
Abstract
In this review we cover key issues, concerned with the effects of cytokines in cardiac tissue and cardiac cells. We discuss the effects of proinflammatory cytokines under non-pathological conditions and their mechanisms dependent and independent of nitric oxide. The role of proinflammatory cytokines is considered in acute myocardial infarction and in heart failure. We also describe proinflammatory cytokines as inductors of arrhythmia. We discuss ionic current alternation as possible mechanisms of cytokines action in heart. We consider TNF-α as a possible player in this signaling cascade. It was shown that TNF-α induced alternation of transmembrane action potentials. Influence of TNF-α on transient outward current (I to), I Kur, I Kr, I Ks, I K1 is also reported. We discuss the interplay between TNF-α and Ca2+ current, influence of TNF-α on SERCA. Then we consider influence of IL-1 on action potentials, I Na, I Ca, I K. We also address the role of IL-2, IL-6, and IL-11. Finally using TNF-α and IL-6 as an example we discuss the effects of cytokines on mechanoelectrical feedback. Perfusion of cardiac tissue with TNF-α containing solution leads to abnormalities in cardiac electrical activity, majorly to prolongation of APD90 and appearance of hump-like depolarization at APD90 level. After reaching E c hump-like depolarization transforms into extra-AP, leading to sustained arrhythmias. TNF-α activates NO cardiomyocyte synthases and the rise of intracellular NO levels opens MGCs, which leads to sodium entry into the cell, which depolarizes cellular membrane, shifting resting potential towards E C. We proposed and proved that TNF-α triggered arrhythmias can be mediated through activation of MGCs. Stretching of preparations removed TNF-α. Perfusion of preparation with IL-6 containing solution leads to fibrillation in response to low levels of stretch. IL-6 mechanisms of action are mediated by NO synthases in cardiomyocytes. The circulating levels of TNF-α and IL-6 were found to be significantly higher in patients suffering from atrial fibrillation. It suggests a positive feedback between inflammation and atrial fibrillation. Proinflammatory cytokines are believed to be markers of atrial fibrillation, and more over, the key element in this positive feedback system.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Heart
- Arrhythmia
- Proinflammatory cytokines
- Cardiomyocytes
- Mechanoelectrical feedback
- Mechanically gated channels
- TNF-α
- IL-2
- IL-6
- IL-11
- Nitric oxide
- Ionic currents
5.1 Introduction
First cytokine related investigations date back to 1950s, but till present a number of questions in this field remain unanswered. For example till late 2000s it was generally accepted that biological effects of cytokines are known. However recently it was found that their effects expand far beyond the immune system and affect many organs and tissues. Latest studies of cardiovascular pathology genesis lead to emergence of a new related field, which can be called “Cardio-immunology”. The role of cytokines in regulation of cardiac function is well described in literature. Unfortunately much fewer studies investigated the cross talk between cytokines effects and effects of myocardium stretch of healthy and pathological tissue. First studies in this field addressed this topic on whole heart level. For example one lab studied the induction of myocardial stretch in an isolated perfused Langendorff preparation by inflation of an intraventricular balloon to an end-diastolic load, which according to their publication resulted in a nearly six fold increase in VEGF message level. They reported a possibility of stretch-induced induction of VEGF expression (Li et al., 1997). Later works focused primarily on cytokine production during stretching of cardiac cells in culture (For example Yokoyama et al., 1999). This field of research remains relatively new and to our knowledge there is only a few number of reviews addressing this topic (for example see Chapter 2 of this Volume). The effects of cytokines on stretched heart and their role in regulation of mechanoelectrical feedback remains unknown. Moreover the role of cytokines in regulation of ion channel activity, their single channel currents and action potentials remain unaddressed. For this reasons we focused our chapter on discussion of the role of main proinflammatory cytokines in regulation of heart bioelectrical activity. In conclusion of our review we analyze the role of a number of cytokines in regulation of mechanoelectrical feedback.
5.2 Physiological Role of Proinflammatory Cytokines
5.2.1 Effects of Proinflammatory Cytokines under Non-pathological Conditions and Their Mechanisms Dependent and Independent of Nitric Oxide
The main and the most obvious physiological function of proinflammatory cytokines is induction of inflammation in the case of tissue damage. However, some of them demonstrate pleiotropic nature, for example IFN-γ is considered a proinflammatory cytokine, but possesses antiviral activity and induces activation of cytotoxic T cells (Dinarello, 2000). The basic mechanism of proinflammatory cytokine-mediated promotion of inflammation is up-regulation of several genes encoding the mediators of inflammation, which are usually not produced in healthy persons. For example, some of these genes code for enzymes that increase the synthesis of leukotrienes, prostanoids and NO. Others are genes of chemokines, the small peptides that facilitate the passage of leukocytes from bloodstream into the tissues, and endothelial adhesion molecules, which are essential for leukocytes adhesion to endothelium (Dinarello, 2000). Of course, proinflammatory action of cytokines is expressed mostly in the damaged tissue, however, as far as proinflammatory cytokines can get to the bloodstream and induce fever and other types of systemic response, they may affect all other organs located far from the nidus of inflammation. Heart is one of their possible targets.
That is why cardiotropic effects of proinflammatory cytokines undoubtedly merit extensive investigation. In the last two decades various effects of proinflammatory cytokines, mostly TNF and IL-1, on contractile and electrical activity of myocardium were demonstrated in diverse in vivo and in vitro studies. These effects may be divided in two groups: the early ones begin to develop immediately after cytokine application and completely develop within the first 3 h, while the delayed response is lasting from hours to days (Prabhu, 2004a).
The early response may be either cardiostimulatory or inhibitory depending on the concentration of cytokine and prevailing cellular and biological milieu. For example, low concentrations of TNF-α (200–500 U/ml) cause transient decrease of AP duration and amplitude of peak Ca2+ transient without affecting I Ca,L in adult mammalian cardiomyocytes (Yokoyama et al., 1993; Sugishita et al., 1999; Stamm et al., 2001a, b). However, high concentrations (≥10,000 U/ml) were observed to produce either inhibition of I Ca,L accompanied with depression of peak Ca2+ transient (Krown et al., 1995) or no changes in Ca2+ turnover despite reduced contractility (Goldhaber et al., 1996). In other studies early positive inotropic effects of TNF-α mediated by increase of peak Ca2+ transient were observed in adult rat cardiomyocytes (Cailleret et al., 2004; Amadou et al., 2002). Other proinflammatory cytokines: IL-1β (Liu and Schreur, 1995; Schreur and Liu, 1997), IL-6 (Sugishita et al., 1999) and IL-2 (Cao et al., 2003a, b) were shown to suppress contractile activity in moderate concentrations (about 1–5 ng/ml). This effect is mediated by I Ca,L depression in the case of IL-1β.
Alteration of Ca2+ cycling is not the only mechanism of immediate cytokine effects. Stimulation of cytokine receptors also leads to rapid induction of NO production by cNOS as well as activation of sphingomyelinase and PLA2-dependent signaling cascades (Prabhu, 2004a; Hedayat et al., 2010). The role of cNOS activation in distribution of early negative inotropic effects was demonstrated for TNF-α (Finkel et al., 1992; Goldhaber et al., 1996), IL-1β (Cain et al., 1999; Kumar et al., 1999), IL-2 (Finkel et al., 1992; McGowan et al., 1994) and IL-6 (Finkel et al., 1992; Kinugawa et al., 1994; Sugishita et al., 1999) in various cardiac preparations and isolated cardiomyocytes. Some of these authors argue that NO-dependent negative inotropic effects of proinflammatory cytokines are produced via activation of soluble guanylate cyclase and subsequent elevation of intracellular cGMP level (Kinugawa et al., 1994; Kumar et al., 1999). On the contrary, several other groups presented serious argumentation against participation of NO-dependent pathway in mediation of acute cytokine effects (Yokoyama et al., 1993; Edmunds et al., 1999; Grandel et al., 2000). Thus, interrelations between cytokines and NO signaling cascade are rather controversial.
It is known that proinflammatory cytokines activate sphingomyelinases in several cell types (Liu et al., 1998; Kolesnick 2002). Sphingomyelinases hydrolyze membrane phospholipide sphingomyeline to form ceramide, which, in turn, can be deacylated by ceramidase to yield sphingosine. This amino alcohol substance and its metabolite sphingosine-1-phosphate act as a second messenger, producing suppression of Ca2+ turnover in cardiomyocytes (Dettbam et al., 1994; Friedrichs et al., 2002). Several studies have indicated the possible contribution of sphyngolipid signaling pathway to early negative inotropic effects of TNF-α in adult feline (Oral et al., 1997) and guinea pig cardiomyocytes (Sugishita et al., 1999), rat ventricular myocardium (Hofmann et al., 2003), isolated rat (Edmunds and Woodward, 1998) and rabbit hearts (Stamm et al., 2001a, b). The role of sphingomyelinase signaling cascade in the contractile effects of other proinflammatory cytokines is sparsely studied, although it was proposed for negative inotropic effect of IL-1β in human atrial trabeculae (Cain et al., 1999). Interestingly, activity of neutral sphingomyelinases is inhibited by glutathione, therefore negative inotropic effect of cytokines exerted via activation of sphingomyelinase signaling cascade depends on the intracellular redox state. In vivo administration of glutathione precursor N-acetylcysteine blocks early cardiodepressant effects of TNF-α, unmasking the positive inotropic effect of this cytokine (Cailleret et al., 2004).
Effects of proinflammatory cytokines attributed to activation of sphingolipid pathway are at least partly dependent on another second messenger – arachidonic acid. TNF-α and IL-1β stimulate phospholipase A2 via activation of their receptors and therefore increase the intracellular level of arachidonic acid (Jayadev et al., 1994; Liu and McHowat, 1998). This messenger activates neutral sphingomyelinases (Robinson et al., 1997), but also has own positive inotropic effect in cardiomyocytes, which is produced via increase of Ca2+ transients (Kang and Leaf, 1994; Damron and Summers, 1997). Therefore, small concentrations of arachidonic acid improve myocardial contractility, while higher concentrations suppress it due to the activation of sphingolipid pathway (Amadou et al., 2002; Damron and Summers, 1997). That is why some researchers explain the dual inotropic effects of TNF-α by such dual action of arachidonic acid (Amadou et al., 2002). Arachidonic acid may also mediate the positive inotropic effect of IL-2 (Bracco et al., 1991).
Delayed effects of proinflammatory cytokines are substantially less controversial than the acute ones. It is generally accepted that sustained application of proinflammatory cytokines, in the first place TNF-α and IL-1β, induces marked decrease of basal myocardial contractility which is aggravated by suppressed responsiveness to stimulation of β-adrenoreceptors (Prabhu, 2004a, b). The latter effect for the first time was demonstrated in rat neonatal myocytes (Gulick et al., 1989; Chung et al., 1990). Authors explained this effect by loss of β-adrenoreceptors ability to activate adenylate cyclase, because cytokines didn’t change the density of receptors and on the other hand direct stimulation of adenylate cyclase by forskolin induced normal cAMP accumulation and augmentation of contractile activity. Subsequent studies confirmed that impairment of signal transduction from receptor to adenylate cyclase, which is mediated by Gs protein, is the main reason of decreased β-adrenoreceptors responsiveness (Rozanski and Witt, 1994; Bick et al., 1997).
Several other groups of investigators believe that suppression of β-adrenoreceptors responsiveness is mediated by sustained upregulation of iNOS leading to increased cGMP intracellular content resulting in decrease of cAMP level due to the activation of PDE2 by cGMP (Balligand et al., 1993, 1994; Balligand, 1999; Ungureanu-Longrois et al., 1995, 1997; Joe et al., 1998). It seems likely that both NO-dependent and NO-independent mechanisms of β-adrenoreceptors responsiveness are decreased by proinflammatory cytokines which are present in the myocardium. Moreover, upregulation of iNOS, which is already shown for most of proinflammatory cytokines in the case of their prolonged application (Hedayat et al., 2010), exerts several other crucial consequences such as S-nitrosylation of thiol residues of important regulatory proteins and formation of peroxynitrite, leading together to prolonged depression of contractile function (Campbell et al., 1996; Massion et al., 2003; Prabhu, 2004a). Obviously, increase in generation of reactive oxygen species (ROS), which is also induced in myocardium by different proinflammatory cytokines (Oyama et al., 1998; Cheng et al., 1999; Panas et al., 1998; Ferdinandy et al., 2000), amplifies the latter effect greatly. For example, in isolated rat heart combination of IL-1β, TNF-α and IFN-γ provokes augmentation of xanthine oxidoreductase and NAD(P)H oxidase activity, which is accompanied with enhanced activity of iNOS and leads to sustained decline in contractility (Ferdinandy et al., 2000). It is important to note that oxidative modifications induced by ROS and peroxynitrite tend to be irreversible (Stamler et al., 2001) and are therefore attributed to serious pathological changes in the structure and function of myocardium.
Thus, the modulation of normal heart function by proinflammatory cytokines is extremely complex and is mediated by diverse signaling mechanisms. However, it becomes even more versatile in the case of cardiac disease. Here we briefly discuss the role of proinflammatory cytokines in pathogenesis of myocardial infarction, heart failure and arrhythmias.
5.2.2 Proinflammatory Cytokines in Acute Myocardial Infarction
The onset of myocardial infarction starts from ischemic and subsequent reperfusion injury of myocardium leading to cardiomyocytes necrosis. Proinflammatory cytokines have significant impact on heart function during all these stages. Although they promote myocardial malfunction during and after ischemic injury, cytokines are able to increase resistance of heart to such injury if added prior to ischemia.
It is accepted that application of TNF-α (Nakano et al., 1998; Eddy et al., 1992), IL-1β (Brown et al., 1990; Maulik et al., 1993) and IL-6 (Smart et al., 2006) prior to induction of ischemia-reperfusion injury exerts clear beneficial cardioprotective effects. The mechanisms of such cytoprotective preconditioning are diverse, but in the case of TNF-α and IL-1β they are undoubtedly associated with induction of manganous superoxide dismutase (MnSOD), which neutralizes the cytotoxic oxygen free radicals, which are generated in the myocardium during ischemia-reperfusion (Chen et al., 1998; Wong and Goeddel, 1988; Maulik et al., 1993). The second substantial protective mechanism is concerned with upregulation of various heat-shock proteins, which attenuate the excessive myocardial production of inflammatory cytokines, following the ischemic and reperfusion injury and having strong deleterious impact on cardiac function (Nakano et al., 1996). IL-1β preconditioning promotes accumulation of leukocytes and generation of H2O2 in myocardium, leading to moderate oxidative stress and induction of cytoprotective mechanisms, which increase resistance of myocardium to ischemic injury (Brown et al., 1990). IL-6 preconditioning is mediated mainly by inhibition of apoptotic signaling pathways in cardiomyocytes via PI3K/Akt-mediated activation of iNOS (Smart et al., 2006).
It is not surprising that during ischemia and subsequent reperfusion proinflammatory cytokines aggravate irreversible injury of myocardium (Schulz, 2008; Schulz and Heusch, 2009). In ischemic myocardium negative effects of TNF-α, such as induction of apoptosis (Haudek et al., 2007) are produced via activation of first type TNF receptors (Flaherty et al., 2008) and can be prevented by treatment with TNF-α antibodies (Belosjorow et al., 2003) or soluble TNF receptors of first type (Sugano et al., 2004), which are capable of binding and inactivation of TNF-α. IL-1 also enhances apoptosis during ischemia and reperfusion (Haudek et al., 2007). This effect is mediated by activation of NO production (Ing et al., 1999) via stimulation of IL-1 receptors and can be blocked by antagonists of these receptors (Abbate et al., 2008). Similarly to IL-1, IL-6 provokes contractile dysfunction in ischemic myocardium via activation of iNOS mediated by JAK2/STAT3 signaling pathway (Yu et al., 2003a, b). In the last years IL-18 was accepted as a new powerful proapoptotic factor, which plays very negative role during ischemia. This cytokine promotes apoptosis in cardiomyocytes via upregulation of death receptors leading to enhancement of extrinsic proapoptotic signaling (Marino and Cardier, 2003; Chandrasekar et al., 2004), activates caspases and suppresses antiapoptotic PI3K/Akt pathway via induction of phosphatase and tensin homolog expression (Chandrasekar et al., 2006).
After the ischemic lesion the myocardium itself becomes the source of proinflammatory cytokines. Obviously, the increased production of cytokines endangers nearby cells altering cardiomyocytes structure and function. However, infarction leads to increased level of proinflammatory cytokines in blood and, as a result, severe systemic inflammatory response syndrome known as the leading cause of death in patients with acute myocardial infarction. TNF-α, IL-6 and, especially, IL-1β play the key role in its pathogenesis (Debrunner et al., 2008). Nevertheless, we will focus on alteration of heart function caused by proinflammatory cytokines, produced in the heart itself after myocardial infarction.
Firstly, all acute cardiotropic effects of proinflammatory cytokines, described in Section 5.2.1, take place after infarction but in much bigger extent than in normal conditions, because concentration of cytokines in the heart are many times higher. Second, described proapoptotic action of cytokines aggravates after infarction. Moreover, other consequences of inflammation, such as infiltration of leukocytes and increase of ROS production take place in the heart and together with proper cytokines’ effects lead to progressive loss of cardiac function. Finally, expression of proinflammatory cytokines provokes hypertrophy of cardiomyocytes and remodeling of extracellular cardiac matrix. These effects potentiate delayed cardiodepressant action of cytokines (see Section 5.2.1) and accelerate development of cardiac failure following acute myocardial infarction (Hedayat et al., 2010).
5.2.3 Role of Proinflammatory Cytokines in Heart Failure
Hypertrophic growth response is shown for all proinflammatory cytokines (Hedayat et al., 2010). Hypertrophic effects of TNF-α, measured in terms of cardiomyocytes size (both length and width), cardiac weight and left ventricular wall thickness (Janczewski et al., 2003) are at least partly mediated by upregulation of angiotensin-converting enzyme and resulting increase in angiotensin II protein level (Flesch et al., 2003). TNF-α stimulates expression of other proinflammatory cytokines in myocardium, which in turn amplify its own hypertrophic action. Hypertrophic effect of IL-1, which was demonstrated in several in vitro (Palmer et al., 1995; Thaik et al., 1995) and in vivo studies (Nishikawa et al., 2006), is NO-independent and appears to be mediated through a tyrosine kinase signaling pathway (Palmer et al., 1995; Thaik et al., 1995). IL-6 also contributes to hypertrophic growth response (Yamauchi-Takihara and Kishimoto, 2000), which is believed to be mediated mainly through JAK/STAT3 signaling pathway (Kunisada et al., 2000). On the contrary to IL-1 and IL-6, hypertrophy of cardiomyocytes induced by IL-18 is mediated through PI3K cascade (Chandrasekar et al., 2005; Colston et al., 2007). Thus, cytokines produced in the heart due to acute infarction induce hypertrophic growth of cardiomyocytes, which primarily helps to regain pumping function, but frequently transforms to maladaptive response leading to progressive heart failure.
Alterations of collagenous extracellular matrix (ECM) providing the physical scaffolding for the spatial organization of cardiomyocytes into cardiac tissue have been demonstrated to play a central role in cardiac remodeling and resulting progressive heart failure (Fedak et al., 2005; Ju and Dixon, 1996). It is shown that TNF-α (Siwik and Colucci, 2004), IL-1β (Siwik et al., 2000) and other proinflammatory cytokines (Hedayat et al., 2010) cause imbalance between synthesis and degradation of ECM through disregulation of degradative enzymes and of matrix metalloproteinases (MMPs) activity. These effects are largely influenced by the duration of exposure, which ranges from increased fibrillar collagen degradation to excessive fibrillar collagen deposition. For example, in short term, activation of MMPs induced by TNF-α leads to enhanced degradation of ECM components which promotes progressive dilation of left ventricle (Sivasubramanian et al., 2001; Li et al., 2000). However, in long term, sustained action of TNF-α results in excessive collagen deposition and increased LV stiffness (Sivasubramanian et al., 2001).
Thus, increased expression of proinflammatory cytokines in the myocardium leads to sustained depression of cardiomyocytes contractile activity (see Section 5.2.1), which is accompanied with even more oppressive irreversible effects: cardiomyocytes hypertrophy and ECM remodeling and results in chronic heart failure.
5.2.4 Proinflammatory Cytokines as Inductors of Arrhythmia
The possibility of proinflammatory cytokines contribution to pathogenesis of cardiac arrhythmias is not so obvious as in the case of myocardial infarction and cardiac failure. Among cardiac arrhythmias, the most common and well-studied is atrial fibrillation (AF). However the specific mechanism of AF initiation remains unknown. It is generally accepted that AF is associated somehow with inflammation. The circulating levels of IL-6 (Marcus et al., 2008), TNF-α (Sata et al., 2004) and IL-18 (Luan et al., 2010) were found to be significantly higher in patients suffering from AF. However, it is unclear whether inflammation is a cause or an effect of AF and, in either case, what mechanistic pathways may be important. One of the hypothesis proposes that AF leads to myocyte calcium overload, promoting atrial myocyte apoptosis, which, in turn, induces local inflammation and complement activation. Tissue damage then ensues and fibrosis sets in (Avilles et al., 2003; Demelis and Panaretou, 2001). Locally released factors of inflammation, including cytokines, can further contribute to membrane dysfunction by inhibiting the calcium cycling machinery. This can eventually lead to the maintenance of AF (Avilles et al., 2003; Demelis and Panaretou, 2001, 2006).
Some experimental data sheds light on mechanism of arrhythmogenic action of proinflammatory cytokines in atrial myocardium. Administrating TNF-α in rats can induce arrhythmias (Krown et al., 1995) and mice with overexpression of TNF-α have a greater incidence of AF (London et al., 2003). The latter is associated with abnormalities in systolic and diastolic Ca2+ handling and may be due to them. In particular, enhanced expression of TNF-α is accompanied by increase in frequency and magnitude of spontaneous Ca2+ releases from the sarcoplasmic reticulum in working cardiomyocytes (Saba et al., 2004). Such releases may lead to induction of delayed afterdepolarizations, which are well known triggers for AF, via activation of reverse Na+/Ca2+ exchanger. In cardiomyocytes from pulmonary veins, which are believed to be important foci of ectopic beats initiating AF or tachycardia (Chen et al., 1999), TNF-α also markedly alters the calcium homeostasis in different ways, increases amplitudes and frequency of delayed afterdepolarizations and thereby increases pulmonary veins ability to initiate AF (Lee et al., 2007).
Thus, there seems to be a positive feedback between inflammation and AF and proinflammatory cytokines are believed to be not only markers of AF, but the key element in this positive feedback system. Moreover, various anti-inflammatory agents, such as statins or angiotensin-converting enzyme inhibitors, are able to suppress atrial remodeling associated with AF and, therefore decrease the possibility of new AF paroxysms (Issac et al., 2007).
The role of proinflammatory cytokines in ventricular arrhythmias induction was also studied just recently. In dogs, etanercept, which binds and inactivates TNF-α, decreased probability of ventricular tachyarrhythmias after coronary artery ligation (Yu et al., 2005a, b), indicating important role of this cytokine in sudden cardiac death. Mechanisms of proarrhythmic effects of cytokines in ventricles seem to be very similar to shown in atrial myocardium. TNF-α (Duncan et al., 2010; Saba et al., 2004) and IL-1β (Duncan et al., 2010) suppress total release of Ca2+ from the sarcoplasmic reticulum induced by AP, but promote local spontaneous Ca2+ releases during diastole. These releases lead to initiation of delayed afterdepolarizations via activation of depolarizing Na+/Ca2+ exchanger current.
5.3 Ionic Current Alternation as a Possible Mechanisms of Cytokines Action in Heart
5.3.1 Tumor Necrosis Factor-α (TNF-α)
Tumor necrosis factor-α (TNF-α) is a proinflammatory cytokine that is implicated in a number of pathophysiological events. It was established that TNF-α affects myocardium through several pathways. TNF-α influences both mechanical and bioelectrical activity of myocardium by alternating membrane potassium and calcium currents and cytoplasmic calcium handling. It was also shown that tumor necrosis factor-α is able to induce rapid as well as slow effects in cardiac muscle.
A lot of studies were carried out to clarify mechanisms of TNF-α action in heart, but often, represented results are contradictory, especially in cases of investigation of cardiac bioelectrical activity. It is necessary to mention that in animal models or patients with heart failure serum TNF-α concentrations belong to pictogram range (up to 10 pg/ml) (Cugno et al., 2000; Testa et al., 1996). While, TNF-α concentration range, utilized to treat heart preparation and cells in electrophysiological studies extend from picogram to hundreds of nanogram per milliliter.
Here some aspects of TNF-α influence on action potentials and ionic currents in heart and TNF-α induced contraction reduction are discussed.
5.3.1.1 TNF-α Induced Alternation of Transmembrane Action Potentials
In isolated guinea pigs papillary muscle (Alloatti et al., 1999) TNF-α in concentration of 1–10 ng/ml induce APD decrease by 10–15% after short term (20–25 min) cytokine administration (Fig. 5.1a). Decreasing of TNF-α induced AP duration was concentration- and time-dependent. APD decreasing was accompanied by reduction of the contractile force. However, as Alloatti reports, the resting membrane potential, the overshoot and the maximum rate of depolarization of the APs were not affected by TNF-α. Recovery of mechanical activity and APs parameters was observes after 20 min of washout by solution free from TNF-α.

TNF-α alternate action potential duration in heart. (a) Effects of TNF-α (10 ng/ml) on action potential duration (APD) in the isolated guinea pig papillary muscle (from Alloatti et al. (1999) with permission of Elsevier and British Cardiac Society from Copyright Clearance Center). (b) Effects of TNF-α (10 ng/ml, 6-h incubation at 4°C) on action potential duration in canine ventricular cells (from Wang et al. (2004) with permission of American Society for Biochemistry and Molecular Biology via Copyright Clearance Center)
Effects of TNF-α on APs were completely abrogated by pretreatment of papillary muscle by the NOS inhibitor – l-NAME or PAF-receptor antagonists (PAF – platelet aggregation factor, CV 3988). Also TNF-α-induced APD decreasing was abolished by chemical (Triton X-100) endocardial endothelium removing. Alloatti et al. suggested that both the generation of NO and PAF production contributes to the TNF-induced alteration of APD. It was also speculated that the production of NO is consequent to the production of PAF.
TNF-α influence on APs of isolated canine ventricular cardiomyocytes was opposite to that, observed in guinea pigs papillary muscle. APD was significantly longer in canine cells pretreated with TNF-α (Fig. 5.1b) then in control cells. Alternation of APD occurred only after 10 h of incubation with 10 ng/ml TNF-α (Wang et al., 2004).
Effects of TNF-α were investigated in experiments with isolated rat ventricular cardiomyocytes. In this study it was shown that after 48 h of exposition of cardiomyocytes to 5 ng/ml TNF-α APD increased more than two times (Fernandez-Velasco et al., 2007).
In ventricular cardiomyocytes, isolated from normal mice, which received chronically NTF-α significant alternation of ionic currents was observed, while AP duration remained unaltered (Grandy and Fiset, 2009). Only slight reduction of AP amplitude was observed. Authors speculate, that the lack of TNF-α-induced APs alterations could be due to counterbalanced alternation of repolarizating and depolarizating currents (resulting in APD preservation).
Transgenic mice that overexpress TNF-α selectively in the heart tissue have been studied as a model of congestive heart failure (Janczewski et al., 2003; Kubota et al., 1997; London et al., 2003; Sivasubramanian et al., 2001). TNF-α- overexpressing mice develop atrial and ventricular arrhythmias. In such mice heart level of TNF-α exceeds cytokine level usually reached in disease state more than ten times (Kubota et al., 1997). Several electrophysiological investigations were realized with such animals. AP duration in ventricles of perfused isolated hearts and in isolated ventricular transgenic mice cardiomyocytes was significantly lengthened (London et al., 2003; Petkova-Kirova et al., 2006) compared to APD in wild type mice.
Thereby, literature data, describing influence of TNF-α to action potential is contradictory. Some authors report decreasing of APs duration, on the contrary to other reports, TNF-α induce prolongation of APs. These discrepancies may occur due to different animals (mice, rat, dog), experimental protocol and wide range of TNF-α concentration used in experiments.
5.3.1.2 TNF-α and Ca2+ Current
Krown and coworkers has shown that in isolated rat ventricular cardiomyocytes TNF-α caused significant inhibition of I Ca which mediated by TNF-α receptors TNFRI (Krown et al., 1995). TNF-α inhibition of the I Ca was dose dependent (Fig. 5.2), but significant inhibition of I Ca was observed in presence of very high concentration of TNF-α (>300 ng/ml), which 1000 times exceeds cytokine level observed in the pathophysiological condition (≈0.01–0.05 ng/ml) (Testa et al., 1996). As follows from I–V curves TNF-α exerts an inhibitory action at all tested potentials. Only partial recovery of I Ca from inhibition was observed after TNF-α washout. Therefore, authors conclude involvement of intracellular second messengers in inhibitory effects of TNF-α.

TNF-α alternate cardiac I Ca,L. (a) L-type channel recordings from a whole cell patch- clamped adult rat cardiac myocyte (5 min, 18,000 U/ml). There was a partial recovery after washout. Control experiments using fatty acid free BSA (data not shown) failed to show a nonspecific protein effect on the channel in the range of protein used in the TNF-α experiments. (b) Current–voltage relationship for I Ca,L in the same cell prior to TNF-α application (open circles), 5 min after TNF-α application (closed circles) and after washout (closed triangles). (c) Average dose-dependent effect of TNF-α on L-type channel current in adult cardiac myocytes demonstrated half-maximal inhibition with 6400 U/ml. Holding potential –50 mV, pulses to 0 mV (from Krown et al. (1995) with permission of Elsevier Limited and FEBS Letters via Copyright Clearance Center)
Therefore, Krown and colleagues assume I Ca suppression as a significant component of mechanism of TNF-α cardiodepressant action in different pathophysiological conditions.
On the contrary several studies reported lack of TNF-α effects on I Ca. In isolated rat and cat ventricle myocytes treated by TNF-α peak I Ca suppression and alternation of I–V relation were not detected (Yokoyama et al., 1993; Fernandez-Velasco et al., 2007).
Same observation was made in cardiomyocytes isolated from NTF-a overexpressing mice (Petkova-Kirova et al., 2006). Measurements of I Ca,L showed no difference in the peak current density between control and TNF-overexpressing myocytes over the range of potentials from –10 to +50 mV. At more negative potentials I Ca,L was slightly larger in TNF-α myocytes. No differences of inactivation and deactivation kinetics of I Ca,L were detected.
Hereby, ability of TNF-α to alternate I Ca in cardiomyocytes remains questionable.
5.3.1.3 Influence of TNF-α on Transient Outward Current (Ito)
Under various pathological conditions, including heart failure, prolongation of action potentials, which occurs due to downregulation or suppression of the potassium currents (I K) (Nuss et al., 1999; Beuckelmann et al., 1993) was observed. Depression of I K and AP prolongation increases probability of arrhythmogenic events in heart (Nattel et al., 2007). Transient outward current (I to) plays an essential role in early repolarization and regulation of duration of APs in myocardium (Hund and Rudy, 2004). In several studies it was shown that failing hearts (in human and in animals model) demonstrate significantly decreased I to (Benitah et al., 1993; Kaab et al., 1998; Li et al., 2004). Isolated cardiomyocytes from transgenic mice overexpressing TNF-α demonstrate prolonged APs (London et al., 2003). Therefore, hypothesis accounting TNF-α as a mediator of I to reduction was elaborated. In turn, TNF-α could modulate I to through mechanisms involving upregulation of iNOS, NO and oxidant species (Fernandez-Velasco et al., 2007).
In study developed by Fernandez-Velasco isolated rat ventricular cardiomyocytes were exposed to TNF-α in concentration of 1–10 ng/ml for 48 h. After exposure to TNF-α amplitude of I to was significantly reduced. Suppression of I to was accompanied by AP prolongation. Inhibitory effect of TNF-α was concentration-dependent. TNF-α affect amplitude and density of current but not voltage dependence. Inactivating curve for I to in cells treated with TNF-α was shifted to negative potentials. Half-maximal inactivation was –49.9 mV in control and –55.6 mV in cells treated with TNF-α. Authors also showed that exposure to TNF-α leads to lowering of I to channel proteins level.
Thereby, depression of I to is caused by several mechanisms – decreasing of current density, lowering level of channels proteins and shifting of inactivating curve to negative potentials.
The decrease of I to induced by TNF-α was fully reversed in cells pretreated by iNOS inhibitors. Similarly, preliminary exposure of cardiomyocytes with antioxidants also reverses TNF-α induced I to decreasing. Discussing these data, authors support the idea that highly reactive oxidant species, such a peroxynitrite, formed by NO and superoxide anions, play a central role in TNF-α-induced ionic currents suppression. This report was hypothesized that posttranslational modification of I to channels by peroxynitrite (nitrosylation, nitration or oxidation) is the main mechanism of TNF-α action (Fernandez-Velasco et al., 2007).
Investigation provided by Kawada and colleagues was focused on TNF-α effect in cultured cardiomyocytes from neonatal (1-day-old) rats (Kawada et al., 2006). Cultured cells were incubated during 48 h with 50 ng/ml TNF-α. Peak I to was significantly reduced in TNF-α – treated myocytes compared with the control cells (17.7 and 3.2 pA/pF respectively). Expression of I to-related mRNA was also reduced.
Study of mice ventricular cardiomyocytes, similar to results obtained in rat and dog cardiomyocytes, demonstrated TNF-α – induced inhibition of I to (by chronic TNF-α treatment) (Grandy and Fiset, 2009). However, depression of I to was not accompanied by lowering of mRNA or protein expression level.
In transgenic mice overexpressing NTF-a, ventricle cardiomyocytes demonstrate significantly decreased I to (Petkova-Kirova et al., 2006). Level of channels proteins responsible for I to was measured in this study. As in rat ventricular cardiomyocytes, transgenic mice show lowered level of I to-related proteins.
Thereby, in studies utilized different animals and experimental protocols it was shown that TNF-α causes reduction of I to. Such convergent results make possible speculation that preferentially I to depression is responsible for AP alternation and plays an important role in TNF-α-induced heart dysfunctions.
5.3.1.4 TNF-α Induced Alternation of IKur
In most species, including human, ultrarapid-activating K+ current (I Kur) was present only in atrial myocardium (Nerbonne and Kass, 2005). Studies focused on electrophysiological effects of NTF-α in heart predominantly deal with ventricular cardiomyocytes. This is, probably, the reason why there is insufficient information about NTF-α influence on the I Kur. TNF-α-induced alternation of I Kur was investigated only in mice. As shown previously, ultrarapid K+ current (also called I K,slow1) is present in adult mice ventricular myocardium (Liu et al., 2011; Brouillette et al., 2004).
In study provided by Grandy and Fiset I Kur parameters were measured in isolated mice ventricular cardiomyocytes after NTF-a chronic (6 week in vivo) administration (Grandy and Fiset, 2009). In cardiomyocytes from TNF-α pretreated mice density of I Kur was significantly smaller than in cell from control animals. I–V curves for I Kur reveal significant difference, especially at positive potentials (Fig 5.3a). Inactivation of I Kur.was similar in myocytes from all animals. Recovery from inactivation was significantly longer in the cells isolated from TNF-α-treated then from control animals. Authors suggested that the prolongation in the recovery from inactivation of I Kur could result in a reduction in current. Also this study revealed that level of mRNA and expression of proteins related to I Kur were not altered by chronic administration of TNFα. Posttranslation modification of channels as result of reactive oxygen species generation in cardiomyocytes is postulated as a main mechanism of TNF-α action.

Reduction of I Kur by TNF-α. (a) I Kur was markedly reduced in myocytes from TNF-α treated mice compared to myocytes from control mice. Left: Representative I Kur traces for a control myocyte (top) and a myocyte from a TNF-α treated mouse (bottom). I Kur was calculated by subtracting currents recorded in the presence of 100 μmol/l 4-AP from currents recorded in the absence of 4-AP (voltage protocol shown in inset). Right: The mean I/V curves show that I Kur was significantly reduced in myocytes from TNF-α treated mice compared to myocytes from control animals at voltages of +20 mV to +50 mV. (b) Slowly inactivating delayed rectifier potassium current I K,slow1 suppressed in TNF-α overexpressing mice. Left: Representative families of I K,slow1 currents in control wild-type cells (top) and TNF cells (bottom). Right: Mean I/V relations for I K,slow1 in cells from TNF mice (squares) and control wild-type mice (triangles) (from Grandy and Fiset (2009) with permission of Academic Press, International Society for Heart Research and Journal of Molecular and Cellular Cardiology via Copyright Clearance Center)
I K,slow1 was measured in transgenic mice, overexpressing TNF-α. In TNF-α cardiomyocytes at potentials positive than –10 mV the density of I K,slow1 was significantly smaller than in cells from wild type mice (Fig. 5.3b). The mean I K,slow1 density measured at +40 mV in TNF-α cardiomyocytes was suppressed approximately two times in comparison to controls (22.6 and 12.5 pA/pF respectively). In contrast with previous, level of channels proteins responsible for I K,slow1 was reduced by more than 40% (Petkova-Kirova et al., 2006).
5.3.1.5 Role of TNF-α in Regulation of K+ Currents – IKr, IKs
Down-regulation of K+ current, including rapid component of delayed-rectifier current – I Kr observed in congestive heart failure is accompanied by prolongation of APs (Nattel et al., 2007). Pore-forming α-subunit of I Kr is encoded by human “ether-a-go-go”-related gene (“HERG channels”). In human ventricular myocardium I Kr is the main contributor to AP repolarization among delayed currents (Virag et al., 2001). Excessive decreasing of I Kr leads to early afterdepolarization and tachyarrhythmias (Rashmi and Hondeghem, 2005; Priebe and Beuckelmann, 1998). As was mentioned above serum level and tissue production of TNF-α in such pathological condition as heart failure is highly increased.
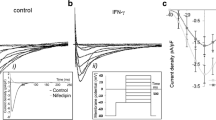
In study provided by Wang et al. (2004) it was shown that short time (15 min) exposure of isolated dog atrial and ventricular cardiomyocytes to TNF-α leads to concentration-dependent decreasing of I Kr (Fig. 5.4a). Amplitude of HERG channels current was decreased by 9 and 35% after exposure to 1 and 100 ng/ml of TNF-α respectively. Effect of NTF-α was larger at more negative potentials. However, I Kr kinetics was unaltered by TNF-α. Depression of I Kr by TNF-α was accompanied by AP duration prolongation (Fig. 5.4b).

Impairment of I Kr function by TNF-α. (a) Three sets of current traces at test potentials between –60 and +60 mV in 10-mV increments. (b) Tail current amplitudes plotted against test potentials. Solid square indicates the control condition; solid circle, exposure to 20 nmol/l ISO; and solid triangle, after addition of 20 ng/ml TNF-α (from Hatada (2006) with permission of Biochemical and Biophysical Research Communications via Copyright Clearance Center)
In this study it was shown that TNF-α -induced decreasing of I Kr was mediated via TNFR1 receptors. Preincubation of cardiomyocytes with inhibitory anti-TNFR1 antibody for 1 h prevents suppression of I Kr induced by short-time or prolonged TNF-α administration. As in case of I to, author suggests intracellular reactive oxygen species generation as a main mechanism of TNF-α-mediated current suppression. Pretreatment of cardiomyocytes with antioxidants (vitamin E, MnTBAP) for 2 h prevented consequent I Kr reduction induced by TNF-α.
Hatada and colleagues examined effects of NTF-α on delayed-rectifier potassium currents in cardiomyocytes isolated from guinea pig ventricle (Hatada et al., 2006). In these experiments TNF-α suppressed I K only after preliminary treatment of cells with isoproterenol, histamine or forskolin. Suppression of I K in normal condition (at basal level) was not observed. Relating to pharmacological analysis, authors conclude that TNF-α preferentially affects slow component of delayed-rectifier (i.e. I Ks) and that NTF-α inhibits potassium currents by reducing intracellular cAMP level.
5.3.1.6 TNF-α in Regulation of IK1
Scarce data regarding influence of TNF-α on inwardly rectifying K+ current (I K1) is present to our knowledge. Only in Grandy and Fiset study (see above) analysis of I K1 after TNF-α administration was performed. In mice ventricular cardiomyocytes TNF-α has no effects on I K1. The current–voltage (I–V) relationships show that the I K1 was comparable in cells from control and TNF-α pretreated mice (Fig. 5.5).

I K1 after chronic TNF-α treatment in mice. The steady-state outward K+ current (I ss) and the inward rectifier K+ current (I K1) were comparable in ventricular myocytes from control and TNF-α treated mice. (a) Representative traces for I ss and I K1 from a control myocyte (top) and a myocyte from a TNF-α treated mouse (bottom). (b) Mean I/V curves show that I ss and I K1 density were similar in control myocytes and myocytes from TNF-α treated mice. The voltage protocol is shown in the inset. The current density for I K1was measured between −110 and −40 mV. To record I ss, the currents were initiated in the presence 100 μmol/l 4-AP and test steps were preceded with a 100 ms step to −40 mV (from Grandy and Fiset (2009) with permission of Academic Press, International Society for Heart Research and Journal of Molecular and Cellular Cardiology via Copyright Clearance Center)
5.3.1.7 TNF-α Induced Stimulation of IKATP
One study was reported on influence of TNF-α on ATP-sensitive potassium channels in cardiomyocytes (El-Ani and Zimlichman, 2003). In cardiac cell line H9c2 pretreated with 50 ng/ml TNF-α for 30 min was caused activation of I KATP. Preincubation of cardiomyocytes with glibenclamide, I KATP blocker, inhibited the effects of TNF-α. Activation of I KATP leads to depression of action potential and contractile force of cardiomyocytes (Zingman et al., 2007). Unfortunately, in this study ionic current was measured with use of radioactive isotope – 86Rb and no electrophysiological data were obtained. Thereby, the role of TNF-α-induced I KATP activation in APD regulating remains questionable.
5.3.1.8 TNF-α as a Regulator of Cytoplasmic Calcium Handling; Influence on SERCA
TNF-α affects contractility in the heart. Two main visions of TNF-α-induced negative inotropic action in cardiomyocytes exists. First one proposes that there is a reduced myofilament responsiveness to [Ca2+]i which is mediated by nitric oxide or sphingosine production (Finkel et al., 1992; Goldhaber et al., 1996; Oral et al., 1997). Second one proposes that intracellular Ca2+ transients are mediated via down-regulation of structures (SERCA, NCX, RyR) relating to Ca2+ handling (Kao et al., 2010; Janczewski et al., 2003).
In study performed on isolated rabbit and guinea pig ventricular myocytes (Goldhaber et al., 1996) TNF-α affected only cell shortening, but not cytoplasmic calcium transients (amplitude or kinetics). Moreover, TNF-α caused a significant increase in diastolic cell length without any change in diastolic and systolic [Ca2+]i. In this investigation inhibition of NO production by l-NAME prevented TNF-α-induced suppression of myocytes contractile activity. The same observation was made in experiments with isolated hamster papillary muscles (Finkel et al., 1992). Contraction alternation without changing of cytoplasmic calcium level may occur due to a reduction in the responsiveness of the myofilaments to [Ca2+]i. Authors suggest that an NO-mediated increase in cGMP could account for decreased myofilament Ca2+ responsiveness.
TNF-α reduces [Ca2+]i transients in isolated rat and feline cardiomyocytes (Krown et al., 1995; Yokoyama et al., 1993) (Fig. 5.6). TNF-α blocks spontaneous contractions of cultured murine cardiomyocytes (Weisensee et al., 1993). In heart of TNF-α overexpressing transgenic mice (“TNF1.6 mice”) authors observed altered intracellular Ca2+ transients with decreased amplitude and peak systolic Ca2+, elevated diastolic Ca2+, and slower kinetics (Janczewski et al., 2003; London et al., 2003). Also in transgenic mice twitch contractions and response to the β-adrenergic stimulation of myocytes were decreased (Janczewski et al., 2003). However, mechanisms of cytoplasmic Ca2+ transients alternation remain unrevealed.

Impairment of Ca2+ transients in cardiomyocites by TNF-α. Indo 1 Ca2+ transients of an adult rat ventricular myocyte are shown. Ca2+ transient measurements were made at room temperature from the cytoplasmic regions of individual cells electrically paced at 0.3 Hz. At the indicated times, 18,000 U/ml recombinant murine TNF-α was added (from Krown et al. (1995) with permission of Elsevier Limited and FEBS Letters via Copyright Clearance Center)
SERCA2a (sarcoplasmic reticulum Ca2+-ATPase) plays an essential role in the cytoplasmic Ca2+ regulation in cardiomyocytes. Heart failure is accompanied with reduction of SERCA2a level (Meyer et al., 1995). Several studies suggested that key mechanisms of TNF-α-induced contraction dysfunction is the down-regulation of SERCA.
TNF-α has been shown to down-regulate the SERCA2a in the expressions of RNA and proteins level in isolated rabbit cardiomyocytes (Lee et al., 2007). Expression of SERCA mRNA and protein were markedly decreased in mouse cardiac muscle cell line – HL-1 treated by TNF-α (50 ng/ml for 24 h) (Kao et al., 2010). Down-regulation of SERCA was accompanied by reduction of [Ca2+]i transients amplitude.
On the one hand in isolated ventricular myocytes from “TNF1.6 mice” transcripts encoding SERCA 2 and phospholamban (PLB) were significantly reduced in comparison with wild type mice (Kubota et al., 1997; Janczewski et al., 2003). Despite reduced mRNA expression SERCA and PLB protein levels were not different in transgenic and wild type mice. Similarly to previous, the molecules related to Ca2+-handling, such as ryanodine receptor, SERCA and NCX (Na/Ca-exchanger) did not show any specific changes in rat cultured cardiomyocytes after treatment with 50 ng/ml TNF-α (Kawada et al., 2006). Thereby, mechanism of TNF-α-induced cytoplasmic calcium transients alternation remain unclear.
5.3.2 Interleukin IL-1
5.3.2.1 INa
Alterations of Na+ channel function can contribute to rhythm disturbances (Grandy et al., 2010). Disease-mediated alterations in Na+ current could be partially responsible for arrhythmias that have been observed HIV patients (Sani and Okeahialam, 2005). Similarly HIV patients and CD4C/HIV mice have elevated levels of the proinflammatory cytokine IL-1β (Monsuez et al., 2007; Grandy et al., 2010). In ventricular myocytes isolated from HIV mice Na+ peak current was significantly reduced. It is possible that alterations in serum proinflammatory cytokines could mediate the reduction of Na+ current and the corresponding changes in the AP waveform as observed in ventricular myocytes from CD4C/HIV mice. Overall, the results suggest that elevated levels of IL-1β may result in a reduction in Na+ current, altering the action potential upstroke and increasing the risk of arrhythmias (Grandy et al., 2010).
5.3.2.2 ICa,L
IL-1β has been suggested to play a role in impaired cardiac performance under these conditions. This suggestion has been supported by in vitro studies showing that IL-1β decreases cardiac contractility (Evans et al., 1993; Hosenpud et al., 1989; Weisensee et al., 1993).
To determine whether cytokines alter the electrical properties of heart cells, the effects of human recombinant IL-1β were examined in excised tissues and dissociated myocytes from guinea pig ventricles (Li and Rozanski, 1993). In excised papillary muscles, IL-1β significantly prolonged action potential duration (measured at 90% repolarization) by 24.2 ± 2.2 (SEM) ms and effective refractory period by 22.9 ± 2.3 ms. Other measured variables were not affected. Treatment of muscles with cyclo-oxygenase inhibitors, indomethacin (10–5 M) or acetyl salicylic acid (2 × 10–4 M), abolished the prolongation of action potential duration elicited by IL-1β. However, the effects of IL-1β were also blocked by the lipoxygenase inhibitor nordihydroguaiaretic acid (2 × 10–5 mol/l) or by treating tissues with the leukotriene receptor blocker, ICI 1986 I5 (10–8 mol/l). In isolated myocytes, 1 ng/ml IL-1β increased I Ca density in 44 of 78 cells by 33.6 ± 7.5% during voltage steps from –40 to 0 mV. Thus IL-1β modifies electrical properties of cardiac cells via lipid second messengers generated by cyclo-oxygenase and lipoxygenase pathways. Voltage clamp analyses suggest that these effects are mediated, at least in part, by changes in the conductance of Ca2+ channels (Li and Rozanski, 1993). In guinea pig ventricular myocytes, IL-1 has been reported to increase I Ca,L (Li and Rozanski 1993). However, these authors in that study GTP was not included in the pipette solution, and only 56% of ventricular myocytes responded to IL-l exposure (Liu and Schreur, 1995). When GTP was included in the pipette solution, authors found that IL-1β inhibited I Ca in all experiments. Therefore, in the presence of GTP, IL-1β suppressed the cardiac I Ca,L by a receptor-coupled G protein-mediated mechanism (Liu and Schreur, 1995). IL-1β attenuates cardiac L-type Ca2+ currents (I Ca,L) via pertussis toxin-insensitive G protein rather than by stimulation of cGMP production. IL-1β caused a concentration-dependent decrease in the peak I Ca,L (Ba2+ as the charge carrier). IL-1β did not significantly alter the voltage dependence of the peak I Ca,L nor the steady-state inactivation and activation, but did slightly slow the rate of inactivation (Liu and Schreur, 1995).
Many studies have suggested that nitric oxide generation via IL-1β-mediated induction of nitric oxide synthase plays a role in the observed effects of IL-1β on cardiac contractility (for example, Evans et al., 1993; Harding et al., 1995). In ferret papillary muscles, IL-1β suppressed the contraction by increasing nitric oxide (NO) production via a dexamethasone-sensitive pathway that consequently increased cellular cGMP (9). However, the NO-mediated negative inotropic effect was not observed when hamster papillary muscles were exposed to IL-1α (Finkel et al., 1992). In cells internally dialyzed with pipette solutions without GTP, the hydrolysis of which is essential for cGMP production, the absence of GTP blocks the suppression of I Ca,L by IL-1β. These results could be explained by an increase in cell cGMP level stimulated by NO synthesis. The reduction of I Ca,L by IL-1β may account for the decreased myocardial contractility associated with pathophysiological conditions, such as immunologically mediated cardiac disorders and myocardial infarction.
Modulation of the β-adrenergic control of the cardiac L-type Ca2+ current (I Ca) by human recombinant IL-1β was examined in guinea pig ventricular myocytes using the whole cell voltage clamp technique (Rozanski and Witt, 1994b). I Ca was evoked in Cs+-loaded myocytes by depolarizing pulses from a holding potential of –40 mV. In the presence of an acidic external solution (pH 5.8), the response of I Ca to isoproterenol was markedly decreased compared with control myocytes studied at pH 7.4. However, when cells were pretreated with 1 ng/ml IL-lβ and then exposed to acid media, β-responsiveness was significantly increased compared with untreated cells. Despite this effect of IL-lβ, maximum I Ca density with 0.01 and 1 μM isoproterenol was still 51 and 58%, respectively, less than that measured at pH 7.4. The enhanced β-responsiveness produced by IL-1β was eliminated by adding amiloride to block Na+/H+ exchange or protein kinase C inhibitors staurosporine (10 nM) and calphostin C (50 nM). However, a direct activator of protein kinase C, phorbol 12-myristate 13-acetate, did not mimic the effects of the cytokine. These data demonstrate that IL-lβ partially restores the β-adrenergic control of cardiac Ca2+ channels suppressed under acidic conditions. Moreover, they suggest that IL-lβ acts by enhancing Na+/H+ exchange through a second messenger pathway that may involve protein kinase C. These cellular mechanisms may play a role in altering ventricular function during cytokine-mediated inflammatory processes that are initiated by myocardial ischemia (Rozanski and Witt, 1994b). Isoproterenol exposed to myocytes pretreated with 1 ng/ml IL-1β evoked a significantly smaller increase in I Ca density compared with control cells. This IL-l-mediated decrease in β-responsiveness was usually observed with pretreatment periods of >l h and varied as a function of the l-arginine concentration of the pretreatment medium. It was prevented by (i) IL-1 receptor antagonist, (ii) substituting d-arginine for l-arginine, or (iii) incubating cells with the nitric oxide synthase inhibitor NG-monomethyl-l-arginine. Thus the present data illustrate that IL-1 significantly alters the β-adrenergic control of cardiac Ca2+ channels by cellular mechanisms that involve the activation of nitric oxide synthase. These mechanisms may play a role in altering ventricular function during cytokine-mediated inflammatory processes affecting the heart (Rozanski and Witt, 1994a).
The possible mechanism by which IL-1β affects β-adrenergic responsiveness of L-type Ca2+ current (I Ca,L) was examined in adult rat ventricular myocytes by use of whole cell patch-clamp techniques (Liu et al., 1999). In the presence of isoproterenol, exposure for 3 min to IL-1β suppressed the isoproterenol-activated I Ca,L. In the presence of IL-1β, the response of I Ca,L to isoproterenol was decreased, and the EC50 for isoproterenol stimulation was increased. However, IL-1β had no effect on basal and isoproterenol-enhanced cAMP content. When I Ca,L was activated by extracellular application of forskolin or 8-(4-chlorophenylthio)-cAMP, a membrane-permeable cAMP analog, or by intracellular dialysis with cAMP, IL-1β had little effect on I Ca,L. In contrast, in the presence of cAMP, IL-1β still suppressed the Iso-enhanced I Ca,L. These results show that the IL-1β -induced decrease in β-adrenergic responsiveness of I Ca,L does not result from inhibition of β-adrenoceptor binding, adenylyl cyclase activity, or cAMP-mediated pathways, suggesting a cAMP-independent mechanism (Liu et al., 1999).
5.3.2.3 Na+–K+ ATPase
Heart failure coupled with ischemia and myocardial hypertrophy were associated in turn with a decrease in the concentration of myocardial Na+–K+ ATPase (Bundgaard and Kjeldsen, 1996) an enzyme located in the T tubules and peripheral sarcolemma (McDonough et al., 1996) and responsible for the generation and maintenance of the electrochemical gradient for Na+ and K+. Many studies have shown that heart diseases are always accompanied with high levels of IL-1β and a decrease in Na+–K+ ATPase concentrations (Harada et al., 1999; Kreydiyyeh et al., 2004). Because in heart failure, changes in the Na+–K+ ATPase activity and expression were noted to accompany increases in circulating levels of IL-1β, the presence of a cause effect relationship between the cytokine and the pump has been investigated (Kreydiyyeh et al., 2004). IL-1 β reduced the protein expression of the α1 subunit of the Na+–K+ ATPase in right and left ventricular homogenates and exerted a significant inhibition of the ATPase activity which correlated with its down-regulatory effect and could thus be ascribed to the decrease in the number of the catalytic a1 subunits. Because the ventricular homogenate is a mixture of different types of cells (endothelial cells, macrophages, fibroblasts), the observed change in the ATPase activity and expression may not reflect necessarily changes in cardiomyocytes. To clarify this point cardiomyocytes were isolated from rats treated with IL-1β, and the activity and protein expression of the ATPase was studied in a homogenate of these isolated cells. IL-1β caused down-regulation of the pump in isolated cardiomyocytes and inhibited its activity. The results suggest that the cytokine acts directly on the cardiomyocytes. Effect of IL-1β is mediated through mitogen-activated protein kinases (MAPK).
Such an inhibition of the Na+–K+ ATPase by IL-1β is expected to decrease the Na+ gradient and Na+-dependent electrolyte movements that regulate excitation–contraction coupling in cardiac myocytes especially calcium movements. Cytosolic Ca2+ increases during contractions and is restored back to normal levels upon relaxation via the Na+–Ca2+ exchanger whose activity is dependent on the Na+ gradient established by the Na+–K+ pump. An inhibition of the pump is thus expected to dissipate the Na+ gradient, inhibit the Na+–Ca2+ antiporter, increase cytosolic Ca2+ and by so doing alter the systolic–diastolic cycle and delay relaxation. In fact, inhibition of the Na+–K+ ATPase has been reported in some cases of heart failure (Paganelli et al., 2001; Long, 2001) and was associated with increases in the levels of pro-inflammatory cytokines. Similarly increases in cytosolic Ca2+ were reported to occur in association with increases in the levels of IL-1β (Kato et al., 1987). The demonstrated inhibitory effect of IL-1β on the pump may thus provide a possible explanation of the previously reported increase in cytosolic Ca2+ levels (Bick et al., 1997, 1999) that occur in myocytes following IL-1β treatment and whose cause remained controversial.
The inhibition of the pump is expected also to alter the function of the Na+/H+ exchanger which regulates intracellular pH and the activity of which is dependent on the Na+ gradient generated by the Na+–K+ ATPase. IL-1β is thus expected to increase intracellular acidity, a process that has already been reported in the ischemic heart where pHi and pHo were noted to reach values as low as 6.0 and 6.5, respectively (Karmazym et al., 1999; Park et al., 1999).
5.3.3 Interleukin-2
IL-2 has been known as a mediator which plays one of the key roles in the immune system via stimulating and coordinating immune responses (Arai et al., 1990). IL-2 and its receptor have been intensively studied on the molecular level. Apart from the cells belonging to the immune system, IL-2 binding sites have been found in various tissues, including rat brain (Araujo et al., 1989), isolated human and rats muscular cells and some other (Smith et al., 1989). Moreover, physiological effects of IL-2, suppressing long-term potentiation of hippocampal neurons and prolonging the open time of extrajunctional acetylcholine receptors in muscle have been described (Trotter et al., 1988).
IL-2 occurs in increased amounts during inflammatory neuronal diseases, such as multiple sclerosis and polyradiculoneuritis. Since the latter is characterized by a severe paralysis, the question arose whether IL-2 may be responsible for a reduction of cellular excitability by blocking excitatory Na+ channels. A preliminary account of some of the results has appeared (Brinkmeier et al., 1992).
Despite direct action as an immune system mediator some other indirect effects of IL-2 such as sodium current inhibition, sarcoplasmic reticulum activity mediation have been described. IL-2 may act either through own IL-2 receptor or through direct interaction with other receptors, including k-opioid receptor (Cao et al., 2003a, b) and sodium channels (Brinkmeier et al., 1992). It has been shown that treatment with IL-2 increasing in a dose-dependent manner the activity of sarcoplasmic reticulum Ca2+-ATPase, but the sarcolemmal Ca2+-ATPase activity is not changing (Cao et al., 2003a, b).
Molecular mechanisms of sarcoplasmic reticulum mediation include cAMP. Treatment with IL-2 at 200 U/ml decreasing the intracellular cAMP concentration in the isolated rat heart, which suggest that cAMP is a target component of IL-2 in the heart. It is also known that cardiac k-opioid receptor stimulation inhibits adenylyl cyclase via Gi/o proteins, leading to a decreased intracellular cAMP level (Zhang and Wong, 1998). Results of decreased sensitivity of SR Ca2+-ATPase to free calcium by IL-2 action, indicates that the activation of k-opioid receptor by IL-2 results in a decreased intracellular cAMP level, which may be responsible for reduced sensitivity of SR Ca2+-ATPase to calcium by affecting the degree of cAMP-dependent phosphorylation of phospholamban.
IL-2 may also act without involvement of second messenger system, directly to the sodium channels in human muscle cells. Clear results demonstrate an inhibitory effect of human IL-2 on the excitatory Na+-channels in skeletal muscle. With IL-2 concentration as those found in body fluids of patients suffering from inflammatory neuronal diseases (Lorenzon et al., 1991) of those used to induce T-lymphocyte activation in vitro. Doubts in direct action of the IL-2 was solved in experiments with anti-IL-2 receptor antibody. This preincubation did not prevent the inhibitory effect of IL-2 on the Na+-channels on the Na+-currents. Furthermore, it is shown that the cytokine blocks the voltage-dependent muscular Na+-channels by keeping the channels in the state of fast inactivation. An IL-2 receptor and a second messenger system are not likely to be involved in this reaction. Thus, the effect of IL-2 is comparable to the action of local anesthetics on Na+-channels (Kaspar et al., 1994).
The same results of the IL-2 was shown on sodium channels in human cardiomyocytes. It is shown that IL-2 has class I antiarrhythmic-like effect with extracellular cytokine concentrations that are also usually reached when patients are under high-dose rIL-2 therapy. According to (Vaughan Williams, 1984) class I antiarrhythmics block sodium channels, shift the steady state inactivation curve in the negative direction, delay recovery of the sodium channels from block and are use dependent. All these properties are possessed by rIL-2.
Human IL-2 consists of 133 amino acids with a molecular weight of 15,000 Da in its active form. Its three-dimensional structure is known. Carboxy-peptidase removes the amino acids one after the other, starting at the C-terminal. Only arginine, proline and hydroxyproline are resistant to its action. The sequence of IL-2 contains an arginine at location 120, so only the last 13 amino acids can be removed. Unless removal of these amino acids changes the structure of the molecule, the sodium channel-binding site of rIL-2 is presumably located within the last 13 amino acids of the molecule. The receptor binding site of IL-2 is located within the first 54 amino acids, near the N-terminus, because blocking of amino acids 59–133 with monoclonal antibodies does not inhibit receptor binding (Proebstle et al., 1995).
Affinity quantitative comparison with established antiarrhythmic drugs shows that the blocking potency of rIL-2 on sodium channels is quite high.
5.3.4 Interleukin-6
IL-6 along with TNF-α is known as one of the main pro-inflammatory cytokines, especially during acute myocardial infarction (AMI). IL-6 exerts acute negative inotropic action without detectable reduction in I ca under basal conditions. Expression of IL-6 and IL-8 genes is increasing in patients assigned to take calcium blockers.
IL-6 exerts an acute negative inotropic action without detectable reduction in I Ca under basal conditions. This sounds as if IL-6 directly inhibited EC-coupling instead of blocking I Ca. However, inhibition of NOS (by l-NMMA) abolished the negative inotropic action of IL-6 (Sugishita et al., 1999). An acute inhibitory effect of NO on I Ca has been shown to be mediated through cGMP-dependent protein kinase in mammalian hearts (Thakkar et al., 1988; Levi et al., 1989; Mery et al., 1991; Ono et al., 1991). The effects of cGMP have been shown to be mediated by phosphodiesterase-dependent mechanisms in lower species such as frogs while cGMP-dependent protein kinase (cGMP-PK) may play a key role in the action of cGMP in rat hearts. Thus, IL-6 may act on L-type calcium channels mediated by activation of cGMP-PK in the ventricular myocytes of the guinea-pig, as used in this study. The NO/cGMP pathway appears to be involved in the reversal of cAMP-stimulated I Ca in guinea-pig ventricular myocytes (Ono et al., 1991; Levi et al., 1994; Wahler et al., 1995). IL-1 also decreased α-adrenergic control of I Ca through NO production in adult guinea-pig ventricular myocytes (Rozanski et al., 1994).
Four different Ca2+-channel blocker, amlodipine, felodipine, isradipine and manidipine, are capable of inducing the activation of genes coding for IL-6 and EL-8 in cultured human VSMC and fibroblasts. The effect occurs at nanomolar concentrations which can be achieved in vivo during therapy. The novel activity of the dihydropyridines cannot be substituted for by two other blood pressure lowering agents such as β-receptor antagonists or diuretics. This suggests an apparently specific activity of the class of drugs affecting inflammatory mechanisms. However, the exact mechanism by which the drugs stimulate remains unclear. Two possible modes of action can be hypothesized; (i) the binding site of the potential operated Ca2+-channels contain an unknown signal transducing element which can be activated by interaction with Ca2+-channel blockers, or (ii) the induction of transcription of IL genes by Ca2+-channel blockers is mediated via an unknown membranous binding site for Ca2+-channel blockers which is linked to a signaling pathway. Further studies should unravel the mechanisms of Ca2+-channel blockers on the interaction of the two ILs with their various receptors (Roedler et al., 1994).
Concentration of pro-inflammatory cytokines (IL-6, TNF-α and hs-CRP) and anti-inflammatory cytokine IL-10 were elevated in AMI patients in comparison with the group with stable angina pectoris, but statistically significant difference was found in IL-6 and hs-CRP level. IL-6:IL-10 ratio is higher in AMI which emphasis the hypothesis of pro-inflammatory to anti-inflammatory cytokine misbalance in AMI. IL-10 may be useful as a marker of myocardial reperfusion in AMI. There is positive linear correlation between IL-6 and IL-10 in acute myocardial infarction and negative linear correlation between HDL and IL-6 (Dizdarević-Hudić et al., 2009).
It is also well known that the cardiac effects of NO are complex and somewhat controversial, probably due to the distinct concentration-dependent sensitivities of its targets and distinguishable signaling mechanisms. NO is known to target the L-type Ca2+ channel and SR in cardiac myocytes through signaling pathways such as cGMP, and/or by directly modifying the proteins (such as by S-nitrosylation/oxidation) (Campbell et al., 1996; Hare, 2003; Massion et al., 2003). Studies in adult ferret ventricular myocytes showed that SIN-1, a NO donor, induces biphasic and bimodal changes in basal I Ca,L (Campbell et al., 1996), which were not observed in frog (Mery et al., 1993), guinea-pig (Wahler and Dollinger, 1995) or rat (Abi-Gerges et al., 2001) ventricular myocytes. We also observed no significant change in basal I Ca,L in ARVM upon acute exposure to 0.1 mM SIN-1 (S.J. Liu, unpublished data: In Yu et al., 2005a, b). Similarly, SIN-1 (up to 0.1 mM) has no significant effect on contractile function of papillary muscle isolated from adult rat (Wyeth et al., 1996). Although we found that cumulative NO production in cell lysates and in culture medium is 2.5- to 3-fold of the basal level after 2 h of IL-6 treatment, it is not surprising that I Ca,L is not significantly altered in our experimental conditions. However, the possibility for alteration in I Ca,L during longer exposure (e.g. >24 h) to IL-6 cannot be excluded.
In summary, exposure to IL-6 for 2 or more hours results in a negative inotropy probably by inhibiting SR Ca2+ reuptake in concomitance with increases in NO in ARVM. The IL-6-induced decrease in PLB phosphorylation is also in accord with its reduction of SR function. The increase in NO production results primarily from expression/activation of iNOS that is mediated via IL-6-induced JAK2/STAT3 activation as shown previously (Yu et al., 2003a, b). Inhibition of iNOS/NO abolishes the IL-6-induced decrease in SR function. Therefore, this leads to our conclusion that IL-6 suppresses SR function via iNOS after chronic exposure. Moreover, such cardiac effects of IL-6 are sustained even after removal of IL-6. This suggests that the transient elevation of IL-6 observed in plasma or ventricular muscle of patients with many cardiac diseases can cause prolonged effects on contractility. An in vivo study in transgenic mice showed that cardiac-specific overexpression of iNOS causes an increase in NO x in hearts (2.5-fold above the wild-type controls) but no severe cardiac dysfunction (Heger et al., 2002). One interpretation for this seeming inconsistency is that the increase in plasma and heart NO x in the transgenic models is moderate so the associated systemic adaptation during development in these transgenic mice results in less detrimental functional consequences. A recent in vivo study reported that IL-6 administration causes heart failure in a dose-dependent manner (Janssen et al., 2005). Thus, IL-6 might play an important role in the pathogenesis of associated chronic heart diseases. Treatment to antagonize the induction and activation of iNOS in the early stage of IL-6-associated heart dysfunction might be beneficial (Yu et al., 2005a, b).
5.3.5 Interleukin-11
There are clinical reports of sudden cardiac fibrillation in cancer patients who were treated with recombinant human IL-11. The question was either that effect is a direct arrhythmogenic action of IL-11 or that was indirect system side effect. The results from (Sartiani et al., 2002) which was performed on single human atrial myocytes using standard patch-clamp technique clearly show the lack of effect on I Ca,L, I K, I to, in agreement with the observation that rhIL-11 does not influence the atrial action potential configuration and particularly it’s duration.
Shortening of atrial action potential is generally associated with shortening of atrial refractory period and hence facilitates induction of atrial flutter/fibrillation (Narayan et al., 1997). Conversely, lengthening of action potential duration is a perquisite for the appearance of early after depolarization, which may trigger arrhythmias (Janse, 1992). Calcium handling by sarcoplasmic reticulum and Na/Ca exchanger is crucial for the changes in atrial action potential duration and the appearance of delayed afterdepolarizations (Tavi et al., 1996, 1998; Benardeau et al., 1996). To avoid any perturbation by the patch pipette of the intracellular milieu and calcium homeostasis, which could have hindered the development of Ca-dependent electrical alterations, action potentials were measured with the perforated-patch technique. Thus, our results allow inferring that superfusion with rhIL-11 does not have direct, acute electrophysiological effects on action potential parameters measured in atrial myocytes. Furthermore, acute exposure of atrial myocytes to rhIL-11 does not affect the pacemaker current I f, nor did it modify the resting potential in these cells. Taken together, these data suggest that it is unlikely that rhIL-11, at least by its direct effects, may cause afterdepolarizations, thought to be the trigger for atrial fibrillation, which is maintained by re-entry (Janse, 1997). We did not find a single direct electrophysiological alteration indicative that rhIL-11 may acutely influence atrial electrical activity. The only electrophysiologic effect of rhIL-11, however not statistically significant, was a small reduction in the I f amplitude. I f is an inward current that is responsible for the diastolic depolarization phase and automatic activity in pacemaker cells (Difrancesco, 1993). This current has been recently described and characterized in human atrial myocytes (Hoppe et al., 1998; Pino et al., 1998; Porciatti et al., 1997). An increase of I f amplitude in association with a reduction in the inward rectifier I K1 or in association with the presence of delayed afterdepolarizations due to a cellular calcium overload has been suggested to potentially trigger premature depolarizations able to generate atrial fibrillation if an appropriate substrate for re-entry is present (Opthof, 1988; Pino et al., 1998). A reduction in I f amplitude should reduce the likelihood for generation of such premature depolarizations.
Thus, it is possible that acute atrial dilatation and consequent atrial stretch rather than direct action on the myocytes by rhIL-11 could be the mechanism by which rhIL-11 might affect cellular atrial electrophysiology and induce atrial arrhythmias. The experimental study in rats presented in the accompanying paper supports this hypothesis. In summary, it seems reasonable to conclude that the lack of effect of rhIL-11 on AP and ionic currents during acute exposure of human atrial myocytes seems to exclude a direct effect of rhIL-11 in the genesis of atrial arrhythmias in patients.
In conclusion, it is very likely that effect of IL-11 is mediated via blockade of sodium channels, water accumulation, edema, atrium dilatation and further mechanically induced fibrillation.
5.4 Cytokines and Mechanosensitivity of the Heart
In recent years many groups investigated the role of cytokines in generation of cardiac stretch effects. This studies employed one of two approaches. Firstly – investigation of cytokine production during stretch of issue or single cells. Those studies are discussed in review of Kovalchuk et al. (Chapter 2, this volume). The second approach – investigation of cytokine effects on cells and tissues during their stretching. We will discuss their findings in this section.
However there is a number of studies addressing cytokine production during tissue stretching (for review see Chapters 2 and 10 of this Volume), investigation of the effects of exogenous cytokines on cells, for example in heart, are limited to those, which were described in previous sections of this chapter. On the other hand there are practically no studies of the regulation of mechanoelectrical feedback in heart by cytokines. Therefore their effects on stretched cardiac tissue remain unknown. For the purpose of investigation of the role of cytokines in regulation of mechanoelectrical feedback we used microelectrode registration of bioelectrical activity of the fragment of the right atria during discrete tissue stretching (Kamkin et al., 2000a).
Several intracellular electrophysiological alterations in the healthy and diseased heart, which were ascribed to mechanoelectrical feedback were reported for the first time about 10 years ago (Kiseleva et al., 2000; Kamkin et al., 2000a). Later it was showed that stretch of isolated cardiomyocytes induced mechanosensitive whole-cell currents which lead to membrane depolarization (Kamkin et al., 2000b, 2003a, b; Zeng et al., 2000; Zhang et al., 2000; Isenberg et al., 2003; Kondratev and Gallitelli, 2003) and elicited stretch-induced depolarizations (SID), observed as prolongation of APD90 (stretch-induced depolarizations: SID) or hump-like SID, that appeared in repolarization phase at APD50 or APD90 level (Kamkin et al., 2003b). Membrane depolarization and SID in action potentials provoke extra-action potentials when these hump-like SID reach a threshold potential. This finding correlated with previous studies of step-wise application of stretch to atrial and ventricular tissue from healthy animals and animals, suffering from infarct, which reported SIDs, emerging as prolongation of APD90 and as typical hump-like SIDs, leading to extra-action potentials (extra-AP), paroxysmal tachycardia and even fibrillation (Kiseleva et al., 2000; Kamkin et al., 2000a). Later they were studied in detail (Lozinsky and Kamkin, 2010; Ward et al., 2010). Thus, activation of mechanically gated channels (MGCs) and the increase of mechanosensitive whole-cell currents, which during cardiomyocyte stretching leads to ion entry carrying the positive charge together compose the cellular mechanisms underlying different mechanically induced alterations and abnormalities.
It is known that certain cytokines induce signaling, which includes for example, transducing signals within intracellular compartments, such as in the case of modification of transcription factors in mitochondria and the nucleus (see for review Calabrese et al., 2009). One of those signaling cascades includes NO-dependent pathway, which is well described in many publications. In the heart NO-synthases play the key role in NO level regulation.
Besides, in response to stretch in cardiac cells NO-synthase activity significantly increases (reviewed in Shah and MacCarthy, 2000; Casadei and Sears, 2003; Seddon et al., 2007). Recently it was shown that NO and NO-synthases as NO source directly regulates mechanically gated channels (MGCs) conductivity. Experimental data in this study was obtained from the isolated ventricular myocytes of mouse, rat and guinea pig by means of patch-clamp method in whole-cell configuration. The data demonstrated that NO donors lead to MGCs activation and appearance of MG-like currents in undeformed ventricular myocytes, whereas, it can also lead to the inactivation of the stretched cells with activation MGCs and inhibit the conductivity of these channels (Kazanski et al., 2010a, 2011). NO scavenger PTIO causes inactivation of all MGCs. In undeformed cells conductance through MGCs is blocked and their activation in the controlled, and on the background of stretch cell the PTIO causes complete MG-current inhibition. Non-selective inhibitors of NO-synthases l-NAME or LNMMA resulted in complete blocking of MGCs. In ventricular myocytes of wild-type mice, NOS1–/– and NOS2–/– the stretching of the cells triggered typical MG-current. On the contrary, in cells from NOS3–/– mice stretch does not cause MG-current. The results discussed on the channel level testify to the NO role in activation and inactivation of MGCs in cardiomyocytes and demonstrate that NOS3 dominates as NO source (Kazanski et al., 2010b, 2011). Therefore the appearance of hump-like SIDs and extra-AP during stretching of atrial tissue stripes is determined by MGCs functioning, which are modulated by NO and NO-synthases.
5.4.1 Tumor Necrosis Factor-α
TNF-α has been implicated in the pathogenesis of cardiovascular diseases, including heart failure, myocarditis, acute myocardial infarction and sepsis-related cardiac dysfunction (Levine et al., 1990; Low-Friedrich et al., 1992; Yokoyama et al., 1993; Latini et al., 1994; Packer, 1995; Kumar et al., 1996). TNF-α also has been shown to be related to the occurrence of cardiac arrhythmias. Mice with a higher TNF-α expression have a greater incidence of atrial and ventricular arrhythmias (London et al., 2003). Administrating TNF-α in rats can induce arrhythmias with a loss of the myocyte inotropy (Krown et al., 1995). TNF-α changes the L-type calcium currents (I Ca,L), and calcium transient in ventricular myocytes (Krown et al., 1995; Goldhaber et al., 1996; Cailleret et al., 2004).
The direct effects of proinflammatory cytokine TNF-α on the contractility of mammalian heart were studied a lot of years ago (Finkel et al., 1992). It was shown, that TNF-α inhibited contractility of isolated hamster papillary muscles in a concentration-dependent, reversible manner. The authors demonstrated, that the nitric oxide synthase inhibitor NG-monomethyl-l-arginine (l-NMMA) blocked these negative inotropic effects. l-Arginine reversed the inhibition by l-NMMA. Removal of the endocardial endothelium did not alter these responses. These findings demonstrate that TNF-α regulated the contractility and the direct negative inotropic effect of cytokines is mediated through a myocardial nitric oxide synthase. The negative inotropic effect of TNF-α was observed in isolated rabbit cells (Goldhaber et al., 1996). This effect was apparent within 20 min of exposure to the cytokine, was similar to that described by Finkel et al. (1992) in a multicellular preparation.
Previous studies have shown that nitric oxide (NO) has important regulatory effects on the cardiovascular system (Moncada et al., 1991; Kelly et al., 1996). NO has been shown to have a role in the development of triggered arrhythmias generated by Ca2+overload (Kubota et al., 2000). Previous study in vivo also showed that NO could suppress trigged activity induced ventricular tachycardia (Chen et al., 2001). It is known that pulmonary veins contain endothelium and smooth muscle which may produce NO through the enzyme of eNOS or iNOS. In addition, cardiac myocytes also express eNOS activity (Massion and Balligand, 2003; Balligand et al., 1994). NO has been shown to regulate PV arrhythmogenesis through mechanoelectrical feedback (Hu et al., 2009). Because PVs was known to induce atrial arrhythmia through the enhancement of triggered activity, it is possible that NO may play a critical role in the pulmonary veins arrhythmogenic activity.
It was shown that cardiac cells themselves produce different quantities of TNF-α during prolonged stretch or during cyclic application of mechanical stretch. Acute stretch caused by volume overload of aorto-caval fistula induces a variety of myocardial responses. These responses can be part of myocardial inflammation dictated by TNF-α, which is elevated after acute aorto-caval fistula. Western blot demonstrated increased local production of TNF-α in the left ventricle and cardiomyocytes. TNF-α produced by cardiomyocytes mediates a predominant inflammatory response to stretch in the early volume overload in the aorto-caval fistula rat, suggesting an important role of TNF-α in initiating pathophysiological response of myocardium to volume overload (Chen et al., 2010). In addition to production of TNF-α by cardiomyocytes during their stretching, cardiac fibroblasts also produce TNF-α during stretch. To determine whether mechanical stress affect the production of TNF-α in the heart, neonatal rat cardiac myocytes and fibroblasts were cultured separately and treated for 6 h with cyclic mechanical stretch. Mechanical stretch stimulated the production of TNF-α in cardiac fibroblasts. Thus mechanical stress, can stimulate production of TNF in cardiac fibroblasts (Yokoyama et al., 1999).
Thus, TNF-α has been shown to be related to the occurrence of cardiac arrhythmias. Moreover, TNF-α regulated the contractility and the direct negative inotropic effect of TNF-α is mediated through a myocardial nitric oxide synthase. This data and studies reporting that NO directly regulates mechanically gated channels (MGCs) (Kazanski et al., 2010a, b, 2011) allowed us to propose that in heart TNF-α would be an effective regulator of MGCs operating and therefore will regulate mechanoelectrical feedback.
In order to test this hypothesis we used the standard microelectrode technique in combination with stretching of the tissue stripe of the right rat atria. Tissue was perfused with standard physiological solution, containing 50 ng of TNF-α per milliliter. Perfusion of tissue with TNF-α lead to appearance of abnormalities in development of bioelectrical activity observed as prolongation of APD90 and appearance of hump-like depolarization at APD90 level. After reaching E c hump-like depolarization transformed into single extra-APs, with following development of sustained arrhythmia. Abnormalities at APD50 level were also observed.
Since TNF-α activates NO synthases of cardiomyocytes (Finkel et al., 1992; Goldhaber et al., 1996; Nakayama et al., 2009; Bougaki et al., 2010) and rise of the concentration of the intracellular NO activates MGCs (Kazanski et al., 2010a, b, 2011), which allows Na ions entry into the cell leading to cellular depolarization and shift of resting potential to E C we proposed, that TNF-α induced arrhythmias can be linked with MGCs activation. This is the most simple mechanism, explaining the appearance of arrhythmias after TNF-α application.
Since the blocker of MGCs Gd3+ at 40 μmol/l eliminates not only hump-like depolarization, but arrhythmias, triggered by TNF-α, as well, it was proposed that TNF-α triggered abnormalities are linked with MGCs activity, which was triggered by increasing of intracellular NO concentration, caused by TNF-α application. In this case they would be similar to stretch induced depolarizations (SID). For testing the hypothesis about the role of NO in regulation of MGCs we used patch-clamp method in whole-cell configuration in combination with stretching of isolated cells (Kamkin et al., 2000b, 2003a; Lozinsky and Kamkin, 2010). It was shown that NO donor SNAP and DEO-NO open MGCs in unstretched cells, and closes them in stretched cells. Therefore MGCs activity is determined by NO concentration. It is known that stretching of cells and tissues stimulates NO synthases activity, which rises intracellular NO concentration (Shah and MacCarthy, 2000; Seddon et al., 2007). Besides that under control conditions tissue stretch induces hump-like depolarizations, which are originating from SIDs (Kamkin et al., 2000a). That’s why we conducted a series of experiments in which we stretched atrial tissue preparations during registration of TNF-α induced hump-like depolarizations or arrhythmias. Tissue stretch completely eliminated mechanoinduced electrical abnormalities, originating in cardiomyocytes in response to TNF-α application. Therefore, TNF-α induces electrical abnormalities in cardiomyocytes, while tissue stretching eliminates them. This effects is likely to be mediated via NO signaling cascades.
If TNF-α acts via an increase in NO concentration after activation of NO-synthase then application of NO donor, for example SNAP sold lead to similar changes in APD90 with following generation of arrhythmias. In order to prove that in absence of any changes in preload we perfused tissue with standard saline solution, containing 3 × 10–4 mol/l of SNAP. Already after 10 min of perfusion we observed appearance of hump-like depolarizations, which later transformed into sustained paroxysmal arrhythmic events, mediated by altered or even normal action potentials. Therefore SNAP caused abnormalities such as hump-like depolarizations, similar to those induced by TNF-α. In order to answer the question whether abnormalities and arrhythmias, induced by SNAP, are similar to those stretch-induced depolarizations (SID), we conducted two series of experiments with 40 μmol/l of Gd3+. In the first series we added Gd3+ to perfusion solution after appearance of SNAP induced hump-like depolarizations or SNAP induced arrhythmias. It was shown that hump-like depolarization in abnormal patterns is completely blocked by 40 μmol/l of Gd3+ within 10 min after its application. SNAP induced arrhythmias were also blocked by 40 μmol/l of Gd3+. In another series Gd3+ was added to the perfusion solution together with SNAP. In this case we did not observe any abnormal action potentials. Since discrete tissue stretching during TNF-α perfusion led to complete elimination of arrhythmias and then blocked electrical abnormalities and taking into consideration that TNF-α effects can be mediated via NO concentration increase we conducted a series of experiments in which we triggered SNAP induced SIDs and arrhythmias, and then stretched cells for further activation of NO-synthases. After appearance of SNAP induced abnormalities or arrhythmias the tissue was stretched. This stretch completely arrhythmias and SI-like depolarization triggered by SNAP application.
In conclusion it was shown that application of SNAP during observation of TNF-α induced abnormalities completely abolishes those abnormalities. For investigation of the functional role of NO-synthases in mediation of TNF-α effects we used their non-selective inhibitor l-NAME, with which we preincubated tissue during 30 min. After that we applied TNF-α for 30 more minutes in combination with l-NAME. Pre incubation of tissue with l-NAME containing solution did not lead to any abnormalities in action potentials even after 30 min of perfusion. More importantly after such pre-incubation TNF-α, applied in combination with l-NAME did not trigger appearance or development of hump-like depolarizations. Neither stretch lead to any hump-like depolarizations.
Therefore arrhythmogenic effect of TNF-α is mediated via MGCs activity and is NO dependent. TNF-α activates NO synthases, which leads to NO concentration increase triggering MGCs activation and Na+ influx to the cell which depolarizes cellular membrane triggering arrhythmias. Tissue stretch completely abolishes mechanoinduced electrical abnormalities, triggered by application of TNF-α to cardiomyocytes probably via additional activation of NO synthases and rise of intracellular NO concentration. Such NO concentration rise would lead to MGCs closing. This data revealing TNF-α NO mediated effects completely correlate with data from patch clamp experiments conducted in combination with single cardiomyocyte stretch. In unstretched cardiomyocytes NO donors trigger MGCs activation, which is abolished by cardiomyocyte stretch.
Thus, these experimental data sheds light on mechanism of arrhythmogenic action of TNF-α in atrial myocardium. We are trying to reveal the link between arrhythmogenic effect of TNF-α and its underlying mechanisms, which we described earlier, data about TNF-α application triggered arrhythmias in animals and clinical studies reporting levels of TNF-α from patients with cardiac arrhythmias and fibrillation. Administrating TNF-α in rats can induce arrhythmias (Krown et al., 1995) and mice with overexpression of TNF-α have a greater incidence of AF (London et al., 2003). The circulating levels of TNF-α (Sata et al., 2004) was found to be significantly higher in patients suffering from AF. In our study we were working with patients’ congestive heart failure (New York Heart Association: Class II, Class III and Class IV). According to NYHA, at congestive heart failure arrhythmias and fibrillation are not considered to be symptoms of the decease. Data from Table 5.1 demonstrates the link between concentration levels of TNF-α and observation of ventricular arrhythmias. Table shows that elevation of TNF-α concentration correlates with increasing percentage of patients with clinical arrhythmic events.
5.4.2 Interleukin-6
IL-6 has been shown to be related to the occurrence of cardiac arrhythmias. IL-6 has been shown to significantly correlate with increased left atrial size (Psychari et al., 2005), an important risk factor for developing atrial fibrillation. In addition, polymorphisms in the promoter region of the IL-6 gene have been associated with post-operative atrial fibrillation (Bittar et al., 2005; Gaudino et al., 2003). IL-6 has not previously been shown to be independently associated with spontaneous (non-post-operative) atrial fibrillation. Polymorphisms of the IL-6 gene were not studied in these patients. Recent studies showed that atrial fibrillation is associated with elevated IL-6 levels and the IL-6 -174CC genotype (Marcus et al., 2008).
In vivo study also showed that elevated IL-6 levels were observed 1–2 h after aortic declamping in cardiopulmonary bypass (Wan et al., 1996). The direct cardiac effect of IL-6 during this period of time remains undefined. De novo synthesis and activation of iNOS induced by IL-6 can be detected in adult rat ventricular myocytes as early as 2 h after exposure (Yu et al., 2003a, b). This study also demonstrated that the IL-6-elicited iNOS activation and decrease in postrest potentiation of contraction in adult rat ventricular myocytes are mediated by activation of Janus kinase (JAK)2/signal transducer and activator of transcription (STAT)3, the upstream mediators of IL-6 signaling (Yu et al., 2003a, b). However, whether iNOS is the downstream mediator of IL-6-induced negative inotropy remains undefined.
In vitro studies on cardiac contractile function have shown that exposure to IL-6 for 2–3 min decreased contractility in papillary muscle isolated from hamster heart (Finkel et al., 1992). Studies with single myocytes also showed that IL-6 suppressed peak systolic [Ca2+]i and cell shortening within 5 min in chick embryonic cardiac myocytes (Kinugawa et al., 1994) and in adult guinea-pig ventricular myocytes (Sugishita et al., 1999). This acute negative inotropic effect of IL-6, accompanied by an increase in cell cGMP production (Kinugawa et al., 1994), was blocked by NG-monomethyl-l-arginine (l-NMMA), an inhibitor of nitric oxide synthase (NOS) (Finkel et al., 1992; Kinugawa et al., 1994; Sugishita et al., 1999). Thus, acute IL-6-induced suppression of cardiac contractility and [Ca2+]i have been suggested to be mediated by a NO-dependent pathway via activation of NOS (Finkel et al., 1992; Kinugawa et al., 1994; Sugishita et al., 1999), probably a constitutive endothelial isoform (eNOS) (Kinugawa et al., 1994). After 2 h incubation IL-6 decreases contractility and Ca2+ o responsiveness with no significant effect on I Ca,L in ARVM. (IL-6 reduces PRP, caffeine responsiveness and the phosphorylation of PLB. The negative inotropic effect is sustained for at least 1 h after removal of IL-6 and IL-6 induces production of NO via activation of iNOS, with inhibition of iNOS abolishing IL-6-induced changes in PRP, caffeine- and Ca2+ o-responsiveness, and PLB phosphorylation (Yu et al., 2005a, b).
In general, IL-6 has been shown to be related to the occurrence of cardiac arrhythmias. Moreover, IL-6 regulated the contractility and the direct negative inotropic effect of IL-6 is mediated through a myocardial nitric oxide synthase. This data and studies reporting that NO directly regulates activity of mechanically gated channels (MGCs) (Kazanski et al., 2010a, b, 2011) allowed us to suppose that in heart IL-6 would be an effective regulator of MGCs activity and therefore will regulate mechanoelectrical feedback.
For testing this hypothesis we used standard microelectrode technique in combination with stretching of right rat atria cardiac tissue preparation. Under control conditions (Kamkin et al., 2000a), during perfusion with standard saline solution stretching of tissue by 1.7 mN caused prolongation of APD90 and appearance of hump-like depolarizations at APD90 level (Fig. 5.7a). Perfusion of tissue with IL-6 containing solution (50 ng/ml) lead to appearance of abnormalities in bioelectrical activity, majorly observed as APD90 prolongation. After 7–10 min of perfusion tissue stretch by 1.5–1.7 mN trigged hump-like depolarizations at APD90 level. After reaching E c hump-like depolarization transformed into single extra-action potential later leading to typical tachycardia, which could be abolished by stretch (Fig. 5.7b).

Response of atrial cardiomyocyte membrane potential to stretch applied by the digital micromanipulator. (a) Control conditions. Stepwise increases in resting force due to stretch (indicated by ↑) up to 1.7 mN significantly increased APD90. Removal of stretch (indicated by ↓) reversed these effects. (b) IL-6 in concentration of 50 ng/ml, 10 min of perfusion. Stepwise increases in resting force due to stretch (indicated by ↑) up to 1.7 mN produced stretch-induced depolarization and tachyarrhythmia. Note: resting force (RF), active force (AF), resting potential (RF), active potential (AP) (the data obtained from the Department of Professor Andre Kamkin)
Continuous perfusion of the preparation with IL-6 containing solution for periods of time longer than 20 min leads to fibrillation triggered by extremely low levels of stretch. Figure 5.8a shows a trace of registration of the resting force (RF), active force (AF), resting potential (RF) and active potential (AP) in control conditions during perfusion of the fragment of right rat atria with standard saline solution. Stretching of tissue by 1.7 mN triggered APD90 prolongation. Figure 5.8b shows effects of tissue stretch after 23 min of perfusion with IL-6 containing solution (50 ng/ml). Even small stretch by 0.3 mN triggered fibrillation, observed as typical bioelectrical activity and absence of AF. Stretch elimination stopped fibrillation. IL-6 effects and their underlying mechanisms are similar to those of TNF-α.

Example of response of cardiomyocyte membrane potential to stretch applied by the digital micromanipulator of right atrial tissue. (a) Control conditions. Stepwise increases in resting force due to stretch (indicated by ↑) up to 1.7 mN produced stretch-induced depolarization (SID) near APD90. (b) IL-6 in concentration of 50 ng/ml, 10 min of perfusion. Stepwise increases in resting force due to stretch (indicated by ↑) only up to 0.3 mN produced fibrillation. Release of the stretch (↓) resulted in complete reversibility of stretch-induced effects. Note: resting force (RF), active force (AF), resting potential (RF), active potential (AP) (the data obtained from the Department of Professor Andre Kamkin)
5.5 Conclusion and Perspectives
All together the data from literature testifies towards hypothesis that studied cytokines regulate action potentials of cardiac cells influencing a number of ion fluxes including I Na, I Ca, I to, I Kur, I Kr, I Ks, I K1. Besides that and possibly even more importantly those compounds regulate mechanoelectrical feedback in heart via activation of NO synthases in cardiomyocytes, primarily NOS3 as is shown on Fig. 5.9 (Prabhu, 2004a). NO imparts cardiac effects via two general signaling modalities: (i) the activation of soluble guanylate cyclase and the formation of cGMP, which in turn activates protein kinase G (PKG) and PKG-dependent phosphorylation events; and (2) the direct oxidation of thiol residues on critical regulatory proteins (S-nitrosylation) (Kojda and Kottenberg, 1999; Massion et al., 2003; Prabhu, 2004a). Rise of intracellular NO concentration opens MGCs, which allows sodium entry into the cell which in turn depolarizes them, shifting resting membrane potential towards E C (Kazanski et al., 2011). This can directly or indirectly lead to arrhythmias triggered by elevated levels of cytokines. A large number of studies, performed in a variety of experimental models, have examined whether NO, derived from NOS3, contributes to early cytokine-induced dysfunction. We already highlighted that cytokine levels go up in different pathological states, including infarct and hypertrophy of cardiac tissue. In combination with data regarding correlation between such electrophysiological manifestations of cardiac pathology as arrhythmias and fibrillation and cytokine blood levels it is possible to propose cytokines as triggers of cardiac arrhythmias and fibrillation. Besides pure theoretical importance such point of view can have clinical application. It would be a valuable approach for creation of test systems for early prediction of cardiac arrhythmias and fibrillation.

Cellular mechanisms underlying immediate cytokine-induced contractile effects. Proinflammatory cytokines modulate several signaling pathways that can ultimately impact contractile function. Rapid activation of NOS3 generates NO, with depression of Ca2+ transients and myofilament Ca2+ sensitivity via the production of cGMP and cGMP-dependent activation of PKG. cGMP/PKG also blunts cAMP/PKA-mediated effects, and NO can also directly depress mitochondrial (Mt) respiration, thus altering mechanoenergetics. In low concentrations, NO can also exert a positive inotropic effect. Cytokines such as TNF-α also deplete GSH and activate membrane-associated neutral sphingomyelinase (NSM), which in turn increases ceramide (C) and sphingosine levels. Sphingosine blunts the inward Ca2+ current (I Ca,L) and Ca2+ release from the ryanodine receptor (RyR) of the SR. Ceramide also blunts I Ca,L. Cytokines also activate cytosolic PLA2 to generate AA, which can, under appropriate conditions, augment the Ca2+ transient and contractility. Cytokines may also acutely impair β-AR coupling to AC via unclear mechanisms. Cross-talk likely occurs between these various pathways. Free fatty acids such as AA have been shown to activate NSM, and sphingosine 1-phosphate (S 1-P) can phosphorylate and activate cNOS. GSH, in turn, inhibits NSM activation. Thus, the ambient oxidant stress and metabolic state, along with the cytokine dose and applicable reflex effects, influences the relative activation of these pathways, resulting in responses that are either cardiostimulatory (with low concentrations and short exposure duration) or cardiodepressant (with higher concentrations and longer exposure time). PLB indicates phospholamban; SERCA, SR Ca2+ ATPase; TnI, troponin I (from Prabhu (2004a) with permission of Lippincott Williams & Wilkins, Circulation Research and American Heart Association via Copyright Clearance Center)
References
Abbate A, Salloum FN, Vecile E et al (2008) Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 117:2670–2683
Abi-Gerges N, Fischmeister R, Mery PF (2001) G protein mediated inhibitory effect of a nitric oxide donor on the L-type Ca2+ current in rat ventricular myocytes. J Physiol (London) 531:117–130
Alloattia G, Pennaa C, De Martinoa A et al (1999) Role of nitric oxide and platelet-activating factor in cardiac alterations induced by tumor necrosis factor-a in the guinea-pig papillary muscle. Cardiovasc Res 41:611–619
Amadou A, Nawrocki A, Best-Belpomme M et al (2002) Arachidonic acid mediates dual effect of TNF-α on Ca2+ transients and contraction of adult rat cardiomyocytes. Am J Physiol 282:C1339–C1347
Arai K, Nishida J, Hayashida K et al (1990) Coordinate regulation of immune and inflammatory responses by cytokines. Rinsho Byori 38(4):347–353
Araujo DM, Lapchak PA, Collier B, Quirion R (1989) Localization of interleukin-2 immunoreactivity and interleukin-2 receptors in the rat brain: interaction with the cholinergic system. Brain Res 498(2):257–266
Aviles R, Martin D, Apperson-Hansen C et al (2003) Inflammation as a risk factor for atrial fibrillation. Circulation 108:3006–3010
Balligand JL (1999) Regulation of cardiac β-adrenergic response by nitric oxide. Cardiovasc Res 43:607–620
Balligand JL, Ungureanu D, Kelly RA, Kozaik L, Pimental D, Michel T, Smith TW (1993) Abnormal contractile function due to induction of nitric oxide synthesis in rat cardiac myocytes follows exposure to activated macrophage-conditioned medium. J Clin Invest 91(5):2314–2319
Balligand JL, Ungureanu-Longrois D, Simmons WW et al (1994) Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. Characterization and regulation of iNOS expression and detection of iNOS activity in single cardiac myocytes in vitro. J Biol Chem 269:27580–27588
Belosjorow S, Bolle I, Duschin A et al (2003) TNF-alpha antibodies are as effective as ischemic preconditioning in reducing infarct size in rabbits. Am J Physiol 284:H927–H930
Benardeau A, Hatem SN, Rucker-Martin C et al (1996) Contribution of Na+/Ca2+ exchange to action potential of human atrial myocytes. Am J Physiol 271:H1151–H1161
Benitah JP, Gomez AM, Bailly P et al (1993) Heterogeneity of the early outward current in ventricular cells isolated from normal and hypertrophied rat hearts. J Physiol (London) 469:111–138
Beuckelmann DJ, Näbauer M, Erdmann E (1993) Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 73(2):379–385
Bick RJ, Liao JP, King TW, LeMaistre A, McMillin JB, Buja LM (1997) Temporal effects of cytokines on neonatal cardiac myocyte calcium transients and adenylate cyclase activity. Am J Physiol 272:H1937–H1944
Bick RJ, Wood DE, Poindexter B, McMillin JB, Karoly A, Wang D et al (1999) Cytokines increase neonatal cardiac myocyte calcium concentrations: the involvement of nitric oxide and cyclic nucleotides. J Interferon Cytokine Res 19:645–653
Bittar MN, Carey JA, Barnard J et al (2005) Interleukin 6 G-174C polymorphism influences outcome following coronary revascularization surgery. Heart Surg Forum 8(3):E140–145
Bougaki M, Searles RJ, Kida K, Yu J, Buys ES, Ichinose F (2010) NOS3 protects against systemic inflammation and myocardial dysfunction in murine polymicrobial sepsis. Shock 34(3):281–290
Bracco MM, Fink SB, Finiasz MR et al (1991) Positive inotropic effect of interleukin-2. Role of phospholipases and protein kinase C. Int J Immunopharm 13:509–515
Brinkmeier H, Kaspar A, Wiethoelter H, Ruedel R (1992) Interleukin-2 inhibits sodium currents in human muscle cells. Eur J Physiol 420:621–623
Brouillette J, Clark RB, Giles WR, Fiset C (2004) Functional properties of K+ currents in adult mouse ventricular myocytes. J Physiol (London) 559(Pt 3):777–798
Brown JM, White CW, Terada LS et al (1990) Interleukin 1 pretreatment decreases ischemia/reperfusion injury. PNAS 87:5026–5030
Bundgaard H, Kjeldsen K (1996) Human myocardial Na,K-ATPase concentration in heart failure. Mol Cell Biochem 163–164:277–283
Cailleret M, Amadou A, Andrieu-Abadie N, Nawrocki A, Adamy C, Ait-Mamar B, Rocaries F, Best-Belpomme M, Levade T, Pavoine C, Pecker F (2004) N-Acetylcysteine prevents the deleterious effect of tumor necrosis factor-(alpha) on calcium transients and contraction in adult rat cardiomyocytes. Circulation 109(3):406–411
Cain BS, Meldrum DR, Dinarello CA et al (1999) Tumor necrosis factor-α and interleukin-1β synergistically depress human myocardial function. Crit Care Med 27:1309–1318
Calabrese V, Cornelius C, Rizzarelli E, Owen JB, Dinkova-Kostova AT, Butterfield DA (2009) Nitric oxide in cell survival: a janus molecule. Antioxid Redox Signal 11:2717–2739
Campbell DL, Stamler JS, Strauss HC (1996) Redox modulation of L-type calcium channels in ferret ventricular myocytes – dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol 108:277–293
Cao C, Xia Q, Bruce Iain C, Zhang X, Fu C, Chen JZ (2003a) Interleukin-2 increases activity of sarcoplasmic reticulum Ca2+-ATPase, but decreases its sensitivity to calcium in rat cardiomyocytes. J Pharmacol Exp Ther 306:572–580
Cao CM, Xia Q, Bruce IC, Shen YL, Ye ZG, Lin GH, Chen JZ, Li GR (2003b) Influence of interleukin-2 on Ca2+ handling in rat ventricular myocytes. J Mol Cell Cardiol 35:1491–1503
Casadei B, Sears CE (2003) Nitric-oxide-mediated regulation of cardiac contractility and stretch responses. Prog Biophys Mol Biol 82(1–3):67–80
Chandrasekar B, Vemula K, Surabhi RM et al (2004) Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem 279:20221–20233
Chandrasekar B, Mummidi S, Claycomb WC, Mestril R, Nemer M (2005) Interleukin-18 is a pro-hypertrophic cytokine that acts through a phosphatidylinositol 3-kinase-phosphoinositide dependent kinase-1-Akt-GATA4 signaling pathway in cardiomyocytes. J Biol Chem 280:4553–4567
Chandrasekar B, Valente AJ, Freeman GL et al (2006) Interleukin-18 induces human cardiac endothelial cell death via a novel signaling pathway involving NF-kappaB-dependent PTEN activation. Biochem Biophys Res Commun 339:956–963
Chen Z, Siu B, Ho YS et al (1998) Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol 30:2281–2289
Chen SA, Hsieh MH, Tai CT et al (1999) Initiation of atrial fibrillation by ectopic beats originating from the pulmonary veins: electrophysiological characteristics, pharmacological responses, and effects of radiofrequency ablation. Circulation 100:1879–1886
Chen YJ, Tsai CF, Chiou CW, Chan P, Chen SA (2001) Effect of nitric oxide on strophanthidin-induced ventricular tachycardia. Pharmacology 62:213–217
Chen Y, Pat B, Zheng J, Cain L, Powell P, Shi K, Sabri A, Husain A, Dell’italia LJ (2010) Tumor necrosis factor-alpha produced in cardiomyocytes mediates a predominant myocardial inflammatory response to stretch in early volume overload. J Mol Cell Cardiol 49(1):70–78
Cheng XS, Shimokawa H, Momii H et al (1999) Role of superoxide anion in the pathogenesis of cytokine-induced myocardial dysfunction in dogs in vivo. Cardiovasc Res 42:651– 659
Chung MK, Gulick TS, Rotondo RE et al (1990) Mechanism of cytokine inhibition of beta-adrenergic agonist stimulation of cyclic AMP in rat cardiac myocytes. Impairment of signal transduction. Circ Res 67:753–763
Colston JT, Boylston WH, Feldman MD, Jenkinson CP, de la Rosa SD, Barton A, Trevino RJ, Freeman GL, Chandrasekar B (2007) Interleukin-18 knockout mice display maladaptive cardiac hypertrophy in response to pressure overload. Biochem Biophys Res Commun 354:552–558
Cugno M, Agostoni P, Brunner HR et al (2000) Plasma bradykinin levels in human chronic congestive heart failure. Clin Sci (London) 99:461–466
Damron DS, Summers BA (1997) Arachidonic acid enhances contraction and intracellular Ca2+ transients in individual rat ventricular myocytes. Am J Physiol 272:H350–H359
Debrunner M, Schuiki E, Minder E et al (2008) Proinflammatory cytokines in acute myocardial infarction with and without cardiogenic shock. Clin Res Cardiol 97:298–305
Dernellis J, Panaretou M (2001) C-reactive protein and paroxysmal atrial fibrillation: evidence of the implication of an inflammatory process in paroxysmal atrial fibrillation. Acta Cardiol 56:375–380
Dernellis J, Panaretou M (2006) Effects of C-reactive protein and the third and fourth components of complement (C3 and C4) on incidence of atrial fibrillation. Am J Cardiol 97:245–248
Dettbarn CA, Betto R, Salviati G et al (1994) Modulation of cardiac sarcoplasmic reticulum ryanodine receptor by sphingosine. J Mol Cell Cardiol 26:229–242
DiFrancesco D (1993) Pacemaker mechanisms in cardiac tissue. Annu Rev Physiol 55:455–472
Dinarello CA (2000) Proinflammatory cytokines. Chest 118:503–508
Dizdarević-Hudić L, Kušljugić Z, Baraković F, Brkić S, Sabitović D, Jahić E, Isabegović M, Smajić E, Hudić I, Divković K (2009) Correlation between interleukin 6 and interleukin 10 in acute myocardial infarction. Bosn J Basic Med Sci 9(4):301–306
Duncan DJ, Yang Z, Hopkins PM et al (2010) TNF-α and IL-1β increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium 47:378–386
Eddy LJ, Goeddel DV, Wong GH (1992) Tumor necrosis factor alpha pretreatment is protective in a rat model of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun 184:1056–1059
Edmunds NJ, Woodward B (1998) Effects of tumour necrosis factor-α on the coronary circulation of the rat isolated perfused heart: a potential role for thromboxane A2 and sphingosine. Br J Pharmacol 124:493–498
Edmunds NJ, Lal H, Woodward B (1999) Effects of tumour necrosis factor-α on left ventricular function in the rat isolated perfused heart: possible mechanisms for a decline in cardiac function. Br J Pharmacol 126:189–196
El-Ani D, Zimlichman R (2003) TNFalpha stimulated ATP-sensitive potassium channels and attenuated deoxyglucose and Ca uptake of H9c2 cardiomyocytes. Ann N Y Acad Sci 1010:716–720
Evans HG, Lewis MJ, Shah AH (1993) Interleukin-1β modulates myocardial contraction via a dexamethasone sensitive production of nitric oxide. Cardiovasc Res 27:1486–1490
Fedak PW, Verma S, Weisel RD, Li RK (2005) Cardiac remodeling and failure from molecules to man (part II). Cardiovasc Pathol 14:49–60
Ferdinandy P, Danial H, Ambrus I et al (2000) Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ Res 87:241–247
Fernandez-Velasco M, Ruiz-Hurtado G, Hurtado O et al (2007) TNF-α downregulates transient outward potassium current in rat ventricular myocytes through iNOS overexpression and oxidant species generation. Am J Physiol 293:H238–H245
Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL (1992) Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 257:387–389
Flaherty MP, Guo Y, Tiwari S et al (2008) The role of TNF-alpha receptors p55 and p75 in acute myocardial ischemia/reperfusion injury and late preconditioning. J Mol Cell Cardiol 45:735–741
Flesch M, Hoper A, Dell’Italia L et al (2003) Activation and functional significance of the renin-angiotensin system in mice with cardiac restricted overexpression of tumor necrosis factor. Circulation 108:598–604
Friedrichs GS, Swillo RE, Jow B et al (2002) Sphingosine modulates myocyte electrophysiology, induces negative inotropy, and decreases survival after myocardial ischemia. J Cardiovasc Pharmacol 39:18–28
Gaudino M, Andreotti F, Zamparelli R et al (2003) The -174G/C interleukin-6 polymorphism influences postoperative interleukin-6 levels and postoperative atrial fibrillation. Is atrial fibrillation an inflammatory complication? Circulation 108(10 Suppl 1):II195–II199
Goldhaber JI, Kim KH, Natterson PD, Lawrence T, Ph Y, Weiss JN (1996) Effects of TNF-α on [Ca2+]i and contractility in isolated adult rabbit ventricular myocytes. Am J Physiol 271:H1449–H1455
Grandel U, Fink L, Blum A et al (2000) Endotoxin-induced myocardial tumor necrosis factor-α synthesis depresses contractility of isolated rat hearts: evidence for a role of sphingosine and cyclooxygenase-2-derived thromboxane production. Circulation 102:2758–2764
Grandy SA, Fiset C (2009) Ventricular K+ currents are reduced in mice with elevated levels of serum TNFα. J Mol Cell Cardiol 47:238–246
Grandy SA, Brouillette J, Fiset C (2010) Reduction of ventricular sodium current in a mouse model of HIV. J Cardiovasc Electrophysiol 21:916–922
Gulick T, Chung MK, Pieper SJ et al (1989) Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte β-adrenergic responsiveness. PNAS 86:6753–6757
Harada E, Nakagawa O, Yoshimura M et al (1999) Effect of interleukin-1 beta on cardiac hypertrophy and production of natriuretic peptides in rat cardiocyte culture. J Mol Cell Cardiol 31:1997–2006
Harding P, Carretero OA, LaPointe MC (1995) Effects of interleukin-1β and nitric oxide on cardiac myocytes. Hypertension 25:421–430
Hare JM (2003) Nitric oxide and excitation–contraction coupling. J Mol Cell Cardiol 35:719–729
Hatada K, Washizuka T, Horie M et al (2006) Tumor necrosis factor-alpha inhibits the cardiac delayed rectifier K current via the asphingomyelin pathway. Biochem Biophys Res Commun 344(1):189–193
Haudek SB, Taffet GE, Schneider MD, Mann DL (2007) TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest 117:2692–2701
Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N (2010) Proinflammatory cytokines in heart failure: double-edged swords. Heart Fail Rev 15:543–562
Heger J, Goedecke A, Flögel U, Merx MW, Molojavyi A, Kühn-VeltenW N, Schrader J (2002) Cardiac-specific overexpression of inducible nitric oxide synthase does not result in severe cardiac dysfunction. Circ Res 90:93–99
Hofmann U, Domeier E, Frantz S et al (2003) Increased myocardial oxygen consumption by TNF-α is mediated by a sphingosine signaling pathway. Am J Physiol 284:H2100–H2105
Hoppe UC, Beuckelmann DJ (1998) Characterization of the hyperpolarization-activated current in isolated human atrial myocytes. Cardiovasc Res 38:788–801
Hosenpud JD, Campbell SM, Mendelson DJ (1989) Interleukin-l-induced myocardial depression in an isolated beating heart preparation. J Heart Transplant 8:460–464
Hu Y, Chen YC, Cheng CC, Higa S, Chen YJ, Chen SA (2009) Fluvastatin reduces pulmonary vein spontaneous activity through nitric oxide pathway. J Cardiovasc Electrophysiol 20:200–206
Hund TJ, Rudy Y (2004) Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation 110:3168–3174
Ing DJ, Zang J, Dzau VJ et al (1999) Modulation of cytokine-induced cardiac myocyte apoptosis by nitric oxide, Bak, and Bcl-x. Circ Res 84:21–33
Isenberg G, Kazanski V, Kondratev D, Gallitelli MF, Kiseleva I, Kamkin A (2003) Differential effects of stretch and compression on membrane currents and [Na+]C in ventricular myocytes. Prog Biophys Mol Biol 82:43–56
Issac TT, Dokainish H, Lakkis NM (2007) Role of inflammation in initiation and perpetuation of atrial fibrillation. J Am Coll Cardiol 50:2021–2028
Janczewski AM, Kadokami T, Lemster B et al (2003) Morphological and functional changes in cardiac myocytes isolated from mice overexpressing TNF-α. Am J Physiol 284:H960–H969
Janse MJ (1992) The premature beat. Cardiovasc Res 26:89–100
Janse MJ (1997) Why does atrial fibrillation occur? Eur Heart J 18(suppl):C12–C18
Janssen SPM, Gayan-Ramirez G, Van Den Bergh A, Herijgers P, Maes K, Verbeken E, Decramer M (2005) Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation 111:996–1005
Jayadev S, Linardic CM, Hannun YA (1994) Identification of arachidonic acid as a mediator of sphingomyelin hydrolysis in response to tumor necrosis factor-α. J Biol Chem 269:5757–5763
Joe EK, Schussheim AE, Longrois D et al (1998) Regulation of cardiac myocyte contractile function by inducible nitric oxide synthase (iNOS): mechanisms of contractile depression by nitric oxide. J Mol Cell Cardiol 30:303–315
Ju H, Dixon IM (1996) Extracellular matrix and cardiovascular diseases. Can J Cardiol 12:1259–1267
Kaab S, Dixon J, Duc J et al (1998) Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation 98:1383–1393
Kamkin A, Kiseleva I, Wagner KD, Leiterer KP, Theres H, Scholz H, Günther J, Lab MJ (2000a) Mechano-electric feedback in right atrium after left ventricular infarction in rats. J Mol Cell Cardiol 32:465–477
Kamkin A, Kiseleva I, Isenberg G (2000b) Stretch-activated currents in ventricular myocytes: amplitude and arrhythmogenic effects increase with hypertrophy. Cardiovasc Res 48:409–420
Kamkin A, Kiseleva I, Isenberg G (2003a) Ion selectivity of stretch-activated cation currents in mouse ventricular myocytes. Eur J Physiol 446(2):220–231
Kamkin A, Kiseleva I, Wagner KD, Bohm J, Theres H, Günther J, Scholz H (2003b) Characterization of stretch-activated ion currents in isolated atrial myocytes from human hearts. Eur J Physiol 446(3):339–346
Kang JX, Leaf A (1994) Effects of long-chain polyunsaturated fatty acids on the contraction of neonatal rat cardiac myocytes. PNAS 91:9886–9890
Kao YH, Chen YC, Cheng CC et al (2010) Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit Care Med 8(1):217–222
Karmazym M, Gan XT, Humphreys RA, Yoshida H, Kusumoto K (1999) The myocardial Na+-H+ exchange structure, regulation, and its role in heart disease. Circ Res 85:777–786
Kaspar A, Brinkmeier H, Riidel R (1994) Local anaesthetic-like effect of interleukin-2 on muscular Na+-channels: no evidence for involvement of the IL-2 receptor. Eur J Physiol 426:61–67
Kato D, Cragoe EJ, Smith TW (1987) Relations among sodium pump inhibition, Na-Ca and Na-H exchange activities, and Ca-H interaction in cultured chick heart cells. Circ Res 60:185–193
Kawada H, Niwano S, Niwano H et al (2006) Tumor necrosis factor-alpha downregulates the voltage gated outward K_ current in cultured neonatal rat cardiomyocytes: a possible cause of electrical remodeling in diseased hearts. Circulation 70:605–609
Kazanski VE, Kamkin AG, Makarenko EY, Lysenko NN, Sutiagin PV, Bo T, Kiseleva IS (2010a) Role of nitric oxide in activity control of mechanically gated ionic channels in cardiomyocytes: NO-donor study. Bull Exp Biol Med 150(1):1–5 English
Kazanski VE, Kamkin AG, Makarenko EY, Lysenko NN, Sutiagin PV, Kiseleva IS (2010b) Role of nitric oxide in the regulation of mechanosensitive ionic channels in cardiomyocytes: Contribution of NO-synthases. Bull Exp Biol Med 150(2):263–267 English
Kazanski V, Kamkin A, Makarenko E, Lysenko N, Lapina N, Kiseleva I (2011) The role of nitric oxide in regulation of mechanically gated channels in the heart. In: Kamkin A, Kiseleva I (eds) Mechanosensitivity in cells and tissues 4. Mechanosensitivity and mechanotransduction. Springer, New York, NY, pp 109–140
Kelly RA, Balligand JL, Smith TW (1996) Nitric oxide and cardiac function. Circ Res 79:363–380
Kinugawa K, Takahashi T, Kohmoto O, Yao A, Aoyagi T, Momomura S, Hirata Y, Serizawa T (1994) Nitric oxide-mediated effects of interleukin-6 on [Ca2+]i and cell contraction in cultured chick ventricular myocytes. Circ Res 75:285–295
Kiseleva I, Kamkin A, Wagner KD, Theres H, Ladhoff A, Scholz H, Günther J, Lab MJ (2000) Mechano-electric feedback after left ventricular infarction in rats. Cardiovasc Res 45:370–378
Kojda G, Kottenberg K (1999) Regulation of basal myocardial function by NO. Cardiovasc Res 41:514–523
Kolesnick R (2002) The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J Clin Invest 110:3–8
Kondratev D, Gallitelli MF (2003) Increments in the concentration of sodium and calcium in cell compartments of stretched mouse ventricular myocytes. Cell Calcium 34:193–203
Kreydiyyeh SI, Abou-Chahine C, Hilal-Dandan R (2004) Interleukin-1beta inhibits Na+-K+ ATPase activity and protein expression in cardiac myocytes. Cytokine 26(1):1–8
Krown KA, Yasui K, Brooker MJ, Dubin AE, Nguyen C, Harris GL, McDonough PM, Glembotski CC, Palade PT, Sabbadini RA (1995) TNF alpha receptor expression in rat cardiac myocytes: TNF alpha inhibition of L-type Ca2+ current and Ca2+ transients. FEBS Lett 376(1–2):24–30
Kubota T, McTiernan CF, Frye CS et al (1997) Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-α. Circ Res 81:627–635
Kubota I, Han X, Opel DJ, Zhao YY, Baliga R, Huang P, Fishman MC, Shannon RP, Michel T, Kelly RA (2000) Increased susceptibility to development of triggered activity in myocytes from mice with raged disruption of endothelial nitric oxide synthase. J Mol Cell Cardiol 32:1239–1248
Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE (1996) Tumor necrosis factor-alpha and interleukin 1-beta are responsible for the in vitro myocardial cell depression induced by human septic shock serum. J Exp Med 183(3):949–958
Kumar A, Brar R, Wang P et al (1999) Role of nitric oxide and cGMP in human septic serum induced depression of cardiac myocyte contractility. Am J Physiol 276:R265–R276
Kunisada K, Negoro S, Tone E et al (2000) Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. PNAS 97:315–319
Latini R, Bianchi M, Correale E, Dinarello CA, Fantuzzi G, Fresco C, Maggioni AP, Mengozzi M, Romano S, Shapiro L, Sironi M, Tognoni G, Turato R, Ghezzi P (1994) Cytokines in acute myocardial infarction: selective increase in circulating tumor necrosis factor, its soluble receptor, and interleukin 1 receptor antagonist. J Cardiovasc Pharmacol 23(1):1–6
Lee SH, Chen YC, Chen YJ et al (2007) Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci 80(19):1806–1815
Levi RC, Alloatti G, Fischmeister R (1989) Cyclic GMP regulates the Ca channel current in guinea pig ventricular myocytes. Eur J Physiol 413:685–687
Levi RC, Alloatti G, Penna C, Gallo MP (1994) Guanylate-cyclase-mediated inhibition of cardiac ICa by carbachol and sodium nitroprusside. Pflugers Arch 426(5):419–426
Levine B, Kalman J, Mayer L, Fillit HM, Packer M (1990) Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 323(4):236–241
Li Y-H, Rozanski GJ (1993) Effects of human recombinant interleukin-1 on electrical properties of guinea pig ventricular cells. Cardiovasc Res 27:525–530
Li J, Hampton T, Morgan JP, Simons M (1997) Stretch-induced VEGF expression in the heart. J Clin Invest 100(1):18–24
Li YY, Feng YQ, Kadokami T et al (2000) Myocardial extracellular matrix remodeling in transgenic mice overexpressing tumor necrosis factor alpha can be modulated by anti-tumor necrosis factor alpha therapy. PNAS 97:12746–12751
Li GR, Lau CP, Leung TK, Nattel S (2004) Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm 1:460–468
Liu SJ, McHowat J (1998) Stimulation of different phospholipase A2 isoforms by TNF-α and IL-1β in adult rat ventricular myocytes. Am J Physiol 275:H1462–H1472
Liu S, Schreur KD (1995) G protein-mediated suppression of L-type Ca2+ current by interleukin-1β in cultured rat ventricular myocytes. Am J Physiol 268:C339–C349
Liu B, Andrieu-Abadie N, Levade T et al (1998) Glutathione regulation of neutral sphingomyelinase in tumor necrosis factor-α-induced cell death. J Biol Chem 273:11313–11320
Liu SJ, Zhou W, Kennedy RH (1999) Suppression of β-adrenergic responsiveness of L-type Ca2+ current by IL-1β in rat ventricular myocytes. Am J Physiol 276:H141–H148
Liu J, Kim KH, London B et al (2011) Dissection of the voltage-activated potassium outward currents in adult mouse ventricular myocytes: Ito,f, Ito,s, IK,slow1, IK,slow2, and Iss. Basic Res Cardiol 106:189–204
London B, Baker LC, Lee JS, Shusterman V, Choi BR, Kubota T, McTiernan CF, Feldman AM, Salama G (2003) Calcium-dependent arrhythmias in transgenic mice with heart failure. Am J Physiol 284:H431–H441
Long CS (2001) The role of interleukin-1 in the failing heart. Heart Fail Rev 6:81–94
Lorenzon P, Ruzzier F, Caratsch CG et al (1991) Interleukin-2 lengthens extrajunctional acetylcholine receptor channel open time in mammalian muscle cells. Pflugers Arch 419(3-4):380–385
Low-Friedrich I, Weisensee D, Mitrou P, Schoeppe W (1992) Cytokines induce stress protein formation in cultured cardiac myocytes. Basic Res Cardiol 87(1):12–18
Lozinsky I, Kamkin A (2010) Mechanosensitive alterations of action potentials and membrane currents in healthy and diseased cardiomyocytes: cardiac tissue and isolated cell. In: Kamkin A, Kiseleva I (eds) Mechanosensitivity in cells and tissues 3. Mechanosensitivity of the heart. Springer, New York, NY, pp 185–238
Luan Y, Guo Y, Li S et al (2010) Interleukin-18 among atrial fibrillation patients in the absence of structural heart disease. Europace 12:1713–1718
Marcus GM, Whooley MA, Glidden DV, Pawlikowska L, Zaroff JG, Olgin JE (2008) Interleukin 6 and atrial fibrillation in patients with coronary disease: data from the heart and soul study. Am Heart J 155(2):303–309
Marino E, Cardier JE (2003) Differential effect of IL-18 on endothelial cell apoptosis mediated by TNF-alpha and Fas (CD95). Cytokine 22:142–148
Massion PB, Balligand JL (2003) Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): lessons from genetically modified mice. J Physiol (London) 546(1):63–75
Massion PB, Feron O, Dessy C, Balligand JL (2003) Nitric oxide and cardiac function: ten years after, and continuing. Circ Res 93:388 –398
Maulik N, Engelman RM, Wei Z et al (1993) Interleukin-1 alpha preconditioning reduces myocardial ischemia reperfusion injury. Circulation 88:387–394
McDonough AA, Zhang Y, Shin V, Frank JS (1996) Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am J Physiol 270:C1221–C1227
McGowan FX, Takeuchi K, del Nido PJ et al (1994) Myocardial effects of interleukin-2. Transplant Proc 26:209–210
Mery PF, Lohmann SM, Walter U, Fischmeister R (1991) Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc Natl Acad Sci USA 88:1197–1201
Mery PF, Pavoine C, Belhassen L, Pecker F, Fischmeister R (1993) Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J Biol Chem 268:26286–26295
Meyer M, Schillinger W, Pieske B et al (1995) Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 92:778–784
Moncada S, Palmer RMJ, Higgs EA (1991) Nitric oxide physiology, pathophysiology. Pharmacol Rev 433:109–142
Monsuez JJ, Escaut L, Teicher E, Charniot JC, Vittecoq D (2007) Cytokines in HIV-associated cardiomyopathy. Int J Cardiol 2:150–157
Nakano M, Knowlton AA, Yokoyama T et al (1996) Tumor necrosis factor-alpha-induced expression of heat shock protein 72 in adult feline cardiac myocytes. Am J Physiol 270:H1231–H1239
Nakano M, Knowlton AA, Dibbs Z, Mann DL (1998) Tumor necrosis factor-alpha confers resistance to hypoxic injury in the adult mammalian cardiac myocyte. Circulation 97:1392–1400
Nakayama H, Bodi I, Correll RN, Chen X, Lorenz J, Houser SR, Robbins J, Schwartz A, Molkentin JD (2009) alpha1G-dependent T-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice. J Clin Invest 119(12):3787–3796
Narayan SM, Cain ME, Smith JM (1997) Atrial fibrillation. Lancet 350:943–950
Nattel S, Maguy A, Le Bouter S, Yeh YH (2007) Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 87:425–456
Nerbonne JM, Kass RS (2005) Molecular physiology of cardiac repolarization. Physiol Rev 85:1205–1253
Nishikawa K, Yoshida M, Kusuhara M et al (2006) Left ventricular hypertrophy in mice with a cardiac-specific overexpression of interleukin-1. Am J Physiol 291:H176–H183
Nuss HB, Kaab S, Kass DA et al (1999) Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol 277:H80–H91
Ono K, Trautwein W (1991) Potentiation by cyclic GMP of b-adrenergic effect on Ca2+ current in guinea pig ventricular cells. J Physiol (London) 443:387–404
Opthof T (1988) The membrane current (If) in human atrial cells: implications for atrial arrhythmias. Cardiovasc Res 38:537–540
Oral H, Dorn GWII, Mann DL (1997) Sphingosine mediates the immediate negative inotropic effects of tumor necrosis factor-α in the adult mammalian cardiac myocyte. J Biol Chem 272:4836–4842
Oyama J, Shimokawa H, Momii H (1998) Role of nitric oxide and peroxynitrite in the cytokine-induced sustained myocardial dysfunction in dogs in vivo. J Clin Invest 101:2207–2214
Packer M (1995) Is tumor necrosis factor an important neurohormoral mechanism in chronic heart failure? Circulation 92(6):1379–1382
Paganelli F, Mougenot R, Maixentt JM (2001) Defective activity and isoform of the Na,K-ATPase in the dilated cardiomyopathic hamster. Cell Mol Biol (Noisy-le-grand) 47:255–260
Palmer JN, Hartogensis WE, Patten M et al (1995) Interleukin-1 beta induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest 95:2555–2564
Panas D, Khadour FH, Szabo C, Schulz R (1998) Proinflammatory cytokines depress cardiac efficiency by a nitric oxide-dependent mechanism. Am J Physiol 275:H1016–H1023
Park CO, Xiao XH, Allen DG (1999) Changes in intracellular sodium and pH in the rat heart during ischaemia: role of the Na+/H+ exchanger. Am J Physiol 276(5Pt2):H1581–H1590
Petkova-Kirova PS, Gursoy E, Mehdi H et al (2006) Electrical remodeling of cardiac myocytes from mice with heart failure due to the overexpression of tumor necrosis factor-α. Am J Physiol 290(5):H2098–H2107
Pino R, Cerbai E, Calamai G et al (1998) Effect of 5-HT4 receptor stimulation on the pacemaker current If in human isolated atrial myocytes. Cardiovasc Res 40:516–522
Porciatti F, Pelzmann B, Cerbai E et al (1997) The pacemaker current If in single human atrial myocytes and the effect of beta-adrenoceptor and A1-adenosine receptor stimulation. Br J Pharmacol 122:963–969
Prabhu SD (2004a) Cytokine-induced modulation of cardiac function. Circ Res 95:1140–1153
Prabhu SD (2004b) Nitric oxide protects against pathological ventricular remodeling: reconsideration of the role of NO in the failing heart. Circ Res 94:1155–1157
Priebe L, Beuckelmann DJ (1998) Simulation study of cellular electric properties in heart failure. Circ Res 82:1206–1223
Proebstle T, Mitrovics M, Scneider M, Hombach V, Ruedel R (1995) Recombinant interleukin-2 acts like a class I antiarrhythmic drug on human cardiac sodium channels. Eur J Physiol 429:462–469
Psychari SN, Apostolou TS, Sinos L, Hamodraka E, Liakos G, Kremastinos DT (2005) Relation of elevated C-reactive protein and interleukin-6 levels to left atrial size and duration of episodes in patients with atrial fibrillation. Am J Cardiol 95(6):764–767
Rashmi RS, Hondeghem LM (2005) Refining detection of drug-induced proarrhythmia: QT interval and TRIaD. Heart Rhythm 2:758–772
Robinson BS, Hii CS, Poulos A, Ferrante A (1997) Activation of neutral sphingomyelinase in human neutrophils by polyunsaturated fatty acids. Immunology 91:274–280
Roedler S, Roth M, Nauck M, Tamm M, Block LH (1994) Ca2+-channel blockers modulate the expression of interleukin-6 and interleukin-8 genes in human vascular smooth muscle cells. J Mol Cell Cardiol 27:2295–2302
Rozanski GJ, Witt RC (1994a) IL-l inhibits β-adrenergic control of cardiac calcium current: role of L-arginine/nitric oxide pathway. Am J Physiol 267:H1753–H1758
Rozanski GJ, Witt RC (1994b) Interleukin-1 enhances β-responsiveness of cardiac L-type calcium current suppressed by acidosis. Am J Physiol 267:H1361–H1367
Saba S, Janczewski AM, Baker LC et al (2004) Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-α. Am J Physiol 289:H1456–H1467
Sani MU, Okeahialam BN (2005) QTc interval prolongation in patients with HIV and AIDS. J Natl Med Assoc 12:1657–1661
Sartiani L, Paoli PD, Lonardo G, Pino R, Conti AA, Cerbai E, Pelleg A, Belardinelli L, Mugelli A (2002) Does recombinant human interleukin-11 exert direct electrophysiologic effects on single human atrial myocytes? J Cardiovasc Pharmacol 39:425–434
Sata N, Hamada N, Horinouchi T et al (2004) C-reactive protein and atrial fibrillation: Is inflammation a consequence or cause of atrial fibrillation? Japan Heart J 45:441–445
Schreur KD, Liu S (1997) Involvement of ceramide in inhibitory effect of IL-1β on L-type Ca2+ current in adult rat ventricular myocytes. Am J Physiol 272:H2591–H2598
Schulz R (2008) TNF-α in myocardial ischemia/reperfusion: damage vs. protection. J Mol Cell Cardiol 45:712–714
Schulz R, Heusch G (2009) Tumor necrosis factor-α and its receptors 1 and 2. Yin and Yang in myocardial infarction? Circulation 119:1355–1357
Seddon M, Shah AM, Casadei B (2007) Cardiomyocytes as effectors of nitric oxide signalling. Cardiovasc Res 75(2):315–326
Shah AM, MacCarthy PA (2000) Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol Ther 86(1):49–86
Sivasubramanian N, Coker ML, Kurrelmeyer KM et al (2001) Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation 104:826–831
Siwik DA, Colucci WS (2004) Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev 9:43–51
Siwik DA, Chang DL, Colucci WS (2000) Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res 86:1259–1265
Smart N, Mojet MH, Latchman DS et al (2006) IL-6 induces PI 3-kinase and nitric oxide dependent protection and preserves mitochondrial function in cardiomyocytes. Cardiovasc Res 69:164–177
Smith MD, Haynes DR, Roberts-Thomson PJ (1989) Interleukin 2 and interleukin 2 inhibitors in human serum and synovial fluid. I. Characterization of the inhibitor and its mechanism of action. J Rheumatol 16(2):149–157
Stamler JS, Lamas S, Fang FC (2001) Nitrosylation. The prototypic redox-based signaling mechanism. Cell 106:675–683
Stamm C, Cowan DB, Friehs I et al (2001a) Rapid endotoxin-induced alterations in myocardial calcium handling: obligatory role of cardiac TNF-alpha. Anesthesiology 95:1396–1405
Stamm C, Friehs I, Cowan DB et al (2001b) Inhibition of tumor necrosis factor-α improves postischemic recovery of hypertrophied hearts. Circulation 104:I350–I355
Sugano M, Hata T, Tsuchida K et al (2004) Local delivery of soluble TNF-alpha receptor 1 gene reduces infarct size following ischemia/reperfusion injury in rats. Mol Cell Biochem 266:127–132
Sugishita K, Kinugawa K, Shimizu T, Harada K, Matsui H, Takahashi T, Serizawa T, Kohmoto O (1999) Cellular basis for the acute inhibitory effects of IL-6 and TNF-α on excitation-contraction coupling. J Mol Cell Cardiol 31:1457–1467
Tavi P, Laine M, Weckstrom M (1996) Effect of gadolinium on stretch-induced changes in contraction and intracellularly recorded action- and after potentials of rat isolated atrium. Br J Pharmacol 118:407–413
Tavi P, Han C, Weckstrom M (1998) Mechanisms of stretch induced changes in [Ca2+]i in rat atrial myocytes: role of increased troponin C affinity and stretch-activated ion channels. Circ Res 83:1165–1177
Testa M, Yeh M, Lee P et al (1996) Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol 28:964–971
Trotter JL, Clifford DB, Anderson CB et al (1988) Elevated serum interleukin-2 levels in chronic progressive multiple sclerosis. N Engl J Med 318(18):1206
Thaik CM, Calderone A, Takahashi N, Colucci WS (1995) Interleukin-1 beta modulates the growth and phenotype of neonatal rat cardiac myocytes. J Clin Invest 96:1093–1099
Thakkar J, Tang SB, Sperelakis N, Wahler GM (1988) Inhibition of cardiac slow action potentials by 8-bromo-cyclic GMP occurs independent of changes in cyclic AMP levels. Can J Physiol Pharmacol 66:1092–1095
Ungureanu-Longrois D, Balligand JL et al (1995) Contractile responsiveness of ventricular myocytes to isoproterenol is regulated by induction of nitric oxide synthase activity in cardiac microvascular endothelial cells in heterotypic primary culture. Circ Res 77:486–493
Ungureanu-Longrois D, Bezie Y, Perret C, Laurent S (1997) Effects of exogenous and endogenous nitric oxide on the contractile function of cultured chick embryo ventricular myocytes. J Mol Cell Cardiol 29:677–687
Vaughan Williams EM (1984) A classification of antiarrhythmic actions reassessed after a decade of new drugs. J Clin Pharmacol 24:129–147
Virag L, Iost N, Opincariu M et al (2001) The slow component of the delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc Res 49:790–797
Wahler GM, Dollinger SJ (1995) Nitric oxide donor SIN-1 inhibits mammalian cardiac calcium current through cGMP-dependent protein kinase. Am J Physiol 268:C45–C54
Wan S, DeSmet JM, Barvais L, Goldstein M, Vincent JL, LeClerc JL (1996) Myocardium is a major source of proinflammatory cytokines in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg 112:806–811
Wang J, Wang H, Zhang Y, Gao H, Nattel S, Wang Z (2004) Impairment of HERG K+ channel function by tumor necrosis factor-alpha: role of reactive oxygen species as a mediator. J Biol Chem 279(14):13289–13292
Ward M-L, David G, Allen DG (2010) Stretch-activated channels in the heart: contribution to cardiac performance. In: Kamkin A, Kiseleva I (eds) Mechanosensitivity in cells and tissues 3. Mechanosensitivity of the heart. Springer, New York, NY, pp 141–167
Weisensee D, Bereiter-Hahn J, Schoeppe W, Low-Friedrich I (1993) Effects of cytokines on the contractility of cultured cardiac myocytes. Int J Immunopharmacol 15:581–587
Wong GH, Goeddel DV (1988) Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science 242:941–944
Wyeth RP, Temma K, Seifen E, Kennedy RH (1996) Negative inotropic actions of nitric oxide require high doses in rat cardiac muscle. Eur J Physiol 432:678–684
Yamauchi-Takihara K, Kishimoto T (2000) Cytokines and their receptors in cardiovascular diseases – role of gp130 signalling pathway in cardiac myocyte growth and maintenance. Int J Exp Pathol 81:1–16
Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL (1993) Cellular basis for the negative inotropic effects of tumor necrosis factor-a in the adult mammalian cardiac myocytes. J Clin Invest 92(5):2303–2312
Yokoyama T, Sekiguchi K, Tanaka T, Tomaru K, Arai M, Suzuki T, Nagai R (1999) Angiotensin II and mechanical stretch induce production of tumor necrosis factor in cardiac fibroblasts. Am J Physiol 276(6 Pt 2):H1968–H1976
Yu X, Kennedy RH, Liu SJ (2003a) JAK2/STAT3, not ERK1/2, mediates interleukin-6-induced activation of inducible nitric oxide synthase and decrease in contractility of adult ventricular myocytes. J Biol Chem 278:16304–16309
Yu X-W, Kennedy RH, Liu SJ (2003b) JAK2/STAT3, not ERK1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes. J Biol Chem 278:16304–16308
Yu X-W, Chen Q, Kennedy RH, Liu SJ (2005a) Inhibition of sarcoplasmic reticular function by chronic interleukin-6 exposure via iNOS in adult ventricular myocytes. J Physiol (London) 566(2):327–340
Yu X, Patterson E, Huang S et al (2005b) Tumor necrosis factor α, rapid ventricular tachyarrhythmias, and infarct size in canine models of myocardial infarction. J Cardiovasc Pharmacol 45:153–159
Zeng T, Bett GCL, Sachs F (2000) Stretch-activated whole cell currents in adult rat cardiac myocytes. Am J Physiol 278:H548–H557
Zhang WM and Wong TM (1998) Suppression of cAMP by phosphoinositol/Ca2+ pathway in the cardiac kappa-opioid receptor. Am J Physiol 274(1 Pt 1):C82–C87
Zhang YH, Youm JB, Sung HK, Lee SH, Ryu SY, Lee S-H, Ho W-K, Earm YE (2000) Stretch-activated and background non-selective cation channels in rat atrial myocytes. J Physiol (London) 523:607–619
Zingman LV, Alekseev AE, Hodgson-Zingman DM, Terzic A (2007) ATP-sensitive potassium channels: metabolic sensing and cardioprotection. J Appl Physiol 103(5):1888–1893
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grant no. 09-04-01277-a). Department of Fundamental and Applied Physiology (Professor and Chairman – Andre Kamkin) was supported by Ministry of Education and Science of the Russian Federation. The Order of Ministry of Education and Science of the Russian Federation No. 743 from 01 July 2010, Supplement, Event 4.4, the Period of Financing 2010–2019.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Kuzmin, V.S. et al. (2012). The Role of Proinflammatory Cytokines in Regulation of Cardiac Bioelectrical Activity: Link to Mechanoelectrical Feedback. In: Kamkin, A., Kiseleva, I. (eds) Mechanical Stretch and Cytokines. Mechanosensitivity in Cells and Tissues, vol 5. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-2004-6_5
Download citation
DOI: https://doi.org/10.1007/978-94-007-2004-6_5
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-2003-9
Online ISBN: 978-94-007-2004-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)