
Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive and usually lethal interstitial lung disease of unknown etiology that is characterized by epithelial cell injury and aberrant activation, expansion of the mesenchymal cell population with the formation of fibroblast/myofibroblast foci, and exaggerated extracellular matrix accumulation. IPF is an aging-related disease, and most patients are over 60 years of age at the time of clinical presentation and diagnosis. Age also influences mortality, and the median survival time is significantly shorter in older individuals compared with younger patients. However, the fundamental mechanisms linking aging to IPF remain unclear. In this chapter, we will discuss some of the modifications naturally occurring in the elderly that may be implicated in the pathogenesis of IPF, including endoplasmic reticulum stress, oxidative stress, mitochondrial dysfunction, dysregulated autophagy, telomere attrition, and a number of epigenetic changes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Aging
Aging is a complex process that is characterized by the progressive decline of the capacity to properly resolve the interaction between injury and repair, leading to progressive multi-organ deterioration and an elevated risk of disease. Aging is associated with the accumulation of damage to molecules, cells, and tissues over a lifetime, which leads to frailty and malfunction [1]. Two theories prevail regarding the aging process; specifically these include the programmed and the damage theories. The programmed concept holds the notion that cells or systems have a biological clock that is responsible for switching on deterioration (programmed longevity, telomere shortening, and immunological changes). The damage theory includes the cumulative effects of oxidative stress caused by free radicals, DNA damage, and other perpetuators. However, no single universal theory fully explains the aging process [2].
Molecular Mechanisms of Aging
Currently, there are multiple modifications that appear to be involved in aging. These include oxidative stress, telomere shortening, heterochromatin loss, autophagy, senescence, and epigenetic changes. Multilevel combinations and interactions of these processes may participate in the normal aging process and explain the development of age-related diseases.
Epigenetic Changes
Epigenetic modifications, including DNA methylation, histone modifications, noncoding RNA, nucleosome positioning, and chromatin arrangement, play central roles in controlling changes in gene expression and genome instability during aging [3].
DNA Methylation
DNA methylation is the epigenetic change most frequently studied. In this process, DNA methyltransferases transfer a methyl group from S-adenosyl-methionine to the C5 position of the pyrimidine ring of cytosine residues in genomic CpG dinucleotides [4]. Hypermethylation in gene promoter or CpG island regions has been shown to repress, while hypomethylation enhances, gene expression [5, 6]. Importantly, DNA methylation does not occur exclusively at CpG islands but also in the so-called CpG island shores (regions of lower CpG density that lie in close proximity (~2 kb) of CpG islands), which are also closely associated with transcriptional inactivation.
Different methylation patterns have far-reaching implications for human biology and age-related diseases. With aging, a global hypomethylation process together with hypermethylation of a number of specific loci has been described. Loss of global DNA methylation over time has also been reported in cancer, but this loss is in repetitive sequences, and its effects on aging are unclear [7]. Likewise, multiple genes from tumor suppressor factors, DNA-binding factors, and transcription factors are increasingly methylated with age [8, 9]. In general, cancer cells are characterized by a massive global loss of DNA methylation, while specific patterns of hypermethylation at the CpG islands of certain promoters are often acquired [10].
Histone Modifications
Histones are basic proteins that interact with DNA, and their posttranslational modifications affect accessibility of diverse transcription factors to the genome. Normally, the nucleosome is composed of a histone octamer with two groups of H2A, H2B, H3, and H4. Histone H1 is located between each nucleosome, which is subject to many types of posttranslational modifications (e.g., methylation, acetylation, phosphorylation, ubiquitination, SUMOylation), especially on their flexible amino-terminal tails [11].
The main histone modifications are acetylation and methylation. Histone hyperacetylation is characterized by a relaxed chromatin structure and active gene transcription, while deacetylation is linked with a compact and inactive chromatin structure. Histones can become mono-, di-, or trimethylated, and the functional consequences depend on the number of methyl groups, the residue itself, and its location within the histone tail. Repression marks could play a pivotal role in aging. Loss of H3K27 trimethylation via downregulation of the histone methyltransferase, EzH2, in humans could be associated with aging [12]. Likewise, H4K16 hypoacetylation, which is caused by the reduced association of a histone acetyltransferase, leads to early onset of cellular senescence [13].
Nucleosome Positioning and Chromatin Arrangement in the Nucleus
Chromatin involves DNA and all associated proteins, but their configuration and distribution along the nucleus are still poorly characterized. The fundamental unit of chromatin is the nucleosome, and ATP-dependent chromatin-remodeling complexes, which alter nucleosome composition and positioning, are necessary to increase DNA accessibility and to carry out transcription as well as DNA repair [14]. In terms of its transcriptional state, euchromatin is transcriptionally active and is characterized by high levels of acetylation and trimethylated H3K4, H3K36, and H3K79. In contrast, heterochromatin contains low levels of acetylation and high levels of H3K9, H3K27, and H4K20 methylation and is related to transcriptional repression.
A heterochromatin loss model of aging has been proposed, which suggests that heterochromatin domains that were established during embryogenesis decline with the aging process [15]. Recent observations indicate a significant interdependence between heterochromatin, epigenetic landscape, and aging [16]. Evidence indicates that epigenetics have a fundamental role in aging; mainly, H3K27me3 and acetylation on H4K16, which are both repressive marks, decline over time [17]. However, detection of numerous noncoding RNAs from heterochromatic regions challenges the concept of heterochromatin as a transcriptionally inactive region [18].
Noncoding RNA
Recent evidence supports the notion that noncoding RNAs play a critical and dynamic role in transcriptional regulation and epigenetic signaling [19]. Based on its length, noncoding RNAs can be divided into at least three groups: short ncRNA, including microRNA (miRNA; 22–23 nts) and piwi-interacting RNA (piRNA; 26–31 nts); medium ncRNA (50–200 nts); and long ncRNA (>200 nts). miRNAs are the best characterized and are primarily involved in posttranscriptional regulation of mRNA [20].
Recent studies demonstrate that diverse miRNAs are differentially expressed during aging. In general, the patterns of miRNA expression during aging appear to be tissue specific. For example, miR-669c and miR-709 levels are increased in mid-age (18-month to 33-month) murine liver tissue, whereas miR-93 and miR-214 are increased in extremely old (33-month) mice compared with 4- or 10-month-old mice [21]. Likewise, upregulation of miR-143 induces senescence in human fibroblasts [22]. Actually, many miRNAs seem to be key modulators of cellular senescence and influence specific senescence-regulatory proteins [23].
Oxidative Stress, Autophagy, and Caloric Restriction
Accumulation of damage contributes to the aging phenotype and to age-related diseases. Three key processes, oxidative stress, autophagy, and caloric restriction, can increase, reduce, or prevent damage that causes cellular dysfunction.
Oxidative Stress
Reactive oxygen species (ROS) are mainly produced in the mitochondria and affect cell function when an imbalance occurs between the production of ROS and the activity of detoxification enzymes such as superoxide dismutases, catalases, glutathione peroxidases, and peroxiredoxins. Strong evidence supports that the average life span is inversely correlated with the rate of mitochondrial superoxide anions and hydrogen peroxide generation. Moreover, the rates of ROS production from mitochondria increase with age in the brain, heart, and kidney of mice [24]. In addition, a wide spectrum of alterations in mitochondria and mitochondrial DNA have been observed with aging, including disorganization of mitochondrial structure, decline in mitochondrial oxidative phosphorylation function, and accumulation of mtDNA mutations [24].
Autophagy
Autophagy is a homeostatic process of self-degradation of cellular components. There are three general types of autophagy: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy. Macroautophagy is the most widely studied process and represents the major pathway of degradation under basal cellular activity. It is usually upregulated by several stimuli that include starvation, hypoxia, microbial infection, endoplasmic reticulum (ER) stress, and oxidative stress [25]. Damaged, superfluous macromolecules or organelles must be isolated from cytosol by autophagosomes. The formation of phagophores requires generation of phospholipid “PtdIns3p” and involvement of two ubiquitin-like systems: LC3 and ATG5-12-16. Phagophores expand to form complete autophagosomes with a double membrane, and the external membrane merges with the lysosome membrane to degrade internal vesicles. CMA is a selective autophagy of soluble proteins that requires unfolding of the cargo protein before entering lysosomes and interacting with a receptor protein, lysosome-associated protein type 2A (LAMP-2A). In microautophagy, the lysosomal membrane itself invaginates to trap the cargo.
Importantly, autophagy declines with age and causes accumulation of toxic metabolites in the cell, which may be due to a specific CMA failure and to an unsatisfactory degradation of lysosomes [26, 27].
Microautophagy represents the specific degradation of mitochondria, which are very susceptible to damage in aging, and it is involved with the unfolding protein response (UPR) in the endoplasmic reticulum that can activate apoptosis [28, 29].
Defects in the cellular machinery that mediate autophagy are present in almost all age-related diseases, including cancer, metabolic disorders, and neurodegenerative diseases. Evidence shows that autophagy activity must be maintained in order to extend life span in various genetically modified organisms, and autophagy-related proteins have been shown to directly mediate longevity pathways [30].
Senescence and Telomere Shortening
Cellular senescence is synonymous with an irreversible arrest of cell growth. In normal replicative senescence, the cell simply enters senescence after a certain number of replications, which is primarily related to a progressive shortening of telomeres [31]. In addition, differential expression of p53 isoforms and of the retinoblastoma tumor suppressor protein and its signaling partners including p16INK4A (a cyclin-dependent kinase inhibitor) has been linked to replicative senescence [32]. However, premature senescence can be induced in the absence of any detectable telomere loss or dysfunction by a variety of stresses. In general, if DNA damage exceeds a certain threshold, cells are destined to undergo either apoptosis or senescence.
Telomere Shortening
Telomeres are tandem arrays of duplex 5′-TTAGGG-3′ repeats located at the ends of eukaryotic chromosomes that protect them from degradation and DNA repair activities. The maintenance of telomeres depends on a specialized ribonucleoprotein, telomerase, which is an RNA-dependent DNA polymerase that can synthesize telomeric repeats and extend telomeres de novo during cell division [33]. Telomerases have two essential components: telomerase reverse transcriptase (TERT) and RNA template (TR). After birth, telomerase is silenced in most somatic cells, and telomeres progressively shorten with aging [31, 34]. Critically short telomeres cannot be repaired by any of the known DNA repair mechanisms, and shortened telomeres consequently trigger a persistent DNA damage response that leads to cellular senescence and/or apoptosis that eventually compromises tissue regenerative capacity and function, which contributes to organismal aging [34].
Aging Lung
Most of the age-related functional changes in the respiratory system involve alterations in the lung itself as well as a decrease in compliance of the chest wall and a decrease in the strength of the respiratory muscles, which affects control of breathing. However, the rate of progression of these changes can differ greatly from person to person.
The aging lung is characterized by decreased static elastic recoil, dilatation of alveolar ducts and alveoli with a loss of gas exchange surface area, and a decline in the number of capillaries per alveolus, which is often referred to as “senile emphysema.” This goes along with a decrease in the diameter of small airways that increases their tendency to close at a given lung volume, which leads to a decrease in expiratory flows and elicits an increase in residual volume at the expense of vital capacity [35]. Concomitantly, there is an increase in lung compliance while chest wall compliance progressively declines, which is presumably related to calcification and other structural changes within the rib cage and its articulations [36].
Extracellular Matrix
The decrease in the lung elastic recoil has been associated with structural and functional alterations in the extracellular matrix (ECM) of the lung parenchyma. Collagens and elastin are the main proteins in the ECM that comprise the scaffold of the alveolar structures and are central in determining the mechanical properties of lung parenchyma. In general, elastic fibers primarily influence lung compliance at the lower pressure range, while collagen fibrils become more important at high lung volumes where inflation becomes limited. Several studies have demonstrated that changes in lung mechanics are associated with structural modifications of the lung ECM. Collagens are the most abundant proteins of the ECM, and collagens represent 15 % to 20 % of the total dry weight of lung tissue. The collagen family is constituted by 28 different types of collagen proteins that together with other ECM components organize a complex network in the lung tissue. Fibrillar types I and III collagens are the most abundant and represent 90 % of the total lung collagen.
Many age-associated alterations of organs and tissues are associated with changes of the ECM proteins. Such changes include differences in posttranslational modifications of glycoproteins such as advanced glycation end-products (AGEs), which in turn influence the turnover of other glycoproteins [37].
Glycation, glycoxidation, and cross-linking of collagens are increased in many aged tissues, which cause changes of physical properties that include fiber stiffness and higher resistance to degradation [37, 38]. Studies in mice have shown that the process of aging contributes to an altered lung ECM, including fibrillar collagens and the AGE load [39].
It is unclear how the total collagen lung content changes with aging. Some biochemical studies in experimental models describe no changes, an increase, or a decrease in collagen proteins in response to lung aging [40–43]. The different results might be related to differences in the methodological procedures used to measure lung collagen content.
Although there is also some debate about the total elastin content in old lung tissue, it appears that functionally intact elastin is reduced with aging, which could also be influenced by an increased modification with AGEs [44, 45].
Immune Response
The aging lung exhibits an increased susceptibility to infections and inflammation, and alterations in both the innate and adaptive arms of the immune system have been implicated.
During the aging process, the lungs usually exhibit some degree of inflammation, even in healthy individuals. Thus, there is evidence for an augmented proinflammatory milieu, with increased levels of cytokines and acute-phase molecules in association with functional decline, a phenomenon that has been termed the “inflamm-aging” [46]. Furthermore, increased levels of interleukin (IL)-1, IL-6, IL-8, IL-18, IL-1 receptor antagonist, and tumor necrosis factor (TNF)-alpha are found in plasma, serum, and mononuclear blood cell culture from elderly subjects [47].
Additionally, a number of alterations in the T-cell-mediated immune response that affect the function and proportions of T-cell subsets are associated with advancing age. Immunosenescence, characterized by a reduction of naive T-cells and a shrinking T-cell repertoire, is a well-recognized phenomenon in humans and animals and is likely responsible for the increased susceptibility to infections and cancer in older individuals [48]. Numerous studies indicate that aging is associated with impaired influenza virus-specific T-cell responses that may be related to a decreased frequency of naive T-cells as well as diminished function of memory and effector T-cells [49]. A decreased memory CD4+ T-cell response to the influenza vaccine has been reported in the elderly, and the CD8+ T-cell response to the influenza virus also diminishes with advancing age [50, 51].
Specifically relevant to the fibrotic response, normal aging is associated with a shift in T lymphocytes from a predominantly Th1 phenotype to a predominantly Th2 phenotype, which is especially evident in frail older people. The Th2-like response promotes the expression of profibrotic factors, and Th2-biased animals are more susceptible to lung injury and fibrosis [52]. Humans with chronic fibrotic lung disease also demonstrate a Th2-biased phenotype [52].
Toll-like receptors (TLRs), initially described as pattern-recognition receptors that identify and protect against microbes, can display impaired function with aging. Specifically, TLR4 function declines with age or cigarette smoke exposure in humans. Furthermore, mice deficient in TLR4 exhibit age-related lung enlargement that is similar, both histologically and functionally, to human lung emphysema. Additionally, TLR4 deficiency is associated with increased reactive oxygen species generation, collectively referred to as oxidant stress, via the upregulation of the NADPH oxidase, Nox3 [53].
Oxidative Stress
Increased oxidative stress, resulting from an imbalance of pro-oxidants and antioxidants, occurs with aging, and the excessive, destructive presence of reactive oxygen species can adversely affect the lung. Senescence of the pulmonary endothelium is implicated in susceptibility to oxidative stress, impaired nitric oxide signaling, and insufficient tissue repair and regeneration [54]. In general, enzymes implicated in the cytoprotective reduction of ROS, such as Cu/Zn superoxide dismutase and NADPH oxidase (among others), tend to have decreased levels in aged pulmonary endothelial cells [55].
Epigenetic Changes in the Aging Lung
Age-related changes in DNA methylation, as described above, have been implicated in cellular senescence and longevity, although the causes and functional consequences in the lungs remain unclear. Lepeule et al. examined the relationships between DNA methylation in nine genes related to inflammation and lung function in a cohort of 756 elderly men (73.3 ± 6.7 years old) [55]. They found that older people had decreased DNA methylation in the carnitine O-acetyltransferase (CRAT), coagulation factor-3 (F3), and Toll-like receptor-2 (TLR2) genes that was significantly associated with lower values for forced vital capacity (FVC) and forced expiratory volume in 1 s (FEV1). This decline in lung function is considered to be related to changes in the large airways. By contrast, decreased methylation in interferon-gamma (IFNγ) and IL-6 genes was paradoxically associated with better lung function. This finding might be explained by the varying roles of IFNγ and IL-6, which may display pro- and anti-inflammatory activities [55].
In summary, the cellular and molecular mechanisms of physiological aging and their association with various lung diseases are still not well understood. Oxidative stress, telomere length regulation, cellular immunosenescence, epigenetic changes, and ECM modifications probably represent some of the key mechanisms that account for declining lung function with advanced age.
Aging in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF)
IPF is a progressive and lethal lung disease of unclear etiology that primarily affects older patients. Symptoms usually occur between ages 50 and 70, and most patients are older than 60 years at the time of clinical presentation and diagnosis. Both the prevalence and incidence of IPF increase markedly with advancing age, particularly after the sixth decade, with a prevalence that has been estimated to exceed 175 cases per 100,000 individuals over 75 years of age [56]. Age also influences mortality, and the median survival time is significantly shorter in older individuals compared with younger patients [57].
Interestingly, predominantly subpleural basal reticular abnormalities on high-resolution computed tomography (HRCT) have been identified in a large number of asymptomatic individuals over 75 years of age, whereas these findings are virtually absent in those under 55 years old [58]. In addition, cysts can be seen in 25 % of subjects in the older age group. Bronchial dilation and wall thickening are also seen significantly more often in older individuals compared to a those in a younger age group. Importantly, all these findings have been demonstrated as independent of pack-year smoking history and indicate that some older individuals may develop interstitial lung abnormalities suggestive of possible UIP without apparent clinical significance. However, uncertainty still remains regarding the relevance of these findings to lung health and whether there are any long-term prognostic implications. In the same context, it has been demonstrated that subclinical interstitial lung disease (ILD) with subpleural distribution is present in a significant proportion of older smokers (56–72 years) screened for the development of COPD [59]. In this study, as compared with participants without interstitial changes on HRCT, those with abnormalities were more likely to have a restrictive lung deficit, suggesting that subclinical ILD may represent an early disease stage for a subset of individuals who will progress to clinically significant ILD.
Mechanisms Linking IPF to Aging
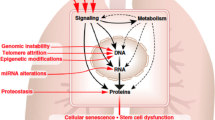
The fundamental molecular mechanism linking aging to IPF is unknown, but several modifications naturally occurring in the elderly may be implicated including oxidative stress, mitochondrial dysfunction, deregulated autophagy, telomere attrition, and others (Fig. 12.1).

Putative mechanisms linking aging to the development of IPF. A variety of modifications naturally occurring in the elderly may affect the behavior of alveolar epithelial cells (in yellow) or fibroblasts (in green), increasing the susceptibility to develop IPF
Oxidative Stress
Reactive oxygen species (ROS) induce cellular dysfunction (such as stress-induced premature senescence), which is believed to contribute to normal aging and play a role in age-related diseases. In aging, systemic imbalance between the antioxidant system (e.g., superoxide dismutases, glutathione) and ROS results in the generation of excess free radicals that can overwhelm cellular antioxidant defenses. Several studies have associated excessive oxidative stress with IPF. Thus, for example, mitochondrial generation of ROS has been suggested to be linked to increased cellular oxidative stress and apoptosis of alveolar epithelial cells [60]. Moreover, there is evidence suggesting that ROS can increase the release of TGF-β from alveolar epithelial cells [61] and can directly activate TGF-β in cell-free systems by disrupting its interaction with latency-associated peptide [62].
Strong evidence has shown that the transcription factor called nuclear factor (erythroid-derived 2)-like 2, or Nrf2, is a “master regulator” in the antioxidant response through the coordinated induction of antioxidant and phase II detoxifying enzymes that are under the regulatory influence of the antioxidant response mechanism [63]. Importantly, however, Nrf2 modulates the expression of hundreds of genes including not only antioxidant enzymes but a large number of genes that control tissue remodeling and fibrosis [63]. Mice that lack Nrf2 are highly susceptible to bleomycin-induced pulmonary inflammation and fibrosis, likely by inducing a Th1 to Th2 switch [64]. However, the expression of Nrf2 has been found to be increased in IPF lungs, which may represent an unsuccessful adaptive attempt to compensate for the increased oxidant burden [65]. Of particular interest, the increased expression of Nrf2 in IPF lungs was localized to alveolar epithelial cells whose chronic injury/activation is a crucial pathogenic event in IPF. More recently, however, decreased expression of nuclear Nrf2 was demonstrated in lung fibroblasts from patients with IPF, which was associated with the appearance of a myofibroblast phenotype [66]. Moreover, Nrf2 inhibition with siRNA induced myofibroblastic differentiation that was associated with increased oxidative stress, while conversely, Nrf2 activation with Keap1 knockdown restored the oxidant/antioxidant balance and reversed the myofibroblastic differentiation [66].
Endoplasmic Reticulum (ER) Stress
Several recent reports have demonstrated that ER stress and apoptosis occur frequently in alveolar epithelial cells (AECs) from IPF lungs [67, 68]. To better understand the putative implication of ER stress in the pathogenesis of lung fibrosis, Lawson et al. developed a transgenic mouse model in which the inducible mutant, L188Q SFTPC, was expressed in type II AECs in the adult mouse. Interestingly, the expression of L188Q SFTPC in type II AECs resulted in ER stress and unfolded protein response (UPR) activation but did not result in fibrotic remodeling. However, after a second profibrotic stimulus (bleomycin) was administered, increased epithelial cell death and fibroblast accumulation with an enhanced lung fibrotic response was found [69]. Similar findings were observed in mice treated with the ER stress-inducing agent tunicamycin. These findings indicate that dysfunctional type II AECs predispose the lung to excessive and dysregulated remodeling after injury.
Importantly, the cell has evolved an adaptive coordinated response to limit accumulation of unfolded proteins in the ER through a series of cell protective responses known collectively as the UPR. With advanced age, however, many of the key components of the UPR (such as the chaperones and enzymes) display reduced expression and activity resulting in ER dysfunction. Moreover, those proteins that remain are more vulnerable to oxidation by ROS [70]. In fact, several recent studies examining the effect of age on the ER stress response support the notion of a diminished protective response and more robust proapoptotic signaling with aging [71].
Therefore, it is possible to speculate that older individuals may have ER stress induced by multiple environmental injuries. Furthermore, because the UP is less efficient in AECs, these cells may respond with apoptotic pathway activation or induced changes in cell phenotype that can occur through epithelial to mesenchymal transition, which may increase the risk to develop IPF.
Autophagy
Autophagy, one mechanism by which the cell rids itself of misfolded proteins, declines with age and contributes to cellular senescence. The role of autophagy in fibrosis has recently being examined, but results from different models have given contradictory results. Therefore, the role of autophagy in disease pathogenesis remains unclear and may involve either impaired or accelerated autophagic activity or imbalances in the activation of autophagic proteins. For example, recent findings suggest that autophagy represents a cytoprotective mechanism that negatively regulates and limits excess collagen accumulation in the kidney, thereby mitigating experimental renal fibrosis [72]. Thus, reduced beclin-1 expression in primary mouse mesangial cells results in increased levels of type I collagen (Col-I). Inhibition of autolysosomal protein degradation by bafilomycin A(1) also increased Col-I protein levels, whereas treatment with trifluoperazine, an inducer of autophagy, results in decreased induction of Col-I levels by TGF-β1 (without alterations in Col-I α1 mRNA). By contrast, autophagy of activated stellate cells has been found to be a necessary requirement for hepatic fibrogenesis in mice [73]. In this case, loss of autophagic function in cultured mouse stellate cells and in mice following hepatic injury was associated with reduced fibrogenesis and matrix accumulation. According to these results, autophagy provides energy that is essential to support stellate cell activation and maintain energy homeostasis in the face of increasing cellular energy demands conferred by fibrogenesis and cell proliferation [73].
It has been shown that genetic or pharmacologic inhibition of Toll-like receptor 4 (TLR4) exacerbates bleomycin-induced pulmonary inflammation and fibrosis by attenuating autophagy-associated degradation of collagen and cell death in fibrotic lung tissues [74]. Moreover, rapamycin, an autophagy activator, reverses the effects of TLR4 antagonism, while attenuation of autophagy by 3-methyladenine reverses the pro-resolving and antifibrotic roles of TLR4 agonists and was associated with reduced survival.
Similarly, lung tissues from patients with IPF demonstrate evidence of decreased autophagic activity as assessed by LC3, p62 protein expression and immunofluorescence, and numbers of autophagosomes [75]. In addition, this report provided evidence that autophagy is not activated in the setting of IPF despite well-described elevations in ER stress, oxidative stress, and (HIF)-1α, which are all known to induce autophagy. Moreover, in vitro experiments demonstrate that the profibrotic mediator, TGF-β1, is likely responsible for the decreased autophagy [75].
Recent evidence has described a potential role of autophagy in aging-associated organ deterioration. Thus, cardiac hypertrophy and fibrosis have been found in aged mice compared with young mice. Levels of beclin-1, Atg5, and the LC3-II/LC3-I ratio were decreased in aged hearts. The involvement of autophagy in cardiac aging was further substantiated by the induction of in vitro autophagy with rapamycin alleviating aging-induced cardiomyocyte mechanical and intracellular Ca2+ derangements [76].
The above findings in various models indicate that the function of autophagy in fibrotic processes remains unclear and may involve either impaired or accelerated autophagic activity. In chronic obstructive pulmonary disease (COPD), another aging-related disease, there is an increase of several autophagic proteins in the lung tissue, while ultrastructural analysis of COPD tissue reveals an increased abundance of autophagosomes relative to normal lung tissue [77]. By contrast, in the case of IPF, the autophagic response seems to be impaired.
Telomere Shortening
A number of age-related pathologies (including that found in the IPF lung and in premature aging syndromes) have been associated with an accelerated rate of telomere shortening. The speed at which telomeres shorten with aging can be influenced by factors considered to accelerate aging and increase the risk of premature death, such as socioeconomic status, perceived stress, smoking, and obesity (all of which have been proposed to negatively affect telomere length) [78]. Studies in patients with familial forms of pulmonary fibrosis have found that the disease appears to be associated with telomere shortening in a subset of patients. This disease susceptibility is provoked by mutations in hTERT or hTR, which underlie the inheritance in 8–15 % of familial cases [79, 80]. In contrast, telomerase mutations are uncommon in patients with sporadic IPF. However, patients with IPF may show telomere lengths below the first percentile for their age in circulating leukocytes, and, importantly, telomeres have been shown to be shortened in alveolar epithelial cells from IPF lungs [81].
However, shortening of telomeres is also observed in COPD [82]. Moreover, telomerase-deficient mice that have sequential shortening of telomeres spontaneously develop emphysema-like lung lesions [83]. Curiously, telomerase deficiency in a murine model leads to telomere shortening, but this does not predispose these animals to enhanced bleomycin-induced lung fibrosis [84].
Epigenetic Changes
As mentioned, environmental factors may contribute to aging-associated diseases through the induction of epigenetic modifications, such as DNA methylation and chromatin remodeling, which may induce alterations in gene expression programs. The definitive corroboration on intraindividual epigenetic variation over time in humans was recently provided in a longitudinal study of DNA methylation patterns in which successive DNA samples were collected more than 10 years apart in more than 100 individuals [7]. Time-dependent changes in global DNA methylation of greater than 20 % were observed within the same individual over an 11- to 16-year span within 8–10 % of individuals in two separate study populations that resided in two widely separated geographic locations. In this study, both losses and gains of DNA methylation were observed over time in different individuals.
In a recent study in IPF, the global methylation pattern was evaluated using human CpG island microarrays [85]. Differential methylation in 625 CpG islands was found in IPF lung tissue samples when compared to control lung tissue samples. Most of these methylation changes were located in intronic, exonic, or intergenic areas, but only 8.8 % were found in gene promoters, where hypomethylation of CpG islands was generally found. The genes with differentially methylated CpG islands in their promoters were associated with biological processes such as cellular assembly and organization, cellular growth and proliferation, cell morphology, cancer, cell signaling, gene expression, and cell death. Interestingly, this study also revealed that the methylation pattern observed in IPF shows great similarity to the methylation pattern of lung cancer.
In another recent study, genome-wide DNA methylation and RNA expression were also examined in IPF and normal control lung tissue [86]. No differences were observed in global DNA methylation, but higher DNMT3a and 3b expression levels were noticed in the IPF lung tissue. Several interesting genes were found to be hyper- or hypomethylated (e.g., TP53INP1, a p53-inducible cell stress response protein that can upregulate genes, and claudin 5 and ZNF167, which are zinc finger proteins that enhance nuclear retention and transactivation of STAT3 that can downregulate genes) [86].
Hypo- or hypermethylation of some specific genes has been reported in IPF [86]. Thus, for example, hypomethylated DNA seems to contribute to the rapid progression of IPF through a Toll-like receptor (TLR) 9-dependent process [87]. Under this mechanism, surgical lung biopsies from rapidly progressive IPF patients clinically exhibit elevated levels of TLR9 gene transcript expression compared to those from stable IPF patients.
Thy-1(CD90) is a cell-surface glycoprotein expressed in normal lung fibroblasts that modulates the profibrotic phenotype of fibroblasts by several mechanisms [88, 89]. In fibroblastic foci of IPF lungs, epigenetic silencing of Thy-1 by promoter region hypermethylation has been demonstrated [90]. After this first report, it was found that treatment with the histone deacetylase inhibitor, trichostatin A, restored Thy-1 expression in Thy-1(−) cells in a time-dependent and concentration-dependent fashion, which was associated with enrichment of histone acetylation [91]. Importantly, restoration of the expression of Thy-1 was associated with both changes in chromatin marks and demethylation of the Thy-1 promoter region. This supports the concept that histone modifications and DNA methylation are coordinately regulated to change the biological behavior of fibrotic lung fibroblasts.
More recently, it has been demonstrated that IPF fibroblasts have reduced expression of the proapoptotic p14ARF due to promoter hypermethylation of CpG islands, which may explain, at least partially, their likely resistance to apoptosis [92]. P14ARF gene expression was restored by treatment with the DNA methyltransferase inhibitor, 5-aza, and this result was further corroborated by using restriction digestion with McrBc, which showed a high level of methylation of the p14ARF promoter in IPF fibroblast primary lines.
Chromatin Structural Changes
Defective histone acetylation is responsible for the repression of COX2 expression, a gene that is likely involved in the antifibrotic response [93]. Using a chromatin immunoprecipitation assay, it was revealed that transcription factor binding to the COX-2 promoter was reduced in IPF fibroblasts compared to that in normal fibroblasts. This effect was dynamically linked to reduced histone H3 and H4 acetylation due to decreased recruitment of histone acetyltransferases and recruitment of the NCoR, CoREST, and mSin3a transcriptional corepressor complexes to the COX-2 promoter [93].
Epigenetic Control of Epithelial to Mesenchymal Transition (EMT)
EMT is a fundamental developmental process that involves actin cytoskeleton reorganization with loss of apical-basal polarity and cell-to-cell contact, resulting in the conversion of epithelial cells to mesenchymal cells [94]. Although there is still some controversy, several studies suggest that an EMT-like process occurs in IPF [95–98]. Recently, global epigenetic reprogramming was observed during TGF-β(beta)-induced EMT in mouse hepatocytes [99]. In this study, the dynamic nature of genome-scale epigenetic reprogramming during EMT induced by this growth factor was demonstrated. Specifically, genome-wide reprogramming of large heterochromatin domains (LOCKs) to a state of reduced H3K9Me2, new LOCK-wide modifications of H3K4Me3 at specific GC-rich LOCKs, and enrichment of H3K36Me3 at LOCK boundaries and numerous EMT-related genes was demonstrated across the genome. This reprogramming appeared to be critical for the EMT induction by TGF-β, because inhibition of bulk chromatin changes by Lsd1 loss of function had marked effects on cell migration and chemoresistance [99].
However, it is unclear if aging affects EMT of alveolar epithelial cells. Interestingly, aging-associated cellular senescence may be involved. Thus, accumulating evidence shows that senescent fibroblasts that acquire a senescence-associated secretory phenotype have the ability to promote tumor progression, in part by inducing EMT in nearby epithelial cells [100]. Moreover, increasing evidence suggests that EMT and senescence, two processes that seem to operate independently, are in fact intertwined [101].
MicroRNAs
As mentioned, miRNAs form a particular class of 21- to 24-nucleotide RNAs that can regulate gene expression posttranscriptionally by affecting the translation and stability of target messenger RNAs. Importantly, some miRNAs have emerged as key regulators during cellular senescence.
A growing body of evidence indicates that dysregulated expression of miRNAs is linked to fibrotic diseases in different organs [102–108]. Recent evidence also supports the notion that miRNAs can regulate cellular plasticity. For example, miR-145 expression facilitates the differentiation of fibroblasts to myofibroblasts suggesting that miRNAs may regulate the plasticity of mesenchymal cells [109].
Regarding lung fibrotic remodeling, several studies have reported disturbed expression of a number of miRNAs. For example, downregulation of mir-29 has been found in bleomycin-induced lung fibrosis in mice. In addition, miR-29 is suppressed by TGF-β1 in lung fibroblasts, and miR-29 levels are inversely correlated with the expression of several profibrotic target genes and with the severity of the fibrosis [110]. By contrast, lungs of mice with bleomycin-induced fibrosis as well as IPF lungs show an upregulation of miR-21 primarily localized to myofibroblasts [105]. Increasing miR-21 levels promote, while reduced levels attenuate, the profibrogenic activity of TGF-β1 in fibroblasts, while miR-21 antisense probes attenuate bleomycin-induced lung fibrosis. Likewise, miR-155 (targeting the angiotensin II type 1 receptor) and keratinocyte growth factor are upregulated in the lungs of mice with bleomycin-induced lung fibrosis [111, 112].
The extent of changes of miRNAs in IPF lungs was recently demonstrated when RNA from IPF and control lungs was extracted and hybridized to miRNA arrays that contained probes for ∼450 miRNAs. In this work, 10 % of the miRNAs on the array were significantly different between IPF and control lungs [104]. One of the downregulated miRNAs, let-7d, was primarily localized in epithelial cells and was directly inhibited by TGF-β. Let-7d regulates EMT in alveolar epithelial cells (at least partially) due to the overexpression of the high-mobility group, AT-hook 2 (HMGA2), a member of the nonhistone chromosomal high-mobility group (HMG) protein family.
Summary
Recent research into the mechanisms of aging and IPF has suggested that they share several common molecular pathways including mitochondrial dysfunction, dysregulated autophagy, telomere attrition, epigenetic changes, and others. However, many studies are still necessary to verify the existing findings in larger cohorts and to establish the mechanisms underlying the putative association between aging and IPF. Identification of the aging-affected signaling pathways that are implicated in the pathogenesis of the pulmonary fibrosis also holds promise in furthering our understanding of IPF. A better knowledge of the age-related changes in lung cells will also help to elucidate the lung aging process itself and eventually to recognize which of these modifications are truly involved in the pathogenesis of IPF.
References
Heemels MT. Ageing. Nature. 2010;464:503.
Berdasco M, Esteller M. Hot topics in epigenetic mechanisms of aging: 2011. Aging Cell. 2012;11:181–6.
Rando TA, Chang HY. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell. 2012;148:46–57.
Lister R, Ecker JR. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res. 2009;19:959–66.
Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–66.
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86.
Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–83.
So K, Tamura G, Honda T, Homma N, Waki T, Togawa N, et al. Multiple tumor suppressor genes are increasingly methylated with age in non-neoplastic gastric epithelia. Cancer Sci. 2006;97:1155–58.
Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20:1164–72.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68.
Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002;14:286–98.
Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci USA. 2006;103:8703–8.
Krishnan V, Chow MZ, Wang Z, Zhang L, Liu B, Liu X, et al. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci USA. 2011;108:12325–30.
Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, Part II: ATP dependent chromatin remodeling. Trends Mol Med. 2007;13:373–80.
Villeponteau B. The heterochromatin loss model of aging. Exp Gerontol. 1997;32:383–94.
Koch CM, Suschek CV, Lin Q, Bork S, Goergens M, Joussen S, et al. Specific age-associated DNA methylation changes in human dermal fibroblasts. PLoS One. 2011;6(2):e16679.
Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23:413–18.
Zaratiegui M, Irvine DV, Martienssen RA. Noncoding RNAs and gene silencing. Cell. 2007;128:763–76.
Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–46.
Krol J, Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610.
Smith-Vikos T, Slack FJ. MicroRNAs and their roles in aging. J Cell Sci. 2012;125(Pt 1):7–17.
Bonifacio LN, Jarstfer MB. MiRNA profile associated with replicative senescence, extended cell culture, and ectopic telomerase expression in human foreskin fibroblasts. PLoS One. 2010;5(9):e12519.
Gorospe M, Abdelmohsen K. MicroRegulators come of age in senescence. Trends Genet. 2011;27:233–41.
Lee HC, Wei YH. Mitochondria and aging. Adv Exp Med Biol. 2012;942:311–27.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9.
Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275:31505–13.
Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp Gerontol. 2005;40:622–33.
Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53.
Woehlbier U, Hetz C. Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci. 2011;36:329–37.
Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–12.
Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–60.
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79.
Londoño-Vallejo JA, Wellinger RJ. Telomeres and telomerase dance to the rhythm of the cell cycle. Trends Biochem Sci. 2012;37(9):391–9.
Vera E, Blasco MA. Beyond average: potential for measurement of short telomeres. Aging (Albany NY). 2012;4:379–92.
Janssens JP, Pache JC, Nicod LP. Physiological changes in respiratory function associated with ageing. Eur Respir J. 1999;13:197–205.
Meyer KC. Aging. Proc Am Thorac Soc. 2005;2:433–9.
Reddy GK. AGE-related cross-linking of collagen is associated with aortic wall matrix stiffness in the pathogenesis of drug-induced diabetes in rats. Microvasc Res. 2004;68:132–42.
DeGroot J, Verzijl N, Budde M, Bijlsma JW, Lafeber FP, TeKoppele JM. Accumulation of advanced glycation end products decreases collagen turnover by bovine chondrocytes. Exp Cell Res. 2001;266:303–10.
Rolewska P, Al-Robaiy S, Navarrete Santos A, Simm A, Silber RE, Bartling B. Age-related expression, enzymatic solubility and modification with advanced glycation end-products of fibrillar collagens in mouse lung. Exp Gerontol. 2013;48:29–37.
Calabresi C, Arosio B, Galimberti L, Scanziani E, Bergottini R, Annoni G, et al. Natural aging, expression of fibrosis-related genes and collagen deposition in rat lung. Exp Gerontol. 2007;42:1003–11.
Huang K, Rabold R, Schofield B, Mitzner W, Tankersley CG. Age-dependent changes of airway and lung parenchyma in C57BL/6J mice. J Appl Physiol. 2007;102:200–6.
Paxson JA, Gruntman A, Parkin CD, Mazan MR, Davis A, Ingenito EP, et al. Age-dependent decline in mouse lung regeneration with loss of lung fibroblast clonogenicity and increased myofibroblastic differentiation. PLoS One. 2012;6:e23232.
Takubo Y, Hirai T, Muro S, Kogishi K, Hosokawa M, Mishima M. Age-associated changes in elastin and collagen content and the proportion of types I and III collagen in the lungs of mice. Exp Gerontol. 1999;34:353–64.
Sherratt MJ. Tissue elasticity and the ageing elastic fibre. Age (Dordr). 2009;31:305–25.
Konova E, Baydanoff S, Atanasova M, Velkova A. Age-related changes in the glycation of human aortic elastin. Exp Gerontol. 2004;39:249–54.
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105.
Ito K. Does lung aging have an impact on chronic obstructive pulmonary disease? J Organ Dysfunction. 2007;3:204–20.
Pawelec G, Larbi A. Immunity and ageing in man: annual review 2006/2007. Exp Gerontol. 2008;43:34–8.
Lee N, Shin MS, Kang I. T-Cell biology in aging, with a focus on lung disease. J Gerontol A Biol Sci Med Sci. 2012;67A:254–63.
Kang I, Hong MS, Nolasco H, Park SH, Dan JM, Choi JY, et al. Age-associated change in the frequency of memory CD4+ T cells impairs long term CD4+ T cell responses to influenza vaccine. J Immunol. 2004;173:673–81.
Mora AL, Woods CR, Garcia A, Xu J, Rojas M, Speck SH, et al. Lung infection with gamma-herpesvirus induces progressive pulmonary fibrosis in Th2-biased mice. Am J Physiol Lung Cell Mol Physiol. 2005;289:L711–21.
Selman M, Rojas M, Mora AL, Pardo A. Aging and interstitial lung diseases: unraveling an old forgotten player in the pathogenesis of lung fibrosis. Semin Respir Crit Care Med. 2010;31:607–17.
Volkova M, Zhang Y, Shaw AC, Lee PJ. The role of Toll-like receptors in age-associated lung diseases. J Gerontol A Biol Sci Med Sci. 2012;67A:247–53.
Jane-Wit D, Chun HJ. Mechanisms of dysfunction in senescent pulmonary endothelium. J Gerontol A Biol Sci Med Sci. 2012;67:236–41.
Lepeule J, Baccarelli A, Motta V, Cantone L, Litonjua AA, Sparrow D, et al. Gene promoter methylation is associated with lung function in the elderly: the normative aging study. Epigenetics. 2012;7:261–9.
Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respi Crit Care Med. 2006;174:810–16.
Noth I, Martinez FJ. Recent advances in idiopathic pulmonary fibrosis. Chest. 2007;132:637–50.
Copley SJ, Wells AU, Hawtin KE, Gibson DJ, Hodson JM, Jacques AE, et al. Lung morphology in the elderly: comparative CT study of subjects over 75 years old versus those under 55 years old. Radiology. 2009;251:566–73.
Washko GR, Hunninghake GM, Fernandez IE, Nishino M, Okajima Y, Yamashiro T, et al. COPDGene investigators. Lung volumes and emphysema in smokers with interstitial lung abnormalities. N Engl J Med. 2011;364:897–906.
Kuwano K, Hagimoto N, Maeyama T, Fujita M, Yoshimi M, Inoshima I, et al. Mitochondria-mediated apoptosis of lung epithelial cells in idiopathic interstitial pneumonias. Lab Invest. 2002;82:1695–706.
Bellocq A, Azoulay E, Marullo S, Flahault A, Fouqueray B, Philippe C, et al. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am J Respir Cell Mol Biol. 1999;21:128–36.
Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol. 1996;10:1077–83.
Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med. 2011;32:234–46.
Kikuchi N, Ishii Y, Morishima Y, Yageta Y, Haraguchi N, Itoh K, et al. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir Res. 2010;11:31.
Markart P, Luboeinski T, Korfei M, Schmidt R, Wygrecka M, Mahavadi P, et al. Alveolar oxidative stress is associated with elevated levels of nonenzymatic low-molecular-weight antioxidants in patients with different forms of chronic fibrosing interstitial lung diseases. Antioxid Redox Signal. 2009;11:227–40.
Artaud-Macari E, Goven D, Brayer S, Hamimi A, Besnard V, Marchal-Somme J, et al. Nrf2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid Redox Signal. 2013;18(1):66–79.
Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–46.
Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–26.
Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA. 2011;108:10562–67.
Naidoo N. ER and aging-protein folding and the ER stress response. Ageing Res Rev. 2009;8:150–9.
Hussain SG, Ramaiah KV. Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem Biophys Res Commun. 2007;355:365–70.
Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J Biol Chem. 2012;287:11677–88.
Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–46.
Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol. 2012;180:275–92.
Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One. 2012;7(7):e41394.
Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM, Ren J. Chronic Akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol. 2011;106:1173–91.
Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3(10):e3316.
Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol. 2007;3:640–9.
Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–26.
Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104:7552–7.
Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105:13051–6.
Savale L, Chaouat A, Bastuji-Garin S, Marcos E, Boyer L, Maitre B, et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179:566–71.
Lee J, Reddy R, Barsky L, Scholes J, Chen H, Shi W, et al. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. Am J Physiol. 2009;296:L57–70.
Degryse AL, Xu XC, Newman JL, Mitchell DB, Tanjore H, Polosukhin VV, et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp Lung Res. 2012;38:124–34.
Rabinovich EI, Kapetanaki MG, Steinfeld I, Gibson KF, Pandit KV, Yu G, et al. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS One. 2012;7(4):e33770.
Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, Crossman DK, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(6):525–35.
Trujillo G, Meneghin A, Flaherty KR, Sholl LM, Myers JL, Kazerooni EA, et al. TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci Transl Med. 2010;2:57ra82.
Rege TA, Hagood JS. Thy-1, a versatile modulator of signaling affecting cellular adhesion, proliferation, survival, and cytokine/growth factor responses. Biochim Biophys Acta. 2006;1763:991–9.
Ramírez G, Hagood JS, Sanders Y, Ramírez R, Becerril C, Segura L, et al. Absence of Thy-1 results in TGF-β induced MMP-9 expression and confers a profibrotic phenotype to human lung fibroblasts. Lab Invest. 2011;91:1206–18.
Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, et al. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39:610–18.
Sanders YY, Tollefsbol TO, Varisco BM, Hagood JS. Epigenetic regulation of thy-1 by histone deacetylase inhibitor in rat lung fibroblasts. Am J Respir Cell Mol Biol. 2011;45:16–23.
Cisneros J, Hagood J, Checa M, Ortiz-Quintero B, Negreros M, Herrera I, et al. Hypermethylation-mediated silencing of p14ARF in fibroblasts from idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;303(4):L295–303.
Coward WR, Watts K, Feghali-Bostwick CA, Knox A, Pang L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol. 2009;29:4325–39.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90.
King Jr TE, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–61.
Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–32.
Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–85.
Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA. 2011;108:E1475–83.
McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. 2011;18:867–74.
Laberge RM, Awad P, Campisi J, Desprez PY. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012;5:39–44.
Smit MA, Peeper DS. Epithelial-mesenchymal transition and senescence: two cancer-related processes are crossing paths. Aging (Albany NY). 2010;2:735–41.
van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–32.
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4.
Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–9.
Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207:1589–97.
Kato M, Zhang J, Wang M, Lanting L, Yuan H, Rossi JJ, Natarajan R. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci USA. 2007;104:3432–37.
Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, Cai B, et al. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res. 2009;83:465–72.
Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104:170–8.
Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–10.
Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, et al. MIR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45:287–94.
Pottier N, Maurin T, Chevalier B, Puisségur MP, Lebrigand K, Robbe-Sermesant K, et al. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: implication in epithelial-mesenchymal interactions. PLoS One. 2009;4(8):e6718.
Martin MM, Lee EJ, Buckenberger JA, Schmittgen TD, Elton TS. MicroRNA-155 regulates human angiotensin II type 1 receptor expression in fibroblasts. J Biol Chem. 2006;281:18277–84.
Acknowledgment
Our research was partially supported by PAPIIT IN214612.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Selman, M., Romero, Y., Pardo, A. (2014). Aging and IPF: What Is the Link?. In: Meyer, K., Nathan, S. (eds) Idiopathic Pulmonary Fibrosis. Respiratory Medicine, vol 9. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-62703-682-5_12
Download citation
DOI: https://doi.org/10.1007/978-1-62703-682-5_12
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-62703-681-8
Online ISBN: 978-1-62703-682-5
eBook Packages: MedicineMedicine (R0)