
Abstract
Iron is essential for basic cellular processes but is toxic when present in excess. Consequently, iron transport into and out of cells is tightly regulated. Most iron is delivered to cells bound to plasma transferrin via a process that involves transferrin receptor 1, divalent metal-ion transporter 1 and several other proteins. Non-transferrin-bound iron can also be taken up efficiently by cells, although the mechanism is poorly understood. Cells can divest themselves of iron via the iron export protein ferroportin in conjunction with an iron oxidase. The linking of an oxidoreductase to a membrane permease is a common theme in membrane iron transport. At the systemic level, iron transport is regulated by the liver-derived peptide hepcidin which acts on ferroportin to control iron release to the plasma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A large number of cellular enzymes depend on iron for their biological function and, consequently, this metal is essential for basic physiological processes. Iron can shuttle between two thermodynamically stable oxidation states, Fe3+ or ferric iron and Fe2+ or ferrous iron. This makes it ideally suited to the catalysis of biochemical reactions, but it also means that it is able to catalyze reactions leading to the production of toxic oxygen radicals, particularly when it is present in excess. Thus, individual cells and whole organisms must tightly regulate their iron influx and efflux to keep iron levels within the physiologically optimal range, providing sufficient for metabolic functions, while preventing the accumulation of potentially toxic excess iron. This review will focus on mechanisms of iron transport and its regulation in mammalian cells; there have been enormous advances in this area in recent years. There have been equally impressive advances in our knowledge of iron transport in lower eukaryotes and prokaryotes, but it is not possible to comprehensively cover these organisms in a limited space. The reader is referred to several excellent recent reviews for a coverage of these areas [1–3].
There are hundreds of proteins that require iron in the cell. Many of these are involved in enzyme catalysis and electron transport, whereas others are needed for oxygen transport and delivery. Major classes of such proteins include those with heme centers (hemoproteins) and those with iron–sulfur (Fe–S) clusters, but many proteins utilize the co-ordination chemistry of iron in quite different ways, and the list of non-heme, non-iron–sulfur proteins is growing steadily. Quantitatively, the most abundant iron-containing proteins in the body are the hemoproteins hemoglobin and myoglobin, which are produced by immature erythroid cells and myocytes, respectively [4]. These proteins carry the oxygen required for cellular respiration. Other hemoproteins, such as the cytochromes, catalases, and cytochrome oxidase, play central roles in basic cellular metabolism. Proteins containing Fe–S clusters play particularly important roles in electron transport and catalysis [5, 6], while proteins with non-heme, non-Fe–S centres include such important enzymes as the lipoxygenases, the amino acid hydroxylases, and the rate limiting enzyme in DNA synthesis, ribonucleotide reductase. Insights into the biology of iron containing proteins can be found in many places (e.g., [7, 8]).
Iron that is taken up by cells and not required immediately for metabolic functions is stored within ferritin [9, 10]. Ferritin consists of a roughly spherical protein shell made up of two types of subunits (H and L) surrounding a central cavity. Iron enters the cavity through pores in the shell and is deposited as ferrihydrite internally. Up to 4,500 atoms of iron can be stored in this way. If cells become heavily iron loaded, another form of storage iron known as hemosiderin may appear [10, 11]. Haemosiderin is derived from the degradation of ferritin aggregates and consists of iron oxide clusters and degraded protein. Both ferritin and hemosiderin iron can be mobilized from cells if the body demand for iron increases.
This review will concentrate on the transport proteins themselves and how they are regulated. A summary of some of the main proteins involved in iron transport is provided in Table 1, and each of these will be considered in more detail below.
Cellular iron uptake
Under normal physiological conditions, mammalian cells acquire most of their iron from the plasma protein transferrin (Tf). Each Tf molecule is able to bind two atoms of ferric iron, and it is this diferric Tf that is most efficiently utilized by cells [12, 13]. However, in certain pathological states, notably iron loading disorders, when the capacity of plasma Tf to bind iron is exceeded, non-transferrin-bound iron (NTBI) can make a major contribution to cellular iron uptake [14, 15]. Iron from circulating heme, hemoglobin and ferritin can also be utilized by some cells, but, like NTBI, these pathways only become significant under pathological conditions.
Transferrin-bound iron uptake
Transferrin is able to deliver its iron to cells via a number of mechanisms, but the high affinity binding of the protein to the plasma membrane protein transferrin receptor 1 (TfR1) is the best characterized [16, 17] (Fig. 1). TfR1 is a disulfide bonded, homodimeric receptor that is found on the surface of most body cells. TfR1 deficiency leads to embryonic lethality in mice, an indication of its physiological essentiality [18]. Cells that are rapidly dividing (e.g., the epithelial cells of the intestinal crypts) or have specialized iron requirements (e.g., immature erythroid cells which require large amounts of iron for hemoglobin synthesis) express particularly high levels of TfR1. The structure of TfR1 ectodomain has been solved to 3.2 Å resolution by X-ray crystallography [19], and the structure of the Tf-TfR1 complex has subsequently been investigated via cryo-electron microscopy [20]. Each TfR1 monomer is a Type II membrane protein that consists of a short, N-terminal cytoplasmic tail, a single transmembrane domain, and a large C-terminal extracellular domain. The cytoplasmic domain contains a consensus internalization motif, while the extracellular domain is able to bind a single molecule of Tf. TfR1 is also heavily modified post-translationally through O- and N-glycosylation, palmitylation and phosphorylation [17].

The transferrin cycle. Plasma ferric iron binds with high affinity to Tf which in turn binds to TfR1 on the plasma membrane. The Tf/TfR1 complex is internalized through clathrin-coated pits by receptor-mediated endocytosis. A proton-pumping ATPase lowers the pH of the endosome to around 5.5. Iron is released at this low pH, a process aided by reduction of the ferric iron by enzymes of the STEAP family. The resulting ferrous iron moves across the endosomal membrane via DMT1 and enters the cytoplasm where it can be utilized for various metabolic functions or stored within ferritin. ApoTf remains bound to TfR1 at the low pH of the endosome and is returned to the extracellular medium after the endosome recycles to the plasma membrane. The trafficking protein Sec15l1 is involved in Tf recycling. Abbreviations: see text
TfR1 carries Tf and consequently Tf-bound iron into the interior of the cell via receptor-mediated endocytosis [16, 17] (Fig. 1). The receptor binds diferric Tf with 10-fold higher affinity than monoferric Tf at physiological pH, and 2,000-fold higher affinity than apotransferrin [21]. The TfR1-diferric Tf complex is endocytosed through clathrin-coated pits, and the resulting vesicles are then uncoated to become endosomes. The endosomes are acidified by a proton pumping ATPase and, at a pH of around 5.5, iron is released from Tf [22]. Since Tf binds iron with such a high affinity, it is likely that iron release also requires reduction [23], and takes advantage of a conformational change in Tf that accompanies its binding to TfR1 [20, 24]. Proteins of the STEAP (six-transmembrane epithelial antigen of the prostate 3) family are candidate endosomal ferric reductases, and STEAP3 has been shown to perform this function in immature erythroid cells [25]. When the gene encoding this protein is mutated, as in the nm1054 mouse strain, a hypochromic, microcytic iron deficiency anemia results [26]. STEAP3 is expressed in a range of other tissues where it might be expected to play a similar role, but there are a number of other members of the STEAP family and they may also act as endosomal reductases, particularly in non-erythroid tissues [27, 28]. Members of the STEAP family are membrane proteins with six predicted transmembrane domains and NAD(P)H- and flavin-dependent ferric reductase activity. They also contain a membrane-associated heme group that facilitates electron transfer. The structure of STEAP3 has recently been determined and it is predicted to function as a dimer [29].
Iron released from internalized diferric Tf (now in its reduced or ferrous form) makes its way across the endosomal membrane and into the cytoplasm through the ferrous iron transporter divalent metal-ion transporter 1 (DMT1) [30], a member of the NRAMP (natural resistance-associated macrophage protein) family of metal ion transporters. These proteins show proton dependent transport of metal ions, so DMT1 is well suited to removing iron from the low pH environment of the endosomes. The NRAMP family will be discussed in more detail in the following section.
After Tf has delivered its iron, it is recycled to the extracellular medium. At the low pH of the endosome, the affinity of TfR1 for apoTf is much higher than at physiological pH, and this explains why the two proteins remain bound as the endosome is recycled to the plasma membrane. However, once the complex reaches the cell surface, the apoTf dissociates [22]. A single round of Tf-mediated iron delivery (the so-called transferrin cycle) is completed in 5–20 min (depending on the cell type), but the half-life of plasma Tf is approximately 8 days in humans [31]. This means that each Tf molecule can potentially undergo hundreds of rounds of iron binding and uptake during its life. Although the basic pathway of Tf recycling has been known for many years, new details continue to be revealed. An example has come from the analysis of the hemoglobin deficit (hbd) mouse [32, 33]. These animals carry a microcytic, hypochromic anemia and it has been known for some time that the delivery of Tf-bound iron to immature erythroid cells is defective in this strain [34, 35]. The gene that is mutated in these animals encodes the exocyst protein Sec15l1. Although the precise role of Sec15l1 in the Tf cycle is not known, it has been co-localized with TfR1 in recycling endosomes [36] and is likely involved in regulating the efficiency of endosome recycling. Whether Sec15l1 homologs play similar roles in non-erythroid tissues is unknown.
While the high affinity TfR1-mediated uptake of diferric-Tf is a very efficient means of delivery iron to cells, it is not the only mechanism by which cells can utilize Tf-bound iron, and low affinity uptake has also been described. In fact, the concentrations of Tf and diferric Tf in the extracellular fluid are so high (25–50 and 15–30 μM, respectively), that under normal circumstances the low affinity process is likely to predominate. The relative contributions of receptor-mediated and non-receptor-mediated uptake of diferric-Tf vary from cell type to cell type. In immature erythroid cells, diferric Tf is taken up essentially entirely through the TfR1 route, whereas for many cells, such as hepatocytes and intestinal enterocytes, both the high and low affinity processes are utilized [13, 37].
The low affinity uptake of diferric Tf has been best studied in liver-derived cells and a number of investigations have clearly shown that the process is not mediated by TfR1 [38, 39]. However, a candidate binding protein is the TfR1 homolog, TfR2, at least in the liver where it is most strongly expressed [40–42]. TfR2, like TfR1, is a homodimeric plasma membrane Tf binding protein, but its affinity for diferric Tf is 25-fold lower than that of TfR1, consistent with a possible role in the low affinity uptake of Tf. Also, like TfR1, Tf taken up by the low affinity process appears to enter an endocytic compartment, but ultimately is recycled to the extracellular medium [38, 43, 44]. Despite this, there may be some differences in the cellular biology of the two uptake processes and much of the Tf taken up by TfR2 appears to remain intracellularly in multivesicular bodies [42]. The demonstration that TfR2 can be detected on both the cell surface and at intracellular sites is consistent with this [45, 46].
While TfR2 appears to be a strong candidate for the low affinity uptake of diferric Tf, other evidence suggests that this may not be the case. Many tissues show low affinity Tf uptake, yet TfR2 has only a limited tissue distribution with particularly high levels being found only in the liver [46]. Some cell types, such as small intestinal crypt enterocytes, do not express TfR2, yet take up much of their Tf-bound iron by the low affinity process [37]. Furthermore, human patients with mutations in TFR2, or mice in which the Tfr2 gene has been deleted, can very effectively deliver iron to their tissues and indeed develop a form of iron loading disease (or hemochromatosis) [47, 48]. In such cases, however, much of the iron taken up may be as NTBI, and not as diferric Tf. Rather than contributing significantly to iron uptake, TfR2 appears to play a more important role in the regulation of body iron homeostasis through its regulation of hepcidin (see below). Other studies have suggested that iron loaded Tf can be taken up by hepatocytes via proteoglycans or fluid phase endocytosis, and by macrophages through surface-bound glyceraldehyde-3-phosphate dehydrogenase [12, 49]. These could also be relevant in other cell types, but whether they represent quantitatively significant pathways of Tf uptake is unclear.
Most work on Tf-bound iron uptake by cells suggests that endocytotic processes are involved, but there is solid evidence that Tf-associated iron can be delivered to cells without endocytosis. This process has been described for several cell types but is most prominent in hepatocytes [50]. Evidence in favor of cell surface uptake includes the demonstration that diferric Tf immobilized on beads too large to be endocytosed is able to deliver its iron to fibroblasts [51], the finding that membrane impermeant iron chelators are able to reduce the uptake of iron from Tf [52, 53] and experiments showing that NTBI can reduce the uptake of Tf-bound iron [54, 55]. This uptake process could also involve a cell surface ferric iron reductase [53], but the mechanism remains poorly characterized.
The NRAMP family of metal-ion transporters
TfR1 will deliver Tf-bound iron to the interior of the cell, but it still remains in the recycling endosomes and is isolated from the cytoplasm and cellular organelles where it is required for metabolic functions. As noted above, the transport of ferrous iron from the endosomes to the cytoplasm is mediated by DMT1, a member of the natural resistance-associated macrophage protein (NRAMP) family [56, 57]. DMT1 is a multispanning membrane protein with 12 predicted transmembrane domains. It efficiently transports ferrous iron but not ferric iron, and this highlights the requirement of a ferric reductase before the ubiquitous environmental ferric iron can be utilized. DMT1 transports not only ferrous iron but a range of other divalent metal irons, including Cu2+, Zn2+, Co2+ and Cd2+ [56]. Despite this relative lack of specificity, the most obvious phenotype resulting from deletion or mutation in the SLC11A2 gene (which encodes DMT1) is iron deficiency anemia [30, 58].
Our understanding of how DMT1 functions as an iron transporter remains incomplete, but a large number of mutagenesis studies have been carried out on both this protein and its homologs, and the effects of these mutations on metal transport have been investigated [57, 59]. Transmembrane (TM) domains 1 and 6 appear to be critical for metal binding and uptake as well as proton coupling. Residues in TM segments 3, 4 and 9 are also required for efficient metal transport, and amino acid residues in several other regions have been shown to play some role in iron transport. No crystal structure of DMT1 is available as yet, but recently an NMR structure of a synthetic peptide corresponding to loop 3 and TM domain 4 has been solved [60]. TM4 is the site of several naturally occurring mutations in both DMT1 and its homolog NRAMP1, so this region clearly plays an important functional role.
Since most cells can take up iron via the Tf cycle, it is not surprising that DMT1 is expressed by most body cells [30, 61]. Tissues with high iron requirements, such as immature erythroid cells and cells of the central nervous system (CNS), have particularly high levels of DMT1 [57]. However, in addition to its widespread ‘housekeeping’ role, DMT1 also plays a specialized role in the uptake of dietary iron across the brush border membrane of intestinal epithelial cells [62] (Fig. 2a). Indeed, it was the search for the protein that mediated the uptake of iron by the intestinal epithelium that initially led to the identification of DMT1 [56, 58]. The placenta must also transport large amounts of iron and it is a prominent site of DMT1 expression, although interestingly it does not appear to be essential for placental iron transport [63]. Iron uptake following TfR2-mediated endocytosis, the surface delivery of Tf-derived iron, and the uptake of NTBI are also likely to utilize DMT1 [12, 54, 64].

Iron uptake in enterocytes and yeast. a Dietary iron is taken up across the brush border membrane of duodenal enterocytes through the iron/proton symporter DMT1. DMT1 is expressed on most body cells, but it plays a specialized role in iron acquisition from the diet. Most dietary iron is in the Fe 3+ or ferric form, but it needs to be reduced to the Fe 2+ or ferrous form before it can be utilized as a substrate by DMT1. DcytB is an iron-regulated reductase of the brush border membrane that is a candidate for this role. However, whether DcytB is the only reductase involved remains to be determined. b The yeast Saccharomyces cerevisiae is a simple eukaryote that is able to utilize iron in a variety of forms. It also must reduce iron before it can be utilized. The reductases Fre1p and Fre2p are the principal plasma membrane enzymes involved in this process, but several others have been described. They can utilize both ionic iron or siderophore-bound iron as substrates. The reduced iron is transported into the cell through a complex of the iron permease Ftr1p and the iron oxidase Fet3p. A variety of siderophores also can be utilized by S. cerevisiae, and these are taken up via members of the ARN and SIT families
The NRAMP family of metal-ion transporters is found in a wide range of organisms from bacteria to mammals, and its members show a high degree of sequence conservation [65]. The first member of the family to be identified in mammals was not DMT1, but the closely related NRAMP1. The gene that encodes NRAMP1 (SLC11A1) was identified as being mutated in a strain of mice with increased susceptibility to infections [66]. It was subsequently shown that NRAMP1 is particularly highly expressed in macrophages and resides in the membrane of the phagosome which is involved in the killing of phagocytosed pathogens [67]. The precise function of the protein is unclear, but, like DMT1, it seems to be involved in the transmembrane transport of iron and other divalent metals [68]. Whether it transports iron into or out of the phagosome is unresolved [69]. An increased concentration of redox active iron inside the phagosome could contribute to the enhanced production of toxic oxygen radicals, whereas a reduction in phagosome iron content could contribute to the killing of pathogens by starving them of essential iron. Based on the knowledge that NRAMP1, like DMT1, transports metals down a proton gradient, the export of iron and/other metals from the phagosome seems more likely. Interestingly, it has recently been demonstrated that NRAMP1 may play a role in iron recycling from senescent erythrocytes [70], further evidence that it is involved in metal export from the phagosome.
The uptake of non-transferrin-bound iron
Transferrin is the major source of iron for most body cells under normal physiological conditions. However, in various pathologic states, other forms of iron can be taken up. One of the most important of these is non-transferrin-bound iron (NTBI). While we normally think of Tf as being required to deliver iron to cells, this is not the case. In fact, in the very rare cases of congenital Tf deficiency in humans, or in mice in which the Tf gene has been deleted, iron delivery to the tissues is extremely rapid and heavy iron loading of the organs develops [71, 72]. It is more appropriate to think of Tf as providing a vehicle for regulating iron delivery to cells.
Since Tf has an exceptionally high affinity for iron (K d 10−23M−1) [73], the amount of NTBI in the plasma under normal circumstances is vanishingly small. Tf is an abundant plasma protein and only approximately 30% of the total iron binding sites in the plasma Tf pool are occupied at any one time under normal conditions. This provides a large excess of Tf iron binding capacity that helps keep potentially toxic NTBI levels very low. However, when the amount of iron entering the plasma increases, a situation most frequently found in a range of iron loading disorders where dietary iron absorption is inappropriately elevated, the capacity of plasma Tf to bind this iron can be exceeded. It can then be measured readily in the circulation and levels up to 10–15 μM, or even higher is some cases, can be detected [14, 74]. Despite much investigation, the precise chemical form of NTBI has proved difficult to define. Much is thought to be chelated by small organic acids such as citrate, but smaller amounts could be bound loosely to proteins such as albumin [15]. Presumably NTBI is present largely in the oxidized or ferric form.
As the studies of atransferrinemia mentioned above clearly demonstrate, NTBI can enter the tissues very rapidly. This has also been clearly demonstrated in animals where Tf was saturated experimentally [75]. Such studies have shown that the liver has a particularly high avidity for NTBI [75, 76], but other tissues such as the pancreas and heart can also take up considerable amounts. Precisely how NTBI makes its way across the plasma membrane and into cells is unclear, but several candidates have been proposed. DMT1 is perhaps the most feasible of these as its role as a major membrane iron transporter has been well characterized. However, mice lacking DMT1 can still accumulate hepatic iron [63], suggesting that DMT1 is not the sole route of NTBI entry. Another candidate is SFT, or stimulator of Fe transport. This protein was isolated as an iron transporter from erythroid leukemia cells and has been found to play a role in both Tf-dependent and independent iron uptake, but its role remains poorly characterized [77–79]. Indeed, the original published sequence was incorrect, and the sequence responsible for conferring iron transport activity consists of several smaller open reading frames [77]. Which of these is responsible for the activity is not known. More recently, the zinc transporter ZIP14 (Zrt-Irt-like protein 14) (SLC39A14) has been shown to facilitate NTBI uptake in hepatocytes [80], so it also potentially plays an important role. Finally, L-type voltage-dependent calcium channels may play a role in iron uptake by cardiac myocytes as blockers of these channels will inhibit iron transport [81]. In reality, NTBI may enter cells via multiple routes with the proportion entering through any one pathway varying from tissue to tissue. Since under most circumstances iron crosses cellular membranes in the reduced or ferrous form, it is likely that ferric iron reduction plays a role in NTBI uptake, and early studies indicated that this may indeed be the case [82].
The utilization of iron from proteins other than transferrin
Most of the iron found in the circulation is not bound to Tf but is found within the hemoglobin of circulating red cells. Under normal physiological conditions, this iron can only be utilized when senescent erythrocytes are phagocytosed by macrophages and broken down. Heme is released from the protein and in turn iron is released from heme by heme oxygenases and exported to the plasma where it can be bound by Tf. Quantitatively, this is by far the most important route of iron entry into the plasma. Under a range of pathological conditions, however, when intravascular hemolysis occurs, free hemoglobin and heme can be found in the circulation. These can be cleared very rapidly by several important scavenging pathways and the iron returned to plasma Tf for reutilization. Mice in which these pathways have been disrupted display a normal phenotype, showing that their scavenging function is not required under normal conditions, but when the animals are placed under hemolytic stress, significant pathology results [83, 84].
Hemoglobin released into the circulation is bound by the liver-derived plasma protein haptoglobin [85], and the resulting complex binds to CD163 on the surface of macrophages [86]. CD163 is expressed on the surface of macrophages throughout the body and delivers its cargo by receptor-mediated endocytosis [87, 88]. Subsequently, the complex is degraded and iron is released. Following hemolysis, free heme can also be found in the plasma and, like hemoglobin, it also rapidly binds to a plasma protein, in this case hemopexin [89]. The heme/hemopexin complex can be taken up by cells through both high affinity and low affinity pathways. In the high affinity pathway, the complex binds to low-density lipoprotein receptor-related protein (LRP)/CD91, a heterodimeric complex found on the surface of hepatocytes, macrophages and some other cell types [90, 91]. Recent evidence suggests that the complex is internalized and subsequently degraded in lysosomes [91], but the recycling of hemopexin to the extracellular medium has also been reported [92]. The identity of the low affinity transporter is unknown. Iron from internalized heme is likely released from the porphyrin ring via heme oxygenase [93].
There are several normal physiological situations where heme and its associated iron must cross cellular membranes. The mitochondrial membranes are one prominent location where this occurs, and this will be considered in more detail below. However, another site where heme transport in prominent is across the brush border membrane of small intestinal epithelial cells. Much of the iron in the diet is in the form of heme (principally derived from myoglobin) and this can be very efficiently absorbed by the intestine [94]. The nature of the heme transport pathway has yet to be fully defined. A candidate heme transporter, HCP1 (heme carrier protein 1), was described recently [95], but subsequent investigations have shown that this protein transports folate more efficiently than heme [96], so whether it is the major enterocyte heme transporter remains unclear. Irrespective of how heme is transported into enterocytes, it is likely that iron is released from the porphyrin ring by heme oxygenase within the cells before being exported to the circulation [97]. However, a recently identified heme export protein, FLVCR (feline leukemia virus subgroup c receptor) [98], is expressed in the duodenum, raising the possibility that some intact heme may be exported from the gut.
Iron in excess of cellular requirements is bound within the iron storage protein ferritin, and the intracellular concentration of the protein can reach quite high levels [9]. Small, yet readily detectable, amounts of ferritin are also secreted into the extracellular medium, and measurements of plasma ferritin represent a reliable indicator of body iron levels in non-inflammatory conditions. This form of the protein has a very low iron content and is unlikely to play a significant role in iron transport, although a role for ferritin in the intrahepatic transfer of iron from Kupffer cells (the resident macrophages) to hepatocytes has been proposed [99]. However, in pathological states, particularly when there is damage to the liver, the major iron storage organ, considerable amounts of iron-rich tissue ferritin can be released. This ferritin can be rapidly cleared from the circulation by the liver [100], but the clearance mechanism is not well defined. A possible ferritin receptor is TIM-2 (T cell immunoglobulin domain and mucin-domain protein 2). This plasma membrane protein is able to bind ferritin and mediate its internalization [101], but its tissue distribution (the bile canaliculi of hepatocytes, bile duct epithelial cells and B cells) seems inconsistent with a role as a ferritin scavenger. More recently, Scara5 (scavenger receptor, member 5) has been demonstrated to bind and internalize ferritin and to deliver ferritin-associated iron to the developing kidney [102]. Scara5 is also expressed in the adult kidney, gonadal epithelia and several other embryonic tissues, and has been shown to preferentially bind L-ferritin, so it likely plays an important role in development and perhaps in adult tissues. Further studies on TIM2, Scara5 and other putative ferritin binding proteins are required. How iron from internalized ferritin is recovered for reutilization is not well characterized, but presumably the protein shell is degraded in lysosomes and the iron released.
Ferric reductases and cellular iron uptake
Most iron in the environment is in the oxidized or ferric form, yet it is the reduced form, ferrous iron, that is more soluble and is more readily transported across membranes. It thus comes as no surprise that ferric iron reductases play a prominent role in cellular iron uptake. Indeed, long before any mammalian cellular iron uptake proteins were identified, cell surface ferric reductase activity had been described [103, 104]. The first eukaryotic ferric reductases were identified in the yeast Saccharomyces cerevisiae and now four such proteins are known to be involved in yeast iron acquisition [2, 105] (Fig. 2b).
The role of the STEAP proteins in reducing iron and facilitating its uptake by DMT1 in mammalian cells was noted above [27], but there are other examples. Ferric reductase activity has been described on the brush border membrane of duodenal enterocytes [106], but no STEAP proteins are present. A ferric reductase that may feed ferrous iron to DMT1 in the gut is DCYTB (duodenal cytochrome B) [107]. This membrane-bound protein has iron reductase activity and its expression is enhanced by stimuli that enhance iron absorption. However, when the gene encoding Dcytb was deleted in mice, the animals have no overt phenotype, showing that Dcytb is not essential [108]. Whether one or more other ferric reductases exist remains to be determined. A mammalian iron reductase is also found in the placenta [109]. Thus, the functional linking of an oxidoreductase to an iron transporter is a common theme in iron metabolism.
Iron efflux from cells
The iron exporter ferroportin 1
Cellular iron transport is by no means a unidirectional process and is not restricted to iron uptake. Most cells have the capacity to release iron into the extracellular medium and some cell types are particularly well adapted for doing this. The best examples are macrophages and duodenal enterocytes. Macrophages process senescent erythrocytes and the iron derived from hemoglobin must be released from heme and returned to the circulation where it can be re-utilized, particularly by the erythroid marrow [110]. This recycling of iron is particularly critical as the amount of iron entering the body through dietary absorption is usually insufficient to meet erythroid requirements. Indeed, the erythroid/macrophage recycling system is responsible for approximately 80% of daily iron traffic in the body. Enterocytes are another excellent example of a cell type that must vectorially transport iron. Dietary iron enters the cells across the brush border membrane via DMT1, but is delivered to the circulation by export across the basolateral membrane of the cells [111]. Small amounts or iron are absorbed under normal circumstances, sufficient to replace obligatory passive body losses, but intestinal absorption assumes a particularly important role when iron recycling through the macrophages is insufficient to cater for erythroid demand. Under these same conditions, stored iron can also be drawn from most body cells, so the capacity to export iron is widespread.
The only plasma membrane iron export protein to be described, and by far the most valuable one, is ferroportin 1 (FPN) (also known as IREG1 or MTP1) [112–114]. FPN is expressed at particularly high levels on the surface of cells with a high capacity to export iron, such as macrophages and enterocytes, but it appears to be present on almost all cells. This is consistent with the known capacity of most body cells to release stored iron to the circulation. FPN is also expressed at high concentrations in the placenta, another organ which demonstrates the directional movement of large amounts of iron [115], and appears to be essential for iron transfer to the fetus [116]. Mutations in the FPN gene in humans or deletion of the gene in mice or zebrafish demonstrate the importance of FPN in iron homeostasis [116, 117]. In humans with reduced FPN function, iron rapidly accumulates in macrophages, such as the Kupffer cells of the liver, and, in time, iron concentrations also increase in parenchymal cells. Intestinal iron absorption is decreased and plasma iron levels fall. This leads to a situation where there is iron loading in the tissues but, paradoxically, there is a systemic anemia as reduced iron export into the plasma means reduced iron supply to erythroid progenitors [117]. In most human subjects with FPN mutations, there remains sufficient FPN activity to allow iron absorption to continue. However, deletion of the Fpn gene solely in the intestinal epithelium in mice is incompatible with postnatal development, confirming the essentiality of the protein in intestinal iron absorption [116].
FPN is a multispanning membrane protein with 12 predicted transmembrane domains and both N and C termini on the cytoplasmic side of the membrane [118]. Like DMT1, it uses ferrous iron as a substrate, but there is very limited information on the mechanism by which the protein transports iron. There remains some debate as to whether FPN functions as a monomer or a dimer. Genetic evidence suggests that a dimer is more likely [119], although recent evidence has shown that purified detergent-solubilized FPN behaves as a monomer [118]. Many mutations in the gene encoding FPN have been identified and these have been well summarized elsewhere [118, 120].
Although FPN is the only known exporter for low molecular weight forms of iron, some iron can also be exported as either heme or ferritin. Feline leukemia virus, subgroup C, receptor (FLVCR) has been shown to act as a heme exporter in mammals, and it is predicted to play a role in reducing excess heme levels in erythroid cell precursors and in heme release from macrophages [98, 121]. As noted above, it could also play a role in other tissues such as the small intestine. Abcg2 (Bcrp) is another heme transport protein in the plasma membrane that appears to facilitate heme efflux from immature erythroid cells [122]. Iron export in the form of ferritin has not been well characterized, but it has been demonstrated that much of the iron released from erythrophagocytosing Kupffer cells (macrophages) is in this form [99].
The requirement of an oxidase for export
Just as efficient cellular iron uptake often requires the concerted actions of a ferrireductase and a ferrous iron transporter, iron efflux also appears to require both a transporter and an enzyme. In this case, the enzyme facilitates iron oxidation rather than reduction. FPN transports iron across the membrane in the ferrous form, but circulating Tf binds ferric iron so oxidation is required. The major plasma ferroxidase has long been known to be the copper-containing protein ceruloplasmin (Cp) [123]. Many years ago, it was shown that animals placed on a copper-deficient diet showed reduced plasma oxidase activity and accumulated iron in their tissues, providing strong evidence that Cp was required for cellular iron efflux. This has since been confirmed with the analysis of human subjects carrying mutations in the Cp gene, or mice in which the Cp gene has been deleted [124, 125]. These subjects have reduced plasma iron levels but increased storage iron in the tissues. While Cp seems to be required for iron release from most tissues, in the small intestine, CNS and placenta, membrane-bound Cp homologs appear to play a similar role. In the small intestine, the Cp homolog hephaestin (HEPH) is required for efficient iron absorption, and animals with defective HEPH function become quite anemic [126, 127]. HEPH consists of an extracellular oxidase domain (which is 50% identical to the Cp amino acid sequence) linked to a single transmembrane domain and a short C terminal tail. Hephaestin is also found in the CNS, and disruption of both Cp and HEPH leads to greater iron accumulation in the brain than deletion of Cp alone [128]. The CNS also expresses a GPI-linked form of Cp [129]. Placenta also contains a Cp homolog which is thought to facilitate iron transfer to the fetus, and it too is presumed to be membrane bound [115]. However, little is known about this protein. It is certainly likely that multiple iron oxidases act on the same tissue to facilitate iron release. The presence of both HEPH and GPI-linked Cp in the CNS supports this concept, as does the demonstration that Cp can contribute to intestinal iron absorption when the stimulus for absorption is particularly high [130]. In the yeast S. cerevisiae, an iron oxidase (Fet3p) also acts in conjunction with an iron transporter (the permease Ftrp1) to move iron across the plasma membrane, but in this case it is required for iron uptake rather than iron export [2] (Fig. 2b).
Precisely how Cp or its homologs facilitate iron release from cells is poorly understood, but a recent study has shed some light on the mechanism. De Domenico and colleagues [131] found that Cp-mediated iron oxidation was required to release the metal from FPN. In the absence of oxidation, ferrous iron remains bound to FPN which is then ubiquitinated, internalised and degraded (Fig. 3). Thus, iron oxidation stabilizes FPN on the plasma membrane. Under normal circumstances, the amount of Cp in the circulation is not rate limiting for iron release. Patients with defects in the copper transporter ATP7B (Wilson disease) have low plasma ferroxidase levels as ATP7B is required to load Cp with essential copper. Only when plasma Cp oxidase levels drop to below approximately 5% [132] of the normal level does iron accumulate in the tissues [133]. Whether HEPH functions in the gut in a similar way to the CNS Cp is unknown.

Model for iron export through FPN. Iron export through FPN requires the action of an iron oxidase and this function is usually fulfilled by either circulating or GPI-linked forms of Cp. De Domenico et al. [130] have proposed a model for the function of Cp whereby the oxidation of FPN-bound ferrous iron by Cp is required for its release into the extracellular medium (a). In the absence of appropriate oxidase activity, ferrous iron remains bound to FPN and the protein is ubiquitinated and targeted for degradation (b). This is only one mechanism by which FPN expression on the plasma membrane can be modulated. The binding of hepcidin to FPN can also facilitate its internalization and degradation
Intracellular iron transport and iron utilization by mitochondria
Cytoplasmic iron trafficking
Iron that is delivered to the cytoplasm, either via endosomes or across the plasma membrane, must be trafficked to intracellular sites where it can be incorporated into the proteins required for various cellular functions. Iron in excess of metabolic needs is incorporated into the iron storage protein ferritin, but can subsequently be accessed if metabolic demand increases [9]. How iron is trafficked from sites of uptake to sites of utilization and storage is very poorly understood. As a redox active metal, it makes sense for iron to be sequestered in some way so that it is not available to catalyze reactions leading to oxygen radical production, but its precise intracellular form is unknown. Iron may be bound to low molecular weight organic acids such as citrate or could be bound loosely to intracellular proteins [134]. A third possibility is that iron is bound to specific chaperone proteins. Chaperones have been well described for copper [135], another redox active metal, but until recently no iron chaperones had been identified. However, it was recently demonstrated that poly (rC) binding protein 1 (PCBP1) was able to facilitate iron loading onto human ferritin expressed in the yeast S. cerevisiae, and it was subsequently shown to perform a similar function in mammalian cell lines [136]. How PCBP1 receives and subsequently delivers iron is not known. PCBP1 appears to be ubiquitously expressed in mammalian cells, so it could be a universal iron chaperone, but further studies are required to investigate its physiological relevance in mammals.
Mitochondrial iron utilization
A large number of cellular proteins require iron as a cofactor and there is considerable diversity in the types of iron centers found within these proteins. Two of the most widespread iron centers are those involving heme and iron–sulfur (Fe–S) clusters, both of which are predominantly synthesized in mitochondria [5, 137]. In view of this, the mitochondrion plays a critical role in cellular iron homeostasis and is a major sink for iron [138]. How iron traffics from the endosome to the mitochondrion and other intracellular sites is not well understood. As noted above, it may be sequestered by low molecular weight ligands or bound either specifically or non-specifically to cytoplasmic protein, and these could deliver iron to the mitochondria iron import proteins. Another possibility has been suggested for immature erythroid cells which take up large amounts of Tf-bound iron and must deliver it to mitochondria for heme synthesis. It has been proposed that the endosomes may make direct contact with mitochondria and thereby transfer their iron [139]. There is some experimental support for this, but how widespread such a process might be is unclear.
However, iron makes its way to the mitochondrion, it must pass through the mitochondrial membranes to the matrix where it can be utilized. The best candidate for mitochondrial iron import is mitoferrin (Mfrn) [140]. Mfrn is a member of the mitochondrial solute carrier family (SLC25) of transport proteins and is located on the inner mitochondrial membrane. When the Mfrn gene is disrupted in mice, the animals show reduced mitochondrial iron uptake and consequently heme synthesis is reduced. However, mice lacking Mfrn remain viable, suggesting some functional redundancy in iron delivery to this organelle. This is not surprising given its critical role in cellular function. Mfrn is predominantly expressed in erythroid cells, but its homolog mitoferrin 2 (Mfrn2)(65% amino acid identity) is found in many non-erythoid tissues and could play a similar role here [141].
Whether low molecular weight forms of iron can exit the mitochondrion is unclear, but it is well established that both heme and Fe–S clusters can leave the organelle. Heme must be able to leave mitochondria as it is incorporated into various proteins in the cytoplasm. A candidate mitochondrial heme exporter is the ATP-binding cassette (ABC) transporter ABCB10, although this process remains quite poorly understood [142]. The outer mitochondrial membrane protein ABCB6 is also able to transport heme, but in this case it likely acts as a heme importer rather than an exporter [142]. The inner mitochondrial membrane protein ABCB7 has emerged as a possible Fe–S cluster exporter [143], although this has yet to be confirmed. When the gene encoding ABCB7 is disrupted in humans, X-linked sideroblastic anemia results and iron accumulates in the mitochondrial matrix [144]. This iron accumulation suggests that the mitochondrion may not be able to divest itself of iron unless it is first incorporated in to Fe–S clusters or heme. Indeed, iron accumulation is also seen in mitochondria when Fe–S cluster synthesis is disrupted, as in the progressive neurodegenerative disorder Friedrich ataxia [145–147]. Friedrich ataxia results from disrupted expression of the protein frataxin which is associated with the inner mitochondrial membrane. Although it has been shown to be linked to the synthesis of Fe–S clusters, the precise function of frataxin remains unknown [145, 146].
Regulation of cellular iron transport
Cellular iron levels must be kept within defined limits. Sufficient iron is required to meet metabolic needs, but excess iron can be toxic. Thus, both iron uptake and efflux are tightly regulated processes. When cellular iron requirements are high, the iron import machinery is expressed at higher levels, while iron export is restricted. Thus, concentrations of TfR1 and DMT1 are increased on the cell surface under iron-deficient conditions, and FPN expression is reduced. Also, the expression of the iron storage protein ferritin is decreased under these conditions as iron is preferentially directed for metabolic functions. When cells are iron replete, the opposite pattern is seen, with reduced TfR1 and DMT1 expression and increased ferritin production. However, this is only what might be described as the “classical” pattern of iron transport regulation. In reality, things are not quite so simple. For example, depending on the tissue and the circumstances, the expression of both DMT1 and FPN can be either increased or decreased. Some of the mechanisms underlying this regulation are considered below.
The IRE/IRP system
The best described system for the regulation of proteins of iron metabolism is the post-transcriptional regulation conferred by the RNA-binding iron regulatory proteins (IRPs) [148–150]. When cellular iron levels are low, iron regulatory protein 1 (IRP1) and iron regulatory protein 2 (IRP2) bind to stem loop structures, known as iron responsive elements (IREs), in the mRNAs encoding a range of proteins of iron metabolism. When the IRE is in the 5′ untranslated region (UTR) of the mRNA, the binding of the IRPs blocks translation, but when the IRE is in the 3′ UTR, IRP binding leads to stabilization of the message and thus allows translation to proceed. The regulation of TfR1 and ferritin provides the classic example of the operation of the IRP/IRE system. The TfR1 mRNA has several IREs in its 3′ UTR, whereas the ferritin mRNA contains a single IRE in the 5′ UTR [10, 17, 151]. When iron requirements are high, the binding of the IRPs stabilizes the TfR1 mRNA but blocks translation of the ferritin message. Thus iron uptake is enhanced and iron storage is reduced. When the cell is iron replete, the opposite response is seen.
TfR1 and ferritin are not the only proteins that possess IREs in their mRNAs. The DMT1 message has two 3′ splice variants, and one of these, like TfR1, has an IRE [56, 152]. Thus, under iron-deficient conditions, DMT1 mRNA is stabilized. The IRE-containing splice variant is particularly abundant in the duodenum where its expression may need to increase rapidly to facilitate enhanced iron absorption, but in other tissues, such as the liver, the two splice forms are expressed at comparable levels [153]. Ferroportin has an IRE in its 5’ UTR and its translation is blocked when cellular iron levels are low [112]. This pattern of regulation is shown in tissues such as the liver where FPN protein levels correlate directly with iron levels. However, in the intestine, FPN levels vary inversely with iron status, suggesting that its regulation is independent of the IRE/IRP system in this tissue. Despite this, the combined deletion of Irp1 and Irp2 in mice leads to increased FPN expression [154]. Thus, it seems likely that both IRP-dependent and independent (presumably transcriptional) regulation of FPN are occurring in the gut, with the non-IRP mechanism dominating under low iron conditions when FPN levels are particularly high. A murine strain in which the FPN IRE is deleted show severely disturbed iron homeostasis highlighting the importance of this regulatory element [155]. The erythroid form of aminolevulinic acid synthase (eALAS), the rate limiting enzyme in heme synthesis, also has a 5′ IRE in its transcript, so when adequate iron is available its expression is increased [156]. This makes physiological sense and ensures that the protoporphyrin biosynthetic pathway is not stimulated when there is insufficient iron to complete the process and produce heme.
IRP1 and IRP2 respond to iron through different mechanisms [149]. Under iron-replete conditions, IRP1 contains an Fe–S cluster and this gives the protein aconitase activity (in this conformation the protein is known as cytosolic aconitase). However, when the supply of iron is insufficient, the Fe–S cluster disassembles and IRP1 is then able to bind IREs. IRP2 on the other hand does not contain a Fe–S cluster and is regulated by synthesis and degradation. When iron is plentiful, the protein is ubiquitinated and degraded via the proteasome, but in the absence of iron, it is stabilized and is able to bind to target mRNAs.
Both IRPs are expressed quite widely in body tissues, although IRP2 has the more restricted expression pattern of the two. It proved somewhat surprising, therefore, that when Irp1 was deleted in mice the phenotype was very mild [157], whereas deletion of Irp2 led to much more severe pathology, including anemia and significant neurological disease [158, 159]. These observations imply that IRP2 is the most important of the IRPs under physiological conditions. A possible explanation for this apparent paradox has come from Rouault and colleagues who showed that, under the low tissue oxygen concentrations typically found in vivo, the Fe–S cluster of IRP1 is very stable and may not be able to respond effectively to changes in cellular iron levels [149, 160]. However, this is not the case for IRP2 which operates by an Fe–S-independent mechanism.
Transcriptional regulation
Not all the proteins involved in iron transport are regulated by the IRE/IRP system, and even those that are show some degree of transcriptional regulation in response to changes in cellular iron status. For example, the expression of the splice variant of DMT1 mRNA that does not contain an IRE is increased under iron-deficient conditions [161, 162], and, as noted above, the FPN gene is likely to be regulated transcriptionally, at least in the small intestine [112]. Similarly, expression of the enterocyte brush border iron reductase DCYTB is increased by iron deficiency, but its mRNA does not contain an IRE, so transcriptional regulation is likely [107]. Iron-dependent transriptional regulation has also been described for the ferritin [163] and TfR1 [164] genes, but the effects are small. The promoter elements required for these genes to respond to iron have not yet been identified.
Genes encoding proteins of iron homeostasis can also be regulated by a number of factors in addition to iron, and these likely exert their effects at the transcriptional level. Two of the most important of these regulatory stimuli are hypoxia and proinflammatory cytokines. Under hypoxic conditions, the expression of a number of iron homeostasis genes is increased [165], and this reflects the physiological requirement to move more iron to the bone marrow for hemoglobin synthesis. The hypoxia-inducible factors appear to be particularly important in modulating the expression of TfR1 [166, 167]. Similarly, inflammatory cytokines have been shown to affect various iron metabolism genes at the transcriptional level [168]. The response of iron homeostasis to inflammatory stimuli likely evolved as an adaptive response to limit iron availability for invading pathogens.
It is also important to recognize that there are significant tissue-dependent variations in the expression of DMT1 and FPN. Under most circumstances, DMT1 expression is increased with iron depletion and decreased as iron levels rise. However, in some tissues such as the kidney and liver, DMT1 has been reported either not to respond to changes in iron status or even to increase when iron levels increase [169, 170]. The mechanisms underlying these responses are not well understood. FPN mRNA levels are increased in the liver in response to iron loading, consistent with IRP-dependent regulation, but in the small intestine they are reduced under these conditions and evidence suggests that both transcriptional and non-transcriptional mechanisms are in operation here.
If iron-dependent transcriptional regulation is poorly characterized in mammals, this is not the case in unicellular eukaryotes. Most of the genes in the yeast S. cerevisiae that encode proteins of iron homeostasis are regulated transcriptionally under the control of the iron-dependent transcription factor Aftp1 [2]. Aftp1 is constitutively expressed, but moves from the cytoplasm to the nucleus when cellular iron levels decline. There, it activates a network of genes encoding proteins involved in iron acquisition, mobilization and utilization.
Alterations in subcellular localization
An important mechanism for altering the membrane transport of iron quickly is to change the location of the transport proteins. This is a much faster regulatory mechanism than altering gene expression, and usually serves as a ‘first line’ response to alterations in iron requirements. Often such changes are supported by longer-term regulation through translational or transcriptional mechanisms. Several examples serve to illustrate how changes in the location of the protein can affect iron transport. (1) DMT1 is expressed on the brush border membrane of enterocytes and remains there as long as the body requires iron. However, if iron transfer across the basolateral membrane decreases and enterocyte iron levels subsequently increase, then DMT1 can be internalized rapidly [171]. This prevents excess iron accumulating in the enterocytes when it is not required. (2) FPN can be rapidly removed from the plasma membrane following the binding of the iron regulatory peptide hepcidin and this prevents iron export [172]. The internalized complex is targeted to the proteasome for degradation. Details of hepcidin and its action are discussed below. (3) The proportion of TfR1 on the cell surface can vary depending on cellular iron status. When iron demand is high, more TfR1 is expressed on the cell surface, and vice versa [16]. And (4) a final example is the stabilization of TfR2 in the presence of diferric Tf and its redirection to recycling endosomes [173]. This may play a role in the regulation of body iron metabolism as TfR2 mediates important regulatory signals.
Despite the importance of processes such as these, we know very little about the mechanisms underlying the changes in subcellular localization and this is a fruitful area for future investigations. DMT1 has been shown to bind to several proteins likely to be involved in membrane protein trafficking and degradation. These include Ndflps1 and 2, proteins of the Nedd4 family that regulate DMT1 levels by acting as adaptors to recruit the ubiquitin ligase WWP2 [174]. In addition, the two major DMT1 isoforms have been shown to exhibit different trafficking patterns [175], consistent with their different physiological roles. The internalization of FPN in response to hepcidin binding involves phosphorylation at either of two adjacent tyrosine residues by Jak2 kinase [176, 177]. After internalization, the protein is dephosphorylated, ubiquitinated and targeted for degradation. Other studies have shown that the membrane-trafficking protein Mon1A is required for efficient transport of FPN to the plasma membrane of macrophages in an iron-dependent manner [178]. However, for all these processes, much remains to be learned.
Systemic iron metabolism
Plasma iron transport
Under normal physiological conditions, essentially all iron in the plasma is bound to transferrin. Only under pathological conditions, when iron entry into the circulation is excessive, do significant amounts of NTBI appear. Tf is one of the most abundant of all plasma proteins and, in a normal adult human, it is present at 25–50 μM (2–4 mg/ml). The great majority of Tf in the plasma is produced by hepatocytes, but locally it can also be synthesized by the CNS and testis (i.e., behind the blood-brain and blood-testis barriers, respectively) [151, 179, 180]. Its abundance means that Tf has substantial iron binding capacity, but under normal conditions it is only approximately 30% saturated with iron. Thus, Tf is able to buffer against a rapid increase in plasma iron and the prevent the accumulation of potentially toxic NTBI.
The structure of Tf has been extensively investigated [7, 181]. It consists of two globular lobes (the N and C terminal lobes), each of which can bind a single iron atom with exceptionally high affinity (K d 10−23 M−1) [78]. This explains why the concentration of plasma NTBI can be kept so low.
As noted above, the role of Tf in the body is not so much to deliver iron to the tissues (as NTBI can be very efficiently taken up by most cells) but to enable it to be delivered in a controlled or regulated way. Since it plays such an essential role, it is not surprising that Tf is synthesized at a constitutively high level that varies only over quite a limited range in response to various stimuli. Tf levels will increase slightly under iron-deficient conditions and can also be stimulated by hypoxia and estrogens [151, 182]. Most regulation of Tf appears to occur at the level of transcription, although there may be some regulation at the translational level [183].
Systemic regulation of iron homeostasis
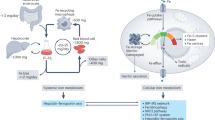
No consideration of iron transport would be complete without mention of the recent major advances in our understanding of the coordinated regulation of body iron homeostasis. These advances have been driven by the demonstration that the liver-derived peptide hepcidin plays a critical role in modulating iron entry into the plasma from macrophages, intestinal enterocytes and other body cells [184–187] (Fig. 4). Hepcidin in its mature form is a 25 amino acid peptide that is secreted by hepatocytes [188]. It acts by binding to FPN and facilitating its internalization and degradation [172, 176], thus reducing iron export from the cell. It is the factors that regulate hepcidin synthesis that lead to an integrated system. Hepcidin levels are relatively high when the body is iron replete, but when iron requirements increase, hepcidin synthesis is reduced and iron entry into the plasma is stimulated. Iron deficiency, hypoxia, increased erythropoiesis and pregnancy are all situations where the body requires more iron and where hepcidin synthesis is reduced [188–190]. High iron levels and inflammation lead to increased hepcidin concentrations with the consequent partitioning of iron to storage sites and a reduction in iron absorption [188, 190].

The regulation of body iron metabolism. The amount of iron entering the plasma is controlled by body iron demand and this in turn reflects a range of stimuli. Low body iron levels, increased erythropoiesis and hypoxia are all stimuli for increased iron export into the circulation from the tissues. Inflammation has the opposite effect and leads to tissue iron withholding. These stimuli exert their effects by acting through a series of proteins on the hepatocyte plasma membrane, including HFE, TfR2, HJV and the IL-6 receptor, to modulate the expression of the iron regulatory hormone hepcidin. Hepcidin in turn binds to FPN on the surface of target cells to influence how much iron they release. Enterocytes and macrophages are major targets, but it is likely that most body cells can respond to hepcidin. An increased hepcidin level leads to reduced FPN on the target membrane and reduced iron release. Iron that enters the plasma is used by all body cells for metabolic functions, but immature erythroid cells have particularly high iron requirements for hemoglobin synthesis. It is the iron requirements of these target cells which complete the cycle
Much effort in recent years has been directed at understanding how hepcidin is regulated. HFE, TfR2 and hemojuvelin (HJV) are all proteins of the hepatocyte plasma membrane that influence hepcidin expression. HJV exerts its effects by signaling through the BMP (bone morphogenetic protein)/SMAD pathway [191–193], and this pathway appears to play a central role in hepdcidin regulation. BMP6, which itself is regulated by iron, is emerging as a particularly important positive regulator of hepcidin [194, 195]. How HFE and TfR2 exert their effects is less clear. There is general agreement, however, that it is these proteins that communicate body iron needs to hepcidin. The nature of the extracellular signals that act on these proteins remains poorly understood, but there is solid evidence that the circulating level of diferric Tf is involved [196, 197]. There is also strong evidence that the hypoxia-inducible transcription factors (HIFs) play an important role in the iron response, and that hypoxia will decrease hepcidin expression [198]. The importance of hepcidin and its upstream regulators in maintaining body iron homeostasis is well demonstrated by the profound changes in iron balance (body iron loading) that are associated with mutations in these genes in humans [199–204]. The pathways involved in hepcidin regulation have been extensively reviewed (e.g., [205, 206]).
Iron transport in specialized mammalian cell types
There are some fundamental aspects of iron handling that are shared by most cells, but, superimposed upon this, each cell type handles iron in its own specific way that is appropriate for its function. Some cells turn over iron rapidly, others store it readily, and yet others, such as immature erythroid cells, utilize most of their iron for a single specific function, i.e., heme synthesis. Some brief summaries of the iron metabolism of some key cells involved in iron homeostasis are described below.
Intestinal enterocytes
In post-partum life, all iron entering the body must come from the diet, and the vast majority of this is absorbed across the mature enterocytes of the proximal small intestine [111]. These specialized absorptive cells can utilize both heme and non-heme iron. The enterocyte brush border membrane expresses high levels of DMT1 and DCYTB, particularly in times of high body iron demand. It also elaborates the protein(s) required for heme uptake, but whether HCP1 or some other protein is involved in this process is unclear. When iron is required by the body, it leaves the enterocyte through a pathway that requires FPN and hephaestin. The enterocyte is one of the most prominent sites of FPN expression and the main site of hephaestin expression. When iron demand is reduced, systemically produced hepcidin binds to FPN on the enterocyte basolateral membrane and causes its internalization and degradation, thus limiting iron efflux to the plasma and iron absorption overall. Iron subsequently accumulates in the enterocytes and this in turn leads to reduced DMT1 synthesis. The accumulated iron is stored within ferritin and is lost when the enterocytes migrate up the villus and are sloughed into the intestinal lumen. This enables the enterocyte to buffer against excessive iron absorption.
The immature enterocytes of the intestinal crypts handle iron quite differently from their mature counterparts. They are not specialized for vectorial iron transport and act rather more like a generic cell. They are rapidly growing and dividing, so they absorb a large amount of Tf-bound iron from the serosal or ‘blood’ side, a characteristic not shown by mature enterocytes. Both TfR1-dependent and TfR1-independent diferric Tf uptake has been demonstrated in crypt enterocytes [37]. As the cells mature and differentiate, they acquire their absorption characteristics but lose the capacity to take up iron from the circulation.
Erythroid cells
Immature erythroid cells have the task of synthesizing hemoglobin and, consequently, they have the highest iron requirements of any cells [207]. They take up iron almost entirely through the Tf-TfR1 pathway, and non-TfR1-mediated iron utilization is negligible. Other proteins required for the delivery of Tf-bound iron, including DMT1 and STEAP3, are strongly expressed in these cells. They have little need to export iron and, consequently, FPN levels are low, although the capacity to export excess heme could be important [121]. Iron uptake in erythroid precursors is particularly prevalent in erythroblasts and reticulocytes [208], but as the cells mature they lose their ability to synthesize new proteins and their capacity to acquire iron. Since the erythroid compartment is quantitatively the most important sink for iron in the body, erythropoiesis-associated signals play a major role in controlling hepcidin expression and, consequently, how much iron is released into the plasma from other cells types.
Macrophages
The critical role of recycling iron in the body is largely carried out by macrophages [110]. They phagocytose senescent red cells, break them down, and recycle the iron. Heme released from degraded globin is the substrate for heme oxygenases 1 and 2 which cleave the ferri-protoporphyrin complex to release iron, carbon monoxide and biliverdin. The iron is then exported from the cells through FPN. Macrophages are perhaps the most prominent site of FPN synthesis of any cells in the body. Although a large amount of erythrocyte-derived iron passes through the macrophages, the great majority of this is not used for the cells’ metabolic functions. Thus, macrophages also possess the capacity to take up Tf-bound iron. They also represent a major storage site for iron, and when iron is not required for hemoglobin synthesis or other functions, it is retained in macrophages in ferritin. Under pathological conditions, macrophages play an important role in scavenging hemoglobin/haptoglobin and heme/hemopexin complexes.
Hepatocytes
Iron homeostasis in hepatocytes is a microcosm of what happens in the body as a whole [209]. Most of the mechanisms of cellular iron uptake that have been described are operational in hepatocytes. They can readily utilize Tf-bound iron (via TfR1, TfR2 or TfR-independent processes) and NTBI. They can also scavenge various other forms of iron such as heme/hemopexin complexes and ferritin. Hepatocytes are the major site of iron storage and sequester iron within ferritin or its degradation product hemosiderin. In addition, hepatocytes are the major site of synthesis of key plasma proteins involved in iron homeostasis, namely Tf, Cp, hemopexin and haptoglobin.
The recognition that the hepatocyte plays the central role in orchestrating systemic iron homeostasis has come from the identification of hepcidin as the key iron metabolism regulator. The hepatocyte is the main site of synthesis of hepcidin, and it is secreted into the circulation and subsequently acts to control iron release from most body cells. The hepatocyte is also the main site of synthesis of the major hepcidin upstream regulators HFE and TfR2, and is a prominent site of HJV synthesis.
Concluding remarks
Over the last 10–15 years, our understanding of iron transport has advanced enormously. Before that time, we had a reasonable understanding of Tf endocytosis, but how iron traversed biological membranes was largely a mystery. Since that time, advances in molecular techniques, the availability of tractable experimental systems, such as knockout mice and yeast, and the analysis of inherited disorders of iron homeostasis have led to the identification of the important iron import and export proteins of the plasma membrane (DMT1 and FPN, respectively), as well as a range of organelle iron transporters and their supporting oxidoreductases. However, the molecular mechanisms by which these proteins transport ‘free’ and bound iron remain relatively poorly understood and represent important areas for ongoing research. This is also true of the cellular biology and, in many cases, the regulation of these proteins. The focus of much research in iron metabolism in recent years has been on how systemic signals for the regulation of body iron homeostasis exert their effects. The demonstration that the liver-derived peptide hepcidin can respond to body iron demand and regulate the release of iron into the plasma by binding to the iron exporter FPN has been a major breakthrough in iron physiology. It has also helped us to understand at the molecular level a range of iron-related pathologies. There is no indication that the rapid pace of research in this field is going to slow anytime soon, and we can look forward to many exciting new discoveries.
References
Crosa JH, Mey AR, Payne SM (eds) (2004) Iron transport in bacteria. American Society for Microbiology, Washington, DC
Philpott CC, Protchenko O (2008) Response to iron deprivation in Saccharomyces cerevisiae. Eukaryot Cell 7:20–27
Sutak R, Lesuisse E, Tachezy J, Richardson DR (2008) Crusade for iron: iron uptake in unicellular eukaryotes and its significance for virulence. Trends Microbiol 16:261–268
Wilson MT, Reeder BJ (2008) Oxygen-binding haem proteins. Exp Physiol 93:128–132
Rouault TA, Tong WH (2005) Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat Rev Mol Cell Biol 6:345–351
Lill R, Mühlenhoff U (2006) Iron-sulfur protein biogenesis in eukaryotes: components and mechanisms. Annu Rev Cell Dev Biol 22:457–486
Crichton R (2001) Inorganic biochemistry of iron metabolism. From Molecular mechanisms to clinical consequences. 2nd edn. Wiley, New York
Castagnetto JM, Hennessy SW, Roberts VA, Getzoff ED, Tainer JA, Pique ME (2002) MDB: the Metalloprotein Database and Browser at The Scripps Research Institute. Nucl Acids Res 30:379–382. http://metallo.scripps.edu
Harrison PM, Arosio P (1996) The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1275:161–203
Koorts AM, Viljoen M (2007) Ferritin and ferritin isoforms I: structure-function relationships, synthesis, degradation and secretion. Arch Physiol Biochem 113:30–54
Iancu TC, Deugnier Y, Halliday JW, Powell LW, Brissot P (1997) Ultrastructural sequences during liver iron overload in genetic hemochromatosis. J Hepatol 27:628–638
Trinder D, Morgan E (2001) Uptake of transferrin-bound iron by mammalian cells. In: Templeton DM (ed) Molecular and cellular iron transport. Marcel Dekker, New York, pp 427–449
Chua AC, Graham RM, Trinder D, Olynyk JK (2007) The regulation of cellular iron metabolism. Crit Rev Clin Lab Sci 44:413–459
Parkes JG, Templeton DM (2001) Transport of non-transferrin-bound iron by hepatocytes. In: Templeton DM (ed) Molecular and cellular iron transport. Marcel Dekker, New York, pp 451–466
Hider RC (2002) Nature of nontransferrin-bound iron. Eur J Clin Invest 32(Suppl 1):50–54
Enns CA (2001) The transferrin receptor. In: Templeton DM (ed) Molecular and cellular iron transport. Marcel Dekker, New York, pp 71–94
Aisen P (2004) Transferrin receptor 1. Int J Biochem Cell Biol 36:2137–2143
Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC (1999) Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet 21:396–399
Lawrence CM, Ray S, Babyonyshev M, Galluser R, Borhani DW, Harrison SC (1999) Crystal structure of the ectodomain of human transferrin receptor. Science 286:779–782
Cheng Y, Zak O, Aisen P, Harrison SC, Walz T (2004) Structure of the human transferrin receptor-transferrin complex. Cell 116:565–576
Tsunoo H, Sussman HH (1983) Characterization of transferrin binding and specificity of the placental transferrin receptor. Arch Biochem Biophys 225:42–54
Klausner RD, Ashwell G, van Renswoude J, Harford JB, Bridges KR (1983) Binding of apotransferrin to K562 cells: explanation of the transferrin cycle. Proc Natl Acad Sci USA 80:2263–2266
Watkins JA, Altazan JD, Elder P, Li C-Y, Nunez M-T, Cui X-X, Glass J (1992) Kinetic characterization of reductant dependent processes in iron mobilization from endocytic vesicles. Biochemistry 31:5820–5830
Bali PK, Zak O, Aisen P (1991) A new role for the transferrin receptor in the release of iron from transferrin. Biochemistry 30:324–328
Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD (2005) Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet 37:1264–1269
Ohgami RS, Campagna DR, Antiochos B, Wood EB, Sharp JJ, Barker JE, Fleming MD (2005) nm1054: a spontaneous, recessive, hypochromic, microcytic anemia mutation in the mouse. Blood 106:3625–3631
Ohgami RS, Campagna DR, McDonald A, Fleming MD (2006) The Steap proteins are metalloreductases. Blood 108:1388–1394
Knutson MD (2007) Steap proteins: implications for iron and copper metabolism. Nutr Rev 65:335–340
Sendamarai AK, Ohgami RS, Fleming MD, Lawrence CM (2008) Structure of the membrane proximal oxidoreductase domain of human Steap3, the dominant ferrireductase of the erythroid transferrin cycle. Proc Natl Acad Sci USA 105:7410–7415
Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC (1998) NRAMP2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci USA 95:1148–1153
Katz JH (1961) Iron and protein kinetics studied by means of doubly labeled human crystalline transferrin. J Clin Invest 40:2143–2152
Lim JE, Jin O, Bennett C, Morgan K, Wang F, Trenor CC 3rd, Fleming MD, Andrews NC (2005) A mutation in Sec15l1 causes anemia in hemoglobin deficit (hbd) mice. Nat Genet 37:1270–1273
White RA, Boydston LA, Brookshier TR, McNulty SG, Nsumu NN, Brewer BP, Blackmore K (2005) Iron metabolism mutant hbd mice have a deletion in Sec15l1, which has homology to a yeast gene for vesicle docking. Genomics 86:668–673
Garrick LM, Edwards JA, Hoke JE, Bannerman RM (1987) Diminished acquisition of iron by reticulocytes from mice with hemoglobin deficit. Exp Hematol 15:671–675
Zhang AS, Sheftel AD, Ponka P (2006) The anemia of “haemoglobin-deficit” (hbd/hbd) mice is caused by a defect in transferrin cycling. Exp Hematol 34:593–598
Zhang XM, Ellis S, Sriratana A, Mitchell CA, Rowe T (2004) Sec15 is an effector for the Rab11 GTPase in mammalian cells. J Biol Chem 279:43027–43034
Anderson GJ, Powell LW, Halliday JW (1994) The endocytosis of transferrin by rat intestinal epithelial cells. Gastroenterology 106:414–422
Trinder D, Zak O, Aisen P (1996) Transferrin receptor-independent uptake of differic transferrin by human hepatoma cells with antisense inhibition of receptor expression. Hepatology 23:1512–1520
Trinder D, Morgan EH, Baker E (1988) The effects of an antibody to the rat transferrin receptor and of rat serum albumin on the uptake of diferric transferrin by rat hepatocytes. Biochim Biophys Acta 943:440–446
Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, Koeffler HP (1999) Molecular cloning of transferrin receptor 2: a new member of the transferrin receptor-like family. J Biol Chem 274:20826–20832
Lee AW, Oates PS, Trinder D (2003) Effects of cell proliferation on the uptake of transferrin-bound iron by human hepatoma cells. Hepatology 38:967–977
Robb AD, Ericsson M, Wessling-Resnick M (2004) Transferrin receptor 2 mediates a biphasic pattern of transferrin uptake associated with ligand delivery to multivesicular bodies. Am J Physiol 287:C1769–C1775
Morgan EH, Smith GD, Peters TJ (1986) Uptake and subcellular processing of 59Fe-125I-labelled transferrin by rat liver. Biochem J 237:163–173
Trinder D, Morgan EH, Baker E (1986) The mechanisms of iron uptake by fetal rat hepatocytes in culture. Hepatology 6:852–858
Fleming RE, Migas MC, Holden CC, Waheed A, Britton RS, Tomatsu S, Bacon BR, Sly WS (2000) Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc Natl Acad Sci USA 97:2214–2219
Deaglio S, Capobianco A, Cali A, Bellora F, Alberti F, Righi L, Sapino A, Camaschella C, Malavasi F (2002) Structural, functional, and tissue distribution analysis of human transferrin receptor-2 by murine monoclonal antibodies and polyclonal antiserum. Blood 100:3782–3789
Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, Majorano N, Totaro A, Gasparini P (2000) The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet 25:14–15
Fleming RE, Ahmann JR, Migas MC, Waheed A, Koeffler HP, Kawabata H, Britton RS, Bacon BR, Sly WS (2002) Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc Natl Acad Sci USA 99:10653–10658
Raje CI, Kumar S, Harle A, Nanda JS, Raje M (2007) The macrophage cell surface glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin receptor. J Biol Chem 282:3252–3261
Thorstensen K, Romslo I (1990) The role of transferrin in the mechanism of cellular iron uptake. Biochem J 271:1–10
Oshiro S, Nakajima H, Markello T, Krasnewich D, Bernardini I, Gahl WA (1993) Redox, transferrin-independent, and receptor-mediated endocytosis iron uptake systems in cultured human fibroblasts. J Biol Chem 268:21586–21591
Cole ES, Glass J (1983) Transferrin binding and iron uptake in mouse hepatocytes. Biochim Biophys Acta 762:102–110
Thorstensen K, Romslo I (1988) Uptake of iron from transferrin by isolated rat hepatocytes. A redox-mediated plasma membrane process? J Biol Chem 263:8844–8850
Trinder D, Morgan E (1997) Inhibition of uptake of transferrin-bound iron by human hepatoma cells by nontransferrin-bound iron. Hepatology 26:691–698
Graham RM, Morgan EH, Baker E (1998) Ferric citrate uptake by cultured rat hepatocytes is inhibited in the presence of transferrin. Eur J Biochem 253:139–145
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA (1997) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388:482–488
Courville P, Chaloupka R, Cellier MF (2006) Recent progress in structure-function analyses of Nramp proton-dependent metal-ion transporters. Biochem Cell Biol 84:960–978
Fleming MD, Trenor CC 3rd, Su MA, Foernzler D, Beier DR, Dietrich WF, Andrews NC (1997) Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 16:383–386
Nevo Y (2008) Site-directed mutagenesis investigation of coupling properties of metal ion transport by DCT1. Biochim Biophys Acta 1778:334–341
Li H, Gu JD, Sun H (2008) Structure, topology and assembly of a 32-mer peptide corresponding to the loop 3 and transmembrane domain 4 of divalent metal transporter (DMT1) in membrane-mimetic environments. J Inorg Biochem 102:1257–1266
Bowen BJ, Morgan EH (1987) Anemia of the Belgrade rat: evidence for defective membrane transport of iron. Blood 70:38–44
Mackenzie B, Garrick MD (2005) Iron imports. II. Iron uptake at the apical membrane in the intestine. Am J Physiol 289:G981–G986
Gunshin H, Fujiwara Y, Custodio AO, Direnzo C, Robine S, Andrews NC (2005) Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J Clin Invest 115:1258–1266
Picard V, Govoni G, Jabado N, Gros P (2000) Nramp 2 (DCT1/DMT1) expressed at the plasma membrane transports iron and other divalent cations into a calcein-accessible cytoplasmic pool. J Biol Chem 275:35738–35745
Nevo Y, Nelson N (2006) The NRAMP family of metal-ion transporters. Biochim Biophys Acta 1763:609–620
Vidal SM, Malo D, Vogan K, Skamene E, Gros P (1993) Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell 73:469–485
Gruenheid S, Pinner E, Desjardins M, Gros P (1997) Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J Exp Med 185:717–730
Goswami T, Bhattacharjee A, Babal P, Searle S, Moore E, Li M, Blackwell JM (2001) Natural-resistance-associated macrophage protein 1 is an H+/bivalent cation antiporter. Biochem J 354:511–519
Wyllie S, Seu P, Goss JA (2002) The natural resistance-associated macrophage protein 1 Slc11a1 (formerly Nramp1) and iron metabolism in macrophages. Microbes Infect 4:351–359
Soe-Lin S, Sheftel AD, Wasyluk B, Ponka P (2008) Nramp1 equips macrophages for efficient iron recycling. Exp Hematol 36:929–937
Bernstein SE (1987) Hereditary hypotransferrinemia with hemosiderosis, a murine disorder resembling human atransferrinemia. J Lab Clin Med 110:690–705
Hayashi A, Wada Y, Suzuki T, Shimizu A (1993) Studies on familial hypotransferrinemia: unique clinical course and molecular pathology. Am J Hum Genet 53:201–213
Aisen P, Leibman A, Zweier J (1978) Stoichiometric and site characteristics of the binding of iron to human transferrin. J Biol Chem 253:1930–1937
Breuer W, Shvartsman M, Cabantchik ZI (2008) Intracellular labile iron. Int J Biochem Cell Biol 40:350–354
Craven CM, Alexander J, Eldridge M, Kushner JP, Bernstein S, Kaplan J (1987) Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: a rodent model for hemochromatosis. Proc Natl Acad Sci USA 84:3457–3461
Wright TL, Brissot P, Ma W-L, Weisinger RA (1986) Characterization of non-transferrin-bound iron clearance by rat liver. J Biol Chem 261:10909–10914
Gutierrez JA, Yu J, Rivera S, Wessling-Resnick M (1997) Functional expression cloning and characterization of SFT, a stimulator of Fe transport. J Cell Biol 139:895–905 [Correction in (1999) J Cell Biol 147, following p 204]
Barisani D, Conte D (2002) Transferrin receptor 1 (TfR1) and putative stimulator of Fe transport (SFT) expression in iron deficiency and overload: an overview. Blood Cells Mol Dis 29:498–505
Gehrke SG, Riedel HD, Herrmann T, Hadaschik B, Bents K, Veltkamp C, Stremmel W (2003) UbcH5A, a member of human E2 ubiquitin-conjugating enzymes, is closely related to SFT, a stimulator of iron transport, and is up-regulated in hereditary hemochromatosis. Blood 101:3288–3293
Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ (2006) Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci USA 103:13612–13617
Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, Yazdanpanah M, Wilson GJ, Schwartz A, Liu PP, Backx PH (2003) L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med 9:1187–1194
Randell EW, Parkes JG, Olivieri NF, Templeton DM (1994) Uptake of non-transferrin-bound iron by both reductive and nonreductive processes is modulated by intracellular iron. J Biol Chem 269:16046–16053
Lim SK, Kim H, Lim SK, bin Ali A, Lim YK, Wang Y, Chong SM, Costantini F, Baumman H (1998) Increased susceptibility in Hp knockout mice during acute hemolysis. Blood 92:1870–1877
Tolosano E, Hirsch E, Patrucco E, Camaschella C, Navone R, Silengo L, Altruda F (1999) Defective recovery and severe renal damage after acute hemolysis in hemopexin-deficient mice. Blood 94:3906–3914
Wassell J (2000) Haptoglobin: function and polymorphism. Clin Lab 46:547–552
Moestrup SK, Moller HJ (2004) CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann Med 36:347–354
Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, Moestrup SK (2001) Identification of the haemoglobin scavenger receptor. Nature 409:198–201
Fabriek BO, Dijkstra CD, van den Berg TK (2005) The macrophage scavenger receptor CD163. Immunobiol 210:153–160
Tolosano E, Altruda F (2002) Hemopexin: structure, function, and regulation. DNA Cell Biol 21:297–306
Herz J, Strickland DK (2001) LRP: a multifunctional scavenger and signaling receptor. J Clin Invest 108:779–784
Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK (2005) Identification of the receptor scavenging hemopexin-heme complexes. Blood 106:2572–2579
Smith A, Farooqui SM, Morgan WT (1991) The murine haemopexin receptor. Evidence that the haemopexin-binding site resides on a 20 kDa subunit and that receptor recycling is regulated by protein kinase C. Biochem J 276:417–425
Maines MD (2005) The heme oxygenase system: update 2005. Antiox Redox Signal 7:1761–1766
Latunde-Dada GO, Simpson RJ, McKie AT (2006) Recent advances in mammalian haem transport. Trends Biochem Sci 31:182–188
Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, Frazer DM, Anderson GJ, Vulpe CD, Simpson RJ, McKie AT (2005) Identification of an intestinal heme transporter. Cell 122:789–801
Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID (2006) Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 127:917–928
West AR, Oates PS (2008) Mechanisms of heme iron absorption: current questions and controversies. World J Gastroenterol 14:4101–4110
Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL (2008) A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 319:825–828
Sibille JC, Kondo H, Aisen P (1988) Interactions between isolated hepatocytes and Kupffer cells in iron metabolism: a possible role for ferritin as an iron carrier protein. Hepatology 8:296–301
Mack U, Cooksley WG, Ferris RA, Powell LW, Halliday JW (1981) Regulation of plasma ferritin by the isolated perfused rat liver. Br J Haematol 47:403–412
Chen TT, Li L, Chung DH, Allen CD, Torti SV, Torti FM, Cyster JG, Chen CY, Brodsky FM, Niemi EC, Nakamura MC, Seaman WE, Daws MR (2005) TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med 202:955–965
Li JY, Paragas N, Ned RM, Qiu A, Viltard M, Leete T, Drexler IR, Chen X, Sanna-Cherchi S, Mohammed F, Williams D, Lin CS, Schmidt-Ott KM, Andrews NC, Barasch J (2009) Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev Cell 16:35–46
Inman RS, Coughlan MM, Wessling-Resnick M (1994) Extracellular ferrireductase activity of K562 cells is coupled to transferrin-independent iron transport. Biochemistry 33:11850–11857
Jordan I, Kaplan J (1994) The mammalian transferrin-independent iron transport system may involve a surface ferrireductase activity. Biochem J 302:875–879
Dancis A, Klausner RD, Hinnebusch AG, Barriocanal JG (1990) Genetic evidence that ferric reductase is required for iron uptake in Saccharomyces cerevisiae. Mol Cell Biol 10:2294–2301
Raja KB, Simpson RJ, Peters TJ (1992) Investigation of a role for reduction in ferric iron uptake by mouse duodenum. Biochim Biophys Acta 1135:141–146
McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ (2001) An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 291:1755–1759
Gunshin H, Starr CN, Direnzo C, Fleming MD, Jin J, Greer EL, Sellers VM, Galica SM, Andrews NC (2005) Cybrd1 (duodenal cytochrome b) is not necessary for dietary iron absorption in mice. Blood 106:2879–2883
Verrijt CE, Kroos MJ, Huijskes-Heins MI, van Eijk HG, van Dijk JP (1998) Non-transferrin iron uptake by trophoblast cells in culture. Significance of a NADH-dependent ferrireductase. Placenta 19:525–530
Knutson M, Wessling-Resnick M (2003) Iron metabolism in the reticuloendothelial system. Crit Rev Biochem Mol Biol 38:61–88
Anderson GJ, Vulpe CD (2001) Regulation of intestinal iron transport. In: Templeton DM (ed) Molecular and cellular iron transport. Marcel Dekker, New York, pp 559–596
Abboud S, Haile DJ (2000) A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 275:19906–19912
Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI (2000) Postional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403:776–781
McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ (2000) A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 5:299–309
McArdle HJ, Andersen HS, Jones H, Gambling L (2008) Copper and iron transport across the placenta: regulation and interactions. J Neuroendocrinol 20:427–431
Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC (2005) The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 1:191–200
Pietrangelo A (2004) Non-HFE hemochromatosis. Hepatology 39:21–29
Rice AE, Mendez MJ, Hokanson CA, Rees DC, Björkman PJ (2009) Investigation of the biophysical and cell biological properties of ferroportin, a multipass integral membrane protein iron exporter. J Mol Biol 386:717–732
De Domenico I, Ward DM, Musci G, Kaplan J (2007) Evidence for the multimeric structure of ferroportin. Blood 109:2205–2209
Wallace DF, Subramaniam VN (2007) Non-HFE haemochromatosis. World J Gastroenterol 13:4690–4698
Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL (2004) Identification of a human heme exporter that is essential for erythropoiesis. Cell 118:757–766
Krishnamurthy P, Schuetz JD (2006) Role of ABCG2/BCRP in biology and medicine. Ann Rev Pharm Toxicol 46:381–410
Hellman NE, Gitlin JD (2002) Ceruloplasmin metabolism and function. Annu Rev Nutr 22:439–458
Harris ZL, Durley AP, Man TK, Gitlin JD (1999) Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci USA 96:10812–10817
Xu X, Pin S, Gathinji M, Fuchs R, Harris ZL (2004) Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Ann N Y Acad Sci 1012:299–305
Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ (1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 21:195–199
Anderson GJ, Frazer DM, McKie AT, Vulpe CD (2002) The ceruloplasmin homolog hephaestin and the control of intestinal iron absorption. Blood Cells Mol Dis 29:367–375
Hadziahmetovic M, Dentchev T, Song Y, Haddad N, He X, Hahn P, Pratico D, Wen R, Harris ZL, Lambris J, Beard J, Dunaief J (2008) Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest Ophthalmol Vis Sci 49:2728–2736
Jeong SY, David S (2003) Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J Biol Chem 278:27144–27148
Cherukuri S, Potla R, Sarkar J, Nurko S, Harris ZL, Fox PL (2005) Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab 2:309–319
De Domenico I, Ward DM, di Patti MC, Jeong SY, David S, Musci G, Kaplan J (2007) Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J 26:2823–2831
Roeser HP, Lee GR, Nacht S, Cartwright GE (1970) The role of ceruloplasmin in iron metabolism. J Clin Invest 49:2408–2417
Shiono Y, Wakusawa S, Hayashi H, Takikawa T, Yano M, Okada T, Mabuchi H, Kono S, Miyajima H (2001) Iron accumulation in the liver of male patients with Wilson’s disease. Am J Gastroenterol 96:3147–3151
Kruszewski M (2003) Labile iron pool: the main determinant of cellular response to oxidative stress. Mut Res 531:81–92
Prohaska JR, Gybina AA (2004) Intracellular copper transport in mammals. J Nut 134:1003–1006
Shi H, Bencze KZ, Stemmler TL, Philpott CC (2008) A cytosolic iron chaperone that delivers iron to ferritin. Science 320:1207–1210
Ajioka RS, Phillips JD, Kushner JP (2006) Biosynthesis of heme in mammals. Biochim Biophys Acta 1763:723–736
Napier I, Ponka P, Richardson DR (2005) Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood 105:1867–1874
Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P (2007) Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 110:125–132
Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH (2006) Mitoferrin is essential for erythroid iron assimilation. Nature 440:96–100
Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J (2009) Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol 29:1007–1016
Krishnamurthy P, Xie T, Schuetz JD (2007) The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol Therapeut 114:345–358
Pondarré C, Antiochos BB, Campagna DR, Clarke SL, Greer EL, Deck KM, McDonald A, Han AP, Medlock A, Kutok JL, Anderson SA, Eisenstein RS, Fleming MD (2006) The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum Mol Gene 15:953–964
Bekri S, Kispal G, Lange H, Fitzsimons E, Tolmie J, Lill R, Bishop DF (2000) Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood 96:3256–3264
Puccio H, Simon D, Cossée M, Criqui-Filipe P, Tiziano F, Melki J, Hindelang C, Matyas R, Rustin P, Koenig M (2001) Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet 27:181–186
Pandolfo M (2003) Friedreich ataxia. Sem Pediatric Neurol 10:163–172
Lodi R, Tonon C, Calabrese V, Schapira AH (2006) Friedreich’s ataxia: from disease mechanisms to therapeutic interventions. Antiox Redox Signal 8:438–443
Pantopoulos K (2004) Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci 1012:1–13
Rouault TA (2006) The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol 2:406–414
Cairo G, Recalcati S (2007) Iron-regulatory proteins: molecular biology and pathophysiological implications. Exp Rev Mol Med 9:1–13
Ponka P, Beaumont C, Richardson DR (1998) Function and regulation of transferrin and ferritin. Sem Hematol 35:35–54
Gunshin H, Allerson CR, Polycarpou-Schwarz M, Rofts A, Rogers JT, Kishi F, Hentze MW, Rouault TA, Andrews NC, Hediger MA (2001) Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett 509:309–316
Tchernitchko D, Bourgeois M, Martin M-E, Beaumont C (2002) Expression of the two mRNA isoforms of the iron transporter Nramp2/DMT1 in mice and function of the iron responsive element. Biochem J 363:449–455
Galy B, Ferring-Appel D, Kaden S, Gröne HJ, Hentze MW (2008) Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab 7:79–85
Mok H, Jelinek J, Pai S, Cattanach BM, Prchal JT, Youssoufian H, Schumacher A (2004) Disruption of ferroportin 1 regulation causes dynamic alterations in iron homeostasis and erythropoiesis in polycythaemic mice. Development 131:1859–1868
Melefors O, Goossen B, Johansson HE, Stripecke R, Gray NK, Hentze MW (1993) Translational control of 5-aminolevulinate synthase mRNA by iron-responsive elements in erythroid cells. J Biol Chem 268:5974–5978
Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Ollivierre-Wilson H, Grinberg A, Love P, Rouault TA (2004) Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J 23:386–395
LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R 3rd, Grinberg A, Love P, Tresser N, Rouault TA (2001) Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet 27:209–214
Galy B, Ferring D, Minana B, Bell O, Janser HG, Muckenthaler M, Schümann K, Hentze MW (2005) Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 106:2580–2589
Meyron-Holtz EG, Ghosh MC, Rouault TA (2004) Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science 306:2087–2090
Frazer DM, Vulpe CD, McKie AT, Wilkins SJ, Trinder D, Cleghorn GJ, Anderson GJ (2001) Cloning and gastrointestinal expression of rat hephaestin: relationship to other iron transport proteins. Am J Physiol 281:G931–G939
Mims MP, Prchal JT (2005) Divalent metal transporter 1. Hematology 10:339–345
Hintze KJ, Theil EC (2006) Cellular regulation and molecular interactions of the ferritins. Cell Mol Life Sci 63:591–600
Casey JL, Di Jeso B, Rao K, Klausner RD, Harford JB (1988) Two genetic loci participate in the regulation by iron of the gene for the human transferrin receptor. Proc Natl Acad Sci USA 85:1787–1791
Peyssonnaux C, Nizet V, Johnson RS (2008) Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 7:28–32
Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G (1999) Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J Biol Chem 274:24142–24146
Lok CN, Ponka P (1999) Identification of a hypoxia response element in the transferrin receptor gene. J Biol Chem 274:24147–24152
Weiss G (2005) Modification of iron regulation by the inflammatory response. Best Pract Res Clin Haematol 18:183–201
Trinder D, Oates PS, Thomas C, Sadlier J, Morgan EH (2000) Localisation of divalent metal transporter 1 (DMT1) to the microvillus membrane of rat duodenal enterocytes in iron deficiency, but to hepatocytes in iron overload. Gut 46:270–276
Scheiber-Mojdehkar B, Sturm B, Plank L, Kryzer I, Goldenberg H (2003) Influence of parenteral iron preparations on non-transferrin bound iron uptake, the iron regulatory protein and the expression of ferritin and the divalent metal transporter DMT-1 in HepG2 human hepatoma cells. Biochem Pharmacol 65:1973–1978
Yeh KY, Yeh M, Watkins JA, Rodriguez-Paris J, Glass J (2000) Dietary iron induces rapid changes in rat intestinal divalent metal transporter expression. Am J Physiol 279:G1070–G1079
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090–2093
Johnson MB, Chen J, Murchison N, Green FA, Enns CA (2007) Transferrin receptor 2: evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol Biol Cell 18:743–754
Foot NJ, Dalton HE, Shearwin-Whyatt LM, Dorstyn L, Tan SS, Yang B, Kumar S (2008) Regulation of the divalent metal ion transporter DMT1 and iron homeostasis by a ubiquitin-dependent mechanism involving Ndfips and WWP2. Blood 112:4268–4275
Lam-Yuk-Tseung S, Gros P (2006) Distinct targeting and recycling properties of two isoforms of the iron transporter DMT1 (NRAMP2, Slc11A2). Biochemistry 45:2294–2301
De Domenico I, Ward DM, Langelier C, Vaughn MB, Nemeth E, Sundquist WI, Ganz T, Musci G, Kaplan J (2007) The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell 18:2569–2578
De Domenico I, Lo E, Ward DM, Kaplan J (2009) Hepcidin-induced internalization of ferroportin requires binding and cooperative interaction with Jak2. Proc Natl Acad Sci USA 106:3800–3805
Wang F, Paradkar PN, Custodio AO, McVey Ward D, Fleming MD, Campagna D, Roberts KA, Boyartchuk V, Dietrich WF, Kaplan J, Andrews NC (2007) Genetic variation in Mon1a affects protein trafficking and modifies macrophage iron loading in mice. Nat Genet 39:1025–1032
Zakin MM (1992) Regulation of transferrin gene expression. FASEB J 6:3253–3258
Sylvester SR, Griswold MD (1994) The testicular iron shuttle: a “nurse” function of the Sertoli cells. J Androl 15:381–385
MacGillivray RTA, Mason AB (2001) Transferrins. In: Templeton DM (ed) Molecular and cellular iron transport. Marcel Dekker, New York, pp 41–69
Cairo G (2001) Regulation of liver iron metabolism. In: Templeton DM (ed) Molecular and cellular iron transport. Marcel Dekker, New York, pp 613–641
Lok CN, Loh TT (1998) Regulation of transferrin function and expression: review and update. Biol Signals Recept 7:157–178
Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S (2001) Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA 98:8780–8785
Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A, Vaulont S (2002) Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA 99:4596–4601
Ganz T (2005) Hepcidin—a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract Res Clin Haematol 18:171–182
Rivera S, Liu L, Nemeth E, Gabayan V, Sorensen OE, Ganz T (2005) Hepcidin excess induces the sequestration of iron and exacerbates tumor-associated anemia. Blood 105:1797–1802
Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loreal O (2001) A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 276:7811–7819
Millard KN, Frazer DM, Wilkins SJ, Anderson GJ (2004) Changes in the expression of intestinal iron transport and hepatic regulatory molecules explain the enhanced iron absorption associated with pregnancy in the rat. Gut 53:655–660
Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S (2002) The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 110:1037–1044
Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, Cooperman S, Eckhaus M, Rouault T, Mishra L, Deng CX (2005) A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2:399–409
Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, Chung RT, Schneyer AL, Woolf CJ, Andrews NC, Lin HY (2006) Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet 38:531–539
Anderson GJ, Frazer DM (2006) Iron metabolism meets signal transduction. Nat Genet 38:503–504
Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP (2009) Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet 41:478–481
Andriopoulos B Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, Knutson MD, Pietrangelo A, Vukicevic S, Lin HY, Babitt JL (2009) BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 41:482–487
Frazer DM, Anderson GJ (2003) The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Mol Dis 30:288–297
Wilkins SJ, Frazer DM, Millard KN, McLaren GD, Anderson GJ (2006) Iron metabolism in the hemoglobin-deficit mouse: correlation of diferric transferrin with hepcidin expression. Blood 107:1659–1664
Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS (2008) Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest 117:1926–1932
Ahmad KA, Ahmann JR, Migas MC, Waheed A, Britton RS, Bacon BR, Sly WS, Fleming RE (2002) Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol Dis 29:361–366
Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, Subramaniam VN, Powell LW, Anderson GJ, Ramm GA (2003) Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 361:669–673
Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D, Camaschella C (2003) Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 33:21–22
Fleming RE, Britton RS, Waheed A, Sly WS, Bacon BR (2004) Pathogenesis of hereditary hemochromatosis. Clin Liver Dis 8:755–773
Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, Andres L, MacFarlane J, Sakellaropoulos N, Politou M, Nemeth E, Thompson J, Risler JK, Zaborowska C, Babakaiff R, Radomski CC, Pape TD, Davidas O, Christakis J, Brissot P, Lockitch G, Ganz T, Hayden MR, Goldberg YP (2004) Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet 36:77–82
Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C (2005) Hepcidin is decreased in TFR2 hemochromatosis. Blood 105:1803–1806
Andrews NC (2008) Forging a field: the golden age of iron biology. Blood 112:219–230
Kemna EH, Tjalsma H, Willems HL, Swinkels DW (2008) Hepcidin: from discovery to differential diagnosis. Haematologica 93:90–97
Wrighting DM, Andrews NC (2008) Iron homeostasis and erythropoiesis. Curr Top Dev Biol 82:141–167
Iacopetta BJ, Morgan EH, Yeoh GC (1982) Transferrin receptors and iron uptake during erythroid cell development. Biochim Biophys Acta 687:204–210
Anderson GJ, Frazer DM (2005) Hepatic iron metabolism. Semin Liver Dis 25:420–432
Acknowledgment
G.J.A. is the recipient of a Senior Research Fellowship from the National Health and Medical Research Council of Australia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Anderson, G.J., Vulpe, C.D. Mammalian iron transport. Cell. Mol. Life Sci. 66, 3241–3261 (2009). https://doi.org/10.1007/s00018-009-0051-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0051-1