
Abstract
Gene selection plays a vital role in understanding the disease progression and further it helps in understanding the therapeutic targets. Most of the genes available in micro array data are not informative for a particular disease of interest. Study of functional analysis and interaction structure of genes plays a vital role in selecting genes associated to complex diseases. This work uses two different network based approaches for gene selection and compares the biological and statistical enrichment of selected genes. Functional modules in the gene expression data are obtained using Gene Correlation Network (GCN) and marker genes in the modules are identified using R package Weighted Gene Co- expression Analysis (WGCNA). WGCNA is considered to be one of the best methods for analysis of global GCN using a suitable threshold that leads to a network with scale free topology. The differentially co-expressed genes are then compared with the existing gene selection approach which integrates the selected co-expressed gene modules with protein-protein interaction (PPI) network. Observation shows that using PPI network which is generated using multitude of high throughput experiments and available in public data bases selects more disease specific genes in comparison to constructed GCN. The study shows that integrative network analysis to find genes may provide greater insight in underlying biological response.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others

Keywords
- Gene correlation network (GCN)
- Protein protein interaction network (PPI)
- Gene selection
- Gene enrichment
- Biological network integration
- Weighted gene correlation network analysis (WGCNA)
1 Introduction
DNA micro array represents the state of a cell at a molecular level and has the capability to analyze thousands of genes in a single experiment. Microarray is a high throughput gene expression data simultaneously monitoring thousands of genes. The study of microarray data is limited by high dimension of features or genes with comparatively less number of samples. The small sample size becomes a limitation in various analysis [1]. Thus, gene selection plays an important role in analysing gene expression and helps to identify the candidate genes that can be further analyzed for disease prognosis [2]. Gene co-expression network which is the collection of co-expressed genes have been found successful to describe the pair wise relationship between gene transcripts [3]. GCN is used to identify modules of genes with similar expression profiles. Apart from focusing on restrictive single data analysis, integrative analysis of biological data at different level provides more reliable and complete information about the genotype as well as the phenotype association. Protein-protein interaction (PPI) combines structural biology and bioinformatics to find the physical interactions among the pair of proteins. Genes that are related to some specific disease need not to be differentially expressed, but may play important role in interconnecting the differentially expressed genes in the PPI network [4].
In this work we have compared two network based approaches for gene selection. Gene co expression network (GCN) is constructed using the tool weighted gene co-expression network analysis and the functional modules in the network are identified. Genes are selected from the modules by ranking. The efficiency of selected genes is then compared with the existing method which integrates co-expression gene clusters with PPI network for selecting the marker genes.
2 Related Work
Individual analysis of biological data at multiple omic level results in incomplete understanding of genetic aetiology of the complex traits. Intergeneration of multiple omic data is expected to compensate for any undependable or noise information in any single data type and is unlikely to lead to false positives [5]. Swarnkar et al. proposed an integrated framework that combines gene expression information with structural facts of PPI networks to identify a set of functionally enriched genes associated with a specific disease [1]. They have identified co-expressed gene modules in the gene expression data set using k-means clustering algorithm which are mapped to PPI network available in standard public data bases in order to find dense sub graph (DSG) in the network.
Biological network gives valuable information in studying system level properties. It can give better insight on disease progression via the identification of perturbed set of genes in different complex diseases. Network biology approach uncovers the underlying mechanisms in disease pathogenesis, identification of new biomarkers, and shed light on personalized therapeutic interventions [6].
WGCNA, a R package [3] for gene co-expression network analysis has been proven to be a well accepted method for global analysis of co-expressed genes and modules. The package provides different R functions environment to study the various aspects of weighted correlation network analysis. Kadarmideen et al. compared two different methods WGCNA and partial code information theory (PCIT) for GCN construction and analysis. They found that WGCNA method is favourable over PCIT method as WGCNA retains biologically relevant hub genes and their connections within sub-networks intact where as PCIT deletes some important edges in the network and hence disrupts the network topology [7].
3 Materials and Methods Used
3.1 GCN
Gene co-expression network (GCN) considered for graphical representation of genes, where each node of the graph is represented as a gene and a pair of nodes is connected with an undirected edge. An undirected edge is found between a pair of genes only if it’s pair-wise expression similarity is above a particular threshold. Construction of co-expression network using the gene expression information is considered as one of the best alternative to the traditional analysis approaches [2]. Large-scale gene co-expression networks analysis shows that the biologically related genes are highly co-expressed across different organisms and across multiple datasets. In GCN, nodes represent genes where node profile xi represents gene expression profile. The Gene Correlation Network is mainly represented using adjacency matrix aij which is constructed considering the co-expression similarity between genes i and j.
3.2 WGCNA
To study the various aspects of weighted correlation network analysis, R provides Weighted Gene Correlation Network Analysis (WGCNA) a software package which comprehends a large collection of R function is being widely used in literature [3].
WGCNA can be used to construct the highly co-expressed gene modules as co-expressed gene cluster from the given gene expression data. Further analysis of these co-expressed gene modules using WGCNA may provide us insight about the representative gene or an eigen gene or an inter modular hub gene in each module. Further it can also provide insight about inter modular connectivity, their relation with external traits and for calculating module membership measures. This analysis of gene correlation network gives further insight in finding candidate biomarkers as therapeutic targets. The approach has been used in various biological context, viz., yeast and mouse genetics, cancer, brain image data analysis etc. [2].
3.3 Datasets
DNA microarray data set for homo-sapiens available in NCBI’s Gene Expression Omnibus (GEO) has been used in our study. We have used prostate cancer gene expression data set for gene selection process. Prostate cancer data consists of 20,000 genes and 104 samples, out of which 34 are normal samples and 70, are cancer related samples. To compare the quality of modules formed using GCN approach with the existing PPI network based approach, we have used cancer gene data set available in NCBI. The cancer data set contains 10807 genes and 17 samples.
3.4 Working Model
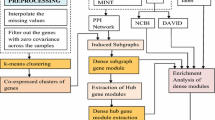
Figure 1 represents the work flow of the model being used to construct the gene correlation network (GCN) from the microarray gene expression data.

Steps of WGCNA based method for gene selection and enrichment
-
1.
We have followed the pre-processing method as described in the existing method [1]. The normalized data from NCBI is collected and missing values in the data are removed by interpolating them using mean or median. Finally the variance across the samples is used for filtering genes from the given dataset. Thus, the size of the data set is reduced to 13,791 genes from 21000 genes and the samples remaining unchanged with 104. Using WGCNA function samples are clustered to identify outlier in the samples. After data cleaning and removing the outlier in the samples we have found 100 samples that are used for further analysis. Figure 2 shows the result of sample clustering.
Fig. 2 
Sample clustering with cut height chosen as 55
-
2.
In GCN modules in the network corresponds to cluster of genes with high absolute co-relation. To construct the network out of the pre-processed data and identify the modules in the network, we have used WGCNA function blockwiseModules () with soft-thresholding power = 4 and taking minimum module size to be 50. Figure 3 represents the scale free topology, as well as the mean connectivity for different soft thresholding powers.
Fig. 3 
Scale free topology to find out the soft threshold power
-
3.
The eigen genes which are the representative genes in each module are identified. Module membership values for all genes with respect to the Eigen genes in the modules are calculated and genes in the modules are ranked according to their module membership values and few top ranked genes are selected. Effectiveness of subset of the selected genes are computed by using different classification techniques.
-
4.
In order to measure the quality of modules, we have compared the modules obtained using our GCN based approach with the modules formed in the existing PPI based approach for gene selection.


3.5 Performance Measures Used
The Matthews coefficient correlation (mcc) is used as a measure of quality of binary classification and is regarded as a balanced measure and can be used for the classes which are of very different sizes [8]. Prostate dataset is having imbalanced ratio between number of samples in positive and negative classes. Thus, to measure the predictive accuracy of the selected genes we are using the mcc. In mcc, overall accuracy, sensitivity, specificity precision and f-measure used for comparison to the known true classes are defined as follows.
where true-positive samples is denoted as tp, tn stands for the count of true-negative samples, fp represents the number of false-positive samples and fn is the number of false-negative samples. The above measures are being used for class performance analysis and comparison with existing methods in the literature. Samples are considered to be divided into two categories, namely diseased samples (positive) and normal samples (negative). The comparative biological significance analysis of the modules obtained in the GCN based approach with the existing PPI network based approach is being made by studying the presence of disease related genes in each of these selected modules.
4 Results and Discussion
We have obtained 18 modules after network construction and module detection. The average module membership of genes in a module taken into consideration, we select 10 modules which are having highly correlated genes shown in the Fig. 5. The highly correlation leads to the higher quality measure. In Figs. 4 and 5 we have shown the percentage of prostate cancer related genes in modules obtained by PPI based gene selection approach and GCN based module selection approach, respectively.

Biological significance study of the PPI interaction approach gene modules in terms of the presence of disease-related genes [1]

GCN based module of biological significance in terms of presence of disease related genes
In Table 1 we take top 10 modules of PPI based approach as well as from GCN based approach. The result states that the percentage of cancer genes with respect to the total number of genes in a module is high in case of PPI integration modules in comparison to WGCNA based GCN approach the percentage of prostate cancer genes with respect to total number of genes in a module is more in few modules of PPI based approach and average in few modules of GCN based approach.
Table 2 shows the percentage of prostate specific genes with respect to cancer related genes in each selected modules. The result shows that except in module 2 the percentage of prostate cancer genes with respect to cancer genes is more in GCN based approach in comparison to PPI based approach. This shows that WGCNA is more efficient in finding co-expressed genes in comparison to cluster based approach used in PPI integration model.
Genes are ranked in decreasing order of their module membership values. On the basis of top few genes are selected for measuring class performance of each module. To evaluate the predictive performance of these genes, we have classified the genes using three different classifiers—K-Nearest Neighbour (knn for k = 3), Support Vector Machine (svm) and Random Forest (rf). The classifiers are applied with 10 fold cross validation. Table 3 summarizes the Sensitivity, Specificity, Precision, F- measure, mcc measures for each of the co-expressed prostate dataset for different classifiers. The result shows that highly correlated modules show higher value lower variance of mcc measure for different classifiers like k-nn, rf and svm. Thus, these selected modules 8, 16, 1 and 6 of GCN for prostate cancer can be further considered for statistical and biological in depth biological analysis. These modules may further give an insight in disease progression and may help in therapeutic analysis.
5 Conclusion and Future Work
It is observed that the network integration based gene modules are more significant in comparison to the traditional expression based gene selection. The study reveals that the module based network integration gene selection is able to find genes which are more discriminative and are found to play vital role in maintaining the interaction among the important genes.
The said property is important for the discovery of disease causing genes. The enrichment achieved by network integration using PPI is found to be stronger compared to that of the GCN based gene selection approach. In few biological analysis done by us it is observed that finding co-expressed gene modules (GCN) is more effective than cluster based approach used in compared PPI integration model. This states that the integration of PPI network with GCN may be studied further in future to find higher label interaction among multiple small co-expressed GCN modules. This may provide more accurate value to the pathway structures and will help in understanding network label biological dynamic in disease progression.
The genes selected using different level biological network integration approaches may be more relevant for the further study of progression of a specific disease. Both the compared approaches, viz., GCN based and PPI network based, can be integrated in different ways for more thorough analysis of candidate gene selection.
References
Swarnkar, T., et al.: Identifying dense subgraphs in protein–protein interaction network for gene selection from microarray data. Netw. Model. Anal. Health Inform. Bioinform. 4(1), 1–18 (2015)
Singh, Rabindra Kumar, Sivabalakrishnan, M.: Feature selection of gene expression data for cancer classification: a review. Proc. Comput. Sci. 50, 52–57 (2015)
Langfelder, P., Horvath, S.: WGCNA: an R package for weighted correlation network analysis. In: BMC Bioinformatics 9.1 (2008)
Chuang, H.Y., et al.: Network‐based classification of breast cancer metastasis. Mol. Syst. Biol. 3(1) (2007)
Ritchie, M.D., et al.: Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 16(2), 85–97 (2015)
Furlong, L.I.: Human diseases through the lens of network biology. Trends Genet. 29, 150–159 (2013)
Kadarmideen, H.N., Watson-Haigh, N.S.: Building gene co-expression networks using transcriptomics data for systems biology investigations: comparison of methods using microarray data. Bioinformation 8(18), 855–861 (2012)
Dao, P., et al.: Optimally discriminative subnetwork markers predict response to chemotherapy. Bioinformatics 27(13), 205–213 (2011)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Nayak, D.S.K., Mahapatra, S., Swarnkar, T. (2018). Gene Selection and Enrichment for Microarray Data—A Comparative Network Based Approach. In: Saeed, K., Chaki, N., Pati, B., Bakshi, S., Mohapatra, D. (eds) Progress in Advanced Computing and Intelligent Engineering. Advances in Intelligent Systems and Computing, vol 564. Springer, Singapore. https://doi.org/10.1007/978-981-10-6875-1_41
Download citation
DOI: https://doi.org/10.1007/978-981-10-6875-1_41
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-6874-4
Online ISBN: 978-981-10-6875-1
eBook Packages: EngineeringEngineering (R0)