
Abstract
Single cell sequencing (SCS) can be harnessed to acquire the genomes, transcriptomes and epigenomes from individual cells. Next generation sequencing (NGS) technology is the driving force for single cell sequencing. scRNA-seq requires a lengthy pipeline comprising of single cell sorting, RNA extraction, reverse transcription, amplification, library construction, sequencing and subsequent bioinformatic analysis. Computational algorithms are essential to fulfill many tasks of interest using scRNA-seq data. scRNA-seq has already enabled researchers to revisit long-standing questions in cancer biology, including cancer metastasis, heterogeneity and evolution. Circulating Tumor Cells (CTC) are not only an important mechanism for cancer metastasis, but also provide a possibility to diagnose and monitor cancer in a convenient way independent of surgical resection of the cancer.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
4.1 Brief Introduction to Single Cell Sequencing
Single cell sequencing (SCS) can be harnessed to acquire the genomes, transcriptomes and epigenomes from individual cells. Next generation sequencing (NGS) technology is the driving force for single cell sequencing. Though various bias could be potentially introduced during the molecule amplification, It has been well recognized that SCS could help detect single nucleotide variations (SNVs) [1], copy number variations (CNVs) [2], structure variations (SVs) [3], gene expression and fusions [4,5,6,7,8], novel transcripts and alternative splicing [9], methylations [10] and chromatin patterns [11, 12] on single cell level with the help of mathematic algorithms and models. SCS also has great potential to reveal novel biological concepts, which have never been investigated. For example, researchers used single cell RNA-seq (scRNA-seq) to uncover new cell types in nervous system [13], immune system and hematopoietic system [14], as well as new insights into the clonal evolution of cancer [15]. Most recently, the accuracy and throughput of SCS have been increased dramatically to be capable of profiling more than thousands of single cells in parallel [5, 16].
4.2 Large-Scale scRNA-seq Library Preparation
scRNA-seq requires a lengthy pipeline comprising of single cell sorting, RNA extraction, reverse transcription, amplification, library construction, sequencing and subsequent bioinformatic analysis. As the key factor to increase the throughput of scRNA-seq study, high-throughput scRNA-seq library preparation technology developed very quickly these few years. FAC sorting-based scRNA-seq library preparation combined with automatic liquid instrument pipeline enable handling 96-well/384-well plate single cells per run [17]. Fluidigm C1 system based on microvalve microfluidic chip that was developed by Quake’s lab enabled to prepare full-length transcripts of 96 single cells in parallel in 2012 [7], and a similar chip with higher throughput up to 800 single cells 3’end transcript preparation was released in 2015. Another type of microfluidic chip, micro-well chip was also used in single-cell RNA amplification. Wu et al. developed an approach called MIRALCS [4], allowing massively parallel single-cell full-length transcripts amplification for 500–1000 single cells based on 5184-well chip. With the same chip, Wafergen Inc. released a single cell preparation system named ICell8, allowing the preparation of 3′ single cell transcripts with throughput up to 1800 cells per run [18]. Taking the advantage of barcode-bead technology, two different groups described microwell chip based method, with the capacity to obtain gene expression from thousands of cells at the single cell level, respectively [19, 20]. In addition, droplet microfluidic technique improves the throughput of single cell 3′ end RNA-seq to million level, and reduce the reaction volume to picoliter [21, 22]. A commercialized instrument with the same strategy was developed by 10X genomics, enabling the preparation of at most 48,000 single cells from eight different samples in parallel. With the development of high-throughput scRNA-seq library preparation technology, the cost has been reduced to less than one dollar per cell, which greatly promotes the studies at the single cell level.
4.3 Computational Analysis of scRNA-seq Data
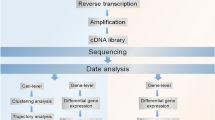
Computational algorithms are essential to fulfill many tasks of interest using scRNA-seq data (Fig. 4.1). There is a general consensus that analyses of scRNA-seq data sets and conventional RNA-seq data have a lot in common. More specifically, the vast majority of the basic pipelines and tools established for sequencing data derived from bulk cell populations are applicable to that from single cells, following steps including read alignment, quality control and gene expression estimation. Whereas more dedicated softwares for use in fields such as identifying and characterizing cellular subpopulations, exploring evolution of cell groups and inferring the transcriptional kinetics are urgently needed owing to the zero-inflated nature and additional functions of scRNA-seq data sources.
-
Quality Control: Single-cell datasets are expected to be extra messy, thus should undertake the quality control step before any downstream analysis. To begin with, FASTQC [23], Qualimap2 [24] and RSeQC [25] are commonly used for assessment of overall sequencing quality. After removal of adapters or noisy data with low quality, raw reads obtained from a well-designed experiment are firstly aligned to the reference genome using tools such as Tophat [26], HISAT [27] and STAR [28]. Subsequently, several indicators are calculated so as to discriminate cells with degraded RNA or substandard sequencing library, for instance, the number of expressed genes, the proportion of reads mapped to endogenous genes and the fraction of external spike-ins in mapped reads [9, 29, 30]. In addition, Treutlein considered normal expression of housekeeping genes a judgment factor of healthy cells [31].

Representative tasks enabled by scRNA-seq. (a) Subpopulation analysis can be performed with various unsupervised clustering algorithms; (b) Pseudotemporal ordering is essential to understand developmental trajectory or disease progression; (c) Differential gene expression analysis is important for the discovery of cell type specific biomarkers; (d) Network inference can be performed to learn regulatory intracellular and intercellular networks; (e) Analysis of alternative splicing offers a new perspective on biology and medicine; (f) Allele specific expression patterns can be addressed using scRNA-seq data
-
Expression estimation and normalization: Gene expression levels of qualified cells can be estimated as count from data without UMIs using HTSeq [32], WemIQ [33] or RSEM [34]. While relative expression including transcripts per million mapped reads (TPM) and reads/fragments per kilobase per million mapped reads (RPKM/FPKM) is widely adopted in downstream analysis. Besides, Islam et al. [29] and Hashimshony et al. [35] provide solutions to UMI-tagged reads. Normalization is essential due to the technical variability in comparison to expression levels between samples. Median normalization or a similar method are popular in many scRNA-seq studies without spike-ins or UMIs [30, 36,37,38,39]. In single cell experiments where spike-ins were applied, technical artifacts can be estimated by difference between their expected and observed expression. Nevertheless, instability arising from inconsistent detection of spike-ins brings about a more current notion of comparing absolute molecular counts of different cells with UMIs, which have greatly reduced the amplification noise by attaching random sequences to cDNA fragments ahead of PCR [21, 29, 40].
-
Identification of subpopulations: Cellular subpopulation identification in heterogenous cells is one of the most exciting areas for exploration in the scRNA-seq experiments. Therefore, various algorithms for clustering have been developed to date. Pollen et al. [41] distinguished different types of cells along lung development using principal component analysis. The study by Li et al. [42] showed transcriptional heterogeneity in colorectal tumors with a novel strategy named reference component analysis (RCA). Following similar lines, self-organizing maps (SOMs) [43], circular a posteriori projection (CAP), ZIFA [44], t-SNE [45] and BackSPIN [46] clustering are approaches developed for differentiating between cells within a biological condition by dimensionality reduction of scRNA-seq data. In addition, RaceID [6] is a computationally efficient tool that relies on k-means clustering, whereas SNN-Cliq [47] clusters individual cells by a graph-based algorithm based on shared nearest neighbor (SNN) similarity measurement. Guo et al. [48] further presented a pipeline for known cell type enrichment that is analogous to gene set enrichment analysis.
-
Differential expression and transcript isoforms across conditions: Once subpopulations are distinguished, differential expression can be applied for cell type characterization. Researchers used to investigate differential expressed genes among cells of different types or stages with bulk RNA-Seq based strategy. However, an abundant zero values on expression matrix from single cells lead to potential fault sets of genes that may have expressed differently resulting from noise. As a consequence, plenty of mixture-model-based methods like MAST [49] and SCDE [50] have been created for accommodation of bimodality in expression levels. Similarly, D3E [51] identify DE genes by comparing two probability distributions on transcriptional bursting model. Korthauer et al. [52] have established a more accurate Bayesian modeling framework, scDD, for differential expression patterns detection under a wide range of circumstances recently. Unlike the traditional methods with a simple mean shift, the scDD model provides posterior probabilities differential distributions (DD) for each gene and classified gene as unimodal distributions (traditional DE), differential modes (DM), differential proportion (DP), or both DM and DE (abbreviated DB).
-
Pseudotemporal ordering: Knowledge of the global expression profile in individual cells provides opportunities for the investigation of dynamic cellular processes such as normal tissue development, stem cell differentiation and tumor progression. A number of computational methods were built on the basis of the theory that differentiation paths can by constructed by reordering unsynchronized cells with gradual changes in gene expression levels at various stages. Similarities to cellular subpopulation identification approaches, most investigators perform pseudotemporal ordering by reducing the dimensionality of gene expression data. Take Monocle [53] as an example, which was the most effective tool to construct the differentiation paths in the infancy of single cell technology. Minimal spanning tree (MST) was built on data processed by independent component analysis (ICA) in Monocle, and the longest path through the MST was considered as a default setting for differentiation. Subsequently, Haghverdi L et al. [54] worked out a diffusion map based method that allows trajectory reconstruction in a single step. Rizvi et al. [55] presented a topology-based algorithm named single-cell topological data analysis (scTDA) for unbiased transcriptional regulation study through a nonlinear and unsupervised statistical framework. Furthermore, when it comes to oscillatory processes, Oscope [56] can be used for oscillatory trajectory reconstruction with co-regulation information among oscillators.
-
Interrogation of spatial information: In spite of looking into the development of cell populations extending in time, scRNA-seq can be applied for spatial reconstruction via the integration of in situ RNA patterns with genome wide gene expression profiles. Satija R et al. [57] has established an accurate spatially resolved tools, Seurat, for mapping cellular localization, with which they inferred cellular localization of cells from dissociated zebrafish (Danio rerio) embryos and generated a transcriptome-wide map of spatial patterning. Meanwhile, another high-throughput approach by Kaia Achim [58] was published online by virtue of a reference gene expression database, which successfully allocates brain cells to precise locations from marine annelid Platynereis dumerilii by comparing specificity-weighted mRNA Profiles. Halpern K B et al. [59] reconstructed a genomic blueprint of mammalian liver by combining landmark genes expression and single-molecule fluorescence in situ hybridization.
-
Network inference: Identification of co-regulated genes with data derived from single cell experiments is superior because it can provide insight into regulatory networks that are hard to be noticed in bulk level. Understanding the transcriptional regulatory networks is of primary interest in a myriad of studies. For convenience, some statistical methods in bulk studies were reused when exploring scRNA-seq data. Weighted correlation network analysis (WGCNA) [60] can be used for gene clustering and summarizing with a comprehensive collection of functions for network construction, module detection, gene selection, calculations of topological properties, data simulation and visualization. Cell-centric statistics (CCs) [61] was invented to model transcriptome dynamics by analyzing aggregated cell-cell statistical distances within biomolecular pathways, for instance, differentially expressed pathways for a single cell of interest. While SCODE [62] inferred the co-regulatory network with ordinary differential equations(ODEs) by integrating the transformation of linear ODEs and linear regression.
-
Differential Splicing: Experimental protocols with full-length transcript coverage to certain sequencing depth provides insight into alternative splicing isoform determination and quantification in scRNA-seq data analysis, which reflects heterogeneity among cells of a biological component from another perspective. A study of heterogeneity in immune cells in 2013 [9] was the first to reveal the dramatic diversity of splicing patterns in mouse bone-marrow-derived dendritic cells(BMDCs). Gokce O et al. [63] used fisher’s exact test for differentially splicing junction definition among cell types and pointed out splice variant expressed in mouse striatum. SingleSplice [64] is the latest tailored method used to detect isoform usage differences in scRNA-seq data, which was applied to mouse embryonic stem cells and eventually shedded insight into the connection between alternative splicing and the cell cycle through a series of analysis.
-
Allelic Expression Patterns: Another subtle point is that allele-specific expression can be accessed for in scRNA-seq to investigate the contribution of parental allele expression. Deng et al. [65] demonstrated an abundant random allele-specific gene expression using train-specific SNPs at single-cell resolution in mouse preimplantation embryos. Reinius B et al. [66] argued in an allele-sensitive scRNA–seq experiment that most patterns of random monoallelic expression of autosomal genes (aRME) are in a decentralized fashion rather than confined to clonally related cells according to previous hypothesis.
4.4 Application of High Throughput scRNA-seq
-
Cancer Biology: scRNA-seq has already enabled researchers to revisit long-standing questions in cancer biology, including cancer metastasis, heterogeneity and evolution. Circulating Tumor Cells (CTC) are not only an important mechanism for cancer metastasis [67], but also provide a possibility to diagnose and monitor cancer in a convenient way independent of surgical resection of the cancer. One landmark study analysed CTC isolated from prostate cancer patients and revealed that the mechanism of resistance to androgen receptor inhibition in recurrent disease is partly due to noncanonical Wnt signaling [68].
A comprehensive picture of cancer heterogeneity is redefined by scRNA-seq. Several studies revealed the heterogeneity of cancer cells [69, 70]. A comprehensive profiling of melanoma using scRNA-seq is a classical example [70]. It was found that two distinct transcriptional signatures were present but they were not mutually exclusive. The melanoma characterized by activation of the transcription factor MITF also harbored a small subpopulation of cells with low MITF activity. The heterogeneity of cancer is not limited to the cell-to-cell variability among cancer cells. More importantly, cancer is itself a heterogeneous tissue comprised of malignant, immune, stromal and endothelial cells [71]. Recently, profiling of the immune cells within the tumor microenvironment is attracting lots of attention [72,73,74,75]. Those studies covered various different cancers and single cell omic technologies. A recent study employed scRNA-seq to analyse T cells isolated from tumor tissues and adjacent normal tissues from hepatocellular carcinoma (HCC) patients, revealing the distinctive functional composition of T cells in HCC and the clonal enrichment of infiltrating Tregs and exhausted CD8 T cells [72].
The clonal evolution of cancer was proposed more than 40 years ago [76]. Longitudinal single cell analysis is now adding new evidence to this widely held concept [77]. Applying single nucleus sequencing to biopsy from primary breast cancer and its liver metastasis, it was suggested that tumor evolution might follow a punctuated expansion mode instead of a gradual progression path [78]. Single cell genome and exome sequencing enabled by MDA further increased the coverage of single cell genome sequencing and rendered the mutation and SNP calling at the single cell possible [79, 80]. The mutation and SNP information for individual cancer cells was valuable for population genetic analysis to understand the clonal evolution of tumor.
-
Developmental Biology: Our understanding of developmental biology has also been dramatically enhanced by scRNA-seq. The identification of rare cell type was realized by the combination of organoid culture, scRNA-seq and development of novel algorithm [6]. This crystalized in the identification of Reg4 as a novel marker for enteroendocrine cells. New markers will then facilitate the investigation of rare cell types. Another recent study focused on the cells in the blood. New types of dendritic cells and monocytes were identified using scRNA-seq [14]. Our understanding of the cell types or subtypes constituting the brain was renewed by single-nucleus RNA sequencing [81] and scRNA-seq [82], while traditionally cell types were defined based on morphology, location and function.
-
The Human Cell Atlas: With the development of high throughput single cell molecular profiling techniques, an international community or network is taking shape rapidly aiming to undertake the ambitious project to identify all cell types in the human body [83]. Single cell omic technologies are situated at the heart of the human cell altlas. Key efforts will be devoted to key organs, such as the liver, the heart, the kidney or the pancreas [84], as well key systems, such as the immune system and the central nervous system [85].
Our understanding of disease will also be greatly refined with the realization of the human cell atlas. In the future biopsy from patients will be routinely assayed with single cell techniques [70, 86] and compared to the normal reference in the human cell atlas. Specific abnormalities will be identified and used to inform both diagnosis and treatment.
The drug industry will benefit dramatically from the human cell atlas. Traditionally, drug discovery and development efforts have been hindered by the challenges that all healthy and diseased tissues are inherently heterogeneous [87]. The emergence and rapid application of single cell analysis tools will pave the way to eventually understand both health and disease at an unprecedented level so that medicine can finally ushers in a new era of personalized healthcare [88, 89].
References
Zafar H, Wang Y, Nakhleh L, Navin N, Chen K (2016) Monovar: single-nucleotide variant detection in single cells. Nat Methods 13:505–507. https://doi.org/10.1038/nmeth.3835
Knouse KA, Wu J, Amon A (2016) Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res 26:376–384. https://doi.org/10.1101/gr.198937.115
Voet T, Kumar P, Van Loo P, Cooke SL, Marshall J, Lin ML, Zamani Esteki M, Van der Aa N, Mateiu L, McBride DJ, Bignell GR, McLaren S, Teague J, Butler A, Raine K, Stebbings LA, Quail MA, D’Hooghe T, Moreau Y, Futreal PA, Stratton MR, Vermeesch JR, Campbell PJ (2013) Single-cell paired-end genome sequencing reveals structural variation per cell cycle. Nucleic Acids Res 41:6119–6138. https://doi.org/10.1093/nar/gkt345
Wu L, Zhang X, Zhao Z, Wang L, Li B, Li G, Dean M, Yu Q, Wang Y, Lin X, Rao W, Mei Z, Li Y, Jiang R, Yang H, Li F, Xie G, Xu L, Wu K, Zhang J, Chen J, Wang T, Kristiansen K, Zhang X, Li Y, Yang H, Wang J, Hou Y, Xu X (2015) Full-length single-cell RNA-seq applied to a viral human cancer: applications to HPV expression and splicing analysis in HeLa S3 cells. GigaSci 4:51. https://doi.org/10.1186/s13742-015-0091-4
Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA (2015) The technology and biology of single-cell RNA sequencing. Mol Cell 58:610–620. https://doi.org/10.1016/j.molcel.2015.04.005
Grun D, Lyubimova A, Kester L, Wiebrands K, Basak O, Sasaki N, Clevers H, van Oudenaarden A (2015) Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 525:251–255. https://doi.org/10.1038/nature14966
Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, Quake SR (2014) Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods 11:41–46. https://doi.org/10.1038/nmeth.2694
Saliba AE, Westermann AJ, Gorski SA, Vogel J (2014) Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res 42:8845–8860. https://doi.org/10.1093/nar/gku555
Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D (2013) Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498:236–240
Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, Kelsey G (2014) Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 11:817–820. https://doi.org/10.1038/nmeth.3035
Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z, Shendure J (2017) Massively multiplex single-cell Hi-C. Nat Methods 14:263–266. https://doi.org/10.1038/nmeth.4155
Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ (2015) Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523:486–490. https://doi.org/10.1038/nature14590
Llorens-Bobadilla E, Zhao S, Baser A, Saiz-Castro G, Zwadlo K, Martin-Villalba A (2015) Single-cell transcriptomics reveals a population of dormant neural stem cells that become activated upon brain injury. Cell Stem Cell 17:329–340. https://doi.org/10.1016/j.stem.2015.07.002
Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N (2017) Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356. https://doi.org/10.1126/science.aah4573
Linnarsson S, Teichmann SA (2016) Single-cell genomics: coming of age. Genome Biol 17:97. https://doi.org/10.1186/s13059-016-0960-x
Vitak SA, Torkenczy KA, Rosenkrantz JL, Fields AJ, Christiansen L, Wong MH, Carbone L, Steemers FJ, Adey A (2017) Sequencing thousands of single-cell genomes with combinatorial indexing. Nat Methods 14:302–308. https://doi.org/10.1038/nmeth.4154
Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I (2014) Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343:776–779. https://doi.org/10.1126/science.1247651
Yuan J, Sims PA (2016) An automated microwell platform for large-scale single cell RNA-Seq. Sci Rep 6:33883. https://doi.org/10.1038/srep33883
Fan HC, Fu GK, Fodor SP (2015) Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science 347:1258367. https://doi.org/10.1126/science.1258367
Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW (2015) Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161:1187–1201. https://doi.org/10.1016/j.cell.2015.04.044
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA (2015) Highly parallel genome-wide expression profiling of individual cells using Nanoliter droplets. Cell 161:1202–1214. https://doi.org/10.1016/j.cell.2015.05.002
Andrews S (2010) https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Okonechnikov K, Conesa A, García-Alcalde F (2015) Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32:292–294
Wang L, Wang S, Li W (2012) RSeQC: quality control of RNA-seq experiments. Bioinformatics 28:2184–2185
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21
Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lönnerberg P, Linnarsson S (2014) Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 11:163–166
Brennecke P, Anders S, Kim JK, Kołodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC (2013) Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods 10:1093–1095
Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR (2014) Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509:371–375
Anders S, Pyl PT, Huber W (2015) HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169
Zhang J, Kuo C-CJ, Chen L (2014) WemIQ: an accurate and robust isoform quantification method for RNA-seq data. Bioinformatics 31:878–885btu757
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinf 12:323
Hashimshony T, Wagner F, Sher N, Yanai I (2012) CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2:666–673
Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O (2015) Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol 33:155–160
Kim JK, Marioni JC (2013) Inferring the kinetics of stochastic gene expression from single-cell RNA-sequencing data. Genome Biol 14:R7
Dueck H, Khaladkar M, Kim TK, Spaethling JM, Francis C, Suresh S, Fisher SA, Seale P, Beck SG, Bartfai T (2015) Deep sequencing reveals cell-type-specific patterns of single-cell transcriptome variation. Genome Biol 16:122
Xue Z, Huang K, Cai C, Cai L, Jiang C-Y, Feng Y, Liu Z, Zeng Q, Cheng L, Sun YE (2013) Genetic programs in human and mouse early embryos revealed by single-cell RNA [thinsp] sequencing. Nature 500:593–597
Grün D, Kester L, Van Oudenaarden A (2014) Validation of noise models for single-cell transcriptomics. Nat Methods 11:637–640
Pollen AA, Nowakowski TJ, Shuga J, Wang X, Leyrat AA, Lui JH, Li N, Szpankowski L, Fowler B, Chen P (2014) Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol 32:1053–1058
Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS (2017) Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet 49(5):708–718
Kim DH, Marinov GK, Pepke S, Singer ZS, He P, Williams B, Schroth GP, Elowitz MB, Wold BJ (2015) Single-cell transcriptome analysis reveals dynamic changes in lncRNA expression during reprogramming. Cell Stem Cell 16:88–101
Pierson E, Yau C (2015) Dimensionality reduction for zero-inflated single cell gene expression analysis. Genome Biology 16:1–10
van der Maaten L, Hinton G (2008) Visualizing data using t-SNE. J Mach Learn Res 9:2579–2605
Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C (2015) Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347:1138–1142
Xu C, Su Z (2015) Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics 31:1974–1980btv088
Guo M, Wang H, Potter SS, Whitsett JA, Xu Y (2015) SINCERA: a pipeline for single-cell RNA-Seq profiling analysis. PLoS Comput Biol 11:e1004575
Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M (2015) MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16:278
Kharchenko PV, Silberstein L, Scadden DT (2014) Bayesian approach to single-cell differential expression analysis. Nat Methods 11:740–742
Delmans M, Hemberg M (2016) Discrete distributional differential expression (D 3 E)-a tool for gene expression analysis of single-cell RNA-seq data. BMC Bioinf 17:110
Korthauer KD, Chu L-F, Newton MA, Li Y, Thomson J, Stewart R, Kendziorski C (2016) A statistical approach for identifying differential distributions in single-cell RNA-seq experiments. Genome Biol 17:222
Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32:381–386
Haghverdi L, Buettner F, Theis FJ (2015) Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31:2989–2998
Rizvi AH, Camara PG, Kandror EK, Roberts TJ, Schieren I, Maniatis T, Rabadan R (2017) Single-cell topological RNA-seq analysis reveals insights into cellular differentiation and development. Nat Biotechnol 35:551–560
Leng N, Chu L-F, Barry C, Li Y, Choi J, Li X, Jiang P, Stewart RM, Thomson JA, Kendziorski C (2015) Oscope identifies oscillatory genes in unsynchronized single-cell RNA-seq experiments. Nat Methods 12:947–950
Satija R, Farrell JA, Gennert D, Schier AF, Regev A (2015) Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33:495–502
Achim K, Pettit J-B, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, Marioni JC (2015) High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat Biotechnol 33:503–509
Halpern KB, Shenhav R, Matcovitch-Natan O, Tóth B, Lemze D, Golan M, Massasa EE, Baydatch S, Landen S, Moor AE (2017) Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542:352–356
Zhang B, Horvath S (2005) A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4:1128
Schissler AG, Li Q, Chen JL, Kenost C, Achour I, Billheimer DD, Li H, Piegorsch WW, Lussier YA (2016) Analysis of aggregated cell–cell statistical distances within pathways unveils therapeutic-resistance mechanisms in circulating tumor cells. Bioinformatics 32:i80–i89
Matsumoto H, Kiryu H, Furusawa C, Ko MS, Ko SB, Gouda N, Hayashi T, Nikaido ISCODE (2017) An efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 33:2314–2321btx194
Gokce O, Stanley GM, Treutlein B, Neff NF, Camp JG, Malenka RC, Rothwell PE, Fuccillo MV, Südhof TC, Quake SR (2016) Cellular taxonomy of the mouse striatum as revealed by single-cell RNA-seq. Cell Rep 16:1126–1137
Welch JD, Hu Y, Prins JF (2016) Robust detection of alternative splicing in a population of single cells. Nucleic Acids Res 44:e73
Deng Q, Ramsköld D, Reinius B, Sandberg R (2014) Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343:193–196
Reinius B, Mold JE, Ramsköld D, Deng Q, Johnsson P, Michaëlsson J, Frisén J, Sandberg R (2016) Analysis of allelic expression patterns in clonal somatic cells by single-cell RNA-seq. Nat Genet 48:1430–1435
Hong Y, Li Z, Zhang Q (2016) A circulating tumor cell cluster-based model for tumor metastasis (Hypothesis). Oncol Lett 12:4891–4895. https://doi.org/10.3892/ol.2016.5358
Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, Desai R, Fox DB, Brannigan BW, Trautwein J, Arora KS, Desai N, Dahl DM, Sequist LV, Smith MR, Kapur R, Wu CL, Shioda T, Ramaswamy S, Ting DT, Toner M, Maheswaran S, Haber DA (2015) RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 349:1351–1356. https://doi.org/10.1126/science.aab0917
Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, Gillespie SM, Dionne D, Luo CC, Ravichandran H, Mylvaganam R, Mount C, Onozato ML, Nahed BV, Wakimoto H, Curry WT, Iafrate AJ, Rivera MN, Frosch MP, Golub TR, Brastianos PK, Getz G, Patel AP, Monje M, Cahill DP, Rozenblatt-Rosen O, Louis DN, Bernstein BE, Regev A, Suva ML (2017) Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355. https://doi.org/10.1126/science.aai8478
Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin JR, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CG, Kazer SW, Gaillard A, Kolb KE, Villani AC, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jane-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA (2016) Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352:189–196. https://doi.org/10.1126/science.aad0501
Mbeunkui F, Johann DJ Jr (2009) Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemother Pharmacol 63:571–582. https://doi.org/10.1007/s00280-008-0881-9
Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, Liu Z, Dong M, Hu X, Ouyang W, Peng J, Zhang Z (2017) Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169:1342–1356. e1316. https://doi.org/10.1016/j.cell.2017.05.035
Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, Meinhof K, Chow A, Kim-Shulze S, Wolf A, Medaglia C, Li H, Rytlewski JA, Emerson RO, Solovyov A, Greenbaum BD, Sanders C, Vignali M, Beasley MB, Flores R, Gnjatic S, Pe’er D, Rahman A, Amit I, Merad M (2017) Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169:750–765., e717. https://doi.org/10.1016/j.cell.2017.04.014
Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, Ryu HS, Kim S, Lee JE, Park YH, Kan Z, Han W, Park WY (2017) Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 8:15081. https://doi.org/10.1038/ncomms15081
Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MAS, Moch H, van den Broek M, Beisel C, Stadler MB, Gedye C, Reis B, Pe’er D, Bodenmiller B (2017) An immune atlas of clear cell renal cell carcinoma. Cell 169:736–749. e718. https://doi.org/10.1016/j.cell.2017.04.016
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194:23–28
Zhao Z, Goldin L, Liu S, Wu L, Zhou W, Lou H, Yu Q, Tsang SX, Jiang M, Li F, McMaster M, Li Y, Lin X, Wang Z, Xu L, Marti G, Li G, Wu K, Yeager M, Yang H, Xu X, Chanock SJ, Li B, Hou Y, Caporaso N, Dean M (2016) Evolution of multiple cell clones over a 29-year period of a CLL patient. Nat Commun 7:13765. https://doi.org/10.1038/ncomms13765
Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M (2011) Tumour evolution inferred by single-cell sequencing. Nature 472:90–94. https://doi.org/10.1038/nature09807
Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, He W, Zeng L, Xing M, Wu R, Jiang H, Liu X, Cao D, Guo G, Hu X, Gui Y, Li Z, Xie W, Sun X, Shi M, Cai Z, Wang B, Zhong M, Li J, Lu Z, Gu N, Zhang X, Goodman L, Bolund L, Wang J, Yang H, Kristiansen K, Dean M, Li Y, Wang J (2012) Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell 148:886–895. https://doi.org/10.1016/j.cell.2012.02.025
Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, Li F, Wu K, Liang J, Shao D, Wu H, Ye X, Ye C, Wu R, Jian M, Chen Y, Xie W, Zhang R, Chen L, Liu X, Yao X, Zheng H, Yu C, Li Q, Gong Z, Mao M, Yang X, Yang L, Li J, Wang W, Lu Z, Gu N, Laurie G, Bolund L, Kristiansen K, Wang J, Yang H, Li Y, Zhang X, Wang J (2012) Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 148:873–885. https://doi.org/10.1016/j.cell.2012.02.028
Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S, Vijayaraghavan R, Wong J, Chen A, Sheng X, Kaper F, Shen R, Ronaghi M, Fan JB, Wang W, Chun J, Zhang K (2016) Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352:1586–1590. https://doi.org/10.1126/science.aaf1204
Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, Rolny C, Castelo-Branco G, Hjerling-Leffler J, Linnarsson S (2015) Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347:1138–1142. https://doi.org/10.1126/science.aaa1934
Regev A, Teichmann S, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, Clevers H, Deplancke B, Dunham I, Eberwine J, Eils R, Enard W, Farmer A, Fugger L, Gottgens B, Hacohen N, Haniffa M, Hemberg M, Kim SK, Klenerman P, Kriegstein A, Lein E, Linnarsson S, Lundeberg J, Majumder P, Marioni J, Merad M, Mhlanga M, Nawijn M, Netea M, Nolan G, Pe’er D, Philipakis A, Ponting CP, Quake SR, Reik W, Rozenblatt-Rosen O, Sanes JR, Satija R, Shumacher T, Shalek AK, Shapiro E, Sharma P, Shin J, Stegle O, Stratton M, Stubbington MJT, van Oudenaarden A, Wagner A, Watt FM, Weissman JS, Wold B, Xavier RJ, Yosef N (2017) The human cell atlas. bioRxiv. https://doi.org/10.1101/121202
Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJ, van Oudenaarden A (2016) A single-cell transcriptome atlas of the human pancreas. Cell Syst 3:385–394. e383. https://doi.org/10.1016/j.cels.2016.09.002
Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, Bertagnolli D, Goldy J, Shapovalova N, Parry S, Lee C, Smith K, Bernard A, Madisen L, Sunkin SM, Hawrylycz M, Koch C, Zeng H (2016) Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci 19:335–346. https://doi.org/10.1038/nn.4216
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344:1396–1401. https://doi.org/10.1126/science.1254257
Marusyk A, Almendro V, Polyak K (2012) Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 12:323–334. https://doi.org/10.1038/nrc3261
Heath JR, Ribas A, Mischel PS (2016) Single-cell analysis tools for drug discovery and development. Nat Rev Drug Discov 15:204–216. https://doi.org/10.1038/nrd.2015.16
Hood L (2013) Systems biology and p4 medicine: past, present, and future. Rambam Maimonides Med J 4:e0012. https://doi.org/10.5041/RMMJ.10112
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Huang, X., Liu, S., Wu, L., Jiang, M., Hou, Y. (2018). High Throughput Single Cell RNA Sequencing, Bioinformatics Analysis and Applications. In: Gu, J., Wang, X. (eds) Single Cell Biomedicine. Advances in Experimental Medicine and Biology, vol 1068. Springer, Singapore. https://doi.org/10.1007/978-981-13-0502-3_4
Download citation
DOI: https://doi.org/10.1007/978-981-13-0502-3_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-0501-6
Online ISBN: 978-981-13-0502-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)