
Abstract
One of the most important tendencies for the development of modern chemistry is related to the new trends in biomedicine and pharmacy. Today, significant advances have been made in the development of biodegradable and biocompatible polymer materials and the synthesis of corresponding polymer nano-micelles. In this work we aimed to synthesize new biodegradable and biocompatible block copolymers based on poly(lactide-co-glycolide) (PLGA) and poly (ethylene glycol) (PEG) with linear (ABA-type) architecture, to investigate their ability to form nanosized micelles and its aptitude to acts as carriers of combretastatin-like antitumor agents. The synthesis of linear PLGA1000-PEG1000-PLGA1000 block copolymer was carried out in line with standard procedures. The size and morphology of the obtained micelles were determined by transmission electron microscopy (TEM) and dynamic light scattering (DLS). The capability of obtained micelles to adsorb and deliver combretastatin-like antitumor agents into living cells was also demonstrated. We showed that the obtained copolymers formed nanosized micelles with the combretastatin-like antitumor agent 16Z. In vitro biocompatibility results denote that all tested blank-nano-micelles are devoid of cytotoxic effects and may be used as non-toxic drug carriers to target cells. Cellular uptake of 16Z-loaded PLGA1000-PEG1000-PLGA1000 micelles by HepG2 cells showed continuous uptake up to 72 h and a delay in the cytotoxic effect of 16Z. In an attempt to design new biomimetic analogs of natural combretastatin A-4 (CA-4) and its synthetic amino-derivatives, we selected benzothiazolone heterocycles as a scaffold for a bioisosteric replacement. In silico, drug-likeness and toxicity predictions showed better properties of benzothioazolone CA-4 analogs. The obtained results are very promising and need future detailed investigations.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Cancer is one of the most life-threatening diseases and the second major cause of death worldwide. A series of changes brought by cancer cells via DNA modifications gives them an advantage over normal cells. Despite the advancement, combating cancer remains to be a challenge and thus, a prime focus nowadays.
The cellular microtubule system is essential for many important cellular processes such as cell division, formation and maintenance of cell shape, regulation of motility, cell signaling, secretion and intracellular transport [1]. Microtubules are generally recognized as an attractive target for the development of potential new anticancer agents [2]. Among the tubulin targeting agents, combretastatin A-4, derived from the bark of the South African tree Combretum caffrum [3, 4], is one of the well-known natural tubulin-binding molecules. CA-4 is a very attractive leading compound because of its simple structure, as well as its high cytotoxicity against a variety of human cancer cell lines, including the multi-drug resistance phenotype [5]. CA-4 strongly inhibits the polymerization of tubulin by binding to the colchicine site and displays selective toxicity toward tumor vasculature by inhibiting their blood supply [6]. However, the application of CA-4 as an anticancer agent is limited by its poor solubility. This failing led to the development of a water-soluble phosphate derivative as a prodrug, which is currently in human clinical trials. The obtained hopeful results encouraged the synthesis of a large number of CA-4 analogs [7].
Recently, we designed, synthesized and studied a series of styrylbenzoxazolones as new analogs of combretastatin A-4 with potential anticancer properties [8]. These styrylbenzoxazolones also have pure water solubility, similar to more than 40% of the new chemical entities developed in the pharmaceutical industry [9]. To overcome the pure water solubility, different nanocarrier-based drug delivery systems continue to gain considerable attention due to their potential to improve the bioavailability and efficacy of the drugs [10]. Several nanocarriers such as biologically erodible microspheres [11], micelles [12], different nanoparticles and quantum dots [13,14,15] have been investigated as potential drug delivery systems.
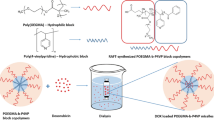
The FDA has approved copolymers containing polyethylene glycol and poly-lactic-glycolic acid blocks for human use. They are attracting great interest both in academia and industry for biomedical applications due to their biodegradable nature and biocompatible properties [16, 17]. In our study, we have used PEG and PLGA as block copolymer compounds for the synthesis of polymeric nano-micelles to make an in-vitro evaluation of the biological responses obtained when micelles are loaded with 16Z, the newly synthesized structural analogs of the CA-4.
2 Experimental
2.1 Preparation of PLGA-PEG-PLGA Micelles and Determination of Critical Micelle Concentration
The synthesis of 16Z and the preparation of PLGA1000-PEG1000-PLGA1000 triblock copolymers are described in detail in Gerova et al. [8] and Ivanova et al. [18], respectively. The obtained PLGA1000-PEG1000-PLGA1000 triblock copolymer was lyophilized (Cryodos, Telstar) to dryness and then dissolved in an appropriate amount of water to obtain a stock solution with a concentration of 500 μM. To achieve the full dissolution of the polymer, the mixture was stirred for 24 h at 4 °C.
The critical micelle concentration (CMC) of PLGA1000-PEG1000-PLGA1000 block copolymers was determined using 1,6-diphenyl-1,3,5-hexatriene (DPH, 98%) as a hydrophobic probe. The obtained series of copolymer solutions (0.250–500 μM) were spiked with a standard solution of DPH in methanol. The final concentration of DPH was 4.0 μM. All samples were kept in the dark at room temperature for 24 h to reach the solubilization equilibrium of DPH in the aqueous phase before analysis with UV-VIS spectroscopy (λ = 285 nm; Beckman L-80 UV–Vis spectrometer, Beckman-Coulter, USA).
2.2 Formation of 16Z-Loaded PLGA-PEG-PLGA Triblock Copolymer Micelles
The loading of 16Z into the polymeric micelles was realized by diluting a stock solution of 16Z with a predetermined volume of aqueous PLGA1000-PEG1000-PLGA1000 triblock copolymer solution. The final concentration of polymer in the micellar solution was kept constant (300 μM) and that of 16Z varied from 0.5 μM to 1.5 μM. The resulting solutions were shaken and kept in the dark at 4 °C for 24 h. Then they were allowed to equilibrate at room temperature for 2 h. The obtained solutions were filtered through 0.22 μm Teflon membrane filters to separate the insoluble matter.
The particle diameter of the formed micelles was measured with Brookhaven instruments 90plus at 37 °C (angle = 90.00, wavelength = 657.0 nm). The morphology of the polymeric micelles was studied by transmission electron microscopy. A sample from the micellar solution was dropped on copper Formvar coated grid and naturally air-dried for several hours at room temperature. The experiments were performed on a JEOL (JEM-100B) microscope with an accelerating voltage of 80 kV. TEM size and morphology of 16Z loaded micelles were analyzed with ImageJ [19].
2.3 Determination of Drug Encapsulation Efficiency and Loading Capacity
16Z-loaded micelles were dissolved in ethanol to measure 16Z concentration through UV spectrophotometry. This method was used to determine the content of 16Z in the micelle solution. The encapsulation efficiency (EE) and drug loading (DL) were calculated according to Chen et al. [20]. Since 16Z is not soluble in water, its content in water was ignored when compared with the loading content of 16Z into the micelles. Thus, the content of free 16Z in water was not included when calculating the loading content and entrapment efficiency.
2.4 In Vitro 16Z Release Study
16Z-loaded micelles and 16Z were dispersed in 2 ml PBS solution in a dialysis membrane and placed at 37 °C incubator. Aliquots of the solution were removed and placed in Eppendorf tubes after 2, 6, 8, 24, 48, 72, and 96 h. Before determination of the drug concentration by HPLC (Proteome Lab PF2D, Beckman-Coulter, USA; C18 column, a linear gradient of acetonitrile in water from 0% to 100%, 30 min), the solution was centrifuged at 10000 rpm at 20 °C for 5 min and the supernatants were tested. The released drug concentrations against time were plotted into a curve.
2.5 Cytotoxicity Assay
Human liver hepatocellular carcinoma cell line (HepG2) was obtained from American Type Culture Collection (HB-8065) and maintained in DMEM (Dulbecco’s Modified Eagle Medium, Applichem, Germany) supplemented with 10% (v/v) FBS (BioWhittaker, USA), penicillin (100 μg/ml), streptomycin (100 μg/ml), and 4.0 mM L-glutamine at 37 °C in a humidified atmosphere of 5.0% CO2 and 95% air.
The cell proliferation inhibitory effect of 16Z-loaded micelles was assayed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. The MTT proliferation assay is based on the ability of live cells to reduce the yellow MTT dye to purple formazan crystals. Logarithmic growing HepG2 cells were plated at a density of 1.0 × 104 cells/well in 100 μl DMEM. Twenty-four hours later, the growth medium was changed with a growth medium containing 16Z-loaded micelle (16Z concentrations of 250, 500, 750 nM, and 150 μM micelles) and the cells were further incubated up to 72 h. Following the incubation period and a change with 100 μl/well of fresh media, 10 μl of MTT solution (5.0 mg/ml) was added to the wells and incubated for further 3 h. 16Z was prepared by dissolving in dimethyl sulfoxide (DMSO) and subsequent dilution with medium to different concentrations in the range of 25 μM to 10 nM. DMSO in the medium never exceeded 0.1%. Cells were also treated with a mixture of 100 μl DMEM with DMSO or 16Z as controls. The formed MTT-formazan product was dissolved in isopropanol and the absorption at 550/630 nm was measured on a vertical spectrophotometer (Bio-Tek Instruments Inc., USA). For each concentration tested, a set of 3 independent experiments of eightplicates was performed.
2.6 In Silico Drug-Likeness and Toxicity Predictions
In an attempt to design new biomimetic analogs of natural CA-4, we selected benzothiazolone heterocycle as a scaffold for a bioisosteric replacement of the CA-4 ring B (Fig. 36.2b). Following the main strategy of bioisosterism, we proffer the synthesis of novel CA-4 benzothiazolone hybrids S19, S21, S23, and RS10 with potential antitumor effects. The in silico drug-likeness and toxicity predictions of the designed CA-4 analogs were carried out using OSIRIS Property Explorer 2017 and the estimated partition coefficients were calculated with the SwissADME program (http://www.swissadme.ch/).
2.7 Statistical Analysis
The data were evaluated by analysis of variance (ANOVA) followed by Tukey’s post-hock test. Differences in the results at the level of p < 0.05 were considered statistically significant. The statistical analysis was carried out using the PASW 18.0 statistical software package (IBM) for Windows.
3 Results and Discussion
3.1 Preparation and Characterization of 16Z-Loaded Micelles
The CMC research was carried out to prove the formation of the PLGA1000-PEG1000-PLGA1000 nano-micelles in aqueous media. DPH was used as a probe in the measurement of CMC. The plot of DPH’s absorption maximum at 285 nm as a function of the polymer content is shown in Fig. 36.1a. The CMC was obtained from the intersection of the tangent to the curve at the inflection with the horizontal tangent through the points at low concentrations. It revealed that the CMC of PLGA1000-PEG1000-PLGA1000 was 100 µM in aqueous solutions, suggesting that when the concentration of PLGA1000-PEG1000-PLGA1000 conjugates was above 100 µM, nanomicelles would spontaneously form in aqueous solutions. This low CMC value indicated that there is a strong capacity to form micelles at a lower concentration.

Characterization of PLGA-PEG-PLGA-based micelles and its 16Z-loaded forms. (a) DPH absorbance at 285 nm vs. lgC for PLGA1000-PEG1000-PLGA1000 in water; (b) TEM micrograph of 16Z-loaded micelles (scale bar 100 nm)
The size distributions are known to affect pharmacokinetics and endocytosis in a polymeric drug delivery system. In our research, particle sizes and size distributions of 16Z loaded PLGA1000-PEG1000-PLGA1000 micelles were measured by particle size analyzer in an aqueous solution at room temperature. Table 36.1 shows the size distributions of the micelles for the observed in batch dispersions. The blank PLGA1000-PEG1000-PLGA1000 micelles showed a narrow size distribution (polydispersity index Pdi: 0.106) with main effective diameter of micelles of ca. 70 nm. An increasing amount of 16Z does not influence the micelles’ size. The dispersions of 16Z-loaded micelles are dominated by a single mode with a peak-size close to 110 nm. In some batch dispersions, there was a second mode (by number) in size-range around 50–70 nm, probably revealing a presence of non-loaded micelles. The size results suggested a possible efficient passive targeting potential to tumor tissue.
TEM image was used to observe further the morphology and size of the micelles. As shown in Fig. 36.1b, the shapes of 16Z loaded PLGA1000-PEG1000-PLGA1000 micelles were almost spherical with diameters ranging from 80 nm to 130 nm, which confirmed the shell–core structure of the polymeric micelles. The smaller sizes of 80 nm observed by TEM as compared to those measured by the particle size analyzer might attribute to evaporation shrinkage of the PEG shell during the drying process of the TEM sample preparation.
Furthermore, we also found that increasing the concentration of 16Z from 0.5 μM to 1.5 μM has a small impact on the encapsulation and loading efficiencies. The encapsulation efficacy increased from 56% to 70%, however, the 16Z’s drug loading efficiencies stayed constant at approximately 25%. These results indicated that the capacity of the triblock polymer influenced drug loading and encapsulation efficiency with only slight increases when drug concentrations increased.
3.2 In Vitro Drug Release Profiles and Effect on HepG2 Viability
To exploit the utility of 16Z-loaded micelles for an effective drug delivery system, we employed the in vitro drug release of 16Z from PLGA1000-PEG1000-PLGA1000 micelles at different time intervals and compared the results to that of 16Z. The release profiles of 16Z micelles were investigated in PBS solution at 37 °C. The cumulative in vitro release of 16Z was significantly different from that of the micelles. Compared to the fast release of 16Z in dissolution medium, 16Z-loaded micelles exhibited a slow release in the initial 8 h (40%). 60% of 16Z was released from the micelles at 24 h, which was followed by the gradual and sustained release of 16Z up to 72 h (80%). The observed slow-release stage might result from a slow degradation of micelles, whereas the sustained release was attributed to the release of the encapsulated 16Z from the micelles. Recently, the two-step release pattern was observed in other drug-loaded micelle formulations [21]. Thus, our results proved that the release of 16Z from the PLGA1000-PEG1000-PLGA1000 micelles in a controlled manner is beneficial for prolonging the drug release behavior.
We determined the anti-proliferative properties of 16Z-loaded micelles in HepG2 cells. As shown in Fig. 36.2a, blank micelles do not show cytotoxicity at 150 μM concentrations (48 and 72 h), which confirms that the PLGA1000-PEG1000-PLGA1000 micelles have good biocompatibility. Moreover, Fig. 36.2a indicates that HepG2 cells exposed to 16Z after 48 h of treatment exhibited significant cytotoxicity in a dose-dependent manner. Although there was a similar cytotoxicity between 16Z and 16Z-loaded micelles at the concentration studied (250 nM, 500 nM, and 750 nM), a notably greater degree of cytotoxicity was observed at a lower dose (250 nM) of 16Z-loaded micelles when compared to 16Z alone. We might speculate that this effect is possibly observed by increased 16Z-loaded micelle solubility and enhanced micelle-mediated cellular uptake at lower concentrations. Probably the dissolution of the polymer into water improves the capacity of the solution to dissolve drugs directly. Furthermore, when concentration increased to 500 or 750 nM, the cytotoxicity of 16Z on HepG-2 cells was higher than that of 16Z-loaded micelles, thus indicating a different mechanism of drug release from the micelles. No differences between the effect of the free 16Z and 16Z-loaded micelles were observed following 72 h of treatment.

In vitro cytotoxicity against HepG2 cells after incubation with 16Z-loaded PLGA1000-PEG1000-PLGA1000 micelles at different concentrations for 48 and 72 h is shown in (a) and schematic representation of the compounds 16Z, CA-4, S19, RS10, S21 and S23 in (b). ∗p < 0.01 versus blank micelles and control group; #p < 0.05 versus 16Z. CA-4: combretastatin A-4; 16Z@micelles: 16Z-loaded micelles
One of the common difficulties with intravenous drug delivery is the low solubility of the drug. In our study, a better solubility of 16Z was accomplished because the drug has a high solubility in the micelle and the PLGA1000-PEG1000-PLGA1000 micelles have a high solubility in the cell culture media. The phase segregation of polymer-blocks into micellar morphology creates an environment , where 16Z can dissolve in the hydrophobic core. PEGs are commonly employed as biocompatible hydrophilic polymers and PLA and PGA degrade over time into acids that can be safely eliminated from a living system, thus allowing us to perform future research for testing the possible anti-cancer effect of 16Z-loaded micelles in vivo.
3.3 In Silico Results of Risks and Drug-Likeness of Ligands
As a part of our ongoing research program to discover novel anticancer agents, we previously reported CA-4 analogs with benzoxazolone scaffold as potent anticancer agents [8]. In continuation of these efforts for development of selective tubulin polymerization inhibitors as potential chemotherapeutic agents, we have designed 4 (S19, RS10, S21, S23) new compounds structurally based on CA-4 and benzothiazolone moiety (Fig. 36.2b). The benzothiazolone was chosen based on its special significance in synthetic chemistry, pharmaceutical chemistry, as well as its anti-tumor properties, and cardiotonic activity.
ChemDraw was used to generate the simplified molecular-input line-entry system further used in OSIRIS Property Explorer [22] for calculation of the drug-likeness as a prediction that determines, whether a particular pharmacological agent has properties consistent with being an orally active drug [23,24,25]. It also provides information on the hydrophilicity of the compound (cLogP), solubility (LogS), molecular weight, drug-likeness and drug score. All four compounds (S19, RS10, S21 and S23) had molecular weights less than 500, which showed that they are likely to be absorbed and can reach the place of action when administered as oral drugs (Fig. 36.2b). All six compounds including 16Z and CA-4 had cLogP values less than 5, suggesting good absorption and permeation across cell membranes [26]. Among the six compounds, CA-4 had the highest value of drug score (0.48). All other compounds had a drug score of less than 0.2, which could be a result of introducing benzoxazolone and benzothiazolone scaffolds. The higher drug-likeness was calculated for S19 (73.3). The LogS prediction of −5.38 (S19), −5.34 (RS10), −5.22 (S21), −4.50 (16Z) and −2.25 (CA-4) indicated that all the compounds should be moderately soluble in water. Low hydrophilicity and therefore high logP values cause poor absorption or permeation. It has been shown that S19, RS10, S21 and S23 compounds have a reasonable probability of well absorbing, since their cLogP value is not greater than 5.0. In general, the drug-likeness value of compound S19 (6.65) is bigger than that of 16Z (−0.53) and CA-4 (4.66). S19 exhibited good in silico ADMET properties but possessed high mutagenic, irritant, and reproductive effective toxicity risks and therefore, it has to be studied in detail in vitro before considering S19 for in vivo drug testing.
References
Downing HK (2000) Annu Rev Cell Dev Biol 16(1):89
Risinger AL, Giles FJ, Mooberry SL (2008) Cancer Treat Rev 35(3):255
Pettit GR, Cragg GM, Herald DL, Schmidt JM, Lohavanijaya P (1982) Can J Chem 60(11):1374
Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall L (1989) Experientia 45(2):209
McGown AT, Fox BW (1990) Cancer Chemother Pharmacol 26(1):79
Lin CM, Ho HH, Pettit GR, Hamel E (1989) Biochemist 28(17):6984
Chaudhary A, Pandeya SN, Kumar P, Sharma PP, Gupta S, Soni N, Verma KK, Bhardwaj G (2007) Mini-Rev Med Chem 7(12):1186
Gerova MS, Stateva SR, Radonova EM, Kalenderska RB, Rusew RI, Nikolova RP, Chanev CD, Shivachev BL, Apostolova MD, Petrov OI (2016) Eur J Med Chem 120:121
Savjani KT, Gajjar AK, Savjani JK (2012) ISRN Pharm 2012:Article ID 195727
Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R, Farokhzad OC (2009) Biomaterials 30(8):1627
Mathiowitz E, Jacob JS, Jong YS, Carino GP, Chickering DE, Chaturvedi P, Santos CA, Vijayaraghavan K, Montgomery S, Bassett M, Morrell C (1997) Nature 386(6623):410
Kraft JC, Freeling JP, Wang Z, Ho RJ (2014) J Pharm Sci 103(1):29
Chen H, Li B, Qiu J, Li J, Jin J, Dai S, Ma Y, Gu Y (2013) Nanoscale 5(24):12409
Chen H, Chi X, Li B, Zhang M, Ma Y, Achilefu S, Gu Y (2014) Biomater Sci 2(7):996
Chen H, Li B, Zhang M, Sun K, Wang Y, Peng K, Ao M, Guo Y, Gu Y (2014) Nanoscale 6(21):12580
Zentner GM, Rathi R, Shih C, McRea JC, Seo MH, Oh H, Rhee BG, Mestecky J, Moldoveanu Z, Morgan M, Weitman S (2001) J Control Release 72(1–3):203
Chen S, Pieper R, Webster DC, Singh J (2005) Int J Pharm 288(2):207
Ivanova L, Popov C, Kolev I, Shivachev B, Karadjov J, Tarassov M, Kulisch W, Reithmaier JP, Apostolova MD (2011) Diam Relat Mater 20(2):165
Rasband WS (1997–2018) ImageJ. US. National Institutes of Health, Bethesda
Chen X, Chen J, Li B, Yang X, Zeng R, Liu Y, Li T, Ho RJY, Shao J (2017) J Colloid Interface Sci 490:542
Hans M, Lowman A (2002) Curr Opin Solid State Mater Sci 6(4):319
Organic Chemistry Portal (2012) http://www.organic-chemistry.org/prog/peo/
Egbert M, Whitty A, Keseru GM, Vajda S (2019) J Med Chem 62(22):10005
Behrouz S, Rad MN, Shahraki BT, Fathalipour M, Behrouz M, Mirkhani H (2019) Mol Divers 23(1):147
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2012) Adv Drug Deliv Rev 64:4
Wu CY, Benet LZ (2005) Pharm Res 22:11
Acknowledgments
The authors are thankful to the National Science Fund of Bulgaria (Project DN 19/13 (2017)) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature B.V.
About this paper

Cite this paper
Atanasov, G., Kolev, I.N., Petrov, O., Apostolova, M.D. (2020). Synthesis of PLGA-PEG-PLGA Polymer Nano-Micelles – Carriers of Combretastatin-Like Antitumor Agent 16Z. In: Petkov, P., Achour, M., Popov, C. (eds) Nanoscience and Nanotechnology in Security and Protection against CBRN Threats. NATO Science for Peace and Security Series B: Physics and Biophysics. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-2018-0_36
Download citation
DOI: https://doi.org/10.1007/978-94-024-2018-0_36
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-2044-9
Online ISBN: 978-94-024-2018-0
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)