DNA damage. This leads to loss of regenerative capacity and eventually to organ failure, including lungs (Sousounis et al. 2014). Understanding the molecular mechanisms of stem cell ageing is critical to elucidating accelerated ageing in the lungs (Oh et al. 2014). Senescent stem cells characteristically show increased generation of mitochondrial ROS, mitochondrial damage and dysfunction, activation of the PI3K-mTOR signaling, DNA damage with double-stranded DNA breaks and a defect in the DNA damage-sensing kinase ATM (ataxia telangiectasia mutated), defective proteostasis and deficiency of anti-ageing molecules, such as sirtuin-1 and FOXO transcription factors. EPCs from COPD patients show activation of PI3K, reduced ATM and reduced sirtuin-1, all of which may be reversible by resveratrol, a sirtuin activator (Paschalaki et al. 2013). Accumulation of various types of DNA damage in stem cells with age may be a major mechanism leading to progressive failure of stem cells to repair damage of organs and thus to slowly progressive organ failure (Behrens et al. 2014). Epigenetic changes in stem cells, including DNA methylation and histone acetylation, also play an important role in stem cell senescence.
MicroRNA and Lung Ageing
MicroRNA (miRNA) are small non-coding single-stranded RNAs of 18-22 nucleotides that regulate the post-transcriptional expression of genes by inhibiting their translation or inducing degradation of mRNAs by binding to complimentary sequences in the 3’-UTR of mRNAs. There is increasing evidence that miRNAs play an important role in in the ageing process and may regulate several key proteins involved in cellular senescence, such as p16 (miR-24), p53 (miR-885-5p), SASP (miR-146) and reduced sirtuin-1 expression (miR-34a) (Gorospe and Abdelmohsen 2011; Munk et al. 2017). Genome-wide assessment of miRNA expression in monocytes of elderly compared to young individuals show that miRNA tend to decrease with age and regulate PI3K-mTOR signaling and DNA repair (Noren Hooten et al. 2010). MiRNAs that have been implicated in cellular senescence are also increased in diseases of accelerated ageing, including COPD (Dimmeler and Nicotera 2013).
MiR-34a, which is known to down-regulate sirtuin-1, shows increased expression in peripheral lungs and epithelial cells of COPD patients and is correlated with increased markers of cellular senescence in lung cells (Baker et al. 2016b). MiR-34a also regulates sirtuin-6, which is reduced in COPD along with sirtuin-1 (Nakamaru et al. 2009). MiR-34a is increased by oxidative stress through activation of PI3K-mTOR signaling pathways and this leads to a parallel reduction in sirtuin-1 and sirtuin-6, whereas other sirtuins are unchanged. Importantly, an antagomir of miR-34a restores sirtuin-1 and -6 in small airway epithelial cells from COPD patients, reduces markers of cellular senescence (p16, p21, p53), reduces the SASP response (IL-1β, IL-6, TNF-α, CCL2, MMP9) and increases the growth of senescent epithelial cells (Baker et al. 2016b). This suggests that blocking specific miRNA may result in rejuvenation of these cells. MiR-34a is also increased in macrophages of COPD patients and this may be associated with the impaired phagocytosis and uptake of apoptotic cells (efferocytosis) that is seen in COPD patients (McCubbrey et al. 2016). MiR-126, which plays a key role in maintain vascular integrity and endothelial cell function, is reduced in endothelial progenitor cells and airway epithelial cells and from COPD patients and plays a key role in regulating the DNA damage response pathway that is linked to cellular senescence (Paschalaki et al. 2018).
MiRNAs are exported from cells in extracellular vesicles, which include microvesicles and exosomes and then may be taken up by other cells (Fujita et al. 2015; Turturici et al. 2014). This might provide a means of spreading senescence from cell to cell within the lung, but also via the circulation to other organs, resulting in accelerated ageing in other systems or multimorbidity (Barnes 2015; Kadota et al. 2018). Thus, targeting miRNAs may be a future therapeutic strategy for treating and preventing cellular senescence and delivery of miRNA via extracellular vesicles and nanoparticles might be a novel way to deliver there therapies (Jiang and Gao 2017).
Epigenetic Mechanisms
There is little evidence that genes have important effects on the ageing process, but increasing evidence that epigenetic mechanisms may be important, with long-term changes in gene expression due to environmental exposures. These epigenetic changes include DNA methylation and modifications in histone proteins (for example, by acetylation, methylation, phosphorylation), which result in increased or decreased gene transcription (Song and Johnson 2018). Chromatin structure changes with age and becomes more “open”, which means it is more transcriptionally active (Feser and Tyler 2011). There is a decline in histone proteins with ageing and consistent changes in histone modifications and associated modifying enzymes, with increased histone acetylation and either increased or decreased histone methylation. Changes in histone methylation with age are likely to be important in accelerated ageing and changes in trimethylation of lysine 4 on histone-3 (H3K4me3) occur during ageing and regulate cellular senescence (Zhang et al. 2014). At present there is little information about histone methylation status in diseases of accelerated ageing, such as COPD. Histone methylation plays an important role in regulating the various proteins involved in autophagy, DNA repair and SASP. The role of histone methyltransferases and demethylases in the regulation of cellular senescence is under intense investigation now that selective enzymes inhibitors are in development. Coactivator-associated arginine methyltransferase-1 (CARM1) deficiency causes cellular senescence in type 2 pneumocytes and accelerates the development of emphysema in mice, indicating a key role for histone methylation in protecting against lung ageing (Sarker et al. 2015). Changes in DNA methylation have been described in COPD cells and may play a role in cellular senescence (Clifford et al. 2018). For example, DNA methylation of the FOXA2 and SPDEF genes in airway epithelial cells of COPD patients has been linked to mucus hypersecretion (Song et al. 2017).
Epigenetic changes associated with ageing are of particular relevance in stem cell populations and lead to dysfunction and depletion (Armstrong et al. 2014). Mesenchymal stem cells from elderly people appear to have less potential for differentiating into different cells types and this is linked to changes in DNA methylation (Horvath 2013).
Immunosenescence
The immune system becomes less effective during ageing, with impaired immune responses to new antigens, a greater susceptibility to infection and an increased tendency to develop autoimmunity. Immunosenescence affects both innate and adaptive immunity resulting in loss of function and has been implicated in chronic inflammatory diseases. Immunosenescence is important in several pulmonary diseases, including COPD, severe asthma and IPF (Murray and Chotirmall 2015). Ageing of innate immunity leads to defective function in innate cells, with impaired cell migration and signaling through pattern recognition receptors, such as Toll-like receptors (TLR) (Hazeldine and Lord 2015). Neutrophils show decreased phagocytosis, chemotaxis and apoptosis and have reduced directional function that is corrected by PI3K inhibition (Sapey et al. 2014). This defect is exaggerated in patients with COPD (Sapey et al. 2011). Macrophages show reduced phagocytosis with age and this is increased to a greater extent in COPD macrophages (Donnelly and Barnes 2012). Dendritic cells produce less interferon and reduced TLR signaling, which is associated with decreased T-cell-mediated immunity, resulting in reduced ability to protect against pathogens in the elderly and an increased risk of carcinogenesis (Mahbub et al. 2011). However in patients with severe COPD there is greater expression of TLR4, which is associated with an amplified inflammatory response to colonizing bacteria (Di Stefano et al. 2017).
Defects in adaptive immunity are also commonly seen in COPD and may be related to ageing. There is a loss of naïve T cells and B cells as the thymus involutes with age, as well as telomere shortening, with consequent reduced responses to new antigens. A characteristic of T cells from elderly individuals is a decrease in CD4+/CD8+ ratios and a loss of the co-stimulatory molecule CD28, with an increase in both CD4+CD28null and CD8+CD28null cells, which have reduced immune and vaccine responses (Arnold et al. 2011). In COPD CD8+CD28null cells have reduced HDAC2 expression and are corticosteroid-resistant (Hodge et al. 2015).
Autoimmunity is also a manifestation of immunosenescence with increased production of autoantibodies that may lead to further tissue damage. In COPD there are increased autoantibodies directed against endothelial cells and against carbonylated proteins formed by exposure to oxidative stress in severe disease (Feghali-Bostwick et al. 2008; Karayama et al. 2010; Kirkham et al. 2011). This increase in autoimmunity has been associated with an imbalance between Th17 and regulatory T cells (Treg). An increased ratio of Th17 to Treg cells is found in sputum In COPD patients (Maneechotesuwan et al. 2013). Treg cells may even transform into Th17 cells under disease conditions with loss of the key Treg transcription factor Foxp3 (Komatsu et al. 2014).
There is evidence for activation of the PI3K signaling pathway. In ageing neutrophils the impaired chemotaxis response is associated with increased PI3K and restored by a PI3K inhibitor (Sapey et al. 2014). A rapamycin analog, everolimus, reduces immunosenescence in normal elderly individuals, with an increased antigenic response to influenza vaccination and a reduction in T cells expressing programmed death-1 (PD-1) receptors that are increased in senescent CD4+ and CD8+ cells (Mannick et al. 2014).
Reduced Anti-ageing Molecules
Many endogenous anti-ageing molecules that counteract the mechanisms of senescence and a reduction in their expression may accelerate the ageing process. Defective anti-ageing molecules have been suggested as a mechanism for accelerated lung ageing in COPD and other pulmonary diseases (Ito and Barnes 2009; Faner et al. 2012; Mercado et al. 2015).
Sirtuins
Silent information regulators or sirtuins are well recognized as anti-ageing molecules that regulate lifespan. Sirtuins are highly conserved NAD+-dependent enzymes that play a role in resistance to stress, genomic stability and energy metabolism (Finkel et al. 2009; Watroba et al. 2017). Of the 7 sirtuins found in mammals most attention has centered on sirtuin-1 and sirtuin-6, as both are associated with prolongation of lifespan in many species, including mammals (Guarente 2011). Sirtuin-1 deacetylates many key regulatory proteins and transcription factors that are known to be involved in DNA repair, inflammation, antioxidant gene expression and cellular senescence, including the PI3K-AKT-mTOR pathway and autophagy (Fig. 3.3). Sirtuin-1 deacetylates the transcription factor FOXO3a, which enhances antioxidant responses (particularly superoxide dismutases) and inhibits p53-induced cellular senescence, inhibits NF-κB leading to suppression of inflammation and activates PGC-1α, which maintains mitochondrial function. Sirtuin-1 is markedly reduced in peripheral lung and circulating PBMC of patients with COPD (Nakamaru et al. 2009; Rajendrasozhan et al. 2008; Baker et al. 2016b). Sirtuin-1 is reduced by oxidative stress via reduction in PTEN and activation of the PI3K-mTOR pathway and in turn sirtuin-1 inhibits mTOR signaling. Sirtuin-1 also activates autophagy by inhibiting mTOR (Lee et al. 2008). The polyphenol resveratrol, found in the skin of red fruits and in red wine, activates sirtuin-1, but is poorly bioavailable, so this has led to the development of more potent sirtuin-1 activating compounds (STACs), which are now in clinical development for the treatment of age-related diseases (Hubbard and Sinclair 2014; Bonkowski and Sinclair 2016). Sirtuin-6 is an ADP-ribosylase as well as protein deacetylase and plays a key role in regulating DNA repair, telomere maintenance, inflammation and metabolic homeostasis and, like sirtuin-1, is linked to extension of lifespan (Kugel and Mostoslavsky 2014). Sirtuin-6 expression is reduced in the lungs of patients with COPD (Nakamaru et al. 2009; Takasaka et al. 2014). Reduced sirtuin-6 is found in airway epithelial cells of COPD patients and is reduced by cigarette smoke exposure, resulting in cellular senescence and impaired autophagy (Takasaka et al. 2014). As discussed above, oxidative stress activates PI3K-mTOR signaling, which increases the expression of miR-34a that directly inhibits sirtuin-1 and sirtuin-6 mRNA and protein expression without any effects on the other sirtuins (Baker et al. 2016b). Circulating sirtuin-1 concentrations are also reduced in patients with COPD (Yanagisawa et al. 2017b).
The role of sirtuins in other lung diseases is less well defined. Reduced sirtuin-1 is found in PBMC from elderly patients with severe asthma and appears to increase IL-4 secretion though increased acetylation of the transcription factor GATA-3 (Colley et al. 2016). Sirtuin-1 knockout in mice enhances lung fibrosis, and sirtuin-1 inhibits TGF-β fibroblast activation. Paradoxically sirtuin-1 is increased in the lungs of IPF patients, perhaps as a compensatory mechanisms (Zeng et al. 2017).
Klotho
Animal models of accelerated ageing have identified other key molecules involved in senescence, Klotho is defective in a mouse model of premature ageing, which has a substantially decreased lifespan, with features of accelerated ageing, including emphysema (Dermaku-Sopjani et al. 2013). Klotho is a transmembrane protein that is a co-receptor of fibroblast growth factor (FGF23) and regulates insulin/IGF signaling, phosphate homeostasis, cell survival and proliferation. It is protective against oxidative stress and decreased expression of Klotho is found in epithelial cells of COPD patients and is reduced by oxidative stress, resulting in increased release of inflammatory cytokines from these cells (Gao et al. 2015). Circulating concentrations of Klotho are also reduced in patients with COPD (Kureya et al. 2016).
SMP-30
Senescence marker protein-30 (SMP30) is an anti-ageing molecule that was identified in a mouse model of ageing, which regulates calcium homeostasis and is also sensitive to oxidative stress (Feng et al. 2004). Reduced expression of SMP30 is found in aged tissues, including lung. In SMP30 knockout mice there is a increased susceptibility to development of emphysema after exposure to cigarette smoke (Sato et al. 2006).
Implications for Therapy
Considerable progress has been made in understanding the molecular mechanisms involved in accelerated ageing in chronic lung diseases. Although it is unrealistic (but not impossible) to expect reversal of the normal ageing process, it may be possible to reduce the mechanisms that accelerate senescence in these diseases. Several novel therapeutic targets that have been identified leading to the development of geroprotectors (Moskalev et al. 2017). Existing drugs (such as metformin and rapamycin) may be repurposed and novel drugs developed by screening and rational drug design. The potential for these drugs is that they may treat multimorbidities, which are diseases of ageing that occur together in the elderly, as there appear to be common pathways (Barnes 2015). Future therapies may also involve lifestyle interventions, such as diet and increased physical activity (de Cabo et al. 2014).
Pharmacological Therapies
Better understanding of cellular senescence mechanisms has identified several potential therapies for inhibiting accelerated ageing in lung disease (Fig. 3.4) (Ito et al. 2012). As discussed above, the PI3K- mTOR pathway plays a key role in cellular senescence and inhibition of autophagy, and inhibitors of this pathway may extend lifespan in several species, including mammals. Rapamycin induces autophagy, increases sirtuin-1 and extends the lifespan of all organisms, including mammals (Bjedov and Partridge 2011; Lamming et al. 2013). Rapamycin is very effective in inhibiting activated mTOR in COPD cells in vitro (Mitani et al. 2016). However, rapamycin and its analogues (rapalogs) have several adverse effects, including mouth ulceration, anaemia, pneumonitis and delayed wound healing, making it unsuitable for long-term use. A pilot study in elderly humans demonstrated its potential in lower doses in reversing immunosenescence and this was not associated with significant adverse effects (Mannick et al. 2014). Metformin is widely used to treat type 2 diabetes and indirectly activates AMPK, resulting in inhibition of mTOR and extension of lifespan in mice, possibly though increasing Nrf2-induced antioxidant gene expression (Anisimov et al. 2008; Martin-Montalvo et al. 2013). Metformin is relatively well-tolerated, it might be a suitable therapy for treating accelerated ageing in COPD and other pulmonary diseases. It has been shown to be well-tolerated in COPD patients (Hitchings et al. 2016). PI3K signaling may also be inhibited by activators of the endogenous inhibitor SHIP-1 (Stenton et al. 2013) and these drugs have already entered into clinical trials for treatment of allergic disease (Leaker et al. 2014).

Targeting cellular senescence in lung disease. The PI3K-mTOR pathway driven by reactive oxygen species (ROS), which activate phosphoinositide-3-kinase (PI3K) and mammalian target of rapamycin (mTOR), which activate microRNA-34a (miR-34a) to inactivate and reduce sirtuin-1 activity, which accelerates the ageing process. There are several endogenous and exogenous inhibitors of this pathway. Increased ROS may be the result of reduced activity of the antioxidant regulating transcription factor Nrf2. There are several ways to modulate this pathway (green boxes). Nrf2 may be activated by small molecule activators, ROS may also be counteracted by antioxidants. PI3K is inhibited by the phosphatases PTEN (phosphatase and tensin homolog) and SHIP-1 (SH2-containing inositol-5'-phosphatase-1). SHIP-1 activators are now in development and there are several PI3K inhibitors. mTOR is inhibited by rapamycin and its less toxic analogs and endogenously by AMP-kinase (AMPK), which may be activated by caloric restriction and by metformin. Increased miR-34a may be targeted specifically by antagomirs. Sirtuin-1 may be activated by resveratrol and novel sirtuin-activating compounds (STACs). Finally senescent cells may be removed by senolytic drugs
Naturally occurring molecules may also be effective and may be obtained via dietary supplementation (neutraceuticals). Quercitin, a polyphenol found in apples, is also an activator of AMPK (Mitani et al. 2017) as well as a senolytic (Schafer et al. 2017). Resveratrol, found in the skins of red fruit and in red wine and increases lifespan in several species, including mice, through the activation of sirtuin-1. In vitro resveratrol inhibits the SASP response in COPD epithelial cells (Culpitt et al. 2003) and is effective against neutrophilic inflammation induced by lipopolysaccharide in mice (Birrell et al. 2005). However, resveratrol and related dietary polyphenols have poor oral bioavailability, rapid metabolism and low potency, which has led to the development of novel potent synthetic analogs known as STACs, which work on an allosteric mechanism to stimulate sirtuin-1 activity (Hubbard and Sinclair 2014; Bonkowski and Sinclair 2016). In mice exposed to cigarette smoke the STAC SRT-2171 prevents the increase in MMP-9 that is associated with neutrophilic inflammation and improves lung function (Nakamaru et al. 2009). In addition, resveratrol reduces senescence in EPCs from COPD patients through an increase in sirtuin-1, indicating that exhausted stem cells may be an important target of STACs (Paschalaki et al. 2013). A resveratrol derivative isorhapontigenin, which is found in Chinese herbal remedies used to treat inflammatory diseases, is more potent than resveratrol at inhibiting the SASP response in COPD cells and has a better oral bioavailability (Yeo et al. 2017).
Oxidative stress is an important driving mechanism leading to accelerated ageing in lung disease, suggesting that antioxidants should be effective therapies. However, existing antioxidants, such as N-acetyl cysteine (NAC), are poorly effective as they are thiol derivatives that become inactivated by oxidative stress. Novel antioxidants include NADPH oxidase (NOX) inhibitors, superoxide dismutase mimetics and activators of Nrf2 (Saso and Firuzi 2014). Since there is strong evidence for mitochondria-derived oxidative stress in age-related lung diseases an intracellular antioxidant may be more effective. The mitochondrial antioxidant SkQ1 reverses ageing-related biomarkers in rats, whereas NAC is ineffective (Kolosova et al. 2012). Nrf2 activators are of particular interest as Nrf2 may be defective in several disease models of accelerated ageing, including COPD. Sulforaphane, which occurs naturally in broccoli, is an Nrf2 activator but is non-specific and toxic in high concentrations, leading to a search for new drugs. Bardoxelone methyl is more potent but also has shown toxicity in clinical studies.
Lifestyle Interventions
Caloric restriction (CR) has been shown to prolong lifespan in species from yeast to mammals, including primates (Colman et al. 2014). This leads to inhibition of PI3K-AKT-mTOR signaling though activation of AMPK and reduces the release of insulin and insulin-like growth factor(IGF)-1 and increases sirtuin-1 (Fontana et al. 2010). Several intermittent fasting regimes that give sufficient CR to activate anti-ageing pathways are now being explored. Periodic fasting has been found to be effective in several animal models of age-related diseases but has not been explored in age-related lung diseases. The Mediterranean diet, is rich in fruit, vegetables, red wine and olive oil, contains flavones, polyphenols and stilbenes that may activate sirtuin-1 and increases healthy life, with reduced incidence of neurodegenerative, cardiovascular and metabolic diseases and cancer (Perez-Lopez et al. 2009). For example, a Mediterranean diet may improve the function of EPCs from elderly patients and thus improve endothelial function in cardiovascular disease (Marin et al. 2013). Several compounds that occur in foodstuffs, as outlined above, may also reduce senescence.
Physical inactivity is a risk factor for the development of diseases of ageing, such as COPD, and is an important determinant of mortality (Gulsvik et al. 2012). Aerobic exercise training provides significant clinical benefits in several age-related biomarkers, including lipid profiles, blood pressure, glucose tolerance, bone density, depression, loss of skeletal muscle (sarcopenia) and quality of life (Fleg 2012). Exercise is the major component of pulmonary rehabilitation in COPD, which results in improved lung function, reduced exacerbations and improved quality of life, although it is not certain whether it is able to reduce mechanisms of accelerated ageing (Casaburi and ZuWallack 2009).
References
Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, Bernard D, Hernando E, Gil J (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133:1006–1018
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15:978–990
Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, Phillips JA 3rd, Lansdorp PM, Loyd JE, Armanios MY (2008) Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A 105:13051–13056
Alder JK, Guo N, Kembou F, Parry EM, Anderson CJ, Gorgy AI, Walsh MF, Sussan T, Biswal S, Mitzner W, Tuder RM, Armanios M (2011) Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med 184:904–912
Alvarez D, Cardenes N, Sellares J, Bueno M, Corey C, Hanumanthu VS, Peng Y, D’Cunha H, Sembrat J, Nouraie M, Shanker S, Caufield C, Shiva S, Armanios M, Mora AL, Rojas M (2017) IPF lung fibroblasts have a senescent phenotype. Am J Phys Lung Cell Mol Phys 313:L1164–l1173
Amsellem V, Gary-Bobo G, Marcos E, Maitre B, Chaar V, Validire P, Stern JB, Noureddine H, Sapin E, Rideau D, Hue S, Le Corvoisier P, Le Gouvello S, Dubois-Rande JL, Boczkowski J, Adnot S (2011) Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 184:1358–1366
Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Yurova MV, Kovalenko IG, Poroshina TE, Semenchenko AV (2008) Metformin slows down aging and extends life span of female SHR mice. Cell Cycle 7:2769–2773
Armanios M (2012) Telomerase and idiopathic pulmonary fibrosis. Mutat Res 730(1-2):52–58
Armstrong L, Al-Aama J, Stojkovic M, Lako M (2014) Concise review: the epigenetic contribution to stem cell ageing: can we rejuvenate our older cells? Stem Cells (Dayton, Ohio) 32(9):2291–2298. https://doi.org/10.1002/stem.1720
Arnold CR, Wolf J, Brunner S, Herndler-Brandstetter D, Grubeck-Loebenstein B (2011) Gain and loss of T cell subsets in old age–age-related reshaping of the T cell repertoire. J Clin Immunol 31(2):137–146
Artal-Sanz M, Tavernarakis N (2009) Prohibitin and mitochondrial biology. Trends Endocrinol Metab 20:394–401
Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, van IWF, Houtsmuller AB, Pothof J, de Bruin RWF, Madl T, Hoeijmakers JHJ, Campisi J, de Keizer PLJ (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169:132–147.e116
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479:232–236
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM (2016a) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530:184–189
Baker J, Vuppusetty C, Colley T, Papaioannou A, Fenwick P, Donnelly L, Ito K, Barnes PJ (2016b) Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci Rep 6:35871
Barnes PJ (2004) Mediators of chronic obstructive pulmonary disease. Pharm Rev 56:515–548
Barnes PJ (2009) Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol 71:451–464
Barnes PJ (2014) Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med 35:71–86
Barnes PJ (2015) Mechanisms of development of multimorbidity in the elderly. Eur Respir J 45:790–806
Barnes PJ (2016) Inflammatory mechanisms in COPD. J Allergy Clin Immunol 138(1):16–27
Barnes PJ, Burney PGJ, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, Wouters EFM (2015) Chronic obstructive pulmonary disease. Nat Rev Primers 1:1–21
Behrens A, van Deursen JM, Rudolph KL, Schumacher B (2014) Impact of genomic damage and ageing on stem cell function. Nat Cell Biol 16:201–207
Belsky DW, Shalev I, Sears MR, Hancox RJ, Lee Harrington H, Houts R, Moffitt TE, Sugden K, Williams B, Poulton R, Caspi A (2014) Is chronic asthma associated with shorter leukocyte telomere length at midlife? Am J Respir Crit Care Med 190:384–391
Birch J, Anderson RK, Correia-Melo C, Jurk D, Hewitt G, Marques FM, Green NJ, Moisey E, Birrell MA, Belvisi MG, Black F, Taylor JJ, Fisher AJ, De Soyza A, Passos JF (2015) DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am J Phys Lung Cell Mol Phys 309:L1124–L1137
Birch J, Victorelli S, Rahmatika D, Anderson RK, Jiwa K, Moisey E, Ward C, Fisher AJ, De Soyza A, Passos JF (2016) Telomere dysfunction and senescence-associated pathways in bronchiectasis. Am J Respir Crit Care Med 193:929–932
Birch J, Barnes PJ, Passos JF (2017) Mitochondria, telomeres and cell senescence: implications for lung ageing and disease. Pharmacol Ther 183:34–49. https://doi.org/10.1016/j.pharmthera.2017.10.005
Birrell MA, McCluskie K, Wong S, Donnelly LE, Barnes PJ, Belvisi MG (2005) Resveratrol, an extract of red wine, inhibits lipopolysaccharide induced airway neutrophilia and inflammatory mediators through an NF-κB-independent mechanism. FASEB J 19:840–841
Bjedov I, Partridge L (2011) A longer and healthier life with TOR down-regulation: genetics and drugs. Biochem Soc Trans 39:460–465
Bonkowski MS, Sinclair DA (2016) Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17:679–690
Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, Duncan SR, Rojas M, Shiva S, Chu CT, Mora AL (2015) PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest 125:521–538
Burney P, Jithoo A, Kato B, Janson C, Mannino D, Nizankowska-Mogilnicka E, Studnicka M, Tan W, Bateman E, Kocabas A, Vollmer WM, Gislason T, Marks G, Koul PA, Harrabi I, Gnatiuc L, Buist S (2014) Chronic obstructive pulmonary disease mortality and prevalence: the associations with smoking and poverty–a BOLD analysis. Thorax 69:465–473
Calhoun C, Shivshankar P, Saker M, Sloane LB, Livi CB, Sharp ZD, Orihuela CJ, Adnot S, White ES, Richardson A, Le Saux CJ (2016) Senescent cells contribute to the physiological remodeling of aged lungs. J Gerontol 71:153–160
Casaburi R, ZuWallack R (2009) Pulmonary rehabilitation for management of chronic obstructive pulmonary disease. N Engl J Med 360:1329–1335
Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM (2010) Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A 107:18880–18885
Chen R, Zhang K, Chen H, Zhao X, Wang J, Li L, Cong Y, Ju Z, Xu D, Williams BR, Jia J, Liu JP (2015) Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J Biol Chem 290:30813–30829
Chilosi M, Carloni A, Rossi A, Poletti V (2013) Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. J Lab Clin Med l16:156–173
Clifford RL, Fishbane N, Patel J, MacIsaac JL, McEwen LM, Fisher AJ, Brandsma CA, Nair P, Kobor MS, Hackett TL, Knox AJ (2018) Altered DNA methylation is associated with aberrant gene expression in parenchymal but not airway fibroblasts isolated from individuals with COPD. Clin Epigenetics 10:32
Colley T, Mercado N, Kunori Y, Brightling C, Bhavsar PK, Barnes PJ, Ito K (2016) Defective sirtuin-1 increases IL-4 expression through acetylation of GATA-3 in patients with severe asthma. J Allergy Clin Immunol 137:1595–1597
Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM (2014) Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun 5:3557. https://doi.org/10.1038/ncomms4557
Correia-Melo C, Passos JF (2015) Mitochondria: are they causal players in cellular senescence? Biochim Biophys Acta 1847:1373–1379
Correia-Melo C, Hewitt G, Passos JF (2014) Telomeres, oxidative stress and inflammatory factors: partners in cellular senescence? Longev Healthsp 3:1
Cottin V, Cordier JF (2012) Combined pulmonary fibrosis and emphysema in connective tissue disease. Curr Opin Pulm Med 18:418–427
Culpitt SV, Rogers DF, Fenwick PS, Shah P, de Matos C, Russell RE, Barnes PJ, Donnelly LE (2003) Inhibition by red wine extract, resveratrol, of cytokine release by alveolar macrophages in COPD. Thorax 58:942–946
de Cabo R, Carmona-Gutierrez D, Bernier M, Hall MN, Madeo F (2014) The search for antiaging interventions: from elixirs to fasting regimens. Cell 157:1515–1526
Dermaku-Sopjani M, Kolgeci S, Abazi S, Sopjani M (2013) Significance of the anti-aging protein Klotho. Mol Membr Biol 30:369–385
Di Stefano A, Ricciardolo FLM, Caramori G, Adcock IM, Chung KF, Barnes PJ, Brun P, Leonardi A, Ando F, Vallese D, Gnemmi I, Righi L, Cappello F, Balbi B (2017) Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur Respir J 49(5). https://doi.org/10.1183/13993003.02006-2016
Dimmeler S, Nicotera P (2013) MicroRNAs in age-related diseases. EMBO Mol Med 5:180–190
Donnelly LE, Barnes PJ (2012) Defective phagocytosis in airways disease. Chest 141:1055–1062
Dunlop EA, Tee AR (2014) mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Sem Cell Devel Biol 36C:121–129
Fadini GP, Losordo D, Dimmeler S (2012) Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res 110(4):624–637. https://doi.org/10.1161/circresaha.111.243386
Faner R, Rojas M, Macnee W, Agusti A (2012) Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 186:306–313
Feghali-Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, Csizmadia E, Zhang Y, Sciurba FC, Duncan SR (2008) Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177:156–163
Feng D, Kondo Y, Ishigami A, Kuramoto M, Machida T, Maruyama N (2004) Senescence marker protein-30 as a novel antiaging molecule. Ann N Y Acad Sci 1019:360–364
Feser J, Tyler J (2011) Chromatin structure as a mediator of aging. FEBS Lett 585(13):2041–2048. https://doi.org/10.1016/j.febslet.2010.11.016
Finkel T, Deng CX, Mostoslavsky R (2009) Recent progress in the biology and physiology of sirtuins. Nature 460:587–591
Fischer BM, Wong JK, Degan S, Kummarapurugu AB, Zheng S, Haridass P, Voynow JA (2013) Increased expression of senescence markers in cystic fibrosis airways. Am J Phys Lung Cell Mol Phys 304:L394–L400
Fleg JL (2012) Aerobic exercise in the elderly: a key to successful aging. Discov Med 13(70):223–228
Fois AG, Paliogiannis P, Sotgia S, Mangoni AA, Zinellu E, Pirina P, Carru C, Zinellu A (2018) Evaluation of oxidative stress biomarkers in idiopathic pulmonary fibrosis and therapeutic applications: a systematic review. Respir Res 19:51
Fontana L, Partridge L, Longo VD (2010) Extending healthy life span–from yeast to humans. Science 328:321–326
Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, Minagawa S, Yumino Y, Ishikawa T, Numata T, Kawaishi M, Hirano J, Odaka M, Morikawa T, Nishimura S, Nakayama K, Kuwano K (2012) Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 1(5):630–641
Fujita Y, Kosaka N, Araya J, Kuwano K, Ochiya T (2015) Extracellular vesicles in lung microenvironment and pathogenesis. Trends Mol Med 21:533–542
Gao W, Yuan C, Zhang J, Li L, Yu L, Wiegman CH, Barnes PJ, Adcock IM, Huang M, Yao X (2015) Klotho expression is reduced in COPD airway epithelial cells: effects on inflammation and oxidant injury. Clin Sci 129:1011–1023
Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Munoz-Canoves P (2016) Autophagy maintains stemness by preventing senescence. Nature 529:37–42
Gorospe M, Abdelmohsen K (2011) MicroRegulators come of age in senescence. Trends Genet 27:23
Guarente L (2011) Sirtuins, aging, and metabolism. Cold Spring Harbor Symp 76:81–90
Gulsvik AK, Thelle DS, Samuelsen SO, Myrstad M, Mowe M, Wyller TB (2012) Ageing, physical activity and mortality–a 42-year follow-up study. Int J Epidemiol 41:521–530
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460:392–395
Hashimoto M, Asai A, Kawagishi H, Mikawa R, Iwashita Y, Kanayama K, Sugimoto K, Sato T, Maruyama M, Sugimoto M (2016) Elimination of p19(ARF)-expressing cells enhances pulmonary function in mice. JCI Insight 1:e87732
Hazeldine J, Lord JM (2015) Innate immunesenescence: underlying mechanisms and clinical relevance. Biogerontology 16:187–201
Hekimi S, Lapointe J, Wen Y (2011) Taking a “good” look at free radicals in the aging process. Trends Cell Biol 21:569–576
Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, Georgilis A, Montoya A, Wolter K, Dharmalingam G, Faull P, Carroll T, Martinez-Barbera JP, Cutillas P, Reisinger F, Heikenwalder M, Miller RA, Withers D, Zender L, Thomas GJ, Gil J (2015) mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17:1205–1217
Hitchings AW, Lai D, Jones PW, Baker EH (2016) Metformin in severe exacerbations of chronic obstructive pulmonary disease: a randomised controlled trial. Thorax 71:587–593
Hodge G, Jersmann H, Tran HB, Roscioli E, Holmes M, Reynolds PN, Hodge S (2015) Lymphocyte senescence in COPD is associated with decreased histone deacetylase 2 expression by pro-inflammatory lymphocytes. Respir Res 16:130
Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, Dijk F, Kalicharan D, Kelders M, Gosker HR, Ten Hacken NH, van der Want JJ, van Oosterhout AJ, Heijink IH (2013) Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res 14:97
Hogg JC, Timens W (2009) The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol 4:435–459
Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14(10):R115
Hosgood HD III, Menashe I, He X, Chanock S, Lan Q (2009) PTEN identified as important risk factor of chronic obstructive pulmonary disease. Respir Med 103:1866–1870
Houben JM, Mercken EM, Ketelslegers HB, Bast A, Wouters EF, Hageman GJ, Schols AM (2009) Telomere shortening in chronic obstructive pulmonary disease. Respir Med 103:230–236
Hubbard BP, Sinclair DA (2014) Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci 35:146–154
Ito K, Barnes PJ (2009) COPD as a disease of accelerated lung aging. Chest 135:173–180
Ito K, Colley T, Mercado N (2012) Geroprotectors as a novel therapeutic strategy for COPD, an accelerating aging disease. Int J Chron Obstruct Pulmon Dis 7:641–652
Janssens JP, Pache JC, Nicod LP (1999) Physiological changes in respiratory function associated with ageing. Eur Respir J 13:197–205
Jiang XC, Gao JQ (2017) Exosomes as novel bio-carriers for gene and drug delivery. Int J Pharm 521:167–175
Johnson SC, Rabinovitch PS, Kaeberlein M (2013) mTOR is a key modulator of ageing and age-related disease. Nature 493:338–345
Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T (2018) Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: insights into the pathophysiology of lung diseases. Mol Asp Med 60:92–103
Karayama M, Inui N, Suda T, Nakamura Y, Nakamura H, Chida K (2010) Antiendothelial cell antibodies in patients with COPD. Chest 138(6):1303–1308
Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss-Coray T, Sierra F (2014) Geroscience: linking aging to chronic disease. Cell 159:709–713
Kirkham PA, Barnes PJ (2013) Oxidative stress in COPD. Chest 144:266–273
Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, Triantaphyllopoulos K, Hussain F, Pinart M, Khan Y, Heinemann L, Stevens L, Yeadon M, Barnes PJ, Chung KF, Adcock IM (2011) Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 184(7):796–802
Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD (2017) The clinical potential of senolytic drugs. J Am Geriatr Soc 65:2297–2301
Kolosova NG, Stefanova NA, Muraleva NA, Skulachev VP (2012) The mitochondria-targeted antioxidant SkQ1 but not N-acetylcysteine reverses aging-related biomarkers in rats. Aging 4:686–694
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H (2014) Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20:62–68
Kubo H (2012) Concise review: clinical prospects for treating chronic obstructive pulmonary disease with regenerative approaches. Stem Cells Transl Med 1(8):627–631
Kugel S, Mostoslavsky R (2014) Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem Sci 39:72–81
Kureya Y, Kanazawa H, Ijiri N, Tochino Y, Watanabe T, Asai K, Hirata K (2016) Down-regulation of soluble alpha-klotho is associated with reduction in serum irisin levels in chronic obstructive pulmonary disease. Lung 194:345–351
Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, Hubbard GB, Ikeno Y, Javors M, Desprez PY, Benz CC, Kapahi P, Nelson PS, Campisi J (2015) MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17:1049–1061
Lamming DW, Ye L, Sabatini DM, Baur JA (2013) Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest 123:980–989
Lange P, Celli B, Agusti A, Boje Jensen G, Divo M, Faner R, Guerra S, Marott JL, Martinez FD, Martinez-Camblor P, Meek P, Owen CA, Petersen H, Pinto-Plata V, Schnohr P, Sood A, Soriano JB, Tesfaigzi Y, Vestbo J (2015) Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med 373:111–122
Leaker BR, Barnes PJ, O'Connor BJ, Ali FY, Tam P, Neville J, Mackenzie LF, MacRury T (2014) The effects of the novel SHIP1 activator AQX-1125 on allergen-induced responses in mild-to-moderate asthma. Clin Exp Allergy 44:1146–1153
Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 105:3374–3379
Lee J, Sandford AJ, Connett JE, Yan J, Mui T, Li Y, Daley D, Anthonisen NR, Brooks-Wilson A, Man SF, Sin DD (2012) The relationship between telomere length and mortality in chronic obstructive pulmonary disease (COPD). PLoS One 7:e35567
Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner M, Wagner DE, Gunther A, Konigshoff M (2017) Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J 50(2):1602367
Li J, Dai A, Hu R, Zhu L, Tan S (2010) Positive correlation between PPARγ/PGC-1α and γ-GCS in lungs of rats and patients with chronic obstructive pulmonary disease. Acta Biochim Biophys Sin 42:603–614
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153(6):1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
Lozano R, Naghavi M, Foreman K (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2095–2128
Mahbub S, Brubaker AL, Kovacs EJ (2011) Aging of the innate immune system: an update. Curr Immunol Rev 7:104–115
Maneechotesuwan K, Kasetsinsombat K, Wongkajornsilp A, Barnes PJ (2013) Decreased indoleamine 2,3-dioxygenase activity and IL-10/IL-17A ratio in patients with COPD. Thorax 68:330–337
Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB (2014) mTOR inhibition improves immune function in the elderly. Sci Transl Med 6:268
Marin C, Yubero-Serrano EM, Lopez-Miranda J, Perez-Jimenez F (2013) Endothelial aging associated with oxidative stress can be modulated by a healthy mediterranean diet. Int J Mol Sci 14:8869–8889
Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M, de Cabo R (2013) Metformin improves healthspan and lifespan in mice. Nat Commun 4:2192
McCubbrey AL, Nelson JD, Stolberg VR, Blakely PK, McCloskey L, Janssen WJ, Freeman CM, Curtis JL (2016) MicroRNA-34a negatively regulates efferocytosis by tissue macrophages in part via SIRT1. J Immunol 196:1366–1375
Meiners S, Eickelberg O, Konigshoff M (2015) Hallmarks of the ageing lung. Eur Respir J 45:807–827
Mercado N, Ito K, Barnes PJ (2015) Accelerated ageing in chronic obstructive pulmonary disease: new concepts. Thorax 70:482–489
Meyer A, Zoll J, Charles AL, Charloux A, de Blay F, Diemunsch P, Sibilia J, Piquard F, Geny B (2013) Skeletal muscle mitochondrial dysfunction during chronic obstructive pulmonary disease: central actor and therapeutic target. Exp Physiol 98:1063–1078
Minagawa S, Araya J, Numata T, Nojiri S, Hara H, Yumino Y, Kawaishi M, Odaka M, Morikawa T, Nishimura SL, Nakayama K, Kuwano K (2011) Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am J Phys Lung Cell Mol Phys 300:L391–L401
Mitani A, Ito K, Vuppusetty C, Barnes PJ, Mercado N (2016) Restoration of corticosteroid sensitivity in chronic obstructive pulmonary disease by inhibition of mammalian target of rapamycin. Am J Respir Crit Care Med 193:143–153
Mitani A, Azam A, Vuppusetty C, Ito K, Mercado N, Barnes PJ (2017) Quercetin restores corticosteroid sensitivity in cells from patients with chronic obstructive pulmonary disease. Exp Lung Res 43:417–425
Mizumura K, Cloonan SM, Haspel JA, Choi AM (2012) The emerging importance of autophagy in pulmonary diseases. Chest 142:1289–1299
Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM (2014) Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124:3987–4003
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451(7182):1069–1075
Monick MM, Powers LS, Walters K, Lovan N, Zhang M, Gerke A, Hansdottir S, Hunninghake GW (2010) Identification of an autophagy defect in smokers’ alveolar macrophages. J Immunol 185:5425–5435
Mora AL, Bueno M, Rojas M (2017) Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin Invest 127:405–414
Moskalev A, Chernyagina E, Kudryavtseva A, Shaposhnikov M (2017) Geroprotectors: a unified concept and screening approaches. Aging Dis 8:354–363
Munk R, Panda AC, Grammatikakis I, Gorospe M, Abdelmohsen K (2017) Senescence-associated MicroRNAs. Int Rev Cell Mol Biol 334:177–205
Munoz-Espin D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15:482–496
Murray MA, Chotirmall SH (2015) The Impact of immunosenescence on pulmonary disease. Mediat Inflamm 2015:692546
Murrow L, Debnath J (2013) Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol 8:105–137
Nakamaru Y, Vuppusetty C, Wada H, Milne JC, Ito M, Rossios C, Elliot M, Hogg J, Kharitonov S, Goto H, Bemis JE, Elliott P, Barnes PJ, Ito K (2009) A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J 23:2810–2819
Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK (2010) microRNA expression patterns reveal differential expression of target genes with age. PLoS One 5:e10724
Oh J, Lee YD, Wagers AJ (2014) Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med 20:870–880
Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, Lin S, Huang L, Chung EJ, Citrin DE, Wang Y, Hauer-Jensen M, Zhou D, Meng A (2017) Inhibition of Bcl-2/xl With ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Phys 9:353–361
Paschalaki KE, Starke RD, Hu Y, Mercado N, Margariti A, Gorgoulis VG, Randi AM, Barnes PJ (2013) Dysfunction of endothelial progenitor cells from smokers and COPD patients due to increased DNA damage and senescence. Stem Cells 31:2813–2826
Paschalaki K, Zampetaki A, Baker J, Birrell M, Starke RD, Belvisi M, Donnelly LE, Mayr M, Randi AM, Barnes PJ (2018) Downregulation of microRNA-126 augments DNA damage response in cigarette smokers and COPD patients. Am J Respir Crit Care Med 197:665–668
Passos JF, Saretzki G, von Zglinicki T (2007) DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res 35:7505–7513
Perez-Lopez FR, Chedraui P, Haya J, Cuadros JL (2009) Effects of the Mediterranean diet on longevity and age-related morbid conditions. Maturitas 64:67–79
Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA (2015) Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep 12:286–299
Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I (2008) SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177(8):861–870
Rutten EP, Gopal P, Wouters EF, Franssen FM, Hageman GJ, Vanfleteren LE, Spruit MA, Reynaert NL (2016) Various mechanistic pathways representing the aging process are altered in COPD. Chest 149:53–61
Salama R, Sadaie M, Hoare M, Narita M (2014) Cellular senescence and its effector programs. Genes Dev 28:99–114
Sapey E, Stockley JA, Greenwood H, Ahmad A, Bayley D, Lord JM, Insall RH, Stockley RA (2011) Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 183:1176–1186
Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, Insall RH, Stockley RA, Lord JM (2014) Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood 123:239–248
Sarker RS, John-Schuster G, Bohla A, Mutze K, Burgstaller G, Bedford MT, Konigshoff M, Eickelberg O, Yildirim AO (2015) Coactivator-associated arginine methyltransferase-1 function in alveolar epithelial senescence and elastase-induced emphysema susceptibility. Am J Respir Cell Mol Biol 53:769–781
Saso L, Firuzi O (2014) Pharmacological applications of antioxidants: lights and shadows. Curr Drug Targets 15:1177–1199
Sato T, Seyama K, Sato Y, Mori H, Souma S, Akiyoshi T, Kodama Y, Mori T, Goto S, Takahashi K, Fukuchi Y, Maruyama N, Ishigami A (2006) Senescence marker protein-30 protects mice lungs from oxidative stress, aging and smoking. Am J Respir Crit Care Med 174:530–537
Savale L, Chaouat A, Bastuji-Garin S, Marcos E, Boyer L, Maitre B, Sarni M, Housset B, Weitzenblum E, Matrat M, Le Corvoisier P, Rideau D, Boczkowski J, Dubois-Rande JL, Chouaid C, Adnot S (2009) Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 179:566–571
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8:14532
Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L (2018) Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res 19:32
Song S, Johnson FB (2018) Epigenetic mechanisms impacting aging: a focus on histone levels and telomeres. Genes 9(4):201
Song J, Heijink IH, Kistemaker LEM, Reinders-Luinge M, Kooistra W, Noordhoek JA, Gosens R, Brandsma CA, Timens W, Hiemstra PS, Rots MG, Hylkema MN (2017) Aberrant DNA methylation and expression of SPDEF and FOXA2 in airway epithelium of patients with COPD. Clin Epigenetics 9:42
Sood A, Assad NA, Barnes PJ, Churg A, Gordon SB, Harrod KS, Irshad H, Kurmi OP, Martin WJ 2nd, Meek P, Mortimer K, Noonan CW, Perez-Padilla R, Smith KR, Tesfaigzi Y, Ward T, Balmes J (2018) ERS/ATS workshop report on respiratory health effects of household air pollution. Eur Respir J 51:1700698
Sorrentino JA, Krishnamurthy J, Tilley S, Alb JG Jr, Burd CE, Sharpless NE (2014) p16INK4a reporter mice reveal age-promoting effects of environmental toxicants. J Clin Invest 124:169–173
Soulitzis N, Neofytou E, Psarrou M, Anagnostis A, Tavernarakis N, Siafakas N, Tzortzaki EG (2012) Downregulation of lung mitochondrial prohibitin in COPD. Respir Med 106:954–961
Sousounis K, Baddour JA, Tsonis PA (2014) Aging and regeneration in vertebrates. Curr Top Dev Biol 108:217–246
Stanley SE, Chen JJ, Podlevsky JD, Alder JK, Hansel NN, Mathias RA, Qi X, Rafaels NM, Wise RA, Silverman EK, Barnes KC, Armanios M (2015) Telomerase mutations in smokers with severe emphysema. J Clin Invest 125:563–570
Stenton GR, Mackenzie LF, Tam P, Cross JL, Harwig C, Raymond J, Toews J, Wu J, Ogden N, MacRury T, Szabo C (2013) Characterization of AQX-1125, a small-molecule SHIP1 activator: Part 1. Effects on inflammatory cell activation and chemotaxis in vitro and pharmacokinetic characterization in vivo. Br J Pharmacol 168:1506–1518
Sureshbabu A, Bhandari V (2013) Targeting mitochondrial dysfunction in lung diseases: emphasis on mitophagy. Front Physiol 4:384
Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E (2006) Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol 8:1291–1297
Takasaka N, Araya J, Hara H, Ito S, Kobayashi K, Kurita Y, Wakui H, Yoshii Y, Yumino Y, Fujii S, Minagawa S, Tsurushige C, Kojima J, Numata T, Shimizu K, Kawaishi M, Kaneko Y, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Nakayama K, Kuwano K (2014) Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol 192:958–968
To Y, Ito K, Kizawa Y, Failla M, Ito M, Kusama T, Elliot M, Hogg JC, Adcock IM, Barnes PJ (2010) Targeting phosphoinositide-3-kinase-δ with theophylline reverses corticosteroid insensitivity in COPD. Am J Respir Crit Care Med 182:897–904
To M, Takagi D, Akashi K, Kano I, Haruki K, Barnes PJ, Ito K (2013) Sputum PAI-1 elevation by oxidative stress-dependent NF-kappaB activation in chronic obstructive pulmonary disease. Chest 144:515–521
Tomita K, Caramori G, Lim S, Ito K, Hanazawa T, Oates T, Chiselita I, Jazrawi E, Chung KF, Barnes PJ, Adcock IM (2002) Increased p21CIP1/WAF1 and B cell lymphoma leukemia-xL expression and reduced apoptosis in alveolar macrophages from smokers. Am J Respir Crit Care Med 166:724–731
Tomita K, Caramori G, Ito K, Lim S, Sano H, Tohda Y, Adcock IM, Barnes PJ (2010) Telomere shortening in alveolar macrophages of smokers and COPD patients. Open Path J 4:23–29
Tsuji T, Aoshiba K, Nagai A (2006) Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med 174:886–893
Turturici G, Tinnirello R, Sconzo G, Geraci F (2014) Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. Am J Phys Cell Physiol 306:C621–C633
van Deursen JM (2014) The role of senescent cells in ageing. Nature 509:439–446
Watroba M, Dudek I, Skoda M, Stangret A, Rzodkiewicz P, Szukiewicz D (2017) Sirtuins, epigenetics and longevity. Ageing Res Rev 40:11–19
Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, Bao W, Pavlidis S, Barnes PJ, Kanerva J, Bittner A, Rao N, Murphy MP, Kirkham PA, Chung KF, Adcock IM (2015) Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 136:769–780
Worby CA, Dixon JE (2014) PTEN. Annu Rev Biochem 83:641–669
Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, Giorgadze N, Jensen MD, LeBrasseur NK, Kirkland JL (2015) JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A 112:E6301–E6310
Yanagisawa S, Baker JR, Vuppusetty C, Fenwick P, Donnelly LE, Ito K, Barnes PJ (2017a) Decreased phosphatase PTEN amplifies PI3K signaling and enhances pro-inflammatory cytokine release in COPD. Am J Phys Lung Cell Mol Phys 313:L230–L239
Yanagisawa S, Papaioannou AI, Papaporfyriou A, Baker J, Vuppusetty C, Loukides S, Barnes PJ, Ito K (2017b) Decreased serum sirtuin-1 in chronic obstructive pulmonary disease. Chest 152(2):343–352
Yeo SC, Fenwick PS, Barnes PJ, Lin HS, Donnelly LE (2017) Isorhapontigenin, a bioavailable dietary polyphenol, suppresses airway epithelial cell inflammation through a corticosteroid-independent mechanism. Br J Pharmacol 174:2043–2059
Zeng Z, Cheng S, Chen H, Li Q, Hu Y, Wang Q, Zhu X, Wang J (2017) Activation and overexpression of Sirt1 attenuates lung fibrosis via P300. Biochem Biophys Res Commun 486:1021–1026
Zhang W, Hu D, Ji W, Yang L, Yang J, Yuan J, Xuan A, Zou F, Zhuang Z (2014) Histone modifications contribute to cellular replicative and hydrogen peroxide-induced premature senescence in human embryonic lung fibroblasts. Free Radic Res 48:550–559
Zheng S, Wang C, Qian G, Wu G, Guo R, Li Q, Chen Y, Li J, Li H, He B, Chen H, Ji F (2012) Role of mtDNA haplogroups in COPD susceptibility in a southwestern Han Chinese population. Free Radic Biol Med 53:473–481
Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, Wren JD, Niedernhofer LJ, Robbins PD, Kirkland JL (2016) Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15:428–435
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Barnes, P.J. (2019). Pulmonary Diseases and Ageing. In: Harris, J., Korolchuk, V. (eds) Biochemistry and Cell Biology of Ageing: Part II Clinical Science. Subcellular Biochemistry, vol 91. Springer, Singapore. https://doi.org/10.1007/978-981-13-3681-2_3
Download citation
DOI: https://doi.org/10.1007/978-981-13-3681-2_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3680-5
Online ISBN: 978-981-13-3681-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)