
Abstract
The assembly and egress of herpes simplex virus (HSV) is a complicated multistage process that involves several different cellular compartments and the activity of many viral and cellular proteins. The process begins in the nucleus, with capsid assembly followed by genome packaging into the preformed capsids. The DNA-filled capsids (nucleocapsids) then exit the nucleus by a process of envelopment at the inner nuclear membrane followed by fusion with the outer nuclear membrane. In the cytoplasm nucleocapsids associate with tegument proteins, which form a complicated protein network that links the nucleocapsid to the cytoplasmic domains of viral envelope proteins. Nucleocapsids and associated tegument then undergo secondary envelopment at intracellular membranes originating from late secretory pathway and endosomal compartments. This leads to assembled virions in the lumen of large cytoplasmic vesicles, which are then transported to the cell periphery to fuse with the plasma membrane and release virus particles from the cell. The details of this multifaceted process are described in this chapter.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Capsid Assembly and Genome Packaging
The first stage of forming new HSV particles is the assembly of the icosahedral capsid. Like all herpesviruses, the HSV capsid is an approximately 125 nm diameter icosahedron with T = 16 symmetry (Schrag et al. 1989), composed of 162 capsomers connected by 320 triplexes (2 copies of VP23 and 1 copy of VP19C) (Newcomb et al. 1993; Okoye et al. 2006). The 162 capsomers include 150 hexons (6 copies of VP5), which make up the edges and faces of the icosahedron, and 11 of the 12 vertices are pentons (5 copies of VP5). The twelfth vertex is the portal complex, a dodecamer of pUL6 arranged in a ring structure, through which the genome is packaged during assembly and released during entry (Newcomb et al. 2001). In addition, 900 copies of the small capsid protein VP26 decorate the outer surface of the capsid, with one copy of VP26 on the tip of each VP5 in the 150 hexons. Furthermore, five copies of a heterodimer composed of pUL17 and pUL25, termed the capsid vertex-specific component (CVSC), associate with each penton. The CVSC is thought to be important for both capsid stability and association with the tegument (Thurlow et al. 2006; Toropova et al. 2011; Trus et al. 2007). One final viral protein component of the capsid is VP24, the protease that processes the scaffold during DNA encapsidation (Sheaffer et al. 2000; Stevenson et al. 1997).
Capsids initially assemble as a procapsid around a scaffold complex composed of ~1900 subunits of two related proteins: pUL26.5 that contains the scaffold core domain and pUL26 that has the viral protease (VP24) fused to the N-terminus of the scaffold core domain via a linker. Approximately 90% of the scaffold is composed of pUL26.5 and 10% is pUL26 (Aksyuk et al. 2015). Both pUL26.5 and pUL26 interact with the major capsid protein VP5 via their identical C-termini. The assembly is thought to initiate with the portal complex associated with scaffold proteins (Newcomb et al. 2005), followed by progressive addition of scaffold-bound VP5 together with preformed triplexes, which produces spherical procapsids containing a single portal complex (Newcomb et al. 1996, 2003; Spencer et al. 1998). Viral DNA, primarily in concatemeric form after synthesis by rolling-circle replication, is packaged into preformed procapsids via the pUL6 portal and requires the action of the terminase complex (pUL15-pUL28-pUL33) (Heming et al. 2014). The terminase complex interacts with packaging signals (pac sequences) in the terminal repeat region at the free end of newly synthesised viral DNA and drives ATP-dependent translocation of the viral genome into the procapsid. Once DNA packaging is complete, the terminase complex cleaves the concatemeric viral DNA at the next terminal repeat to separate the packaged single genome length of DNA from the rest of the concatemer (Tong and Stow 2010). As viral DNA begins to be packaged into the procapsid, the protease domain of pUL26 scaffold protein is activated, causing its autocatalytic release from the N-terminus of pUL26 to become VP24, the free protease protein. VP24 cleaves both pUL26 and pUL26.5 near their C-termini releasing the core scaffold domains, termed VP21 and VP22a, respectively, from their bound VP5. The majority of the cleaved scaffold protein products are released from the capsid providing the space for the viral genome, although at least some of the VP24 protease domain is retained inside the capsid (McClelland et al. 2002; Sheaffer et al. 2000). This whole process leads to large structural changes resulting in the rearrangement of the spherical procapsid into the stable icosahedral capsid containing the viral genome (often termed C-capsids or nucleocapsids) (Heymann et al. 2003; Roos et al. 2009). Two other forms of stable icosahedral capsids that lack DNA, A-capsids and B-capsids, are also produced during this process; A-capsids contain little or no scaffold protein, whereas B-capsids retain an inner shell of processed scaffold (Cardone et al. 2012a). Both A-capsids and B-capsids are thought to be dead-end products that result from defective or abortive DNA encapsidation, and these DNA-less capsids rarely exit the nucleus.
2 Nuclear Egress
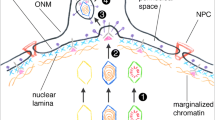
Upon completion of capsid assembly and genome packaging, the resulting nucleocapsids need to escape the confines of the nuclear envelope. The nuclear envelope is a formidable barrier, being composed of two phospholipid bilayers – the inner nuclear membrane facing the nucleoplasm and the outer nuclear membrane facing the cytoplasm. There are numerous pores within the nuclear envelope, where the inner nuclear membrane and outer nuclear membrane fuse, which are filled by nuclear pore complexes that tightly regulate the transport of cargo between the cytoplasm and nucleus. The size exclusion of these pores is typically around 39 nm diameter or less (Pante and Kann 2002) and is thus too small to accommodate herpesvirus capsids, which are ~125 nm. Instead, transport of nucleocapsids into the cytoplasm is achieved by budding of nucleocapsids at the inner nuclear membrane to form primary enveloped particles (also termed perinuclear virions) within the perinuclear space, followed by fusion of primary enveloped particles with the outer nuclear membrane to release nucleocapsids into the cytoplasm. While many details of this process are still unclear, much progress has been made recently in understanding this unusual mode of intracellular transport.
To gain access to the inner nuclear membrane, the underlying nuclear lamina must be penetrated. The nuclear lamina is a dense mesh of intermediate filament-type proteins (lamins) and associated proteins, which interacts with chromatin and aids the structural integrity of the nucleus. Local disruption of the nuclear lamina to enable nucleocapsid access to the inner nuclear membrane is facilitated through phosphorylation of lamins and associated proteins by viral and cellular kinases, including pUS3 and PKC isoforms (Bjerke and Roller 2006; Leach and Roller 2010).
The budding of nucleocapsids at the inner nuclear membrane is driven by the nuclear egress complex (NEC), a heterodimer of pUL31 and a tail-anchored membrane protein pUL34. The NEC recruits PKC isoforms (Park and Baines 2006) and is itself a target for phosphorylation by pUS3 (Kato et al. 2005), which facilitates the correct localisation of the NEC to the nuclear membrane (Reynolds et al. 2001). The other HSV protein kinase, pUL13, can also regulate the localisation of the NEC, either by phosphorylation of pUS3 or by a pUS3-independent mechanism (Kato et al. 2006).
The recruitment of capsids to the inner nuclear membrane involves the interaction of the NEC with pUL25, part of the heterodimeric CVSC present on the vertices of capsids. DNA-filled capsids (C-capsids/nucleocapsids) have higher levels of occupancy of the CVSC on their vertices than either A- or B-capsids (Newcomb et al. 2006; Sheaffer et al. 2001), providing a mechanism by which DNA-filled capsids can be selected for nuclear export (O’Hara et al. 2010; Yang and Baines 2011).
Recent structural studies have begun to uncover the molecular mechanisms by which the NEC mediates primary envelopment of herpesvirus capsids. The NEC oligomerises on the inner nuclear membrane to form a hexagonal scaffold that coats the inner surface of the budding membrane and links the membrane to the nucleocapsid (Bigalke and Heldwein 2017; Zeev-Ben-Mordehai et al. 2015). The NEC has an intrinsic activity to deform membranes and cause membrane scission, suggesting the NEC alone is sufficient for forming perinuclear enveloped virus particles (Hagen et al. 2015). However, other viral proteins may be involved in regulating nuclear egress, including pUL16 (Gao et al. 2017), pUL21 (Le Sage et al. 2013), pUL47 (Liu et al. 2014) and pUS3 (Reynolds et al. 2002), as well as the nonstructural proteins pUL24 (Lymberopoulos et al. 2011) and ICP22 (Maruzuru et al. 2014). One protein with a well-established, although enigmatic, role in nuclear egress is the viral kinase pUS3. Deletion of the US3 gene or introduction of an inactivating mutation in the kinase domain of pUS3 results in the accumulation of primary enveloped virions in the perinuclear space, often observed as bulges protruding into the nucleoplasm termed herniations (Reynolds et al. 2002; Ryckman and Roller 2004). This suggests a role for pUS3 kinase activity in regulating the fusion of primary enveloped virions with the outer nuclear membrane or dissociation of the nucleocapsid from the NEC (Newcomb et al. 2017). However, this appears to be a more facilitatory, non-essential function because US3 deletion viruses are viable and nucleocapsids are still able to gain access to the cytoplasm, undergo secondary envelopment and release infectious virions from cells. It is possible that pUL13, or host kinases, can at least partially compensate for loss of pUS3 function.
The precise composition of perinuclear virions is unknown, with much uncertainty about the presence of various tegument and envelope proteins. Regarding tegument proteins, immuno-electron microscopy studies have suggested that both pUS3 and VP16 are present in perinuclear virions (Naldinho-Souto et al. 2006; Reynolds et al. 2002), and proteomics analysis of partially purified perinuclear virions identified pUL49 as a component of these particles (Padula et al. 2009). However, for many of the viral proteins proposed to regulate nuclear egress, it is unclear if they become components of primary enveloped particles or indeed whether their roles in nucleocapsid transport across the nuclear envelope are direct or indirect. Recent cryo-electron microscopy studies of primary enveloped virions have also demonstrated limited space between the NEC and nucleocapsid, suggesting few tegument proteins are likely to be incorporated to a significant level during nuclear egress and the majority of tegument is acquired in the cytoplasm (Newcomb et al. 2017).
Whether there are any viral membrane proteins that are specifically incorporated into primary enveloped virions and what functional roles they play in nuclear egress is also unclear. Several viral membrane proteins localise to the nuclear envelope in infected cells and thus could be incorporated into perinuclear virions, including gB, gD, gH and gM (Baines et al. 2007; Farnsworth et al. 2007b; Wills et al. 2009). Given the need for perinuclear virions to fuse with the outer nuclear membrane, the presence of the viral entry proteins gB, gD and gH could indicate a potential role of these proteins in the outer nuclear membrane fusion event. Indeed, some evidence suggests gB is involved in HSV-1 nuclear egress, possibly in a redundant manner with gH, and that this activity of gB in nuclear egress is regulated by pUS3 (Farnsworth et al. 2007b; Wisner et al. 2009; Wright et al. 2009). However, gB deletion viruses still efficiently assemble and release virions from infected cells, albeit lacking gB, suggesting any role of this glycoprotein in nuclear egress is facilitatory rather than essential (Farnsworth et al. 2007b). Furthermore, in pseudorabies virus, a related alphaherpesvirus, deletion of gB and gH does not affect nuclear egress (Klupp et al. 2008).
Following de-envelopment, nucleocapsids detach from the NEC leaving the NEC proteins behind in the outer nuclear membrane in a process that may partially rely on pUS3 kinase activity (Newcomb et al. 2017; Reynolds et al. 2002). Once released, the nucleocapsid must recruit then tegument and undergo secondary envelopment to form an infectious virion.
3 Tegument Assembly
The tegument is a complex proteinaceous layer that connects the nucleocapsid to the viral envelope, which in HSV contains up to 24 different viral proteins (Table 2.1). As well as performing a structural role within the virion, the tegument is also a reservoir for proteins that modulate host cell function, such as the ubiquitin ligase ICP0 and the virion host shut-off protein (Vhs/pUL41), which are important for antagonising antiviral host responses (Boutell and Everett 2013; Smiley 2004). Unlike the icosahedral herpesvirus capsid, the structure of the tegument is rather poorly defined; the lack of symmetry within the tegument prevents high-resolution single-particle structural analysis by cryo-electron microscopy. Tegument proteins are often broadly subdivided into ‘inner’ and ‘outer’ tegument, with inner tegument proteins more tightly associated with the nucleocapsid and outer tegument proteins weakly associated with the nucleocapsid and/or associated with the inner surface of the envelope. These definitions mainly come from biochemical experiments investigating how labile the association of tegument proteins with nucleocapsids is to increasing salt concentration, following disruption of the viral enveloped with detergent. Therefore, such designations do not necessarily provide information of the structural organisation of these proteins within virions. Recently, some details of tegument organisation have begun to be uncovered by modern techniques in fluorescence microscopy analysis of single virus particles (Bohannon et al. 2013; Laine et al. 2015).
The tegument protein that is most tightly associated to nucleocapsids is pUL36 (also termed VP1/2), the C-terminal domain of which has been shown to interact with pUL25, part of the heterodimeric CVSC present on nucleocapsid pentons (Coller et al. 2007). Single-particle analysis of nucleocapsids obtained from purified virions that have been stripped of their envelope and all tegument proteins except pUL36 identified extra density protruding from capsid vertices, suggesting that at least part of pUL36, most likely its C-terminus, is the one tegument protein that does display some icosahedral symmetry (Cardone et al. 2012b). More recently, it has been shown that the presence of pUL36 is necessary for the CVSC to form suggesting that pUL36 residues may contribute to the observed CVSC density in cryo-EM reconstructions of nucleocapsids or that pUL36 is required to stabilise the structure of the pUL17 and pUL25 heterodimer (Fan et al. 2015).
As well as being tightly associated with nucleocapsids, pUL36 is one of the few tegument proteins, along with pUL37 and VP16, that are essential for HSV assembly; loss of functional pUL36 leads to accumulation of non-enveloped nucleocapsids in the cytoplasm, suggesting a failure of tegument to associate with nucleocapsids in the absence of pUL36 (Desai 2000; Roberts et al. 2009). Furthermore, pUL36 is the largest protein in herpesviruses, being >3000 amino acids in HSV, and has been shown to interact with the two other essential assembly proteins pUL37 and VP16 (Mijatov et al. 2007; Svobodova et al. 2012). This has led to the logical suggestion that pUL36 could serve as a platform or central organiser for subsequent assembly of the rest of the tegument. In addition to pUL36, pUL37 and pUS3 are usually classified as inner tegument proteins, whereas most of the other tegument proteins are generally considered outer tegument proteins. However, the association properties of most tegument proteins with nucleocapsids remain undefined, with investigations hampered due to low copy numbers within virions as well as a lack of suitable detection reagents for many tegument proteins.
The location where the tegument first begins to associate with nucleocapsids during virion assembly is unclear, with disagreement in the literature regarding the association of tegument proteins with capsids in the nucleus. Given the tight association of the inner tegument pUL36 with nucleocapsids and the importance of pUL36 for association of pUL37 and VP16 with nucleocapsids and subsequent virion assembly, it is reasonable to suggest pUL36 will be one of the first tegument proteins to interact with nucleocapsids during virion morphogenesis. However, there is evidence both for and against pUL36 associating with capsids in the nucleus (Bucks et al. 2007; Fan et al. 2015; Henaff et al. 2013; Radtke et al. 2010). Furthermore, it doesn’t appear that pUL36 has an important role in the nuclear egress stage of the assembly pathway because deletion of UL36 does not prevent transport of nucleocapsids from the nucleus into the cytoplasm (Desai 2000; Roberts et al. 2009). The observations that some tegument proteins are important for efficient nuclear egress, as described above, could suggest association of these tegument proteins with nucleocapsids before or during primary envelopment, and indeed pUS3 has been detected in primary enveloped virions (Henaff et al. 2013; Reynolds et al. 2002). However, it is also possible tegument proteins could function in a regulatory manner during nuclear egress but not physically associate with nucleocapsids at this stage and then become incorporated into assembling virions by interaction with nucleocapsid and/or other tegument or envelope proteins in the cytoplasm. An example of the complexities in interpreting when tegument proteins become incorporated into assembling virions is the major tegument protein VP16. It is well established that VP16 is imported into the nucleus for its role during immediate-early viral gene expression (Campbell et al. 1984), VP16 has been observed in perinuclear virions by immuno-EM studies (Naldinho-Souto et al. 2006) and VP16 interacts with the inner tegument protein pUL36 (Svobodova et al. 2012), suggesting potential association with capsids in the nucleus. However, VP16-negative capsids can be readily observed in the cytoplasm of infected cells, VP16 deletion inhibits virion assembly but does not appear to affect nuclear egress, and VP16 interacts with the cytoplasmic domain of gH as well as several other ‘outer’ tegument proteins (pUL41, pUL46, pUL47 and VP22), suggesting VP16 incorporation into virions occurs in the cytoplasm (Elliott et al. 1995; Gross et al. 2003; Mossman et al. 2000; Smibert et al. 1994; Svobodova et al. 2012; Vittone et al. 2005). It should also be born in mind that the tegument assembly process could be somewhat flexible, whereby a few copies of some tegument proteins can associate with nucleocapsids in the nucleus and be carried across the nuclear envelope, but then further copies of these tegument proteins assemble onto nucleocapsids in the cytoplasm. Alternatively, nuclear-localised tegument proteins may transiently associate with nucleocapsid prior to or during nuclear egress and then dissociate once the nucleocapsid reaches the cytoplasm before reacquisition later during virion assembly. Future development of imaging technologies that allow direct observation of the dynamics of virion assembly at the single-particle level will hopefully shed light on this topic.
Regardless of whether individual tegument proteins can or do associate with nucleocapsids before they exit the nucleus, it is clear that the majority of the tegument assemble s in the cytoplasm. To form the complex tegument layer, a network of protein-protein interactions with significant redundancy is thought to occur, including tegument-tegument, tegument-capsid and tegument-envelope interactions (Lee et al. 2008; Vittone et al. 2005). The inner tegument proteins pUL36, pUL37 and pUS3 are presumably recruited to nucleocapsids before the outer tegument proteins, many of which may assemble during the secondary envelopment process by virtue of interacting with the cytoplasmic domains of viral envelope proteins. Several tegument proteins have been shown to be important for efficient virion assembly, and so they may facilitate the formation of the complex tegument layer, although as mentioned above only three, pUL36, pUL37 and VP16 appear to be essential for virion assembly, suggesting reasonable flexibility within the assembly process that can compensate for the loss of one or more ‘non-essential’ components.
VP16, pUL47 and pUL49 are the most prevalent proteins in the tegument, with copy numbers estimated to be ca. 500–1500 per virion for each of these proteins (Clarke et al. 2007; Newcomb et al. 2012). These proteins may be central organisers of the tegument structure: VP16 has been shown to interact with pUL41 (Vhs), pUL46, pUL47 and pUL49, as well as pUL36 and the cytoplasmic domain of gH (Elliott et al. 1995; Gross et al. 2003; Smibert et al. 1994; Svobodova et al. 2012; Vittone et al. 2005); pUL47 also interacts with pUL17 providing another link between the tegument and nucleocapsids (Scholtes et al. 2010); pUL49 also interacts with pUL16, ICP0 and the cytoplasmic domains of gD, gE and gM (Farnsworth et al. 2007a; Maringer et al. 2012; Starkey et al. 2014).
In addition to the essential pUL36 and pU37, there are six other tegument proteins that are conserved throughout the herpesvirus family, and while not ‘essential’ these tegument proteins are also important for virion assembly. Firstly, there is pUL11, pUL16 and pUL21, which have been shown to form a tripartite complex that associates with membranes via the lipid anchors present on pUL11 and through interaction with the cytoplasmic domain of gE (Han et al. 2012). There is also evidence that both pUL16 and pUL21 interact with capsids, suggesting the pUL11-pUL16-pUL21 complex can directly connect the envelope with the nucleocapsid (de Wind et al. 1992; Meckes and Wills 2007). Secondly, there are pUL7, pUL14 and pUL51 which may also form a complex. pUL7 and pUL51 have recently been shown to form a complex through direct protein-protein interaction, and pUL14 has also been shown to interact with pUL51 (Albecka et al. 2017; Oda et al. 2016; Roller and Fetters 2015). As yet it is unclear if a tripartite complex of pUL7-pUL14-pUL51 forms or if there are independent pUL7-pUL51 and pUL14-pUL51 complexes. Deletion of each of these three proteins leads to defects in cytoplasmic virion assembly, and the stability of pUL7 and pUL51 relies on each other (Albecka et al. 2017; Oda et al. 2016). Similar to pUL11, pUL51 is associated with membranes via a lipid anchor providing additional links between the tegument and envelope. Therefore, it appears there are at least two independent protein complexes that can link the envelope to the underlying tegument and the nucleocapsid that are conserved throughout the herpesvirus family.
While many interactions between tegument proteins have been identified, it is important to note that such an extensive and seemingly redundant network of interactions between these proteins makes it problematic to investigate the precise roles of individual components or interactions during virion assembly. It is often difficult to know whether identified interactions are direct or indirect and whether they occur within the virion structure or during other, non-assembly activities of these multifunctional virus proteins. Elucidating the molecular details of tegument protein structures, both in isolation and in complex with each other, will be needed to shed further light on these complex assembly events.
4 Secondary Envelopment
The final stage of assembling mature HSV particles is secondary envelopment, sometimes referred to as final envelopment, the process by which nucleocapsids with a full complement of tegument proteins are encased within a lipid bilayer containing all the viral envelope proteins. This occurs at cytoplasmic membranes resulting in HSV particles inside the lumen of cytoplasmic compartments. To orchestrate this process, a complex series of interactions must occur between viral capsid, tegument and envelope proteins, as well as with cellular membranes and host proteins involved in regulating membrane modulation and transport.
The cellular compartment from which the final envelope of HSV is derived is somewhat unclear. It often stated that the trans-Golgi network (TGN), a major sorting station for membrane proteins of the secretory and endocytic pathways, is the site of HSV secondary envelopment. Evidence for this comes from the observation of co-localisation of several viral envelope glycoproteins with markers of the TGN (Beitia Ortiz de Zarate et al. 2004; Crump et al. 2004; Foster et al. 2004b; McMillan and Johnson 2001). Furthermore, it has been shown that appending localisation signals from cellular TGN resident proteins onto HSV-1 glycoproteins leads to incorporation into virions (Whiteley et al. 1999), and inhibitors that perturb Golgi/TGN function, such as brefeldin A, block HSV-1 assembly (Cheung et al. 1991; Koyama and Uchida 1994), supporting the notion of envelopment of virions by TGN membrane. However, there is also evidence for endosomes being the sites of HSV-1 secondary envelopment. Electron microscopy studies have clearly shown the presence of endocytic tracers within membrane compartments that are wrapping cytoplasmic nucleocapsids (Hollinshead et al. 2012). Furthermore, blocking the function of specific Rab GTPases that are associated with endosomal trafficking inhibits HSV-1 assembly (Johns et al. 2011; Zenner et al. 2011), and inhibition of endocytosis prevents the incorporation of viral glycoproteins into virions (Albecka et al. 2016). These data point to endosomal compartments being a major source of membrane during HSV secondary envelopment. However, because of the highly dynamic and fluid nature of the secretory and endocytic pathways in cells, it can be difficult to accurately interpret cellular membrane compartment identity, particularly in infected cells where the cytopathic effects of HSV infection are known to perturb membrane traffic and cellular organelle structure (Henaff et al. 2012). Furthermore, the majority of cellular proteins that are defined as markers of particular organelles frequently undergo rapid transport between various secretory and endocytic compartments, and so in the context of HSV infection defining the identity of a membrane by the presence of such cellular markers can be fraught with difficulties. It must also be considered that the secondary envelopment of HSV could involve membranes that originate from more than one cellular compartment, including TGN and endosomes. Given the size of HSV particles, several vesicles containing viral envelope proteins may need to fuse to provide sufficient membrane in order to wrap the large nucleocapsid/tegument complex that makes up the internal structure of the virus particle.
Despite the uncertainties about secondary envelopment compartment identity, there is little doubt that this occurs at cellular membranes that are derived from late secretory pathway (e.g. the TGN) and/or endosomal pathway compartments, and not at early secretory pathway membranes such as the endoplasmic reticulum (ER) or Golgi apparatus. Artificially targeting viral envelope proteins to the ER prevents their incorporation into virions (Browne et al. 1996; Whiteley et al. 1999), and blocking ER-Golgi transport inhibits virus assembly and leads to accumulation of viral envelope proteins in the ER (Zenner et al. 2011).
As with the rest of the virion structure, the envelope of HSV is also highly complex, containing up to 16 different viral transmembrane proteins (Table 2.1). Therefore, all of these different membrane proteins need to be accumulated in the appropriate cellular compartment(s) so that they can be incorporated into mature virions. Due to the highly dynamic and interconnected nature of the late secretory and endocytic pathways, cellular membrane proteins are often localised to discrete compartments through an active retrieval mechanism, whereby proteins that leave the ‘home’ compartment are recycled back through vesicle transport. Therefore, it is of little surprise that many viral membrane proteins have adopted a similar mechanism by encoding targeting motifs in their cytoplasmic domains that mimic equivalent cellular motifs for packaging into transport vesicles. For example, tyrosine-based targeting motifs in glycoprotein B (gB) and glycoprotein E (gE) have been shown to direct endocytosis and intracellular targeting of these viral envelope proteins (Alconada et al. 1999; Beitia Ortiz de Zarate et al. 2004). However, not all HSV envelope proteins possess recognised targeting motifs, including the essential entry proteins gD and gH-gL. These envelope proteins appear to rely on interaction with other HSV proteins including gM and the gK-pUL20 complex to mediate their endocytosis and correct localisation to intracellular assembly compartments during infection (Crump et al. 2004; Lau and Crump 2015; Ren et al. 2012).
To help drive efficient virion assembly, several interactions between the tegument and envelope are thought to occur in a co-operative and partially redundant manner. The redundant nature of these envelope-tegument interactions is demonstrated by the observations that single deletion of gB, gD or gE results in little or no attenuation of secondary envelopment, whereas the combined deletion of gB and gD or gD and gE causes a dramatic inhibition of secondary envelopment leading to the accumulation of large aggregates of non-enveloped cytoplasmic capsids (Farnsworth et al. 2003; Johnson et al. 2011). Similar observations have been made for gM and the lipid-anchored tegument protein pUL11, with only minor defects in assembly caused by the loss of either protein individually, whereas deletion of both gM and pUL11 causes a profound inhibition of secondary envelopment (Leege et al. 2009). These observations support the notion that numerous interactions between envelope proteins and the underlying tegument occur to facilitate secondary envelopment, although loss of some of these interactions can be tolerated by the virus.
Two envelope proteins that appear to be particularly important for secondary envelopment are gK and pUL20. These multifunctional membrane proteins form a complex, and their correct subcellular localisation is reliant on each other (Foster et al. 2004b). Loss of either gK or pUL20 leads to significant defects in secondary envelopment (Foster et al. 2004a; Jayachandra et al. 1997; Melancon et al. 2004). The gK-pUL20 complex has been shown to interact with the pUL37 tegument protein as well as being important for the subcellular localisation of gD and gH-gL suggesting important functions of the gK-pUL20 complex during secondary envelopment in organising viral envelope proteins and mediating interactions with the tegument (Jambunathan et al. 2014; Lau and Crump 2015).
Consistent with the idea of a complex and redundant series of tegument-glycoprotein interactions helping to drive secondary envelopment, several tegument proteins have been reported to interact with the cytoplasmic domains of envelope proteins; VP16 with gH (Gross et al. 2003); VP22 with gD, gE and gM (Farnsworth et al. 2007a; Maringer et al. 2012); pUL11 with gD and gE (Farnsworth et al. 2007a; Han et al. 2011); and pUL37 with gK (Jambunathan et al. 2014). In fact, envelope-tegument interactions appear to be sufficient to drive secondary envelopment because this process can occur in the absence of nucleocapsids, forming the so-called light particles that contain most, if not all, tegument and envelope proteins but lack a nucleocapsid (Rixon et al. 1992; Szilagyi and Cunningham 1991).
The final stage of secondary envelopment is membrane scission to separate the virus from the host cell membrane, giving rise to a virion contained within the lumen of a vesicle. Similar to many enveloped viruses, HSV utilises the membrane scission activity of the cellular endosomal sorting complex required for transport (ESCRT) machinery for this final stage of assembly (Crump et al. 2007; Pawliczek and Crump 2009). The ESCRT machinery is a series of protein complexes that are normally involved in remodelling host membranes with the same topology as virus budding, for example, budding of material from the cytoplasm into the lumen of multivesicular bodies (Henne et al. 2011). Currently it is unclear how HSV recruits and regulates the ESCRT machinery at sites of secondary envelopment. Other viruses are known to recruit ESCRT complexes via their matrix proteins (Votteler and Sundquist 2013), and so tegument protein(s) appear the most likely candidates to directly or indirectly interact with ESCRT proteins. Indeed HSV-1 pUL36 has been shown to interact with the ESCRT-I subunit TSG101 (Calistri et al. 2015). However, TSG101 does not appear to be essential for HSV-1 assembly as siRNA depletion of this cellular protein does not inhibit virus replication (Pawliczek and Crump 2009). Given the complexity and redundancy within viral protein interactions during secondary envelopment, recruitment of the ESCRT machinery could be mediated by several viral proteins in a similarly redundant fashion, providing more than one mechanism to engage this important cellular machinery.
5 Virion Transport and Release
The completion of secondary envelopment results in virions being contained within large intracellular vesicles. To release viruses to the extracellular environment, these virion-containing vesicles need to be transported to the cell periphery and fuse with the plasma membrane. Currently there is little understanding of which cellular pathways are used during virus release and how they are controlled, although undoubtedly many host regulators of secretion are involved.
Given their size and the distances involved, it is likely that most if not all virion-containing vesicles require transport along microtubules via the activity of plus-end-directed kinesin motor proteins to travel from sites of secondary envelopment to the cell periphery. This will be particularly important upon reactivation of HSV from latency due to the long distances between the cell body and axon termini in neurons. There is some disagreement in the literature about the nature of virus particles that are transported along the axons during virus egress. Two extreme models are (1) HSV particles that undergo secondary envelopment in the cell body of neurons and are then transported along the axons as mature virions in large transport vesicles and (2) nucleocapsids and membrane compartments containing envelope proteins that are transported separately along the axons and then secondary envelopment occurs at axon termini (Cunningham et al. 2013; Kratchmarov et al. 2012; Taylor and Enquist 2015). There is evidence for both these models, although it is also possible that a combination of different types of transport occurs, with fully assembled virions in transport vesicles, partially enveloped virions and membrane-less nucleocapsids all capable of being transported along the axons. Despite these uncertainties, there can be little doubt that kinesin motors must be recruited and activated to enable efficient virus egress. Kinesin-1, also known as conventional kinesin, has been shown to interact with the envelope protein pUS9 and with the tegument protein pUS11, giving two potential mechanisms for recruitment and regulation of this microtubule motor (Diefenbach et al. 2015, 2002). Interestingly pseudorabies virus (PRV) appears to recruit kinesin-3 via the combined activities of pUS9 and the gE-gI heterodimer (Kratchmarov et al. 2013). In HSV-1, deletion of pUS9 and gE has also been shown to inhibit egress of virions from the cell body into the axons in neuronal cells, lending weight to the hypothesis that pUS9 and gE-gI promote microtubule-based transport during virus egress, although defects in secondary envelopment are also observed in the absence of pUS9 and gE (DuRaine et al. 2017). Furthermore, the lack of pUS9 does not appear to affect HSV egress in epithelial cells, suggesting different mechanisms are required for virion transport to the cell periphery in neuronal and non-neuronal cells.
The gE-gI complex is well established as being important for cell-to-cell spread of HSV in epithelial cells. Loss of gE-gI function causes a substantial decrease in the spread of infection between cells in monolayer cultures, which is thought to be due to reduced targeting of virus secretion to cell junctions (Dingwell et al. 1994; Dingwell and Johnson 1998; Johnson et al. 2001). The targeting of virus secretion to cell junctions is thought to rely on the activity of specific sorting signals in the cytoplasmic domains of both gE and gI (Farnsworth and Johnson 2006; McMillan and Johnson 2001). However, whether this is related to kinesin motor activity or interactions with some other cellular transport regulators is unclear.
Upon reaching the cell periphery, the large virion-containing vesicles will encounter cortical actin, a dense mesh of cytoskeleton underlying the plasma membrane. This could pose a significant barrier to secretion of HSV from infected cells, by restricting access of the large virion-containing transport vesicles to the plasma membrane. A similar problem is faced by large cellular secretory vesicles, such as melanosomes, and is overcome through the action actin motors such as Myosin Va. HSV may well utilise a similar mechanism; Myosin Va has been shown to be important for efficient HSV-1 secretion from epithelial cells (Roberts and Baines 2010), although the details of how this actin-based motor is recruited or regulated by the virus is unclear.
The final stage of HSV egress is the fusion of the virion-containing transport vesicle with the plasma membrane to release the newly synthesised virus from the producing cell. Such vesicle fusion events within cells rely on the action of several proteins including Rab GTPases, membrane tethering complexes and SNARE fusion proteins. There are many different pathways for secretion of proteins from cells that are regulated by different subsets of membrane traffic regulators, and it is currently unclear which cellular Rab, tether or SNARE proteins are involved in HSV secretion. Some Rab proteins, namely, Rab3A, Rab6A, Rab8A and Rab11A, as well as the SNARE protein SNAP-25, have been shown to localise to vesicles containing virus particles or viral tegument and glycoproteins (Hogue et al. 2014; Miranda-Saksena et al. 2009), although whether they have any functional role in HSV egress is unknown.
It is conceivable that secretion of the newly formed viruses is controlled entirely by endogenous cellular factors that are normally recruited to the membranes that make up the secondary envelopment compartments. However, it seems more likely that these processes are co-opted and controlled by the virus. Viral proteins that recruit or regulate the transport, docking and fusion of virion-containing vesicles would need to be present on the cytoplasm-facing domain of such vesicles. The most obvious candidates for recruiting the necessary host cell factors are viral envelope proteins and membrane-associated tegument proteins that could remain on the vesicle membrane, in addition to being incorporated into virions. Both pUS9 and gE-gI are good candidates to be present on the vesicle membrane as this would position their cytoplasmic domains to be available for recruiting and regulating host trafficking proteins such as kinesins. The tegument protein pUL51, which is membrane-associated due to a lipid anchor modification, has been shown to be important for virus cell-to-cell spread and so is also a good candidate for being retained on virion-containing exocytic vesicle membranes (Albecka et al. 2017; Roller et al. 2014). However, which specific subset of viral proteins remain on the cytoplasmic surface of the vesicle membranes, how they are retained or recruited and how they function to control transport, docking and fusion of these large virion-containing vesicles with the plasma membrane remain to be discovered.
6 Summary
As described in this chapter, the assembly and egress of herpesviruses is a complex multistage process that requires the co-ordinated activities of numerous viral and cellular factors. While we know many details about the structure and assembly of the capsid and the components of mature virions, there is still much to discover. Future research to analyse the process of HSV assembly at the single-particle level in real time and to uncover the detailed mechanisms of host factor involvement at specific stages of virus morphogenesis will be important to shed new light on this fascinating subject.
References
Aksyuk AA, Newcomb WW, Cheng N, Winkler DC, Fontana J, Heymann JB, Steven AC (2015) Subassemblies and asymmetry in assembly of herpes simplex virus procapsid. MBio 6(5):e01525–e01515. https://doi.org/10.1128/mBio.01525-15
Albecka A, Laine RF, Janssen AF, Kaminski CF, Crump CM (2016) HSV-1 glycoproteins are delivered to virus assembly sites through dynamin-dependent endocytosis. Traffic 17(1):21–39. https://doi.org/10.1111/tra.12340
Albecka A, Owen DJ, Ivanova L, Brun J, Liman R, Davies L, Ahmed MF, Colaco S, Hollinshead M, Graham SC, Crump CM (2017) Dual function of the pUL7-pUL51 tegument protein complex in herpes simplex virus 1 infection. J Virol 91(2). https://doi.org/10.1128/JVI.02196-16
Alconada A, Bauer U, Sodeik B, Hoflack B (1999) Intracellular traffic of herpes simplex virus glycoprotein gE: characterization of the sorting signals required for its trans-Golgi network localization. J Virol 73(1):377–387
Baines JD, Wills E, Jacob RJ, Pennington J, Roizman B (2007) Glycoprotein M of herpes simplex virus 1 is incorporated into virions during budding at the inner nuclear membrane. J Virol 81(2):800–812. https://doi.org/10.1128/JVI.01756-06
Beitia Ortiz de Zarate I, Kaelin K, Rozenberg F (2004) Effects of mutations in the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B on intracellular transport and infectivity. J Virol 78(3):1540–1551
Bigalke JM, Heldwein EE (2017) Have NEC coat, will travel: structural basis of membrane budding during nuclear Egress in herpesviruses. Adv Virus Res 97:107–141. https://doi.org/10.1016/bs.aivir.2016.07.002
Bjerke SL, Roller RJ (2006) Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 347(2):261–276. https://doi.org/10.1016/j.virol.2005.11.053
Bohannon KP, Jun Y, Gross SP, Smith GA (2013) Differential protein partitioning within the herpesvirus tegument and envelope underlies a complex and variable virion architecture. Proc Natl Acad Sci U S A 110(17):E1613–E1620. https://doi.org/10.1073/pnas.1221896110
Boutell C, Everett RD (2013) Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94(Pt 3):465–481. https://doi.org/10.1099/vir.0.048900-0
Browne H, Bell S, Minson T, Wilson DW (1996) An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: evidence for reenvelopment during egress. J Virol 70(7):4311–4316
Bucks MA, O’Regan KJ, Murphy MA, Wills JW, Courtney RJ (2007) Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361 (2):316–324. S0042-6822(06)00880-4 [pii] https://doi.org/10.1016/j.virol.2006.11.031
Calistri A, Munegato D, Toffoletto M, Celestino M, Franchin E, Comin A, Sartori E, Salata C, Parolin C, Palu G (2015) Functional interaction between the ESCRT-I component TSG101 and the HSV-1 tegument ubiquitin specific protease. J Cell Physiol 230(8):1794–1806. https://doi.org/10.1002/jcp.24890
Campbell ME, Palfreyman JW, Preston CM (1984) Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J Mol Biol 180(1):1–19
Cardone G, Heymann JB, Cheng N, Trus BL, Steven AC (2012a) Procapsid assembly, maturation, nuclear exit: dynamic steps in the production of infectious herpesvirions. Adv Exp Med Biol 726:423–439. https://doi.org/10.1007/978-1-4614-0980-9_19
Cardone G, Newcomb WW, Cheng N, Wingfield PT, Trus BL, Brown JC, Steven AC (2012b) The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J Virol 86(8):4058–4064. https://doi.org/10.1128/JVI.00012-12
Cheung P, Banfield BW, Tufaro F (1991) Brefeldin A arrests the maturation and egress of herpes simplex virus particles during infection. J Virol 65(4):1893–1904
Clarke RW, Monnier N, Li H, Zhou D, Browne H, Klenerman D (2007) Two-color fluorescence analysis of individual virions determines the distribution of the copy number of proteins in herpes simplex virus particles. Biophys J 93(4):1329–1337. https://doi.org/10.1529/biophysj.107.106351
Coller KE, Lee JI, Ueda A, Smith GA (2007) The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J Virol 81 (21):11790–11797. JVI.01113-07 [pii] https://doi.org/10.1128/JVI.01113-07
Crump CM, Bruun B, Bell S, Pomeranz LE, Minson T, Browne HM (2004) Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J Gen Virol 85(Pt 12):3517–3527. https://doi.org/10.1099/vir.0.80361-0
Crump CM, Yates C, Minson T (2007) Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J Virol 81 (14):7380–7387. JVI.00222-07 [pii] https://doi.org/10.1128/JVI.00222-07
Cunningham A, Miranda-Saksena M, Diefenbach R, Johnson D (2013) Letter in response to: making the case: married versus separate models of alphaherpes virus anterograde transport in axons. Rev Med Virol 23(6):414–418. https://doi.org/10.1002/rmv.1760
de Wind N, Wagenaar F, Pol J, Kimman T, Berns A (1992) The pseudorabies virus homology of the herpes simplex virus UL21 gene product is a capsid protein which is involved in capsid maturation. J Virol 66(12):7096–7103
Desai PJ (2000) A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol 74(24):11608–11618
Diefenbach RJ, Miranda-Saksena M, Diefenbach E, Holland DJ, Boadle RA, Armati PJ, Cunningham AL (2002) Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J Virol 76(7):3282–3291
Diefenbach RJ, Davis A, Miranda-Saksena M, Fernandez MA, Kelly BJ, Jones CA, LaVail JH, Xue J, Lai J, Cunningham AL (2015) The basic domain of herpes simplex virus 1 pUS9 recruits Kinesin-1 to facilitate egress from neurons. J Virol 90(4):2102–2111. https://doi.org/10.1128/JVI.03041-15
Dingwell KS, Johnson DC (1998) The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J Virol 72(11):8933–8942
Dingwell KS, Brunetti CR, Hendricks RL, Tang Q, Tang M, Rainbow AJ, Johnson DC (1994) Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J Virol 68(2):834–845
DuRaine G, Wisner TW, Howard P, Williams M, Johnson DC (2017) Herpes simplex virus gE/gI and US9 promote both envelopment and sorting of virus particles in the cytoplasm of neurons, two processes that precede anterograde transport in axons. J Virol 91(11). https://doi.org/10.1128/JVI.00050-17
Elliott G, Mouzakitis G, O'Hare P (1995) VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J Virol 69(12):7932–7941
Fan WH, Roberts AP, McElwee M, Bhella D, Rixon FJ, Lauder R (2015) The large tegument protein pUL36 is essential for formation of the capsid vertex-specific component at the capsid-tegument interface of herpes simplex virus 1. J Virol 89(3):1502–1511. https://doi.org/10.1128/JVI.02887-14
Farnsworth A, Johnson DC (2006) Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J Virol 80(7):3167–3179. https://doi.org/10.1128/JVI.80.7.3167-3179.2006
Farnsworth A, Goldsmith K, Johnson DC (2003) Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J Virol 77(15):8481–8494
Farnsworth A, Wisner TW, Johnson DC (2007a) Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J Virol 81 (1):319–331. JVI.01842-06 [pii] https://doi.org/10.1128/JVI.01842-06
Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC (2007b) Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104(24):10187–10192. https://doi.org/10.1073/pnas.0703790104
Foster TP, Melancon JM, Baines JD, Kousoulas KG (2004a) The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J Virol 78(10):5347–5357
Foster TP, Melancon JM, Olivier TL, Kousoulas KG (2004b) Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J Virol 78(23):13262–13277. https://doi.org/10.1128/JVI.78.23.13262-13277.2004
Gao J, Hay TJM, Banfield BW (2017) The product of the herpes simplex virus 2 UL16 gene is critical for the egress of capsids from the nuclei of infected cells. J Virol 91(10). https://doi.org/10.1128/JVI.00350-17
Gross ST, Harley CA, Wilson DW (2003) The cytoplasmic tail of herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 317(1):1–12
Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bauerlein FJ, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC, Grunewald K (2015) Structural basis of vesicle formation at the inner nuclear membrane. Cell 163(7):1692–1701. https://doi.org/10.1016/j.cell.2015.11.029
Han J, Chadha P, Meckes DG Jr, Baird NL, Wills JW (2011) Interaction and interdependent packaging of tegument protein UL11 and glycoprotein e of herpes simplex virus. J Virol 85(18):9437–9446. https://doi.org/10.1128/JVI.05207-11
Han J, Chadha P, Starkey JL, Wills JW (2012) Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc Natl Acad Sci U S A 109(48):19798–19803. https://doi.org/10.1073/pnas.1212900109
Heming JD, Huffman JB, Jones LM, Homa FL (2014) Isolation and characterization of the herpes simplex virus 1 terminase complex. J Virol 88(1):225–236. https://doi.org/10.1128/JVI.02632-13
Henaff D, Radtke K, Lippe R (2012) Herpesviruses exploit several host compartments for envelopment. Traffic 13(11):1443–1449. https://doi.org/10.1111/j.1600-0854.2012.01399.x
Henaff D, Remillard-Labrosse G, Loret S, Lippe R (2013) Analysis of the early steps of herpes simplex virus 1 capsid tegumentation. J Virol 87(9):4895–4906. https://doi.org/10.1128/JVI.03292-12
Henne WM, Buchkovich NJ, Emr SD (2011) The ESCRT pathway. Dev Cell 21(1):77–91. https://doi.org/10.1016/j.devcel.2011.05.015
Heymann JB, Cheng N, Newcomb WW, Trus BL, Brown JC, Steven AC (2003) Dynamics of herpes simplex virus capsid maturation visualized by time-lapse cryo-electron microscopy. Nat Struct Biol 10(5):334–341. https://doi.org/10.1038/nsb922
Hogue IB, Bosse JB, Hu JR, Thiberge SY, Enquist LW (2014) Cellular mechanisms of alpha herpesvirus egress: live cell fluorescence microscopy of pseudorabies virus exocytosis. PLoS Pathog 10(12):e1004535. https://doi.org/10.1371/journal.ppat.1004535
Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, Elliott G (2012) Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J 31(21):4204–4220. https://doi.org/10.1038/emboj.2012.262
Jambunathan N, Chouljenko D, Desai P, Charles AS, Subramanian R, Chouljenko VN, Kousoulas KG (2014) Herpes simplex virus 1 protein UL37 interacts with viral glycoprotein gK and membrane protein UL20 and functions in cytoplasmic virion envelopment. J Virol 88(11):5927–5935. https://doi.org/10.1128/JVI.00278-14
Jayachandra S, Baghian A, Kousoulas KG (1997) Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J Virol 71(7):5012–5024
Johns HL, Gonzalez-Lopez C, Sayers C, Hollinshead M, Elliott G (2011) A role for human Rab6 in Herpes Simplex Virus Morphogenesis. In: 36th international Herpesvirus workshop, Gdansk, Poland
Johnson DC, Webb M, Wisner TW, Brunetti C (2001) Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J Virol 75(2):821–833. https://doi.org/10.1128/JVI.75.2.821-833.2001
Johnson DC, Wisner TW, Wright CC (2011) Herpes simplex virus glycoproteins gB and gD function in a redundant fashion to promote secondary envelopment. J Virol 85(10):4910–4926. https://doi.org/10.1128/JVI.00011-11
Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y (2005) Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J Virol 79(14):9325–9331. https://doi.org/10.1128/JVI.79.14.9325-9331.2005
Kato A, Yamamoto M, Ohno T, Tanaka M, Sata T, Nishiyama Y, Kawaguchi Y (2006) Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J Virol 80(3):1476–1486. https://doi.org/10.1128/JVI.80.3.1476-1486.2006
Klupp B, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC (2008) Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J Virol 82(13):6299–6309. https://doi.org/10.1128/JVI.00386-08
Koyama AH, Uchida T (1994) Inhibition by Brefeldin A of the envelopment of nucleocapsids in herpes simplex virus type 1-infected Vero cells. Arch Virol 135(3–4):305–317
Kratchmarov R, Taylor MP, Enquist LW (2012) Making the case: married versus separate models of alphaherpes virus anterograde transport in axons. Rev Med Virol 22(6):378–391. https://doi.org/10.1002/rmv.1724
Kratchmarov R, Kramer T, Greco TM, Taylor MP, Ch'ng TH, Cristea IM, Enquist LW (2013) Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J Virol 87(17):9431–9440. https://doi.org/10.1128/JVI.01317-13
Laine RF, Albecka A, van de Linde S, Rees EJ, Crump CM, Kaminski CF (2015) Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat Commun 6:5980. https://doi.org/10.1038/ncomms6980
Lau SY, Crump CM (2015) HSV-1 gM and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Virus 7(3):915–938. https://doi.org/10.3390/v7030915
Le Sage V, Jung M, Alter JD, Wills EG, Johnston SM, Kawaguchi Y, Baines JD, Banfield BW (2013) The herpes simplex virus 2 UL21 protein is essential for virus propagation. J Virol 87(10):5904–5915. https://doi.org/10.1128/JVI.03489-12
Leach NR, Roller RJ (2010) Significance of host cell kinases in herpes simplex virus type 1 egress and lamin-associated protein disassembly from the nuclear lamina. Virology 406(1):127–137. https://doi.org/10.1016/j.virol.2010.07.002
Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ (2008) Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378 (2):347–354. S0042-6822(08)00390-5 [pii] https://doi.org/10.1016/j.virol.2008.05.035
Leege T, Fuchs W, Granzow H, Kopp M, Klupp BG, Mettenleiter TC (2009) Effects of simultaneous deletion of pUL11 and glycoprotein M on virion maturation of herpes simplex virus type 1. J Virol 83(2):896–907. https://doi.org/10.1128/JVI.01842-08
Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y (2014) Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88(9):4657–4667. https://doi.org/10.1128/JVI.00137-14
Loret S, Guay G, Lippe R (2008) Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82 (17):8605–8618. JVI.00904-08 [pii] https://doi.org/10.1128/JVI.00904-08
Lymberopoulos MH, Bourget A, Ben Abdeljelil N, Pearson A (2011) Involvement of the UL24 protein in herpes simplex virus 1-induced dispersal of B23 and in nuclear egress. Virology 412(2):341–348. https://doi.org/10.1016/j.virol.2011.01.016
Maringer K, Stylianou J, Elliott G (2012) A network of protein interactions around the herpes simplex virus tegument protein VP22. J Virol 86(23):12971–12982. https://doi.org/10.1128/JVI.01913-12
Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, Arii J, Kato A, Kawaguchi Y (2014) Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol 88(13):7445–7454. https://doi.org/10.1128/JVI.01057-14
McClelland DA, Aitken JD, Bhella D, McNab D, Mitchell J, Kelly SM, Price NC, Rixon FJ (2002) pH reduction as a trigger for dissociation of herpes simplex virus type 1 scaffolds. J Virol 76(15):7407–7417
McMillan TN, Johnson DC (2001) Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J Virol 75(4):1928–1940. https://doi.org/10.1128/JVI.75.4.1928-1940.2001
Melancon JM, Foster TP, Kousoulas KG (2004) Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J Virol 78(14):7329–7343. https://doi.org/10.1128/JVI.78.14.7329-7343.2004
Mijatov B, Cunningham AL, Diefenbach RJ (2007) Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology 368(1):26–31. https://doi.org/10.1016/j.virol.2007.07.005
Miranda-Saksena M, Boadle RA, Aggarwal A, Tijono B, Rixon FJ, Diefenbach RJ, Cunningham AL (2009) Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J Virol 83 (7):3187–3199. JVI.01579-08 [pii] https://doi.org/10.1128/JVI.01579-08
Mossman KL, Sherburne R, Lavery C, Duncan J, Smiley JR (2000) Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J Virol 74(14):6287–6299
Naldinho-Souto R, Browne H, Minson T (2006) Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J Virol 80(5):2582–2584. https://doi.org/10.1128/JVI.80.5.2582-2584.2006
Newcomb WW, Trus BL, Booy FP, Steven AC, Wall JS, Brown JC (1993) Structure of the herpes simplex virus capsid. Molecular composition of the pentons and the triplexes. J Mol Biol 232(2):499–511. https://doi.org/10.1006/jmbi.1993.1406
Newcomb WW, Homa FL, Thomsen DR, Booy FP, Trus BL, Steven AC, Spencer JV, Brown JC (1996) Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid formation. J Mol Biol 263(3):432–446. https://doi.org/10.1006/jmbi.1996.0587
Newcomb WW, Juhas RM, Thomsen DR, Homa FL, Burch AD, Weller SK, Brown JC (2001) The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J Virol 75(22):10923–10932. https://doi.org/10.1128/JVI.75.22.10923-10932.2001
Newcomb WW, Thomsen DR, Homa FL, Brown JC (2003) Assembly of the herpes simplex virus capsid: identification of soluble scaffold-portal complexes and their role in formation of portal-containing capsids. J Virol 77(18):9862–9871
Newcomb WW, Homa FL, Brown JC (2005) Involvement of the portal at an early step in herpes simplex virus capsid assembly. J Virol 79(16):10540–10546. https://doi.org/10.1128/JVI.79.16.10540-10546.2005
Newcomb WW, Homa FL, Brown JC (2006) Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J Virol 80(13):6286–6294. https://doi.org/10.1128/JVI.02648-05
Newcomb WW, Jones LM, Dee A, Chaudhry F, Brown JC (2012) Role of a reducing environment in disassembly of the herpesvirus tegument. Virology 431(1–2):71–79. https://doi.org/10.1016/j.virol.2012.05.017
Newcomb WW, Fontana J, Winkler DC, Cheng N, Heymann JB, Steven AC (2017) The primary enveloped virion of herpes simplex virus 1: its role in nuclear egress. MBio 8(3). https://doi.org/10.1128/mBio.00825-17
Oda S, Arii J, Koyanagi N, Kato A, Kawaguchi Y (2016) The interaction between herpes simplex virus 1 tegument proteins UL51 and UL14 and its role in Virion morphogenesis. J Virol 90(19):8754–8767. https://doi.org/10.1128/JVI.01258-16
O'Hara M, Rixon FJ, Stow ND, Murray J, Murphy M, Preston VG (2010) Mutational analysis of the herpes simplex virus type 1 UL25 DNA packaging protein reveals regions that are important after the viral DNA has been packaged. J Virol 84(9):4252–4263. https://doi.org/10.1128/JVI.02442-09
Okoye ME, Sexton GL, Huang E, McCaffery JM, Desai P (2006) Functional analysis of the triplex proteins (VP19C and VP23) of herpes simplex virus type 1. J Virol 80(2):929–940. https://doi.org/10.1128/JVI.80.2.929-940.2006
Padula ME, Sydnor ML, Wilson DW (2009) Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J Virol 83(10):4757–4765. https://doi.org/10.1128/JVI.01927-08
Pante N, Kann M (2002) Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol Biol Cell 13(2):425–434. https://doi.org/10.1091/mbc.01-06-0308
Park R, Baines JD (2006) Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80(1):494–504. https://doi.org/10.1128/JVI.80.1.494-504.2006
Pawliczek T, Crump CM (2009) Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J Virol 83 (21):11254–11264. JVI.00574-09 [pii] https://doi.org/10.1128/JVI.00574-09
Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B (2010) Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog 6(7):e1000991. https://doi.org/10.1371/journal.ppat.1000991
Ren Y, Bell S, Zenner HL, Lau SY, Crump CM (2012) Glycoprotein M is important for the efficient incorporation of glycoprotein H-L into herpes simplex virus type 1 particles. J Gen Virol 93(Pt 2):319–329. https://doi.org/10.1099/vir.0.035444-0
Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ (2001) U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75(18):8803–8817
Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD (2002) Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76(17):8939–8952
Rixon FJ, Addison C, McLauchlan J (1992) Assembly of enveloped tegument structures (L particles) can occur independently of virion maturation in herpes simplex virus type 1-infected cells. J Gen Virol 73(Pt 2):277–284. https://doi.org/10.1099/0022-1317-73-2-277
Roberts KL, Baines JD (2010) Myosin Va enhances secretion of herpes simplex virus 1 virions and cell surface expression of viral glycoproteins. J Virol. JVI.00732-10 [pii] https://doi.org/10.1128/JVI.00732-10
Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D (2009) Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J Virol 83 (1):105–116. JVI.01032-08 [pii] https://doi.org/10.1128/JVI.01032-08
Roller RJ, Fetters R (2015) The herpes simplex virus 1 UL51 protein interacts with the UL7 protein and plays a role in its recruitment into the virion. J Virol 89(6):3112–3122. https://doi.org/10.1128/JVI.02799-14
Roller RJ, Haugo AC, Yang K, Baines JD (2014) The herpes simplex virus 1 UL51 gene product has cell type-specific functions in cell-to-cell spread. J Virol 88(8):4058–4068. https://doi.org/10.1128/JVI.03707-13
Roos WH, Radtke K, Kniesmeijer E, Geertsema H, Sodeik B, Wuite GJ (2009) Scaffold expulsion and genome packaging trigger stabilization of herpes simplex virus capsids. Proc Natl Acad Sci U S A 106(24):9673–9678. https://doi.org/10.1073/pnas.0901514106
Ryckman BJ, Roller RJ (2004) Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J Virol 78(1):399–412
Scholtes LD, Yang K, Li LX, Baines JD (2010) The capsid protein encoded by U(L)17 of herpes simplex virus 1 interacts with tegument protein VP13/14. J Virol 84(15):7642–7650. https://doi.org/10.1128/JVI.00277-10
Schrag JD, Prasad BV, Rixon FJ, Chiu W (1989) Three-dimensional structure of the HSV1 nucleocapsid. Cell 56(4):651–660
Sheaffer AK, Newcomb WW, Brown JC, Gao M, Weller SK, Tenney DJ (2000) Evidence for controlled incorporation of herpes simplex virus type 1 UL26 protease into capsids. J Virol 74(15):6838–6848
Sheaffer AK, Newcomb WW, Gao M, Yu D, Weller SK, Brown JC, Tenney DJ (2001) Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J Virol 75(2):687–698. https://doi.org/10.1128/JVI.75.2.687-698.2001
Smibert CA, Popova B, Xiao P, Capone JP, Smiley JR (1994) Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J Virol 68(4):2339–2346
Smiley JR (2004) Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J Virol 78(3):1063–1068
Spencer JV, Newcomb WW, Thomsen DR, Homa FL, Brown JC (1998) Assembly of the herpes simplex virus capsid: preformed triplexes bind to the nascent capsid. J Virol 72(5):3944–3951
Starkey JL, Han J, Chadha P, Marsh JA, Wills JW (2014) Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J Virol 88(1):110–119. https://doi.org/10.1128/JVI.02555-13
Stevenson AJ, Morrison EE, Chaudhari R, Yang CC, Meredith DM (1997) Processing and intracellular localization of the herpes simplex virus type 1 proteinase. J Gen Virol 78(Pt 3):671–675. https://doi.org/10.1099/0022-1317-78-3-671
Svobodova S, Bell S, Crump CM (2012) Analysis of the interaction between the essential herpes simplex virus 1 tegument proteins VP16 and VP1/2. J Virol 86 (1):473–483. JVI.05981-11 [pii] https://doi.org/10.1128/JVI.05981-11
Szilagyi JF, Cunningham C (1991) Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J Gen Virol 72(Pt 3):661–668
Taylor MP, Enquist LW (2015) Axonal spread of neuroinvasive viral infections. Trends Microbiol 23(5):283–288. https://doi.org/10.1016/j.tim.2015.01.002
Thurlow JK, Murphy M, Stow ND, Preston VG (2006) Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J Virol 80(5):2118–2126. https://doi.org/10.1128/JVI.80.5.2118-2126.2006
Tong L, Stow ND (2010) Analysis of herpes simplex virus type 1 DNA packaging signal mutations in the context of the viral genome. J Virol 84(1):321–329. https://doi.org/10.1128/JVI.01489-09
Toropova K, Huffman JB, Homa FL, Conway JF (2011) The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J Virol 85(15):7513–7522. https://doi.org/10.1128/JVI.00837-11
Trus BL, Newcomb WW, Cheng N, Cardone G, Marekov L, Homa FL, Brown JC, Steven AC (2007) Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol Cell 26(4):479–489. https://doi.org/10.1016/j.molcel.2007.04.010
Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ (2005) Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol 79 (15):9566–9571. 79/15/9566 [pii] https://doi.org/10.1128/JVI.79.15.9566-9571.2005
Votteler J, Sundquist WI (2013) Virus budding and the ESCRT pathway. Cell Host Microbe 14(3):232–241. https://doi.org/10.1016/j.chom.2013.08.012
Whiteley A, Bruun B, Minson T, Browne H (1999) Effects of targeting herpes simplex virus type 1 gD to the endoplasmic reticulum and trans-Golgi network. J Virol 73(11):9515–9520
Meckes DG, Jr., Wills JW (2007) Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J Virol 81 (23):13028–13036. doi:https://doi.org/10.1128/JVI.01306-07
Wills E, Mou F, Baines JD (2009) The U(L)31 and U(L)34 gene products of herpes simplex virus 1 are required for optimal localization of viral glycoproteins D and M to the inner nuclear membranes of infected cells. J Virol 83(10):4800–4809. https://doi.org/10.1128/JVI.02431-08
Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC (2009) Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J Virol 83(7):3115–3126. https://doi.org/10.1128/JVI.01462-08
Wright CC, Wisner TW, Hannah BP, Eisenberg RJ, Cohen GH, Johnson DC (2009) Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J Virol 83(22):11847–11856. https://doi.org/10.1128/JVI.01397-09
Yang K, Baines JD (2011) Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci U S A 108(34):14276–14281. https://doi.org/10.1073/pnas.1108564108
Zeev-Ben-Mordehai T, Weberruss M, Lorenz M, Cheleski J, Hellberg T, Whittle C, El Omari K, Vasishtan D, Dent KC, Harlos K, Franzke K, Hagen C, Klupp BG, Antonin W, Mettenleiter TC, Grunewald K (2015) Crystal structure of the herpesvirus nuclear egress complex provides insights into inner nuclear membrane remodeling. Cell Rep 13(12):2645–2652. https://doi.org/10.1016/j.celrep.2015.11.008
Zenner HL, Yoshimura S, Barr FA, Crump CM (2011) Analysis of Rab GTPase-activating proteins indicates that Rab1a/b and Rab43 are important for herpes simplex virus 1 secondary envelopment. J Virol 85 (16):8012–8021. JVI.00500-11 [pii] https://doi.org/10.1128/JVI.00500-11
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Crump, C. (2018). Virus Assembly and Egress of HSV. In: Kawaguchi, Y., Mori, Y., Kimura, H. (eds) Human Herpesviruses. Advances in Experimental Medicine and Biology, vol 1045. Springer, Singapore. https://doi.org/10.1007/978-981-10-7230-7_2
Download citation
DOI: https://doi.org/10.1007/978-981-10-7230-7_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-7229-1
Online ISBN: 978-981-10-7230-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)