
Abstract
Adenosine A3 receptors are widely distributed in the central nervous system but are expressed at a low level and have lower affinity for adenosine in comparison to the A1 and A2A receptors. Nevertheless, they appear to tonically modulate motor activity as pointed out in A3 receptor-deleted mice.
The role of A3 receptor in several pathophysiological conditions is often controversial. In conditions such as seizures or ischemia, when extracellular concentrations of adenosine increase, A3 receptors may contribute to neurotransmission and cell damage. A pro-convulsant effect of A3 receptor is feasible, especially in the immature brain, thus raising the possibility that A3 receptor might facilitate seizure-induced neuronal damage and activity-dependent plastic changes. Most data support a pro-nociceptive role of A3 receptor involving both central nervous system and pro-inflammatory effects at peripheral tissues. The outcome of A3 receptor stimulation on synaptic transmission during hypoxic/ischemic phenomena appears to depend on the duration and intensity of the ischemic episode. While A3 receptor may play a protective role in the first phase of ischemia by decreasing synaptic transmission, prolonged A3AR stimulation by high adenosine concentrations could be pivotal in transforming the A3AR-mediated effects from protective to injurious. Detrimental effects of A3AR activation may be due, at least in part, to increased excitoxicity. Glial A3AR stimulated by high adenosine levels caused by a prolonged central trauma may well be implicated in neuroinflammatory tissue responses.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
In 1972 Ginsborg and Hirst (1972) described for the first time that the nucleoside adenosine inhibited acetylcholine release at the rat-diaphragm neuromuscular junction and that this effect was abolished by theophylline. This observation was soon supported by Ribeiro and Walker (1975) using the frog neuromuscular junction preparation. Authors discussed these results in relation to the capability of adenosine to increase cyclic AMP (cAMP), an effect that was abolished by theophylline as previously demonstrated by Sattin and Rall (1970). In 1979 van Calker et al. (1979) proposed that adenosine regulates the accumulation of cAMP in cultured brain cells, via two different types of adenosine receptors and in 1980, Londos et al. (1980) proposed subclasses of membrane adenosine receptors: the A1/Ri (that inhibited adenylate cyclase) and the A2/Ra (that activated adenylate cyclase). In 1984 Ribeiro and Sebastião (1984) in an attempt to characterize the type of adenosine receptors involved in the inhibitory action of adenosine at the frog neuromuscular junction, suggested that the adenosine receptors at the frog neuromuscular junction should not be classified as A1/A2 because the potency profile of adenosine agonists did not fit the pharmacological profile proposed for either A1 or A2 receptors. As a consequence, Ribeiro and his Ph.D. student, Ana M. Sebastião, were requested by the editors of Progress in Neurobiology to write a review on adenosine receptors. In reviewing the available pharmacological information, three different adenosine receptor entities emerged based on the affinity of different adenosine agonists: A1 with a pharmacological profile with L-PIA, CHA>CADO>D-PIA, NECA (high stereoselectivity for the PIA isomers) and negatively coupled to adenylate cyclase; the A2 with NECA>CADO>L-PIA, CHA, D-PIA with low stereoselectivity for the PIA isomers (L-PIA∼D-PIA) and positively coupled to adenylate cyclase. A third entity was then proposed, an A3 adenosine receptor (A3AR) with an agonist profile with L-PIA, CHA, NECA>CADO, and D-PIA usually less potent than CADO, possibly linked to calcium (Ribeiro and Sebastião 1986).
After cloning, it was clarified that rat A3AR is homologous with the adenosine A1 and A2A receptors and belongs to the G-protein-coupled receptor family (Meyerhof et al. 1991a, b). After cellular expression of cloned A3AR and pharmacological characterization, rat A3AR emerged as having a very low affinity for xanthine-based adenosine receptor antagonists such as theophylline. Various specie homologues of this receptor have been cloned, including the human A3AR (Salvatore et al. 1993). It emerged that A3AR cloned from different species show different pharmacological properties. Cloning and expression of the human A3AR stably expressed in Chinese hamster ovary cells pointed out that, contrary to rat A3AR, human A3AR is xanthine sensitive.
2 Distribution of A3 AR in the Central Nervous System (CNS)
The first binding studies on solubilized membranes from rat brain demonstrated the presence of a low affinity adenosine receptor with characteristics of the A3 subtype (Oliveira et al. 1991). However, in situ hybridization studies in the rat indicated the presence of A3AR mRNA only in the testis (Meyerhof et al. 1991a; Zhou et al. 1992; Rivkees 1994) and not in the CNS (Rivkees et al. 2000). Similarly, no expression of A3AR in the brain of mice or in the hippocampi of humans was detected (Rivkees et al. 2000). However, by reverse transcription–polymerase chain reaction (RT-PCR) A3AR expression was found distributed in the rat heart, lung and widespread in the rat and mouse brain (Zhou et al. 1992; Dixon et al. 1996; von Arnim et al. 2000). Low levels were detected by A3AR binding in various regions of the mouse brain (Jacobson et al. 1993), more than five times below that of the A1 receptor (Cunha et al. 1995) or of the A2A receptor (Cunha et al. 1996a).
There are significant brain regional differences in the levels of A3AR mRNA. In the mice, there is evidence for the expression of the A3AR in the hippocampus, thalamus and hypothalamus (Yaar et al. 2002). It is generally accepted that A3AR has species-specific tissue distribution. In humans and sheep, A3AR is significantly expressed in many peripheral tissues with lower levels in the CNS and testis (Linden et al. 1993; Salvatore et al. 1993).
As to A3AR localization in the different cell type of the CNS, the presence of A3AR in neurons, primarily at presynaptic sites, was demonstrated by PCR of laser dissected hippocampal neurons and by western blotting in rat hippocampal nerve terminal membranes (Lopes et al. 2003). Moreover, A3AR mRNA is expressed in microglia (Fiebich et al. 1996) and has been identified by Northern blot analysis in mouse astrocytes (Zhao et al. 1999).
3 The Roles of A3AR in the CNS
The endogenous neuromodulator, adenosine, controls and integrates a wide range of brain functions; its extracellular levels vary according to behavioral state and pathophysiological condition. Dysfunction of the adenosine system is involved in pathologies ranging from epilepsy to neurodegenerative disorders and psychiatric conditions. Less is known about the contribution of the low-affinity A3AR to the regulation of brain function and neuropathological conditions if compared to high-affinity A1 and A2A adenosine receptors. Its role in several pathophysiological conditions is often enigmatic and controversial.
3.1 Role of A3 AR in Memory and Cognition
Phenomena of synaptic plasticity, long-term potentiation (LTP) and long-term depression (LTD) are the likely cellular substrates for learning and memory. It is possible to induce either LTP or LTD according to the magnitude of the transient calcium levels attained at restricted synaptic spine domains. Smaller calcium increases predominantly activate protein phosphatases, leading to LTD, whereas higher calcium levels activate protein kinases, causing LTP (Dudek and Bear 1993).
Adenosine affects synaptic plasticity phenomena acting on both A1 and A2A receptors (de Mendonca and Ribeiro 2000; Rebola et al. 2008). The estimated affinity of rat A3AR for the endogenous ligand, adenosine (Ki = 1 μM), is considerably lower compared to that of A1 receptors (Ki = 10 nM) or A2A receptors (Ki = 30 nM) (Jacobson et al. 1995). Since the extracellular concentration of endogenous adenosine does not exceed 300 nM (Latini and Pedata 2001) it is difficult to detect a physiological role of A3AR in the CNS. However, conditions of stimulation that elicit synaptic plasticity can also transiently raise the extracellular adenosine concentration three to tenfold over basal levels (Cunha et al. 1996b). A role for A3AR on LTP and LTD has also been reported in the hippocampus (Costenla et al. 2001). Activation of A3AR essentially attenuates LTD (Costenla et al. 2001; Huang et al. 2007) and allows induction of LTP. The facilitating effects of the A3AR agonist, (1-[2-chloro-6-[[(3-iodophenyl)methyl]amino]-9H-purin-p-yl]-1-deoxy-N-methyl-beta-D-ribofuranuronamide (Cl-IB-MECA) on LTP were observed with weak subliminal θ-burst induction conditions but not with high frequency stimulation, which would elicit marked postsynaptic depolarization and Ca2+ increase, thus overcoming the facilitory effect of A3AR activation. This effect is consistent with a modification in the threshold for the induction of long-term synaptic changes, and might be due to coupling of A3AR to the phospholipase C transducing pathway (Abbracchio et al. 1995), thus promoting a shift to increased Ca2+ levels and protein kinase activation. The Cl-IB-MECA effect is prevented by the selective A3AR antagonist, MRS1191, indicating a genuine A3AR mediated response. However, the A3AR antagonist, MRS1191, does not by itself modify θ-burst-induced LTP, suggesting that tonically released adenosine is not able to activate A3AR in order to modulate LTP.
Behavioral studies have confirmed the involvement of A3AR in spatial learning and memory. In female Swiss mice, 1-deoxy-1-[6-[[(3-iodophenyl)-methyl]amino]-9H-purin-9-yl]-N-methyl-beta-D-ribofuranuronamide (IB-MECA), administered i.p. 20 min before tests, diminished scopolamine- and MK-801-induced impairment of spontaneous alternation in Y-maze and learning abilities in a passive avoidance task indicating that A3AR stimulation may ameliorate spatial memory and long term memory impairments in terms of cholinergic and glutamatergic deficits (Rubaj et al. 2003). It must be mentioned, however, that in the same animal species, Borowicz et al. (1997), using the passive avoidance task, demonstrated that a non selective A3AR agonist, N(6)-2-(4 aminophenyl)ethyladenosine (APNEA), administered i.p. 30 min before behavioral tests, impaired long-term memory.
When looking at A3AR-mediated behavioral effects, it should be remembered that A3AR agonists have depressant effects on locomotor activity (Jacobson et al. 1993).
3.2 Role of A3AR in Locomotion
By studying locomotor activity in an open field, Jacobson et al. (1993) demonstrated that intraperitoneal administration of 3-IB-MECA in mice induces a depression of motor activity. These results are consistent with the most recent experiments performed in A3AR-deleted mice. Significant increases in some aspects of motor function were observed in A3AR-deleted mice by using three different tests: activity in the open field; number of arm entries in the elevated-plus maze; and number of transitions in the light/dark box (Fedorova et al. 2003). The change in motor activity appears selective without evidence of ataxia. The increase in motor activity was attributed to disinhibition of cortical neurons because selective A3AR stimulation inhibits excitatory neurotransmission in rat cortical neurons (Brand et al. 2001). Most recently it was confirmed that both adolescent (21-day old) and adult A3AR knockout (KO) mice showed an increase of motor activity in the open field (Bjorklund et al. 2008a). Moreover, a reduced response to the motor-stimulating effect of caffeine or amphetamine was found in A3AR KO mice. These data are surprising in view of the poor affinity of A3AR for caffeine (Bjorklund et al. 2008a).
Work reviewed above indicates that even though A3AR are expressed at low level in the central nervous system, they play a tonic role in modulating motor activity.
3.3 Role of A3AR Receptors in Convulsions
Adenosine was identified as an endogenous anticonvulsant in the brain more than 20 years ago (Dunwiddie 1980; Lee et al. 1984; Dragunow et al. 1985; Dragunow 1991) and it was suggested that dysfunction of the adenosine-based neuromodulatory system might contribute to epileptogenesis (Boison 2007, 2008). Many experimental convulsive procedures led to a considerable rise in extracellular levels of adenosine (for a review see: Boison 2008). Clear-cut evidence that this really occurs in epileptic patients was provided by During and Spencer (1992). Their studies, with microdialysis probes implanted in the hippocampi of epileptic patients with intractable complex partial epilepsy, revealed that extracellular levels of adenosine in the dialysate were elevated by six- to 31-fold during seizures.
Recently, the therapeutic potential of stem cells engineered to release adenosine as a local source to augment endogenous adenosinergic functions was assayed in two cell transplantation experiments (Li et al. 2007, 2008; Boison 2008). Most of the studies about the role of adenosine as an anticonvulsant emphasize the preeminent involvement of A1 adenosine receptors. However, several studies using different experimental models of epilepsy have investigated the role of adenosine A2A and A3 receptors in this condition.
A first report conducted in an in vivo seizure model in mice indicates that stimulation of A3AR protects from seizures (von Lubitz et al. 1995). Acute systemic administration of IB-MECA protects against chemically-induced (NMDA injection) but not electrically-induced seizures while a protective effect of chronically administered IB-MECA is evident in both chemically- and electrically-evoked seizures (von Lubitz et al. 1995). The protective effect of acute administration of IB-MECA is attributed to both arteriolar constriction and severe hypotension (von Lubitz et al. 1994), which can reduce the final intracerebral concentration of the chemoconvulsant NMDA (von Lubitz et al. 1995). In a study conducted on seizure-sensitive DBA/2 mice, an animal model of generalized reflex epilepsy, the intraperitoneal administration of A3AR agonist IB-MECA is without effect (De Sarro et al. 1999). More recently, Vianna et al. (2005) demonstrated in adult rats in status epilepticus (SE) induced by pilocarpine, that pretreatment with the A3AR antagonist MRS1220 does not alter the incidence of SE but reduces the latency to develop it.
Although the above reviewed evidence would support the concept that A3AR exerts a protective role against seizures, in some cases data indicates that A3AR stimulation by endogenous adenosine may aggravate epileptic activity, as determined in CA1 area of rat immature hippocampal slices in which seizure activity was induced by Mg2+ deprivation together with brief high frequency stimulation of Shaffer collaterals (Etherington and Frenguelli 2004). Such an A3AR-mediated excitatory effect is in agreement with the notion that activation of A3AR in the adult hippocampus exerts excitatory effects, that is increases high-threshold calcium currents (Fleming and Mogul 1997), desensitizes the inhibitory A1 receptors (Dunwiddie et al. 1997) and inhibits presynaptic inhibitory metabotropic glutamate receptors (Macek et al. 1998). In addition, 2-Cl-IB-MECA increases the amplitude of electrically evoked and the frequency of spontaneous epileptiform field potentials recorded during GABAA receptor blockade in CA3 area of rat immature hippocampal slices (Laudadio and Psarropoulou 2004). An enhanced amplitude of evoked responses associated with the increase in frequency of spontaneous discharges may facilitate seizure-induced neuronal damage. The frequency of spontaneous discharges recorded during GABAA and A1 or A2A receptor blockade is not affected by the A3AR antagonist, MRS1220 (Laudadio and Psarropoulou 2004). This indicates that adenosine, tonically released from immature slices, is unlikely to increase the rate of spontaneous discharges. However, the high affinity uptake blocker NBTI (Deckert et al. 1988), which increases extracellular adenosine, induced spontaneous discharges in a subset of rat immature slices (Laudadio and Psarropoulou 2004). Two lines of evidence suggest that this effect is mediated by A3AR. First, 2-Cl-IB-MECA has no additional excitatory effect following an NBTI-induced excitation, and secondly, the NBTI-induced increase in the frequency of spontaneous discharges (in the presence of the A1 antagonist DPCPX) was reversed by the A3AR antagonist MRS1220. The NBTI-induced excitation in immature slices is a novel finding which may be peculiar to the developing hippocampus. In fact in the adult hippocampus NBTI has an inhibitory effect on evoked or spontaneous discharges; this is in line with the consistent A1-mediated inhibitory effects of adenosine (Sanderson and Scholfield 1986).
The variable effects of NBTI in the immature and mature hippocampus might be ascribed to changes in the levels of endogenous adenosine (Park et al. 1987) and/or in the proportion of A1 and A3AR.
In conclusion, the excitatory effects of A3AR activation on synchronous epileptiform discharges in vitro suggest that an increase of endogenous adenosine in conditions of stress, that is seizures or hypoxia, may enhance synaptic activity in the immature brain. In addition, they raise the possibility that the A3AR subtype may play a role in the establishment of activity-dependent plastic changes.
The blockade of A3AR with the selective antagonist, MRS1334, improves the stability of GABAAergic neurotransmission, as assessed in a patch clamp study on GABAA receptor isolated from neurosurgically resected epileptic human nervous tissues and microtransplanted into Xenopus oocytes, and on human epileptic slices obtained from neurosurgical resection (Roseti et al. 2008).
Therefore, antagonism of A3AR may increase the inhibitory efficacy of GABAA receptor in some forms of human epilepsy, pointing towards new therapeutic targets to fight epilepsy.
3.4 Role of A3AR in Nociception
Adenosine exerts complex influences on pain transmission by different mechanisms in the brain and spinal cord, as demonstrated in a broad spectrum of animal pain models (for a review see: Sawynok 1998). Several therapeutic approaches to pain and inflammation based on mimicking or modulating the effects of endogenous adenosine are currently under preclinical and clinical investigation. These include the use of adenosine itself, the use of direct-acting adenosine receptor agonists and the use of agents designed to modulate the levels and therefore the actions of adenosine in the extracellular space (adenosine-kinase (AK) inhibitors) (see: Gao and Jacobson 2007). Much evidence indicates that adenosine receptor activation in the spinal cord produces antinociception due to stimulation of A1 receptors. This effect is attributed to presynaptic inhibition of excitatory neurotransmitter release with subsequent reduction of substance P concentration, as detected in cerebrospinal fluid (Sjolund et al. 1997) and therefore due to postsynaptic inhibition of the glutamate effects (DeLander and Wahl 1988).
A3AR ko have decreased nociception, as assessed by the hot-plate test (Fedorova et al. 2003), probably due to a decrease in the supraspinal processing and “recognition” of painful stimuli. This is consistent with the localization of A3AR in thalamic nuclei (Yaar et al. 2002) where they may play a role in processing nociceptive information.
Following carrageenan-induced inflammation in the hind paw, heat hyperalgesia, plasma extravasation and edema were significantly reduced in A3AR-deleted mice compared to wild type mice (Wu et al. 2002) suggesting that A3AR plays a role in generating the localized inflammatory response which is in agreement with previous evidence that adenosine A3AR activation produces pain behaviors secondary to mast cell degranulation and release of histamine and 5-hydroxytryptamine (5-HT) that exert nociceptive actions at sensory nerve terminal (Sawynok et al. 1999). Subcutaneous administration of adenosine A3AR agonists produces nociceptive behavior (Sawynok et al. 1997).
The above data support the pro-nociceptive role of A3AR involved in both central nervous system effects and pro-inflammatory effects on peripheral tissues.
It must be mentioned, however, that intrathechal administration of IB-MECA does not exhibit an antinociceptive profile in acute nociception as assessed in the early phase pain response of the formalin test, but it does depress the late phase of prolonged pain that measures hyperalgesia related to formalin-induced inflammation (Yoon et al. 2005, 2006). This suggests an involvement of spinal adenosine A3AR in protection from nociception and is in agreement with the observation that A3AR agonists control the in vitro release of pain-related neuropeptides from the rat spinal cord (Mauborgne et al. 2002).
3.5 Role of A3AR in Mood and Affects
Miller and Hoffman (1994) demonstrated that activation of A3AR results in increased 5-HT-uptake in rat basophilic leukemia cells. Likewise, but in rat central nervous system, the activation of adenosine A3AR by the agonist APNEA during adenosine A1 receptor blockade, decreases hippocampal extracellular 5-HT levels in freely moving rats (Okada et al. 1997). The inhibitory effect of the A3AR agonist on extracellular 5-HT levels is abolished by inhibition of 5-HT reuptake activity with DU24565 and fluoxetine (Okada et al. 1999). It was later established using rat basophilic leukemia 2H3 cells, that activation of A3AR, via both PKG and p38 MAPK, stimulates the activity of 5-HT transporter (SERT) (Zhu et al. 2004) that is the main responsible for inactivation of synaptic 5-HT and a main target of antidepressant drugs which inhibit SERT activity.
More recently, it has been reported that the A3AR agonist IB-MECA rapidly (10 min) and selectively stimulates 5-HT transport in mouse midbrain, hippocampal, and cortical synaptosomes (Zhu et al. 2007). IB-MECA-induced stimulation of 5-HT uptake is blocked by the selective A3AR antagonist MRS1191 and is absent in synaptosomes prepared from A3AR-knockout mice (Zhu et al. 2007). In view of these results, a possible antidepressant effect of A3AR antagonists could be envisaged. However such extrapolation is not supported by behavioral depression tests evaluated in A3AR deleted mice. A3AR ko mice show an increase in the amount of time spent immobile in two tests of behavioral depression, the forced-swim test and the tail-suspension test, respectively (Fedorova et al. 2003). This response is probably not attributable to a decrease in motor activity, mainly because of the increased locomotion expressed by the A3AR deleted genotype (Fedorova et al. 2003).
Although there is evidence that A3AR ko mice are more prone to depressive behavior, it is also reported that they have increased performance in the elevated-plus maze and light/dark box suggestive of reduced anxiety but this is most probably a consequence of the increase in exploratory activity due to increased motor activity (Fedorova et al. 2003).
3.6 A3AR and Cerebral Blood Flow Regulation
One important issue not always sufficiently clarified is whether modification of blood pressure contributes to some effects of systemically administered A3AR agonists. Von Lubitz et al. (1994) have shown that preischemic administration of IB-MECA results in a significant delay in the return of postischemic blood flow, and this may affect neuronal survival (see Section 9.4.1). The hypotensive response observed following adenosine A3AR activation in the anaesthetized rat may involve mediator release from mast cells (Fozard and Carruthers 1993; Fozard et al. 1996). Indeed A3AR activation results in rapid mast cell degranulation in the anaesthetized rat. Moreover, a direct central regulation of arterial blood pressure is suggested by the study of Stella et al (1998).
4 Role of A3AR in Neurodegeneration
There is major evidence regarding the role of A3AR in neurodegenerative phenomena from in vivo and in vitro studies in hypoxia/ischemia models.
Although at relatively low levels in comparison to A1 and A2A receptors, A3AR mRNA was detected in the rat and mouse brain by RT-PCR (Dixon et al. 1996) and radioligand binding (Jacobson et al. 1993). This receptor is widespread in the rat and mouse brain (see: Gessi et al. 2008). In comparison to A1 and A2A receptors, A3AR has less affinity for adenosine (10–30 nM versus 1 µM). However, since extracellular adenosine concentrations increase dramatically during ischemia (Hagberg et al. 1987), evidence now indicates that all three adenosine receptors are potential targets for therapeutic treatment of stroke.
It is well accepted that by stimulation of A1 receptors, adenosine exerts a protective role in ischemia by reducing Ca2+ influx by counteracting the presynaptic release of excitatory neurotransmitters (Corradetti et al. 1984). Moreover, by directly increasing the K+ and Cl− ion conductance, adenosine stabilizes neuronal membrane potentials, thus reducing neuronal excitability (Greene and Haas 1991). Furthermore, adenosine, through A1 receptor activation, inhibits NMDA receptor-mediated currents (de Mendonca et al. 1995) and the NMDA receptor component of synaptic potentials during hypoxia (Sebastião et al. 2001). Reductions in synaptic transmission, in cellular metabolism and in energy consumption as well as moderate lowering of the body/brain temperature are protective in ischemia. Although data converge in demonstrating a neuroprotective effect of adenosine through A1 receptors during ischemia, the use of selective A1 agonists is hampered by unwanted peripheral effects, i.e. sedation, bradycardia, hypotension (Kafka and Corbett 1996). More recently, the role of A2A receptors in ischemic neuroprotection has been studied. Most data report a beneficial effect evoked by A2A antagonists that is attributed to reduced excitotoxicity and lower production of intracellular mediators involved in transcription mechanisms that may be relevant to neurodegeneration (Chen et al. 2007; Chen and Pedata 2008).
4.1 Role of A3AR in Hypoxia/Ischemia
The studies currently in the literature concerning the role of A3AR in the pathophysiology of cerebral ischemia are rather contradictory and have been matter of discussion in several review papers (Jacobson 1998; Jacobson et al. 1999; Baraldi et al. 2000; von Lubitz 1999; von Lubitz et al. 1999, 2001).
An early in vivo study in the model of global forebrain ischemia in the gerbil showed that a selective agonist of A3AR, IB-MECA, acutely administered 15 min prior to ischemia, impaired post-ischemic blood flow, increased mortality and exacerbated the loss of hippocampal neurons (von Lubitz et al. 1994). IB-MECA administration 20 min prior to transient middle cerebral ischemia also resulted in a significant increase in infarct size(von Lubitz et al. 2001).
In agreement with a possible noxious role of A3AR in ischemia, it has been more recently shown in a model of in vitro ischemic preconditioning, that the selective A3AR antagonist, 5-propyl-2-ethyl-4-propyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate (MRS1523) applied before and during oxygen–glucose deprivation (OGD), facilitated the full recovery of CA1 hippocampal neurotransmission after a severe (7 min), irreversible OGD period (Pugliese et al. 2003). The harmful role of A3AR during in vitro OGD is confirmed by the observation that the A3AR selective antagonists MRS1523, the new antagonists, LJ1251 ((2R,3R,4S)-2-(2-chloro-6-(3-iodobenzylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol), the 2-arylpyrazolo[3,4-c]quinoline and 4-modified-2-aryl-1,2,4-triazolo[4,3-a]quinoxalin-1-one derivatives and the 4-bismethanesulfonylamino-2-phenyl-1,2,4-triazolo[4,3-a]quinoxalin-1-one compound, consistently abolish or delay the occurrence of anoxic depolarization (AD) and significantly prevent the irreversible disruption of excitatory neurotransmission caused by a severe (7 min) ischemic episode (Pugliese et al. 2006, 2007; Colotta et al. 2007, 2008). The appearance of AD (Pugliese et al. 2006; Tanaka et al. 1997) is strictly correlated with the extent of brain damage during ischemia both in vivo and in vitro (Somjen 2001) and alterations in AD characteristics caused by A3AR antagonists may be attributable to their actions on glutamate-mediated cellular responses (see: Pugliese et al. 2006, 2007). NMDA receptors are essential to AD initiation and propagation (Somjen 2001). The block of A3AR, by removing A3AR-mediated impairment of the feedback inhibition of glutamate release exerted by specific metabotropic glutamate receptor subtypes (Macek et al. 1998) may reduce the participation of glutamate in triggering the AD. Interestingly, Cl-IB-MECA facilitates epileptiform discharges in the CA3 area of immature rat hippocampal slices (Laudadio and Psarropoulou 2004), suggesting that following a rise of endogenous adenosine, as occurs during convulsions associated with hypoxia, A3AR facilitates excitation, thus limiting the known protective inhibitory effect of adenosine in the brain. An A3AR direct regulation of glutamate efflux appears unlikely on the basis of in vitro results showing that neither A3AR activation with Cl-IB-MECA nor its blockade with MRS1191 modify neurotoxicity caused by kainate and cyclothiazide in cultured neurons (Rebola et al. 2005).
Contrary to the above information, Hentschel et al. (2003) demonstrated that under 5 min hypoxic conditions (95% N2–5% CO2) in vitro, selective activation of A3AR by a brief (5 min) application of IB-MECA, inhibits excitatory neurotransmission on cortical neurons. Such effect is blocked by the selective A3AR antagonist MRS1220. These data indicate that A3AR may sustain inhibition of synaptic activity during hypoxia and therefore mediate neuroprotection. Furthermore, IB-MECA acutely administered 20 min after transient (30 min) focal cerebral ischemia decreases the infarct volume (von Lubitz et al. 2001). A possible protective role of A3AR is supported by the observation that mice deleted for A3AR showed more pronounced hippocampal pyramidal neuron damage following repeated episodes of moderate hypoxia (Fedorova et al. 2003) and an increase in cerebral infarction after transient ligation of the middle cerebral artery (Chen et al. 2006). In addition, intracerebroventricular or repeated intravenous administration (i.e., at 165 and 15 min before transient ligation of the middle cerebral artery) of Cl-IB-MECA decrease cerebral infarction assessed 2 days later. Cl-IB-MECA decreased the size of infarction in the wild-type controls, but not in the A3AR knockout animals, confirming that Cl-IB-MECA-induced protection was mediated through the A3AR (Chen et al. 2006). We must notice that the selective A3AR antagonist, MRS1191, administered intracerebroventricularly 30 min before transient (60 min) middle cerebral artery occlusion (MCAo) did not modify the extent of infarction (Shen et al. 2005). This observation is in agreement with the poor role of A3AR in normal physiological transmission. MRS1523 and LJ1251 do not in fact modify synaptic transmission in the CA1 area of the hippocampus under normoxic conditions (Pugliese et al. 2006, 2007).
These conflicting results on the excitatory or inhibitory role of A3AR on synaptic activity under hypoxia/ischemia may be reconciled by recent data reported by Pugliese et al. (2007). The A3AR antagonist, MRS1523, applied before a brief (2 min) OGD reduces the depression of CA1 hippocampal neurotransmission. This result indicates an inhibitory role of A3 AR on synaptic transmission during brief OGD. Indeed a depression of synaptic activity such as that brought about by adenosine A1 receptors during ischemia is considered neuroprotective. In fact, antagonists of A1 receptors reduce inhibition of synaptic transmission, impair the recovery of synaptic potentials (Sebastião et al. 2001) and shorten the onset of AD induced by hypoxia in the CA1 region of hippocampal slices (Lee and Lowenkopf 1993). Results by Brand et al. (2001), Lopes et al (2003) and Pugliese et al (2007) indicate no interplay between A3AR and A1 receptors on the inhibition of excitatory transmission. The decrease in synaptic depression brought about by A3AR antagonists after hypoxia or a brief OGD period suggests that A3AR as A1 receptor inhibit fEPSP amplitude during the first few minutes of OGD, therefore sharing a neuroprotective role with A1 receptors.
Results suggest that in the first phase of ischemia A3AR, by decreasing synaptic transmission, play a protective synergistic role with A1 receptors. However, recovery of transmission after ischaemia may be influenced by several processes, some eventually unrelated to the level of synaptic transmission during hypoxia. Prolonged ischemic conditions could play a pivotal role in switching the effects of A3AR stimulation from A1-like inhibition to potentiation of an excitotoxic glutamate effect. The activation of phospholipase C (PLC) by A3AR has been reported in striatal and hippocampal slices (Abbracchio et al. 1995). Rat cortical neurons exposed to hypoxia in vitro show an increase in activation of protein kinase C (PKC) after selective A3AR stimulation (Nieber and Hentschel 2006). Similarly to what is described in the heart, PKC-dependent activation of KATP channels may enhance adenosine protection (Liang and Jacobson 1998), but if OGD is applied long enough to be considered severe, PKC activation induced by A3AR could account for an increase in intracellular calcium, which may participate in increasing tissue excitability and thus lead to irreversible synaptic failure.
Taking into account that during ischemic conditions, adenosine is released from hippocampal slices, reaching concentrations up to 30 µM after 5-min OGD (Latini et al. 1998; Pearson et al. 2006) and that A3AR are stimulated by µM concentrations of adenosine (Fredholm et al. 2001) we may speculate that A3AR mediated effects would become particularly deleterious during ischemia, when high levels of adenosine are reached extracellularly and detrimental effects of A3AR activation may be due, at least in part, to increased excitoxicity.
Altogether these data suggest that the outcome of A3AR stimulation on synaptic transmission during hypoxic/ischemic phenomena depends on the duration and severity of the ischemic episode. Although A3AR may play a protective role in the first phase of ischemia, prolonged A3AR stimulation by high adenosine concentrations could be pivotal in transforming the A3AR-mediated effects from protective to injurious.
Interestingly, in the in vitro OGD model in hippocampal slices, it was found that a long application (before and during OGD) of 5′-N-methylcarboxamidoadenosine derivatives Cl-IB-MECA and of new selective A3 agonists (Volpini et al. 2002, 2007): VT72 (N6-methoxy-2-phenylethynyl), VT158 (N6-methoxy-2-phenylethynyl), VT160 (N6-methoxy-2-(2-pyridinyl)-ethynyl), VT163 (N6-methoxy-2-p-acetylphenylethynyl) and AR132 (N6-methyl-2-phenylethynyladenosine) have effects similar to those of antagonists upon the OGD-induced depression of synaptic transmission and on the appearance of AD after the severe (7 min) OGD period (Pugliese et al. 2007). These effects may be attributed to desensitization of A3AR. In fact, both human and rat A3AR are desensitized within a few minutes after agonist exposure (Palmer et al. 1995; Trincavelli et al. 2002). A stimulation as massive as that reached in the presence of endogenous adenosine plus exogenous A3AR agonists might induce substantial A3AR plastic adjustments such as desensitization. These in vitro results probably concord with the observation in the model of global forebrain ischemia in the gerbil (von Lubitz et al. 1994), that chronic administration (10-day pre-ischemic) of IB-MECA improves post-ischemic cerebral flow circulation, survival and neuronal preservation (von Lubitz et al. 1994, 1999) and that repeated intravenous administration of Cl-IB MECA before MCAo ligation increases locomotor activity and decreases cerebral infarction (Chen et al. 2006). Chronic preischemic administration of IB-MECA also results in significant preservation of ischemia-sensitive microtubule-associated protein 2 (Map-2), enhancement of the expression of glial fibrillary acidic protein (GFAP) and depression of nitric oxide synthase in ischemic brain tissue (von Lubitz 1999).
Summarizing the above discussed evidence, the protective/injurious effects of A3AR during ischemia appear to depend on time after the onset of the ischemic insult which is consistent with the cascade of events described after ischemia (Dirnagl et al. 1999), some of which may also relate to inflamation. Initially, massive excitoxicity may be controlled by A3AR and later, an inflammatory cascade could be potentiated by prolonged A3AR stimulation. Timing of treatment with respect to the onset of the ischemic insult may therefore account for the different effects of pre- versus post-ischemia administration of A3AR agonists. Moreover, A3AR desensitization may account for the different effects of acute versus chronic agonist treatments reported in different studies.
These observations raise the question of the time-related utility of A3AR antagonists/agonists for treatment of ischemia. It may be speculated that after ischemia, prolonged treatment with A3AR agonists first protects by reducing glutamate-mediated excitotoxicity thus supporting a depression of neuronal activity and energy save; later after ischemia because it desensitizes A3AR, avoiding late onset deleterious A3AR influences. This last-described protective effect could also be ascribed to antagonists administered late with respect to the onset of ischemia. However, the fact that preischemic stimulation of A3AR in the in vivo model of global forebrain ischemia in the gerbil aggravates cerebral damage (von Lubitz et al. 1994) dictates caution in using A3AR agonists to protection from ischemia-induced brain damage. Certainly further studies aimed at verifying the effect of agonists versus antagonists at different times after in vivo ischemia will help clarify the utility of this class of drugs in ischemia.
4.2 A3AR and Neuroinflammation
The evidence summarized in the previous section suggests that A3AR may control ischemic brain injury by controlling excitoxicity. Although excitotoxicity is invoked in the pathophysiology of most neurodegenerative central diseases, how A3AR directly contribute to modulation of brain injury is largely unknown. Under neurodegenerative conditions involving ischemia, trauma, excitoxicity and bioenergetic dysfunctions, the interplay of resident glial cells with infiltrating peripheral bone marrow-derived cells produces neuroinflammation. An important role for A3AR in modifying the inflammatory response was pointed out by first studies in mast cells where activation of these receptors appears to be responsible for release of allergic mediators contributing to inflammatory expansion (Ramkumar et al. 1993;Fozard et al. 1996; Gao et al. 2001) and mast cell degranulation (Reeves et al. 1997; Salvatore et al. 2000; Tilley et al. 2000; Zhong et al. 2003). On the other hand, exposure of blood peripheral cell lines to selective A3AR agonists resulted in both anti- and proinflammatory effects (see: Gessi et al. 2008).
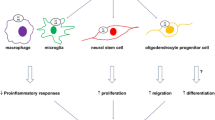
Microglia, astrocytes and oligodendrocytes are cell type sensors responding to neurodegenerative phenomena in the CNS. Evidence suggests that central A3AR exert an important role in brain injury by affecting not only neurons but also glial function controlling important intracellular signaling pathways that are involved in neuroinflammation (Fig. 9.1). Below is a review of A3AR influence on glial functions.

Schematic diagram illustrating A3 adenosine receptor localization in the brain. ADO: adenosine; ADA: adenosine deaminase; ATP: adenosine triphosphate, AMP: adenosine monophospate; AKA: adenosine kinase; T: bidirezional nucleoside transporter; NPTDase: family of ecto-nucleotidases, including NPTDase 1,2,3. During cerebral ischemia, extracellular ADO concentration increases acting on A3 adenosine receptors located on different cell type
4.2.1 Effects of A3AR in Astrocytes
A3AR mRNA has been identified by Northern blot analysis in mouse astrocytes (Zhao et al. 1999). Early evidence indicates that A3AR on astrocytes mediate both protection and cell death, depending on A3AR agonist concentration (Abbracchio et al. 1997; Yao et al. 1997; Jacobson et al. 1999; Di Iorio et al. 2002).
On astroglial cell lines (human astrocytoma ADF cells) low (nM) concentrations of the selective A3AR agonist Cl-IB-MECA induced a marked reorganization of cell cytoskeleton accompanied by induction of expression of small GTP-binding protein of the Rho family that is involved in control of actin cytoskeleton and by changes of intracellular distribution of the antiapoptotic protein Bcl-XL (Abbracchio et al. 1997, 2001).
In addition, stimulation of cultured murine astrocytes with Cl-IB-MECA induces the release of CCL-2, a chemokine which may exert neuroprotective effects (Wittendorp et al. 2004). Recently, in human D384 astrocytoma cells, Cl-IB-MECA at relatively low concentration (0.8 μM), reduced ATP depletion and apoptosis caused by hypoxic conditions. Furthermore, primary astrocytes prepared from A3AR KO mice were more affected by hypoxia than those prepared from WT mice (Bjorklund et al. 2008b). In vivo, in the ischemia model of MCAo (transient, 30 min occlusions), IB-MECA administered after ischemia proved to decrease the intensity of reactive gliosis involving microglia and astrocytes as evaluated 7 days after ischemia. (von Lubitz et al. 2001). However some data have indicated no effect of A3AR selective stimulation. In rat primary cultures, IB-MECA (1 μM) failed to modulate intracellular calcium signaling ([Ca2+]i) elicited by ATP (Alloisio et al. 2004) and no evidence was found that A3AR affects intracellular calcium levels in acutely isolated rat astrocytes (Pilitsis and Kimelberg 1998).
Conversely, a high concentration (μM) of IB-MECA induced apoptosis of various cell lines including astrocytes (Yao et al. 1997; Jacobson et al. 1999). In agreement with this evidence, in primary cultures of rat astrocytes and in C6 glial cells, it was shown that treatment with the A3AR agonist Cl-IB-MECA (10 μM) induced apoptosis and reduced the expression of endogenous Bcl-2, whereas it did not affect the expression of Bax. this suggests that intense activation of A3AR is pro-apoptotic in glial cells via bcl2 and caspase-3 dependent pathways. (Appel et al. 2001). In primary cultures of mouse astrocytes, adenosine caused an increase in [Ca(2+)i] most probably by acting on A3AR (Chen et al. 2001). In rat cultured astrocytes, apoptosis caused by adenosine was significantly reduced by the selective A3AR antagonist MRS1523 (Di Iorio et al. 2002).
On the whole, data are supportive that A3AR may exert a cytoprotective or noxious effect in astrocytes, depending on the intensity of receptor stimulation.
4.2.2 Effects of A3AR in Microglia
Several possible antiinflammatory effects are directly mediated by A3AR on microglial cells. A3AR mRNA is expressed in microglia (Fiebich et al. 1996) where it mediates several effects. Lee et al. (2006) have demonstrated that Cl-IB-MECA suppresses LPS-induced NF-kappaB activation and TNF-alpha production in mouse BV2 microglial cells. In primary mouse microglial cells and N13 microglia cell line, A3AR stimulation increases both ERK1/2 and p38 MAPK phosphorylation via phosphatidylinositol-3′-kinase (Hammarberg et al. 2003, 2004). Interestingly, a concentration-dependent effect was noticed, that is high ERK1/2 phosphorylation occurred at low A3AR agonist concentration, decreasing with increasing agonist concentration (Hammarberg et al. 2003).
Knowledge of the intracellular networks activated by adenosine A3AR may help to elucidate the pathophysiological role of this receptor. Reactive gliosis, in response to central trauma, hypoxia/ischemia and neurodegeneration, includes phenotypical alterations of microglia and astrocytes and increased astrocyte number, occurs. Oligodendrocytes first encounter damage and death. After a central trauma, reactive gliosis is generally regarded as beneficial at first but, if prolonged, it may enhance tissue damage by production of deleterious factors (Neary and Snowden 1996). Glial A3AR stimulated by high adenosine levels caused by a prolonged central trauma may well be implicated in neuroinflammatory tissue responses.
Plastic adjustments of A3AR induced by brain injury might also be relevant to the modulation of intracellular pathways and cell safety. A3AR appear to be very sensitive to prolonged stress in vitro. An up-regulation of A3AR mRNA was observed 1 h after 3-nitropropionate exposure in hippocampal slices, normalization ensued 24 h later (von Arnim et al. 2000). A3AR up-regulation was also reported in the hippocampus of a transgenic mouse model of Alzheimer’s disease (APP23tg) where impaired oxidative phosphorylation was detected prior to amyloid deposition (von Arnim et al. 2006).
5 Conclusions and Perspectives
Adenosine A3 receptors are widely distributed in the CNS but are expressed at a low level and have lower affinity for adenosine in comparison to the A1 and A2A receptors. Nevertheless, they appear to tonically modulate motor activity as pointed out in A3AR-deleted mice. The role of A3AR in several pathophysiological conditions is often controversial. In an attempt to synthesize the major evidence in the literature, it emerges that in conditions that create an extracellular increase of adenosine such as seizures or ischemia, A3AR may contribute to neurotransmission and cell damage. Recently a pro-convulsant effect of A3AR stimulation emerged, especially in the immature brain, thus raising the possibility that A3AR might facilitate seizure-induced neuronal damage. This also raises the possibility that the A3AR subtype plays a role in the establishment of activity-dependent plastic changes. Moreover, most data support a pro-nociceptive role of A3AR involving both central nervous system and pro-inflammatory effects at peripheral tissues.
Major evidence for A3AR in neurodegenerative phenomena emerges from studies performed in in vivo and in vitro models of hypoxia/ischemia. Data from the current literature suggest that the outcome of A3AR stimulation on synaptic transmission during hypoxic/ischemic phenomena depends on the duration and intensity of the ischemic episode. It has been hypothesized that while A3AR play a protective role in the first phase of ischemia by decreasing synaptic transmission, prolonged A3AR stimulation by high adenosine concentrations could be pivotal in transforming the A3AR-mediated effects from protective to injurious. Detrimental effects of A3AR activation may be due, at least in part, to increased excitoxicity. Glial A3AR stimulated by high adenosine levels caused by a prolonged central trauma may well be implicated in neuroinflammatory tissue responses. Moreover, A3AR appear very sensitive to prolonged stress in vitro and plastic adjustments of A3AR induced by brain injury and pharmacological treatment with agonists might be relevant to their final role. All these observations raise the question of the time-related utility of A3AR antagonists/agonists for treatment of ischemia. Certainly further studies aimed at verifying the effect of agonists versus antagonists at different times after in vivo ischemia will help clarify the utility of this potent class of drugs in ischemia and in different neurodegenerative diseases.
Abbreviations
- AK:
-
Adenosine-kinase
- AD:
-
Anoxic depolarization
- aCSF:
-
Artificial cerebrospinal fluid
- APNEA:
-
N(6)-2-(4 Aminophenyl)ethyladenosine
- AR:
-
Adenosine Receptor
- AR132:
-
N6-methyl-2-Phenylethynyladenosine
- CADO:
-
2-Chloroadenosine
- CNS:
-
Central nervous system
- CCL-2:
-
Chemokine (C-C motif) ligand 2
- cAMP:
-
Cyclic AMP
- CHA:
-
N6-cyclohexyladenosine
- Cl-IB-MECA:
-
1-[2-Chloro-6-[[(3-iodophenyl)methyl]amino]-9H-purin-p-yl]-1-deoxy-N-methyl-beta-D-ribofuranuronamide
- EHNA:
-
Erythro-9-(2-hydroxy-3-nonyl)adenine hydrochloride
- ERK1/2:
-
Extracellular signal-regulated kinases
- NECA:
-
5-N-Ethylcarboxyamidoadenosine
- D-PIA:
-
D(-)N(6)-(2-Phenylisopropyl)adenosine
- DPCPX:
-
8-Cyclopentyl-1,3-dipropylxanthine
- GABAA :
-
Gamma-aminobutyric acid A
- GFAP:
-
Glial fibrillary acidic protein
- KO:
-
Knockout
- 5-HT:
-
5-Hydroxytryptamine
- IB-MECA:
-
1-Deoxy-1-[6-[[(3-iodophenyl)-methyl]amino]-9H-purin-9-yl]-N-methyl-beta-D-ribofuranuronamide
- LJ1251:
-
(2R,3R,4S)-2-(2-chloro-6-(3-iodobenzylamino)-9H-purin-9-yl)tetrahydrothiophene-3,4-diol
- L-PIA:
-
L(-)N(6)-(2-Phenylisopropyl)adenosine
- LPS:
-
Lipopolysaccharide
- LTD:
-
Long-term depression
- LTP:
-
Long-term potentiation
- Map-2:
-
Microtubule-associated protein 2
- MCAo:
-
Middle cerebral artery occlusion
- MAPK:
-
Mitogen activated protein kinase
- MRS1191:
-
3-ethyl-5-benzyl-2-methyl-6-phenyl-4-phenylethynyl-1,4-(+/−)-dihydropyridine-3,5-dicarboxylate
- MRS1220:
-
9-Chloro-2-(2-furanyl)-5-((phenylacetyl)amino)-[1,2,4]triazolo[1,5-c]quinazoline
- MRS1340:
-
1,4-Dihydro-2-methyl-6-phenyl-4-(phenylethynyl)-3,5-pyr idinedicarboxylic acid 3-ethyl-5-[(3-nitrophenyl)methyl] ester
- MRS1523:
-
5-Propyl-2-ethyl-4-propyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate
- NMDA:
-
N-Methyl-D-aspartate
- NBTI:
-
S-(4-Nitrobenzyl)-6-theoinosine
- OGD:
-
Oxygen–glucose deprivation
- PKC:
-
Protein kinase C
- PKG:
-
cGMP-dependent protein kinase
- PLC:
-
Phospholipase C
- RT-PCR:
-
Reverse transcription–polymerase chain reaction
- SERT:
-
Serotonin-selective reuptake transporter
- TNF alpha:
-
Tumour necrosis factor alpha
- VT72:
-
N6-Methoxy-2-phenylethynyl
- VT158:
-
N6-Methoxy-2-phenylethynyl
- VT160:
-
N6-Methoxy-2-(2-pyridinyl)-ethynyl
- VT163:
-
N6-Methoxy-2-p-acetylphenylethynyl
References
Abbracchio MP, Brambilla R, Ceruti S, Kim HO, von Lubitz DK, Jacobson KA, Cattabeni F (1995) G protein-dependent activation of phospholipase C by adenosine A3 receptors in rat brain. Mol Pharmacol 48(6):1038–1045
Abbracchio MP, Ceruti S, Brambilla R, Franceschi C, Malorni W, Jacobson KA, von Lubitz DK, Cattabeni F (1997) Modulation of apoptosis by adenosine in the central nervous system: a possible role for the A3 receptor. Pathophysiological significance and therapeutic implications for neurodegenerative disorders. Ann N Y Acad Sci 825:11–22
Abbracchio MP, Camurri A, Ceruti S, Cattabeni F, Falzano L, Giammarioli AM, Jacobson KA, Trincavelli L, Martini C, Malorni W, Fiorentini C (2001) The A3 adenosine receptor induces cytoskeleton rearrangement in human astrocytoma cells via a specific action on Rho proteins. Ann N Y Acad Sci 939:63–73
Alloisio S, Cugnoli C, Ferroni S, Nobile M (2004) Differential modulation of ATP-induced calcium signalling by A1 and A2 adenosine receptors in cultured cortical astrocytes. Br J Pharmacol 141(6):935–942
Appel E, Kazimirsky G, Ashkenazi E, Kim SG, Jacobson KA, Brodie C (2001) Roles of BCL-2 and caspase 3 in the adenosine A3 receptor-induced apoptosis. J Mol Neurosci 17(3):285–292
Baraldi PG, Cacciari B, Romagnoli R, Merighi S, Varani K, Borea PA, Spalluto G (2000) A(3) adenosine receptor ligands: history and perspectives. Med Res Rev 20(2):103–128
Bjorklund O, Halldner-Henriksson L, Yang J, Eriksson TM, Jacobson MA, Dare E, Fredholm BB (2008a) Decreased behavioral activation following caffeine, amphetamine and darkness in A3 adenosine receptor knock-out mice. Physiol Behav 95(5):668–676
Bjorklund O, Shang M, Tonazzini I, Dare E, Fredholm BB (2008b) Adenosine A1 and A3 receptors protect astrocytes from hypoxic damage. Eur J Pharmacol 596(1–3):6–13
Boison D (2007) Adenosine as a modulator of brain activity. Drug News Perspect 20(10):607–611
Boison D (2008) The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol 84(3):249–262
Borowicz KK, Kleinrok Z, Czuczwar SJ (1997) N6-2-(4-aminophenyl)ethyl-adenosine enhances the anticonvulsive activity of antiepileptic drugs. Eur J Pharmacol 327(2–3):125–133
Brand A, Vissiennon Z, Eschke D, Nieber K (2001) Adenosine A(1) and A(3) receptors mediate inhibition of synaptic transmission in rat cortical neurons. Neuropharmacology 40:85–95
Chen GJ, Harvey BK, Shen H, Chou J, Victor A, Wang Y (2006) Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J Neurosci Res 84(8):1848–1855
Chen JF, Pedata F (2008) Modulation of ischemic brain injury and neuroinflammation by adenosine A2A receptors. Curr Pharm Des 14(15):1490–1499
Chen JF, Sonsalla PK, Pedata F, Melani A, Domenici MR, Popoli P, Geiger J, Lopes LV, de Mendonca A (2007) Adenosine A2A receptors and brain injury: broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog Neurobiol 83(5):310–331
Chen Y, Rathbone MP, Hertz L (2001) Guanosine-induced increase in free cytosolic calcium concentration in mouse astrocytes in primary cultures: does it act on an A3 adenosine receptor? J Neurosci Res 65(2):184–189
Colotta V, Catarzi D, Varano F, Capelli F, Lenzi O, Filacchioni G, Martini C, Trincavelli L, Ciampi O, Pugliese AM, Pedata F, Schiesaro A, Morizzo E, Moro S (2007) New 2-arylpyrazolo[3,4-c]quinoline derivatives as potent and selective human A3 adenosine receptor antagonists. Synthesis, pharmacological evaluation, and ligand–receptor modeling studies. J Med Chem 50(17):4061–4074
Colotta V, Catarzi D, Varano F, Lenzi O, Filacchioni G, Martini C, Trincavelli L, Ciampi O, Traini C, Pugliese AM, Pedata F, Morizzo E, Moro S (2008) Synthesis, ligand–receptor modeling studies and pharmacological evaluation of novel 4-modified-2-aryl-1,2,4-triazolo[4,3-a]quinoxalin-1-one derivatives as potent and selective human A3 adenosine receptor antagonists Bioorg Med Chem 16(11):6086–6102
Corradetti R, Lo CG, Moroni F, Passani MB, Pepeu G (1984) Adenosine decreases aspartate and glutamate release from rat hippocampal slices. Eur J Pharmacol 104(1–2):19–26
Costenla AR, Lopes LV, de Mendonca A, Ribeiro JA (2001) A functional role for adenosine A3 receptors: modulation of synaptic plasticity in the rat hippocampus. Neurosci Lett 302(1):53–57
Cunha RA, Constantino MC, Sebastião AM, Ribeiro JA (1995) Modification of A1 and A2a adenosine receptor binding in aged striatum, hippocampus and cortex of the rat. Neuroreport 6(11):1583–1588
Cunha RA, Johansson B, Constantino MD, Sebastião AM, Fredholm BB (1996a) Evidence for high-affinity binding sites for the adenosine A2A receptor agonist [3H] CGS 21680 in the rat hippocampus and cerebral cortex that are different from striatal A2A receptors. Naunyn Schmiedebergs Arch Pharmacol 353(3):261–271
Cunha RA, Vizi ES, Ribeiro JA, Sebastião AM (1996b) Preferential release of ATP and its extracellular catabolism as a source of adenosine upon high- but not low-frequency stimulation of rat hippocampal slices. J Neurochem 67(5):2180–2187
de Mendonca A, Sebastião AM, Ribeiro JA (1995) Inhibition of NMDA receptor-mediated currents in isolated rat hippocampal neurones by adenosine A1 receptor activation. Neuroreport 6(8):1097–1100
de Mendonca A, Ribeiro JA (2000) Long-term potentiation observed upon blockade of adenosine A1 receptors in rat hippocampus is N-methyl-D-aspartate receptor-dependent. Neurosci.Lett 291(2):81–84
De Sarro G, De Sarro A, Di Paola ED, Bertorelli R (1999) Effects of adenosine receptor agonists and antagonists on audiogenic seizure-sensible DBA/2 mice. Eur J Pharmacol 371(2–3):137–145
Deckert J, Morgan PF, Daval JL, Nakajima T, Marangos PJ (1988) Ontogeny of adenosine uptake sites in guinea pig brain: differential profile of [3H]nitrobenzylthioinosine and [3H]dipyridamole binding sites. Brain Res 470(2):313–316
DeLander GE, Wahl JJ (1988) Behavior induced by putative nociceptive neurotransmitters is inhibited by adenosine or adenosine analogs coadministered intrathecally. J Pharmacol Exp Ther 246(2):565–570
Di Iorio P, Kleywegt S, Ciccarelli R, Traversa U, Andrew CM, Crocker CE, Werstiuk ES, Rathbone MP (2002) Mechanisms of apoptosis induced by purine nucleosides in astrocytes. Glia 38(3):179–190
Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22(9):391–397
Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ, Freeman TC (1996) Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol 118(6):1461–1468
Dragunow M, Goddard GV, Laverty R (1985) Is adenosine an endogenous anticonvulsant? Epilepsia 26(5):480–487
Dragunow M (1991) Adenosine and seizure termination. Ann Neurol 29(5):575
Dudek SM, Bear MF (1993) Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci 13(7):2910–2918
Dunwiddie TV (1980) Endogenously released adenosine regulates excitability in the in vitro hippocampus. Epilepsia 21(5):541–548
Dunwiddie TV, Diao L, Kim HO, Jiang JL, Jacobson KA (1997) Activation of hippocampal adenosine A3 receptors produces a desensitization of A1 receptor-mediated responses in rat hippocampus. J Neurosci 17(2):607–614
During MJ, Spencer DD (1992) Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol 32(5):618–624
Etherington LA, Frenguelli BG (2004) Endogenous adenosine modulates epileptiform activity in rat hippocampus in a receptor subtype-dependent manner. Eur J Neurosci 19(9):2539–2550
Fedorova IM, Jacobson MA, Basile A, Jacobson KA (2003) Behavioral characterization of mice lacking the A3 adenosine receptor: sensitivity to hypoxic neurodegeneration. Cell Mol Neurobiol 23(3):431–447
Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, Bauer J, Gebicke-Haerter PJ (1996) Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia 18(2):152–160
Fleming KM, Mogul DJ (1997) Adenosine A3 receptors potentiate hippocampal calcium current by a PKA-dependent/PKC-independent pathway. Neuropharmacology 36(3):353–362
Fozard JR, Carruthers AM (1993). Adenosine A3 receptors mediate hypotension in the angiotensin II-supported circulation of the pithed rat. Br J Pharmacol 109(1):3–5
Fozard JR, Pfannkuche HJ, Schuurman HJ (1996). Mast cell degranulation following adenosine A3 receptor activation in rats. Eur J Pharmacol 298(3):293–297
Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53(4):527–552
Gao Z, Li BS, Day YJ, Linden J (2001) A3 adenosine receptor activation triggers phosphorylation of protein kinase B and protects rat basophilic leukemia 2H3 mast cells from apoptosis. Mol Pharmacol 59(1):76–82
Gao ZG, Jacobson KA (2007) Emerging adenosine receptor agonists. Expert Opin Emerg Drugs 12(3):479–492
Gessi S, Merighi S, Varani K, Leung E, Mac Lennan S, Borea PA (2008) The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol Ther 117(1):123–140
Ginsborg BL, Hirst GD (1972) The effect of adenosine on the release of the transmitter from the phrenic nerve of the rat. J Physiol 224(3):629–645
Greene RW, Haas HL (1991) The electrophysiology of adenosine in the mammalian central nervous system. Prog Neurobiol 36(4):329–341
Hagberg H, Andersson P, Lacarewicz J, Jacobson I, Butcher S, Sandberg M (1987) Extracellular adenosine, inosine, hypoxanthine, and xanthine in relation to tissue nucleotides and purines in rat striatum during transient ischemia. J Neurochem 49(1):227–231
Hammarberg C, Schulte G, Fredholm BB (2003) Evidence for functional adenosine A3 receptors in microglia cells. J Neurochem 86(4):1051–1054
Hammarberg C, Fredholm BB, Schulte G (2004) Adenosine A3 receptor-mediated regulation of p38 and extracellular-regulated kinase ERK1/2 via phosphatidylinositol-3’-kinase. Biochem Pharmacol 67(1):129–134
Hentschel S, Lewerenz A, Nieber K (2003) Activation of A(3) receptors by endogenous adenosine inhibits synaptic transmission during hypoxia in rat cortical neurons. Restor Neurol Neurosci 21(1–2):55–63
Huang CC, Yang PC, Lin HJ, Hsu KS (2007) Repeated cocaine administration impairs group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. J Neurosci 27(11):2958–2968
Jacobson KA, Nikodijevic O, Shi D, Gallo-Rodriguez C, Olah ME, Stiles GL, Daly JW (1993) A role for central A3-adenosine receptors. Mediation of behavioral depressant effects. FEBS Lett 336(1):57–60
Jacobson KA, Siddiqi SM, Olah ME, Ji XD, Melman N, Bellamkonda K, Meshulam Y, Stiles GL, Kim HO (1995) Structure–activity relationships of 9-alkyladenine and ribose-modified adenosine derivatives at rat A3 adenosine receptors. J Med Chem 38(10):1720–1735
Jacobson KA (1998). Adenosine A3 receptors: novel ligands and paradoxical effects. Trends Pharmacol Sci 19(5):184–191
Jacobson KA, Hoffmann C, Cattabeni F, Abbracchio MP (1999) Adenosine-induced cell death: evidence for receptor-mediated signalling. Apoptosis 4(3):197–211
Kafka SH, Corbett R (1996) Selective adenosine A2A receptor/dopamine D2 receptor interactions in animal models of schizophrenia. Eur J Pharmacol 295(2–3):147–154
Latini S, Bordoni F, Corradetti R, Pepeu G, Pedata F (1998) Temporal correlation between adenosine outflow and synaptic potential inhibition in rat hippocampal slices during ischemia-like conditions. Brain Res., 794, (2) 325-328.
Latini S, Pedata F (2001) Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem 79(3):463–484
Laudadio MA, Psarropoulou C (2004) The A3 adenosine receptor agonist 2-Cl-IB-MECA facilitates epileptiform discharges in the CA3 area of immature rat hippocampal slices. Epilepsy Res 59(2–3):83–94
Lee KS, Schubert P, Heinemann U (1984) The anticonvulsive action of adenosine: a postsynaptic, dendritic action by a possible endogenous anticonvulsant. Brain Res 321(1):160–164
Lee KS, Lowenkopf T (1993) Endogenous adenosine delays the onset of hypoxic depolarization in the rat hippocampus in vitro via an action at A1 receptors. Brain Res 609(1–2):313–315
Lee JY, Jhun BS, Oh YT, Lee JH, Choe W, Baik HH, Ha J, Yoon KS, Kim SS, Kang I (2006) Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of PI 3-kinase/Akt and NF-kappaB activation in murine BV2 microglial cells. Neurosci Lett 396(1):1–6
Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D (2008) Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest 118(2):571–582
Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brustle O, Boison D (2007) Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain 130 Pt 5:1276–1288
Liang BT, Jacobson KA (1998) A physiological role of the adenosine A3 receptor: sustained cardioprotection. Proc Natl Acad Sci U S A 95(12):6995–6999
Linden J, Taylor HE, Robeva AS, Tucker AL, Stehle JH, Rivkees SA, Fink JS, Reppert SM (1993) Molecular cloning and functional expression of a sheep A3 adenosine receptor with widespread tissue distribution. Mol Pharmacol 44(3):524–532
Londos C, Cooper DM, Wolff J (1980) Subclasses of external adenosine receptors. Proc Natl Acad Sci U S A 77(5):2551–2554
Lopes LV, Rebola N, Pinheiro PC, Richardson PJ, Oliveira CR, Cunha RA (2003) Adenosine A3 receptors are located in neurons of the rat hippocampus. Neuroreport 14 (12):1645–1648
Macek TA, Schaffhauser H, Conn PJ (1998) Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. J Neurosci 18(16):6138–6146
Mauborgne A, Polienor H, Hamon M, Cesselin F, Bourgoin S (2002) Adenosine receptor-mediated control of in vitro release of pain-related neuropeptides from the rat spinal cord. Eur J Pharmacol 441(1–2) 47–55
Meyerhof W, Muller-Brechlin R, Richter, D (1991a) Molecular cloning of a novel putative G-protein coupled receptor expressed during rat spermiogenesis. FEBS Lett 284(2):155–160
Meyerhof W, Paust HJ, Schonrock C, Richter D (1991b) Cloning of a cDNA encoding a novel putative G-protein-coupled receptor expressed in specific rat brain regions. DNA Cell Biol 10(9):689–694
Miller KJ, Hoffman BJ (1994) Adenosine A3 receptors regulate serotonin transport via nitric oxide and cGMP. J Biol Chem 269(44):27351–27356
Neary D, Snowden J (1996) Fronto-temporal dementia: nosology, neuropsychology, and neuropathology. Brain Cogn 31(2):176–187
Nieber K, Hentschel S (2006) Signalling pathways of the adenosine A3 receptors in rat cortical neurons. In: Proceedings of the 8th international symposium on adenosine and adenine nucleotides, Ferrara, Italy, 24–28 May 2006
Okada M, Kawata Y, Kiryu K, Mizuno K, Wada K, Tasaki H, Kaneko S (1997) Effects of adenosine receptor subtypes on hippocampal extracellular serotonin level and serotonin reuptake activity. J Neurochem 69(6):2581–2588
Okada M, Kawata Y, Murakami T, Wada K, Mizuno K, Kondo T, Kaneko S (1999) Differential effects of adenosine receptor subtypes on release and reuptake of hippocampal serotonin. Eur J Neurosci 11(1):1–9
Oliveira JC, Sebastião AM, Ribeiro JA (1991) Solubilized rat brain adenosine receptors have two high-affinity binding sites for 1,3-dipropyl-8-cyclopentylxanthine. J Neurochem 57(4):1165–1171
Palmer TM, Benovic JL, Stiles GL (1995) Agonist-dependent phosphorylation and desensitization of the rat A3 adenosine receptor. Evidence for a G-protein-coupled receptor kinase-mediated mechanism. J Biol Chem 270(49):29607–29613
Park TS, Van Wylen DG, Rubio R, Berne RM (1987) Interstitial fluid adenosine and sagittal sinus blood flow during bicuculline-seizures in newborn piglets. J Cereb Blood Flow Metab 7(5):633–639
Pearson T, Damian K, Lynas RE, Frenguelli BG (2006) Sustained elevation of extracellular adenosine and activation of A1 receptors underlie the post-ischaemic inhibition of neuronal function in rat hippocampus in vitro. J Neurochem 97(5):1357–1368
Pilitsis JG, Kimelberg HK (1998) Adenosine receptor mediated stimulation of intracellular calcium in acutely isolated astrocytes. Brain Res 798(1–2):294–303
Pugliese AM, Latini S, Corradetti R, Pedata F (2003) Brief, repeated, oxygen–glucose deprivation episodes protect neurotransmission from a longer ischemic episode in the in vitro hippocampus: role of adenosine receptors. Br J Pharmacol 140(2):305–314
Pugliese AM, Coppi E, Spalluto G, Corradetti R, Pedata F (2006) A3 adenosine receptor antagonists delay irreversible synaptic failure caused by oxygen and glucose deprivation in the rat CA1 hippocampus in vitro. Br J Pharmacol 147(5):524–532
Pugliese AM, Coppi E, Volpini R, Cristalli G, Corradetti R, Jeong LS, Jacobson KA, Pedata F (2007) Role of adenosine A3 receptors on CA1 hippocampal neurotransmission during oxygen–glucose deprivation episodes of different duration. Biochem Pharmacol 74(5)768–779
Ramkumar V, Stiles GL, Beaven MA, Ali H (1993) The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J Biol Chem 268(23):16887–16890
Rebola N, Rodrigues RJ, Oliveira CR, Cunha RA (2005) Different roles of adenosine A1, A2A and A3 receptors in controlling kainate-induced toxicity in cortical cultured neurons. Neurochem Int 47(5):317–325
Rebola N, Lujan R, Cunha RA, Mulle C (2008) Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 57(1):121–134
Reeves JJ, Jones CA, Sheehan MJ, Vardey CJ, Whelan CJ (1997) Adenosine A3 receptors promote degranulation of rat mast cells both in vitro and in vivo. Inflamm Res 46(5):180–184
Ribeiro JA, Walker J (1975) The effects of adenosine triphosphate and adenosine diphosphate on transmission at the rat and frog neuromuscular junctions. Br J Pharmacol 54(2):213–218
Ribeiro JA, Sebastião AM (1984) Enhancement of tetrodotoxin-induced axonal blockade by adenosine, adenosine analogues, dibutyryl cyclic AMP and methylxanthines in the frog sciatic nerve. Br J Pharmacol 83(2):485–492
Ribeiro JA, Sebastião AM (1986) Adenosine receptors and calcium: basis for proposing a third (A3) adenosine receptor. Prog Neurobiol 26(3):179–209
Rivkees SA (1994) Localization and characterization of adenosine receptor expression in rat testis. Endocrinology 135(6):2307–2313
Rivkees SA, Thevananther S, Hao H (2000) Are A3 adenosine receptors expressed in the brain? Neuroreport 11(5):1025–1030
Roseti C, Martinello K, Fucile S, Piccari V, Mascia A, Di Gennaro G, Quarato PP, Manfredi M, Esposito V, Cantore G, Arcella A, Simonato M, Fredholm BB, Limatola C, Miledi R, Eusebi F (2008) Adenosine receptor antagonists alter the stability of human epileptic GABAA receptors. Proc Natl Acad Sci U S A 105(39):15118–15123
Rubaj A, Zgodzinski W, Sieklucka-Dziuba M (2003) The influence of adenosine A3 receptor agonist: IB-MECA, on scopolamine- and MK-801-induced memory impairment. Behav Brain Res 141(1):11–17
Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG (1993) Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci U S A 90(21):10365–10369
Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA (2000) Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem 275(6):4429–4434
Sanderson G, Scholfield CN (1986) Effects of adenosine uptake blockers and adenosine on evoked potentials of guinea-pig olfactory cortex. Pflugers Arch 406(1):25–30
Sattin A, Rall TW (1970) The effect of adenosine and adenine nucleotides on the cyclic adenosine 3′, 5′-phosphate content of guinea pig cerebral cortex slices. Mol Pharmacol 6(1):13–23
Sawynok J, Zarrindast MR, Reid AR, Doak GJ (1997) Adenosine A3 receptor activation produces nociceptive behaviour and edema by release of histamine and 5-hydroxytryptamine. Eur J Pharmacol 333(1):1–7
Sawynok J (1998) Adenosine receptor activation and nociception. Eur J Pharmacol 347(1):1–11
Sawynok J, Reid AR, Esser MJ (1999) Peripheral antinociceptive action of amitriptyline in the rat formalin test: involvement of adenosine. Pain 80(1–2):45–55
Sebastião AM, de Mendonca A, Moreira T, Ribeiro JA (2001) Activation of synaptic NMDA receptors by action potential-dependent release of transmitter during hypoxia impairs recovery of synaptic transmission on reoxygenation. J Neurosci 21(21):8564–8571
Shen H, Chen GJ, Harvey BK, Bickford PC, Wang Y (2005) Inosine reduces ischemic brain injury in rats. Stroke 36(3):654–659
Sjolund KF, Sollevi A, Segerdahl M, Lundeberg T (1997) Intrathecal adenosine analog administration reduces substance P in cerebrospinal fluid along with behavioral effects that suggest antinociception in rats. Anesth Analg 85(3):627–632
Somjen GG (2001) Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81(3):1065–1096
Stella L, de Novellis V, Marabese I, Berrino L, Maione S, Filippelli A, Rossi F (1998) The role of A3 adenosine receptors in central regulation of arterial blood pressure. Br J Pharmacol 125(3):437–440
Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H (1997) Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol 78(2):891–902
Tilley SL, Wagoner VA, Salvatore CA, Jacobson MA, Koller BH (2000) Adenosine and inosine increase cutaneous vasopermeability by activating A(3) receptors on mast cells. J Clin Invest 105(3):361–367
Trincavelli ML, Tuscano D, Marroni M, Falleni A, Gremigni V, Ceruti S, Abbracchio MP, Jacobson KA, Cattabeni F, Martini C (2002) A3 adenosine receptors in human astrocytoma cells: agonist-mediated desensitization, internalization, and down-regulation. Mol Pharmacol 62(6):1373–1384
van Calker D, Muller M, Hamprecht B (1979) Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. J Neurochem 33(5):999–1005
Vianna EP, Ferreira AT, Dona F, Cavalheiro EA, Silva Fernandes MJ (2005) Modulation of seizures and synaptic plasticity by adenosinergic receptors in an experimental model of temporal lobe epilepsy induced by pilocarpine in rats. Epilepsia 46(Suppl 5):166–173
Volpini R, Costanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G (2002) N(6)-alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A(3) receptor and a starting point for searching A(2B) ligands. J Med Chem 45(15):3271–3279
Volpini R, Dal Ben D, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G (2007) N6-methoxy-2-alkynyladenosine derivatives as highly potent and selective ligands at the human A3 adenosine receptor. J Med Chem 50(6):1222–1230
von Arnim CA, Timmler M, Ludolph AC, Riepe MW (2000) Adenosine receptor up-regulation: initiated upon preconditioning but not upheld. Neuroreport 11(6):1223–1226
von Arnim CA, Spoelgen R, Peltan ID, Deng M, Courchesne S, Koker M, Matsui T, Kowa H, Lichtenthaler SF, Irizarry MC, Hyman BT (2006) GGA1 acts as a spatial switch altering amyloid precursor protein trafficking and processing. J Neurosci 26(39):9913–9922
von Lubitz DK (1999) Adenosine and cerebral ischemia: therapeutic future or death of a brave concept? Eur J Pharmacol 371(1):85–102
von Lubitz DK, Lin RC, Popik P, Carter MF, Jacobson KA (1994) Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol 263(1–2):59–67
von Lubitz DK, Carter MF, Deutsch SI, Lin RC, Mastropaolo J, Meshulam Y, Jacobson KA (1995) The effects of adenosine A3 receptor stimulation on seizures in mice. Eur J Pharmacol 275(1):23–29
von Lubitz DK, Ye W, McClellan J, Lin RC (1999) Stimulation of adenosine A3 receptors in cerebral ischemia. Neuronal death, recovery, or both? Ann N Y Acad Sci 890:93–106
von Lubitz DK, Simpson KL, Lin RC (2001) Right thing at a wrong time? Adenosine A3 receptors and cerebroprotection in stroke. Ann N Y Acad Sci 939:85–96
Wittendorp MC, Boddeke HW, Biber K (2004) Adenosine A3 receptor-induced CCL2 synthesis in cultured mouse astrocytes. Glia 46(4):410–418
Wu WP, Hao JX, Halldner-Henriksson L, Xu XJ, Jacobson MA, Wiesenfeld-Hallin Z, Fredholm BB (2002) Decreased inflammatory pain due to reduced carrageenan-induced inflammation in mice lacking adenosine A3 receptors. Neuroscience 114(3):523–527
Yaar R, Lamperti ED, Toselli PA, Ravid K (2002) Activity of the A3 adenosine receptor gene promoter in transgenic mice: characterization of previously unidentified sites of expression. FEBS Lett 532(3):267–272
Yao Y, Sei Y, Abbracchio MP, Jiang JL, Kim YC, Jacobson KA (1997) Adenosine A3 receptor agonists protect HL-60 and U-937 cells from apoptosis induced by A3 antagonists. Biochem Biophys Res Commun 232(2):317–322
Yoon MH, Bae HB, Choi JI (2005) Antinociception of intrathecal adenosine receptor subtype agonists in rat formalin test. Anesth Analg 101(5):1417–1421
Yoon MH, Bae HB, Choi JI, Kim SJ, Chung ST, Kim CM (2006) Roles of adenosine receptor subtypes in the antinociceptive effect of intrathecal adenosine in a rat formalin test. Pharmacology 78(1):21–26
Zhao Z, Francis C, Ravid K (1999) Characterization of the mouse A3 adenosine receptor gene: exon/intron organization and promoter activity. Genomics 57(1):152–155
Zhong H, Shlykov SG, Molina JG, Sanborn BM, Jacobson MA, Tilley SL, Blackburn MR (2003) Activation of murine lung mast cells by the adenosine A3 receptor. J Immunol 171(1):338–345
Zhou QY, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O (1992) Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc Natl Acad Sci U S A 89(16):7432–7436
Zhu CB, Hewlett WA, Feoktistov I, Biaggioni I, Blakely RD (2004) Adenosine receptor, protein kinase G, and p38 mitogen-activated protein kinase-dependent up-regulation of serotonin transporters involves both transporter trafficking and activation. Mol Pharmacol 65(6):1462–1474
Zhu CB, Steiner JA, Munn JL, Daws LC, Hewlett WA, Blakely RD (2007) Rapid stimulation of presynaptic serotonin transport by A(3) adenosine receptors. J Pharmacol Exp Ther 322(1):332–340
Acknowledgements
The laboratory work of the authors is supported by research grants from Fundação para a Ciência e Tecnologia (FCT), Gulbenkian Foundation, PRIN-MIUR Ministero dell’Istruzione, dell’Università e della Ricerca, Ente Cassa di Risparmio, Firenze, Italy and European Union (COST B30).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Pedata, F., Pugliese, A.M., Sebastião, A.M., Ribeiro, J.A. (2010). Adenosine A3 Receptor Signaling in the Central Nervous System. In: Borea, P. (eds) A3 Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics. Springer, Dordrecht. https://doi.org/10.1007/978-90-481-3144-0_9
Download citation
DOI: https://doi.org/10.1007/978-90-481-3144-0_9
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-90-481-3143-3
Online ISBN: 978-90-481-3144-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)