
Abstract
Krüppel-type or C2H2 zinc fingers represent a dominant DNA-binding motif in eukaryotic transcription factor (TF) proteins. In Krüppel-type (KZNF) TFs, KZNF motifs are arranged in arrays of three to as many as 40 tandem units, which cooperate to define the unique DNA recognition properties of the protein. Each finger contains four amino acids located at specific positions, which are brought into direct contact with adjacent nucleotides in the DNA sequence as the KZNF array winds around the major groove of the alpha helix. This arrangement creates an intimate and potentially predictable relationship between the amino acid sequence of KZNF arrays and the nucleotide sequence of target binding sites. The large number of possible combinations and arrangements of modular KZNF motifs, and the increasing lengths of KZNF arrays in vertebrate species, has created huge repertoires of functionally unique TF proteins. The properties of this versatile DNA-binding motif have been exploited independently many times over the course of evolution, through attachment to effector motifs that confer activating, repressing or other activities to the proteins. Once created, some of these novel inventions have expanded in specific evolutionary clades, creating large families of TFs that are lineage- or species-unique. This chapter reviews the properties and their remarkable evolutionary history of eukaryotic KZNF TF proteins, with special focus on large families that dominate the TF landscapes in different metazoan species.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Introduction
The C2H2 zinc finger motif, first identified in studies of the Xenopus TF TIFIIA [1] is by far the most common protein domain in metazoan TFs (see Chapter 3 ). Most versions of this abundant motif correspond to a subtype called the “Krüppel-type”, named for the Drosophila Krüppel protein, a developmentally active TF that bears the canonical C2H2 zinc-binding structure [1, 2]. The C2H2 zinc finger motif was originally thought to be specific to eukaryotes, but a very similar structural domain has been identified in some bacterial TF genes, hinting at more ancient origins [3]. The most striking and characteristic feature of these 28 amino acid motifs is a secondary structure that is dependent upon the coordination of a single zinc atom by paired cysteine (C) and histidine (H) residues (Fig. 4.1). This zinc-dependent structure is required for the interaction between the finger motif and nucleic acids; in the absence of zinc, or if elements of the conserved C2H2 structure are abolished through mutation, zinc fingers lose their ability to fold properly and to bind DNA [1, 4–6].

Tandem Krüppel-type zinc finger structure displaying the C2H2 motif. Individual zinc ions interact with paired cysteine (C) and histidine (H) residues, stabilizing protein fold structure within zinc fingers. Each finger consists of two β-sheets and one α-helix, the latter of which contains residues that make up the DNA-binding interface (at positions –1, 2, 3, and 6 relative to the helix) as indicated in the figure. The common structure of a finger sequence motif is represented, with X denoting an amino acid residue of any type with the subscript representing the number (X 2–4 represents between 2 and 4 non-specified amino acid residues). The consensus sequence, TGEKP(Y/F), is a highly conserved “H/C link” region between consecutive fingers
In addition to the paired cysteine and histidine residues, Krüppel-type zinc finger (KZNF) motifs contain a highly conserved inter-finger “spacer”, or H/C link sequence, a seven amino acid segment with the consensus sequence TGEKP(Y/F) (Fig. 4.1). KZNF proteins carry out many different kinds of molecular functions, including protein-protein interactions, RNA binding, and sequence-specific binding to DNA. Some DNA-binding KZNFs are now known to carry out functions related to meiotic recombination and chromosome segregation [7–11], or maintenance of DNA methylation marks [12, 13]. Additional functions related to chromosome structure and maintenance may be found as new research is completed. However, most KZNFs with specific DNA recognition capabilities are thought to function as TFs, and this latter class of proteins is the primary focus of this chapter.
Typically, DNA binding KZNF proteins contain 3 or more zinc-finger motifs, which are arranged in tandem within the protein (Figs. 4.1 and 4.2). These multifingered, or “polydactyl” KZNF proteins include many of the best-known TFs in eukaryotes, including yeast, plants, invertebrate and vertebrate species. TF proteins with as many as 40 tandem KZNF motifs can be found in most vertebrate genomes and long polydactyl KZNF proteins are also found in plants [14]. The tandem arrangement of KZNF motifs permits the adjacent fingers to interact and stabilize DNA binding of the protein at specific sites, as will be discussed in more detail in the following sections. While zinc-fingers define binding site specificity and stability for KZNF proteins, most TFs of this type also require one or more “effector” motifs to translate site-specific DNA binding into gene regulatory activities impacting neighboring genes.

Exon and protein structure of a typical KZNF TF protein. Many KZNF genes, including those of the KRAB, SCAN, BTB/POZ, ZAD and other families, contain one or more exons encoding a specific N-terminal effector domain, and a second exon encoding a “spacer” or “tether” region and an array of 3–40 zinc finger motifs. The tandem arrangement of KZNF-encoding sequences, which contain highly conserved structural elements, has enabled rapid evolution of proteins of this type
Over the course of evolution, exons encoding tandem KZNF arrays have become associated with coding sequences for a wide variety of different effector domains, to generate proteins with novel structures and activities. Many of these novel KZNF proteins have arisen in, and remain exclusive to, particular evolutionary lineages; some of these species-specific genes have expanded through repeated duplication events to form large families of lineage-specific genes. While this same process has occurred for many gene types, the lineage-specific expansion of KZNF genes is a striking and extraordinary story. This chapter will focus on basic functions of the KZNF motifs, the types of TFs that rely on their highly specific targeting abilities, and their remarkable evolutionary trajectory.
4.2 Zinc Finger–DNA Interactions
The structural elements that control the interaction between KZNF motifs and DNA “target sites” include the paired cysteine and histidine residues, as well as the amino acids surrounding them. The arrangement and spacing of elements within the finger motif, including the H/C link, are critical to maintaining the zinc-finger structure, and are therefore very highly conserved [1]. Most importantly for DNA binding, residues near the C-terminal end of each finger fold into an alpha helix, positioning specific amino acids within the helix to interact directly with DNA (Figs. 4.1 and 4.3). In particular, positions –1, 2, 3 and 6 (relative to the alpha helix) play a critical role in DNA interaction: together, the amino acids at these four positions in each finger are thought largely to determine DNA binding specificity of the protein [15, 16].

KZNF motif DNA-binding interactions. The alpha helices of KZNF motifs contain amino acid residues that bind to DNA nucleotides (at the –1, 2, 3, and 6 sites as shown at top). The relationship between fingers and nucleotides is not one-to-one, as the amino acid at the +2 position will interact with the nucleotide complementary to the neighboring finger’s +6 binding site. In this fashion, fingers wind around the major groove of the DNA molecule (illustrated in the lower panel of the figure)
The array of multiple, adjacent fingers in these proteins winds around the DNA double strand within the major groove, wrapping around the DNA molecule in an intimate spatial relationship that places the DNA-contacting residues of each finger in register with nucleotides within a turn of the helix. The interaction between the four DNA contacting amino-acid residues in each finger and nucleotides at the DNA binding site is not a simple 1:1 relationship, as there is some overlap between nucleotides bound by adjacent fingers [6, 17–19]. However, the arrangement is such that each finger defines binding specificity at a net of 3 adjacent nucleotides, while exerting some influence over the binding specificity of neighboring KZNF motifs (Fig. 4.3).
4.2.1 Predicting a Zinc-Finger Code
This precisely structured relationship between nucleotides in a binding site and specific amino acids in the DNA-contacting portion of each C2H2 finger implies the existence of a zinc-finger DNA binding “code”, and the possibility that a KZNF protein’s binding preferences might be predicted de novo from its amino acid sequence. In fact, several different groups have designed mathematical formulas and informatics tools that predict KZNF binding codes [19–21]. These programs are built upon knowledge derived from in vitro DNA binding experiments and structural data, together with calculations of predicted energies of interaction between specific amino acids and nucleotides. Although these methods have proved successful in designing custom KZNF proteins to bind with maximum efficiency to unique sites both in vitro and in vivo [22] it is still unclear if they can accurately predict the binding preferences of KZNF proteins as they exist in normal cellular contexts.
There are several reasons why KZNF arrays, and especially long polydactyl proteins, might not behave in living systems as in vitro models would predict. Firstly, most in vitro studies have focused on predetermined libraries of zinc-finger triplets that are selected for maximal binding to naked DNA under non-biological conditions; by contrast, natural selection in living systems operates in a much more complex milieu, and has taken full advantage of the combinatorial possibilities to produce KZNF protein repertoires of remarkable diversity. Since zinc-finger DNA binding is known to be context-dependent, extrapolations from in vitro experiments with specific KZNF triplets to the behavior of the highly diverse KZNF proteins in metazoan cells are fraught with uncertainties.
Secondly, there is some evidence to suggest that some KZNF proteins might be modified post-translationally in a cell-type specific way that could alter their DNA recognition specificity. For example, phosphorylation of the DNA binding domains in the KZNF protein, Yin yang 1 (YY1) has been shown to affect the protein’s ability to bind DNA targets [23]. The extent to which most KZNF proteins are modified in vivo is unknown.
Thirdly, it is not clear that all fingers in polydactyl proteins need be necessarily engaged simultaneously, or ever engaged at all, in DNA binding. Indeed, several proteins have been described in which the same KZNF motifs can act as DNA binding elements in some instances, and serve alternative, unrelated functions in other circumstances. For example, in the yeast KZNF protein, ZAP1, two of the five zinc-fingers can serve alternatively as DNA-binding or zinc-response elements [24]. In mammals, two C-terminal fingers in the KZNF protein, ZAC, can either participate in DNA binding or be sequestered for interactions with protein partner, p300 [25]. Through differential use of specific subsets of its seven KZNF motifs, ZAC can recognize two distinct sets of high-affinity binding sites [26]. Similarly, a 30-fingered protein, OAZ, can use subsets of fingers to recognize more than one DNA binding site, and use others to mediate dimer formation or interactions with protein co-factors [27]. Similar dual-purpose activities have been implicated for a large number of KZNF proteins in many species [28]. There is therefore good reason to suspect that many polydactyl proteins will act in this way, utilizing subsets of fingers alternatively for various functions in a range of biological contexts.
4.2.2 Experimental Data on KZNF–DNA Interactions
Much current knowledge regarding the interactions between polydactyl KZNF proteins and DNA binding sites is based on in vitro experiments; the in vivo functions of most members of this abundant protein class remain a mystery. The picture should be clarified significantly in the next several years, as in vivo DNA binding sites for more polydactyl KZNF proteins are mapped through unbiased methods, in particular, through chromatin-immunoprecipitation followed by high-throughput sequencing (“ChIP-seq”). To date, only a small number of proteins have been examined using these unbiased methods. The conserved polydactyl KZNF protein, RE1-silencing TF (REST), was one of the first TFs to be mapped using ChIP-seq methods [29]. REST in vivo binding sites had been studied extensively on a gene-by-gene level, and the results of ChIP-seq studies, while fascinating, largely confirmed what was known about the binding site and types of preferred gene targets for this regulatory protein.
However, the analysis revealed significant levels of protein binding to REST “half sites”, representing 5′ or 3′ segments of the strong, well characterized 21 base-pair consensus sequence, referred to as “NRSE” (for neuron-restrictive silencer element) that correlates well with the predicted binding site for the 8-fingered REST protein (Table 4.1). The levels of half-site binding indicate that in some contexts, REST may use only a portion of its fingers to recognize DNA, thereby significantly increasing the potential regulatory repertoire of this abundant transcriptional regulator [29]. Binding sites for a second polydactyl protein, the SCAN-KRAB protein ZNF263, have also recently been identified using ChIP-seq; the single 24 nucleotide consensus binding site predicted in these studies suggests that this protein uses most of its 9 zinc-fingers for DNA binding [30]. The binding site predicted for ZNF263 bears some similarity to the site that would be computationally predicted from the protein’s amino acid sequence computationally, as well as some striking differences.
Additional insights have also been provided through earlier ChIP experiments coupled to microarrays (“ChIP-chip”) for a small number of additional KZNF proteins, including the multifunctional protein, CTCF [31, 32]. However, despite this progress, a remarkably tiny fraction of this exceptionally large and versatile protein family has known regulatory functions, gene targets, or DNA binding sites. For that reason, most of what we know about their functional properties comes from “special case” stories focused on the products of single, possibly unrepresentative, KZNF genes. This picture should change dramatically, with the advent of “next-generation” sequencing technologies and their coupling to chromatin-binding assays, in the next few years.
4.3 Evolutionary History: The Rise and Fall of Lineage-Specific KZNF Families
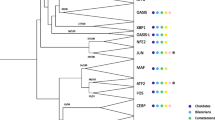
The polydactyl KZNF TF family includes hundreds of members in many eukaryotic species (Fig. 4.4), many of which have highly been conserved over the course of evolution [33]. An example includes the mammalian Krüppel-like factor (KLF) family, a group of 17 three-fingered genes related distantly to the ancient TF, SP1 [34]. The Drosophila genome contains 4 related Klf genes that share many properties, including developmental expression and key roles in differentiation and development, with the mammalian proteins [33, 34]. The KLF family exemplifies the features typical

Phylogenetic distribution of polydactyl KZNF protein families. The distribution and number of polydactyl KZNF proteins in different families is shown in this phylogeny of all eukaryotic model systems. The gain and loss of genes over evolution can be seen along the tree for all polydactyl C2H2 (red) BTB/POZ (orange), ZAD (green), KRAB (blue), and SCAN (purple) KZNF families. The numbers of genes in each family are shown in the accompanying table. Information was compiled from the PFAM Database [76], unless otherwise noted as coming from [1][79], [2][77], [3][39], [4][42] or [5][41]
of many KZNF family groups: most of these proteins contain short KZNF arrays, and have been well conserved in metazoan species.
However, most genomes contain subfamilies of KZNF genes with very different evolutionary histories and fates. Over the course of evolution, distinct KZNF families have emerged independently in different lineages, through exon shuffling events that bring DNA sequences encoding polydactyl KZNF arrays together with different types of protein-interaction or chromatin-modifying “effector” domains (see Chapter 12 for a general overview of TF effector domains). New versions of KZNF proteins, coupling long polydactyl arrays with different types of activation, repression, or protein-interaction effectors, have arisen in different evolutionary lineages. Some of the genes encoding these novel constructs have subsequently expanded by repeated duplication events into large gene families; these in turn have either been integrated into key regulatory networks and conserved, or lost and replaced by other KZNF families in subsequent lineages.
4.3.1 An Ancient Family: BTB/POZ
One of the most ancient families of this type, in which arrays of KZNF fingers are attached to an N-terminal BTB/POZ motif, is represented in most eukaryotic species. As with many families, the numbers of BTB/POZ proteins has varied throughout evolution, changing through whole genome duplications, single-gene duplications, and gene loss (Fig. 4.4). The BTB/POZ domain (named BTB because of its presence in Drosophila Broad Complex, tramtrack and bric a brac genes, and POZ for “poxviruses and zinc finger”) is found associated with several types of proteins, including but not limited to those containing KZNF array. The primary function of BTB/POZ appears to be protein dimerization, although the activities of the proteins in which this domain are found suggest a more specific functional role. Several BTB/POZ-KZNF proteins are found in Drosophila, where they play key roles in both local gene regulation and higher-order chromatin structure, often in the context of embryonic development [35]. Similar developmental functions have been attributed to BTB/POZ-KZNF proteins in humans and mice. Whereas the originally discovered BTB/POZ genes function mainly as transcriptional repressors, these proteins can operate as agents of chromosome decondensation and gene activation as well [36].
4.3.2 Lineage-Specific Inventions
In addition to this older family of genes, most metazoan genomes appear to carry surprisingly large numbers of lineage-specific KZNF genes. Typically, these genes represent novel constructs, in which exons encoding specific N-terminal effector domains are spliced to one or more exons encoding adjacent elements of a C-terminal KZNF array (Fig. 4.2). Most of these proteins also contain a region between the effector and KZNF motifs, usually referred to as a tether or spacer sequence. This typical structure is found in KZNF proteins of several different types, which are restricted to certain evolutionary lineages and have expanded by duplication into large TF families.
In Drosophila, 98 genes are found that encode KZNF attached to an N-terminal repressive motif called ZAD, whose function is as-yet poorly characterized [37]. Like BTB/POZ, the ZAD domain facilitates protein-protein interactions. A single ZAD-like gene, ZNF276, exists in vertebrate species, but an expanded ZAD family is found only in insects with the largest numbers found in higher homometabolous species (i.e. those that go through metamorphosis) (Fig. 4.4) [38]. Like the largest KZNF families in other species, ZAD-KZNF genes are found clustered together on insect chromosomes, reflecting the fact that the families arose through repeated rounds of tandem segmental duplications [39]. Although most ZAD-KZNF genes are of unknown function, most are expressed in the female germline and a few have been linked to developmental mutations in Drosophila [38]. The lineage-specific expansion of this class of KZNF proteins, phenotypes associated with certain family members, and their developmental expression make it likely that the ZAD-KZNF proteins play a role in species-specific developmental processes. A role for these genes in regulating developmental pathways could explain the dramatic expansion of the ZAD-ZNF family particularly in metamorphic species.
In vertebrate genomes, two other KZNF families have expanded into large gene families that are limited to certain evolutionary clades. In proteins of the SCAN-KZNF family, a C-terminal protein-interacting SCAN domain is attached through a tether sequence of varying length to N-terminal KZNF arrays [40]. SCAN domains are found in most vertebrates and are associated with a variety of other protein motifs, but the combination of SCAN with KZNF arrays has only been detected in mammals [40, 41]. After SCAN-KZNF genes arose, they expanded into a small family in most mammalian species, with a total of 57 protein-coding genes in the human genome (Fig. 4.4). Like the ZAD-KZNF genes in insects, SCAN-KZNF coding genes are frequently found in clusters, with related family members located adjacent to each other at specific chromosomal sites. The primary expansion of this family through segmental duplications must have occurred relatively early in mammalian evolution, since most SCAN-KZNF gene clusters, and the genes that are resident within them, are represented by orthologs in the different mammalian species. Nevertheless, a small number of lineage specific SCAN-KZNF gene duplicates have also been identified in comparisons between the gene sets of human, dog and mouse [40, 42]. A small number of mammalian SCAN-containing KZNF proteins also include a KRAB motif (see below), and SCAN- and KRAB-containing KZNF genes are sometimes found together in chromosomal clusters [41, 42]. These data indicate some intermingling of genes of these two types over the course of evolutionary history.
4.3.3 A Case Study: The KRAB-ZNF Family
A second major KZNF family has diverged rapidly and dramatically in gene copy number in different mammalian lineages. The KRAB-A, or Krüppel-associated box, type A domain is a 41-residue element that interacts with a ubiquitous co-factor, called KAP-1, to attract histone deacetylase complexes to specific DNA sites [43–47] (also see Chapter 12 ). A single gene, called Meisetz or Prdm9, was formed through association of an exon encoding a KRAB domain, together with sequences encoding a second effector, the SET domain, to an exon encoding tether sequence and polydactyl KZNF array, in early metazoan history [48]. A recognizable Prdm9 ortholog can be found in echinoderms, protochordates, and vertebrate species. However both KZNF-motif number and sequence of the DNA-binding amino acids in the PRDM9 protein vary widely between species, exhibiting signs of strong positive selection [49]. In addition to its predicted role in transcriptional regulation, Prdm9 has recently been shown to play a key role in marking hotspots for meiotic recombination in mammals [9, 10, 50].
Whereas Prdm9 and its close relatives form a very small family in most vertebrates, a revised version of this protein type, containing one or more KRAB domains and a KZNF array but lacking the SET domain, has undergone dramatic expansion especially in mammalian lineages. Over 400 KRAB-KZNF genes exist in the human genome, and similar numbers are found in all mammalian genomes that have been examined [42]. By their sheer numbers, this single family of KZNF proteins dominates the mammalian transcription-factor landscape, comprising up to one-fourth of that total number of predicted human TF genes [51]. Most intriguingly, although all mammals have roughly equal numbers of proteins of this type, the number of 1:1 orthologous pairs is remarkably small. For example, although both human and mouse possess hundreds of KRAB-ZNF genes, only 112 genes represent convincing orthologs that are shared by these two species [42]. About one-third of human KRAB-ZNF genes are primate-specific, and a similar number of mouse genes can be found only in other rodents. For example, a cluster of mouse genes on chromosome 13 (chr13), including genes involved in regulating the sex-limited expression of target genes, contains many KRAB-ZNF coding sequences that are restricted to the Mus lineage [52]. Similarly, about 30 human genes of this type have arisen through segmental duplication since the divergence of old world monkeys, creating novel transcriptional regulators that exist only in higher primates [53].
The tendency toward tandem segmental duplication may help explain why KRAB-KZNF genes have been gained and lost so frequently over the course of vertebrate evolution. Tandem segmental duplications, like those found in the KZNF gene clusters, are known to be hotspots of copy number variation (CNV) both between and within species, driving duplications and deletions through illegitimate recombination events [54, 55]. If the duplication units include a full-length gene, each recombination event can give rise to versions of the chromosome with one less or one additional gene, respectively. Recent studies have confirmed that many protein-coding genes are copy-number variant in the human population, and genes located in segmental duplications rank among those most likely to lost or gained in certain human individuals. Not surprisingly, many KRAB-KZNF loci are found among recently generated segmental duplications [53] and among these copy-number-variant genes.
As these data show, the KRAB-KZNF gene family has evolved rapidly, and still is evolving, with novel genes created through duplication, and even some conserved genes displaying sequence changes that reflect the influence of positive selection. Recent studies show that as new duplicates arise, they can change rapidly in function through two different routes. First, the newly duplicated genes can change in expression pattern, diverging from the parental gene copy in tissue-specific sites and levels of gene expression [53]. Although KRAB-KZNF genes reside in closely packed gene clusters, neighboring genes do not often share similar expression patterns, even when the two genes are closely related [42, 53, 56–58]. These data suggest that (1) the genes are typically duplicated along with the regulatory elements needed to drive their tissue-specific expression patterns, and (2) that neighbors are probably shielded from the influence of enhancers or repressive elements controlling the surrounding genes. Whatever the mechanism, the ability to quickly adapt unique expression patterns after duplication has provided a rapid path to functional divergence for KRAB-KZNF genes.
The second route through which new KRAB-KZNF paralogs can diverge rapidly from parental genes is through sequence changes that affect the DNA binding properties of the encoded proteins. This divergence occurs through two different mechanisms. First, paralogous gene copies can acquire non-synonymous mutations in the DNA-binding amino acids in the finger motifs; for many KRAB-KZNF gene paralogs, the acquisition of novel DNA-binding sequences has occurred under the influence of positive selection [41, 53, 56, 59, 60]. An alternative path to paralog divergence involves a mechanism that is unique to proteins like the polydactyl KZNFs, which contain multiple, similar motifs that are encoded in a single exon (Fig. 4.3). The sequences encoding these protein motifs are essentially tandem repeat sequences, and are prone to the same types of duplications and deletions observed for microsatellites and other simple genomic repeats. As a result, paralogous KRAB-KZNF proteins often differ from each other in KZNF motif number, often due to the deletion or duplication of one or more zinc-fingers from the middle of the KZNF array [59, 60]. This process can occur rapidly, giving rise to proteins that are otherwise nearly identical, but contain different numbers and arrangements of tandem KZNF motifs [53]. Because of the intimate relationship that exists between an ordered array of amino acids in the KZNF alpha-helical region and the nucleotide sequence at target sites, deletion or duplication of fingers from within an array is expected to have significant impact on DNA binding, target-site preference, and stability of KZNF association with DNA.
4.3.4 A General Path to Rapid Divergence for Polydactyl KZNF Genes
Although these paths to paralog divergence are best described for mammalian KRAB-ZNF genes, the pattern of divergence also follows for SCAN-KZNF subfamily [42] and our recent studies indicate a similar pattern for primate BTB/POZ-KZNF proteins [53]. There is no reason to believe that similar patterns of divergence would not have defined the growth of KZNF gene families of other types and in other species as well. In fact, a recent survey of KZNF genes in multiple species detected lineage-specific family members in virtually every genome analyzed, and showed that positive selection acting to diversify DNA-binding capabilities of KZNF proteins of many different types [41]. The key feature that drives duplication and deletion of KZNF motifs in KRAB-KZNF genes is the occurrence of multiple, tandemly arranged finger-encoding repeats in a single exon; this kind of structure is present in a large fraction of KZNF genes in every species (Fig. 4.3). Whether finger deletion and duplication are driven by illegitimate recombination between the adjacent repeats, or a mechanism such as replication slippage, remains to be determined. However, the high frequency of these events and the relatively rapid pace in which they have occurred in divergence of recent primate duplicates argue for the latter mechanism, which is known to drive a similar pace of genomic divergence at microsatellites and other simple sequence repeats [61].
The ability to create new DNA binding capabilities through binding-sequence divergence or zinc finger number and arrangement is likely to underlie much of this gene family’s remarkable growth and success. However, despite similarities in structure, with N-terminal effectors and KZNF arrays encoded intact on separate exons, the different families of polydactyl KZNF genes display very different evolutionary histories. Why have the older BTB/POZ-KZNF genes and the vertebrate-specific SCAN-KZNF families not exploded in numbers, as the KRAB-KZNFs have done? What drove the expansion of ZAD-KZNF genes in insects, and the expansion of a less-characterized family, the FAX/FAD-KZNFs [62] in amphibian genomes?
In considering the functions of the major mammalian KZNF effectors, we may find a clue to this mystery. Whereas BTB and SCAN appear to be concerned primarily with protein homo- and hetero-dimerization, KRAB is thought to play a very different role. Whereas future studies of KRAB domain function may still hold some surprises, it is thought primarily to interact directly with a single, abundant, and ubiquitous co-factor, KAP-1 [63, 64]. Because KAP-1 is so abundant, and serves as a “universal” KRAB co-factor, new KRAB proteins can arise with little effect on other interaction partners. KRAB does not mediate dimer formation, and KRAB-KZNF proteins appear to bind to target sites without the need for partners to stabilize their interaction with DNA. The long polydactyl KZNF arrays that are found in most mammalian proteins of this type probably underlie the independence of proteins of this type. Human KRAB-KZNF proteins contain an average of 12 KZNF motifs; an array of this length could theoretically specific a binding site of 36 bp, an extraordinary length compared to the binding sites of most known TFs. In reality, most binding sites that have been determined for polydactyl KZNFs range from 6 to 27 bp; some examples of well established binding sites are shown as “motif logos” (illustrations that represent the probability of finding a particular nucleotide at a position within the binding site) in Table 4.1. For proteins with binding sites on the longer end of this range, the binding between DNA sequence and the KZNF protein, wound with precision around the double-stranded DNA, would be predicted to be unusually specific and stable.
In contrast to KRAB-KZNFs, the average SCAN-KZNF and BTB/POZ-KZNF proteins contain a smaller number of zinc fingers [42], consistent with the idea that these proteins need to dimerize with other, similar proteins for secure binding to DNA. The potential combinatorial action of these dimerizing proteins provides a way to achieve functional diversity far beyond that implied by the numbers of individual genes. However, their predicted dependence on other proteins for activity may also constrain the ability of new genes to evolve, and established genes to be lost in these gene families. These concepts may help explain why genes encoding BTB/POZ and SCAN-containing KZNF genes have been more restrained than their ZAD-KZNF and KRAB-KZNF cousins in their tendency to gain and lose members over evolutionary time. Because they cannot diverge without affecting the functions of interacting proteins, TFs that require partners for activity tend to be more conserved, and more likely to be locked in to larger regulatory pathways, than independently acting proteins might be.
These basic tenets of protein evolution allow some predictions for the functions of effectors for non-mammalian KZNF effectors, like the insect effector, ZAD. Although the exact functions of the ZAD are not yet know, the prolific expansion of ZAD-KZNF genes in insect genomes, and the differences in ZAD-KZNF repertoires observed in comparisons of different insect genomes [38, 65], suggest that, like KRAB, this effector might function in concert with a ubiquitous co-factor; in analogy to KAP-1, this co-factor might be predicted to correspond to a protein or complex that interfaces with the chromatin remodeling machinery.
4.4 Challenges and Future Directions
Although the KRAB-KZNF family is by far the most dynamic group in mammalian genomes, the larger KZNF family has clearly played a significant role in the evolution of all eukaryotic clades. The versatile building block provided by the ancient C2H2 motif, its ability to assemble into long arrays for stable DNA binding, and the sheer diversity in DNA recognition capabilities that can be achieved by their combinatorial action, have made them a mainstay of TF repertoires and a dominant component of all eukaryotic genomes.
Despite their dominance, and the molecular “recognition code” that is believed to underlie their DNA binding capabilities, the functions of only a tiny fraction of KZNF proteins in any genome is known, and indeed it is not known whether predicted sequence specificities are generally correct. This lack of functional knowledge is especially acute for the polydactyl KZNF genes, due in part to their lack of interspecies conservation and their duplicative histories, which ensure some degree of functional redundancy. In vitro studies of purified polydactyl KZNF proteins are hampered by their low solubility, due to the high cysteine content of the proteins; in vivo studies are complicated by the high degree of similarity between paralogous proteins. And because of the extreme evolutionary diversity of KRAB-KZNFs in vertebrates and similar lineage-specific families, repertoires of such proteins have been fully counted only a small number of completely sequenced genomes [41].
However, the emergence of new technologies is beginning to shine new light on this shadowy component of the metazoan regulatory machinery. Microarrays bearing double-stranded oligonucleotides are currently being used to map binding-site preferences for a large number of GST-tagged TF proteins, including some KZNFs [66]; this method offers a significant advantage in terms of effort and time required to map binding sites compared to previous in vitro methods. However, polydactyl KZNFs have presented unique challenges to methods such as this, which depend on availability of soluble tagged proteins.
Some progress has been made through strategies such as tagging short peptides containing overlapping subsets of fingers from a longer KZNF array (T.R. Hughes, personal communication). Binding sites for other proteins have been successfully determined using established methods, such as “Systematic Evolution of Ligands by Exponential Enrichment” (or SELEX, [67–69] (Table 4.1)), and more recently through the application of bacterial one-hybrid selection systems [70]. However, ultimately, an understanding of the binding properties of long polydactyl KZNF proteins, of the prevalence of finger “multitasking”, and of the functional consequences of their unique patterns of evolutionary divergence, will require methods that fully report their binding-site occupancy in living cells. Because of paralog sequence similarity and other factors, mapping binding sites of KRAB-KZNF proteins and the member of other, similar lineage-specific protein families presents a special challenge. However, new strategies including the use of “designer” KZNF recombinases [71, 72] to facilitate in vivo TF tagging, in combination with high-throughput sequencing, hold significant promise to unlock the long-standing mysteries regarding the functions of these abundant eukaryotic TFs. The true impact of the KZNF family’s dynamic evolutionary history on speciation, interspecies divergence, and individual differences in gene regulation eventually will only be deciphered when their binding sites, regulatory activities, and interactions with other chromatin proteins are known.
References
Miller J, McLachlan AD, Klug A (1985) Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J 4:1609–1614
Ollo R, Maniatis T (1987) Drosophila Krüppel gene product produced in a baculovirus expression system is a nuclear phosphoprotein that binds to DNA. Proc Natl Acad Sci USA 84:5700–5704
Bouhouche N, Syvanen M, Kado CI (2000) The origin of prokaryotic C2H2 zinc finger regulators. Trends Microbiol 8:77–81
Frankel AD, Berg JM, Pabo CO (1987) Metal-dependent folding of a single zinc finger from transcription factor IIIA. Proc Natl Acad Sci USA 84:4841–4845
Lee MS, Gippert GP, Soman KV, Case DA, Wright PE (1989) Three-dimensional solution structure of a single zinc finger DNA-binding domain. Science 245:635–637
Pavletich NP, Pabo CO (1991) Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252:809–817
Arya GH, Lodico MJ, Ahmad OI, Amin R, Tomkiel JE (2006) Molecular characterization of teflon, a gene required for meiotic autosome segregation in male Drosophila melanogaster. Genetics 174:125–134
Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327:836–840
Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, Donnelly P (2010) Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327:876–879
Parvanov ED, Petkov PM, Paigen K (2010) Prdm9 controls activation of mammalian recombination hotspots. Science 327:835
Phillips CM, Dernburg AF (2006) A family of zinc-finger proteins is required for chromosome-specific pairing and synapsis during meiosis in C. elegans. Dev Cell 11:817–829
Dickson J, Gowher H, Strogantsev R, Gaszner M, Hair A, Felsenfeld G, West AG (2010) VEZF1 elements mediate protection from DNA methylation. PLoS Genet 6:e1000804
Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC (2008) A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell 15:547–557
Englbrecht CC, Schoof H, Bohm S (2004) Conservation, diversification and expansion of C2H2 zinc finger proteins in the Arabidopsis thaliana genome. BMC Genomics 5:39
Choo Y, Klug A (1994) Toward a code for the interactions of zinc fingers with DNA: selection of randomized fingers displayed on phage. Proc Natl Acad Sci USA 91:11163–11167
Wuttke DS, Foster MP, Case DA, Gottesfeld JM, Wright PE (1997) Solution structure of the first three zinc fingers of TFIIIA bound to the cognate DNA sequence: determinants of affinity and sequence specificity. J Mol Biol 273:183–206
Elrod-Erickson M, Pabo CO (1999) Binding studies with mutants of Zif268. Contribution of individual side chains to binding affinity and specificity in the Zif268 zinc finger-DNA complex. J Biol Chem 274:19281–19285
Elrod-Erickson M, Rould MA, Nekludova L, Pabo CO (1996) Zif268 protein-DNA complex refined at 1.6 A: a model system for understanding zinc finger-DNA interactions. Structure 4:1171–1180
Kaplan T, Friedman N, Margalit H (2005) Ab initio prediction of transcription factor targets using structural knowledge. PLoS Comput Biol 1:e1
Liu J, Stormo GD (2008) Context-dependent DNA recognition code for C2H2 zinc-finger transcription factors. Bioinformatics 24:1850–1857
Persikov AV, Osada R, Singh M (2009) Predicting DNA recognition by Cys2His2 zinc finger proteins. Bioinformatics 25:22–29
Klug A (2010) The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem 79:213–231
Rizkallah R, Hurt MM (2009) Regulation of the transcription factor YY1 in mitosis through phosphorylation of its DNA-binding domain. Mol Biol Cell 20:4766–4776
Bird AJ, Zhao H, Luo H, Jensen LT, Srinivasan C, Evans-Galea M, Winge DR, Eide DJ (2000) A dual role for zinc fingers in both DNA binding and zinc sensing by the Zap1 transcriptional activator. EMBO J 19:3704–3713
Hoffmann A, Barz T, Spengler D (2006) Multitasking C2H2 zinc fingers link Zac DNA binding to coordinated regulation of p300-histone acetyltransferase activity. Mol Cell Biol 26:5544–5557
Hoffmann A, Ciani E, Boeckardt J, Holsboer F, Journot L, Spengler D (2003) Transcriptional activities of the zinc finger protein Zac are differentially controlled by DNA binding. Mol Cell Biol 23:988–1003
Hata A, Seoane J, Lagna G, Montalvo E, Hemmati-Brivanlou A, Massague J (2000) OAZ uses distinct DNA- and protein-binding zinc fingers in separate BMP-Smad and Olf signaling pathways. Cell 100:229–240
Brayer KJ, Segal DJ (2008) Keep your fingers off my DNA: protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem Biophys 50:111–131
Johnson DS, Mortazavi A, Myers RM, Wold B (2007) Genome-wide mapping of in vivo protein–DNA interactions. Science 316:1497–1502
Frietze S, Lan X, Jin VX, Farnham PJ (2010) Genomic targets of the KRAB and SCAN domain-containing zinc finger protein 263. J Biol Chem 285:1393–1403
Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B (2007) Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128:1231–1245
Smith ST, Wickramasinghe P, Olson A, et al. (2009) Genome wide ChIP-chip analyses reveal important roles for CTCF in Drosophila genome organization. Dev Biol 328:518–528
Knight RD, Shimeld SM (2001) Identification of conserved C2H2 zinc-finger gene families in the Bilateria. Genome Biol 2:RESEARCH0016
Pearson R, Fleetwood J, Eaton S, Crossley M, Bao S (2008) Kruppel-like transcription factors: a functional family. Int J Biochem Cell Biol 40:1996–2001
Albagli O, Dhordain P, Deweindt C, Lecocq G, Leprince D (1995) The BTB/POZ domain: a new protein-protein interaction motif common to DNA- and actin-binding proteins. Cell Growth Differ 6:1193–1198
Kelly KF, Daniel JM (2006) POZ for effect – POZ-ZF transcription factors in cancer and development. Trends Cell Biol 16:578–587
Jauch R, Bourenkov GP, Chung HR, Urlaub H, Reidt U, Jäckle H, Wahl MC (2003) The zinc finger-associated domain of the Drosophila transcription factor grauzone is a novel zinc-coordinating protein-protein interaction module. Structure 11:1393–1402
Chung HR, Löhr U, Jäckle H (2007) Lineage-specific expansion of the zinc finger associated domain ZAD. Mol Biol Evol 24:1934–1943
Chung HR, Schafer U, Jäckle H, Böhm S (2002) Genomic expansion and clustering of ZAD-containing C2H2 zinc-finger genes in Drosophila. EMBO Rep 3:1158–1162
Edelstein LC, Collins T (2005) The SCAN domain family of zinc finger transcription factors. Gene 359:1–17
Emerson RO, Thomas JH (2009) Adaptive evolution in zinc finger transcription factors. PLoS Genet 5:e1000325
Huntley S, Baggott DM, Hamilton AT, Tran-Gyamfi M, Yang S, Kim J, Gordon L, Branscomb E, Stubbs L (2006) A comprehensive catalog of human KRAB-associated zinc finger genes: insights into the evolutionary history of a large family of transcriptional repressors. Genome Res 16:669–677
Abrink M, Ortiz JA, Mark C, Sanchez C, Looman C, Hellman L, Chambon P, Losson R (2001) Conserved interaction between distinct Krüppel-associated box domains and the transcriptional intermediary factor 1 beta. Proc Natl Acad Sci USA 98:1422–1426
Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ 3rd. (1996) KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 10:2067–2078
Lorenz P, Koczan D, Thiesen HJ (2001) Transcriptional repression mediated by the KRAB domain of the human C2H2 zinc finger protein Kox1/ZNF10 does not require histone deacetylation. Biol Chem 382:637–644
Margolin JF, Friedman JR, Meyer WK, Vissing H, Thiesen HJ, Rauscher FJ 3rd. (1994) Kruppel-associated boxes are potent transcriptional repression domains. Proc Natl Acad Sci USA 91:4509–4513
Witzgall R, O’Leary E, Leaf A, Onaldi D, Bonventre JV (1994) The Krüppel-associated box-A (KRAB-A) domain of zinc finger proteins mediates transcriptional repression. Proc Natl Acad Sci USA 91:4514–4518
Birtle Z, Ponting CP (2006) Meisetz and the birth of the KRAB motif. Bioinformatics 22:2841–2845
Thomas JH, Emerson RO, Shendure J (2009) Extraordinary molecular evolution in the PRDM9 fertility gene. PLoS One 4:e8505
Baudat F, Buard J, Grey C, de Massy B (2010) [Prdm9, a key control of mammalian recombination hotspots]. Med Sci (Paris) 26:468–470
Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM (2009) A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10:252–263
Krebs CJ, Larkins LK, Khan SM, Robins DM (2005) Expansion and diversification of KRAB zinc-finger genes within a cluster including Regulator of sex-limitation 1 and 2. Genomics 85:752–761
Nowick K, Hamilton AT, Zhang H, Stubbs L (2010) Rapid sequence and expression divergence suggests selection for novel function in primate-specific KRAB-ZNF genes. Mol Biol Evol 27:2606–2617
Cooper GM, Nickerson DA, Eichler EE (2007) Mutational and selective effects on copy-number variants in the human genome. Nat Genet 39:S22–29
Stankiewicz P, Lupski JR (2010) Structural variation in the human genome and its role in disease. Annu Rev Med 61:437–455
Hamilton AT, Huntley S, Kim J, Branscomb E, Stubbs L (2003) Lineage-specific expansion of KRAB zinc-finger transcription factor genes: implications for the evolution of vertebrate regulatory networks. Cold Spring Harb Symp Quant Biol 68:131–140
Nowick K, Stubbs L (2010) Lineage-specific transcription factors and the evolution of gene regulatory networks. Brief Funct Genomics 9:65–78
Shannon M, Ashworth LK, Mucenski ML, Lamerdin JE, Branscomb E, Stubbs L (1996) Comparative analysis of a conserved zinc finger gene cluster on human chromosome 19q and mouse chromosome 7. Genomics 33:112–120
Hamilton AT, Huntley S, Tran-Gyamfi M, Baggott DM, Gordon L, Stubbs L (2006) Evolutionary expansion and divergence in the ZNF91 subfamily of primate-specific zinc finger genes. Genome Res 16:584–594
Shannon M, Hamilton AT, Gordon L, Branscomb E, Stubbs L (2003) Differential expansion of zinc-finger transcription factor loci in homologous human and mouse gene clusters. Genome Res 13:1097–1110
Hardwick RJ, Tretyakov MV, Dubrova YE (2009) Age-related accumulation of mutations supports a replication-dependent mechanism of spontaneous mutation at tandem repeat DNA Loci in mice. Mol Biol Evol 26:2647–2654
Nietfeld W, Conrad S, van Wijk I, Giltay R, Bouwmeester T, Knochel W, Pieler T (1993) Evidence for a clustered genomic organization of FAX-zinc finger protein encoding transcription units in Xenopus laevis. J Mol Biol 230:400–412
Kim SS, Chen YM, O’Leary E, Witzgall R, Vidal M, Bonventre JV (1996) A novel member of the RING finger family, KRIP-1, associates with the KRAB-A transcriptional repressor domain of zinc finger proteins. Proc Natl Acad Sci USA 93:15299–15304
Moosmann P, Georgiev O, Le Douarin B, Bourquin JP, Schaffner W (1996) Transcriptional repression by RING finger protein TIF1 beta that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res 24:4859–4867
Duan J, Xia Q, Cheng D, Zha X, Zhao P, Xiang Z (2008) Species-specific expansion of C2H2 zinc-finger genes and their expression profiles in silkworm, Bombyx mori. Insect Biochem Mol Biol 38:1121–1129
Badis G, Berger MF, Philippakis AA, et al. (2009) Diversity and complexity in DNA recognition by transcription factors. Science 324:1720–1723
Ellington AD, Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346:818–822
Oliphant AR, Brandl CJ, Struhl K (1989) Defining the sequence specificity of DNA-binding proteins by selecting binding sites from random-sequence oligonucleotides: analysis of yeast GCN4 protein. Mol Cell Biol 9:2944–2949
Tuerk C, Gold L (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510
Noyes MB, Meng X, Wakabayashi A, Sinha S, Brodsky MH, Wolfe SA (2008) A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res 36:2547–2560
Dekelver RC, Choi VM, Moehle EA, et al. (2010) Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res 20:1133–1142
Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS (2009) Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19:1279–1288
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K (2007) High-resolution profiling of histone methylations in the human genome. Cell 129:823–837
Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133:1106–1117
Delwel R, Funabiki T, Kreider BL, Morishita K, Ihle JN (1993) Four of the seven zinc fingers of the Evi-1 myeloid-transforming gene are required for sequence-specific binding to GA(C/T)AAGA(T/C)AAGATAA. Mol Cell Biol 13:4291–4300
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunesekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A (2010) The PFAM protein families database. Nucl Acids Res 38 (Database Issue):D211–D222
Materna SC, Howard-Ashby M, Gray RF, Davidson EH (2006) The C2H2 zinc finger genes of Strongylocentrotus purpuratus and their expression in embryonic development. Devel Biol 300:108–120
Portales-Casamar E, Kirov S, Lim J, Lithwick S, Swanson MI, Ticoll A, Snoddy J, Wasserman WW (2007) PAZAR: a framework for collection and dissemination of cis-regulatory sequence annotation. Genome Biol 8:R207
Riechmann JL, Heard J, Martin G, Reuber L, Jiang CZ, Keddie J, Adam L, Pineda O, Ratcliffe OJ, Samaha RR, Creelman R, Pilgrim M, Broun P, Zhang JZ, Ghandehari D, Sherman BK, Yu GL (2000) Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290:2105–2110
Schaub M, Myslinski E, Schuster C, Krol A, Carbon P (1997) Staf, a promiscuous activator for enhanced transcription by RNA polymerases II and III. EMBO J 16:173–181
Shrivastava A, Calame K (1994) An analysis of genes regulated by the multi-functional transcriptional regulator Yin Yang-1. Nucl Acids Res 22:5151–5155
Thiagalingam A, De Bustros A, Borges M, Jasti R, Compton D, Diamond L, Mabry M, Ball DW, Baylin SB, Nelkin BD (1996) RREB-1, a novel zinc finger protein, is involved in the differentiation response to Ras in human medullary thyroid carcinomas. Mol Cell Biol 16:5335–5345
Tsai RY, Reed RR (1998) Identification of DNA recognition sequences and protein interaction domains of the multiple-Zn-finger protein Roaz. Mol Cell Biol 18:6447–6456
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Stubbs, L., Sun, Y., Caetano-Anolles, D. (2011). Function and Evolution of C2H2 Zinc Finger Arrays. In: Hughes, T. (eds) A Handbook of Transcription Factors. Subcellular Biochemistry, vol 52. Springer, Dordrecht. https://doi.org/10.1007/978-90-481-9069-0_4
Download citation
DOI: https://doi.org/10.1007/978-90-481-9069-0_4
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-90-481-9068-3
Online ISBN: 978-90-481-9069-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)