
Abstract
Glutamate, an amino acid, is the principal excitatory neurotransmitter within the vertebrate nervous system. Glutamate is involved in most aspects of normal brain function including cognition, memory and learning, and also plays major roles in the development of the central nervous system, including synapse induction and elimination, and cell migration, differentiation and death. Glutamate further plays a signaling role in peripheral organs and tissues, such as the heart, kidney, intestine, lungs, muscles, liver, ovary, testis, bone, pancreas and the adrenal, pituitary and pineal glands.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Systemic Lupus Erythematosus
- Metabotropic Glutamate Receptor
- Experimental Autoimmune Encephalomyelitis
- Chemotactic Migration
- AMPA GluR3
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Extended Summary
Glutamate, an amino acid, is the principal excitatory neurotransmitter within the vertebrate nervous system. Glutamate is involved in most aspects of normal brain function including cognition, memory and learning, and also plays major roles in the development of the central nervous system, including synapse induction and elimination, and cell migration, differentiation and death. Glutamate further plays a signaling role in peripheral organs and tissues, such as the heart, kidney, intestine, lungs, muscles, liver, ovary, testis, bone, pancreas and the adrenal, pituitary and pineal glands.
In this review, we discuss the involvement of glutamate and its receptors in the immune system, and argue that glutamate is in fact not only a neurotransmitter, but a ‘Neuro-Immuno-Transmitter’ – a novel term coined herein, since four major criteria are undoubtedly met.
-
First: Glutamate receptors (GluRs), both ionotropic and metabotropic, are highly expressed in various immune cells, among them T cells, B cells, macrophages, and dendritic cells. Interestingly, different GluRs, or different levels of certain GluRs, are expressed in resting and activated T cells.
-
Second: Glutamate by itself, as well as glutamate agonists and antagonists, bind GluRs expressed in many types of immune cells, and can either trigger or suppress key immune functions. The exact glutamate-induced effect is determined by the context, and especially by glutamate’s concentration and whether the cells are naïve/resting or rather activated. There is a marked difference between the response of naïve/resting and activated T cells to glutamate.
-
Third: Glutamate is produced by immune cells of several types, among them neutrophils, monocytes/macrophages and activated microglia. Furthermore, immune-derived glutamate has functional consequences on various target cells, and can for example bind GluRs expressed in human brain and dermal microvascular endothelial cells, resulting in decreased cell permeability.
-
Fourth: Glutamate seems to contribute to certain immune diseases, among them hematological cancers: T-leukemia and T-lymphoma, autoimmune diseases like Multiple Sclerosis, and human immunodeficiency virus (HIV) type 1 infection. On top of all that, GluR antibodies (Abs), i.e. anti-AMPA GluR3, anti-NMDA NR1 and anti-NMDA NR2A/B Abs, seem to play a role in ‘Autoimmune Epilepsy’, Encephalitis and Sytemic Lupus Eerythematosus. GluR Abs are found in serum and/or cerebrospinal fluid of patients, and induce detrimental effects on glutamate signaling, and on the viability of neuronal and glial cells in-vitro and in-vivo. On top of all that, we speculate that in the coming years new evidence would be revealed, showing that glutamate and/or its receptors are in fact ‘guilty’ in a kaleidoscope of additional immune diseases. This, of course, may open new avenues for medical interventions.
To conclude, glutamate, GluRs and GluR Abs seems to play an active, broad, potent and important role in both the nervous system and the immune system under physiological and pathological conditions. All the above mentioned topics are discussed in this book chapter.
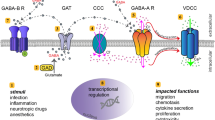
4.2 Glutamate
Glutamate (Fig. 4.1) is an amino acid that functions as an excitatory neurotransmitter. The excitatory action of glutamate in the mammalian brain and spinal cord has been known since the 1950s, but only during the late 1970s it became widely recognized that glutamate is the principal excitatory neurotransmitter within the vertebrate nervous system (Meldrum 2000). Glutamate is in fact involved in most aspects of normal brain function including cognition, memory and learning, and also plays major roles in the development of the central nervous system (CNS), including synapse induction and elimination, as well as cell migration, differentiation and death (Danbolt 2001; Foster and Fagg 1984; Komuro and Rakic 1993; Mayer and Westbrook 1987). Glutamate also plays a signaling role in peripheral organs and tissues, among them the heart, kidney, intestine, lungs, muscles, liver, ovary, testis, bone and pancreas, and the adrenal, pituitary and pineal glands. In these tissues, glutamate may be important in mediating cardio-respiratory, endocrine and reproductive functions, which include hormone regulation, heart rhythm, blood pressure, circulation and reproduction (for detailed reviews see Nedergaard et al. 2002; Hinoi et al. 2004; Gill and Pulido 2001). Importantly, although highly regulated, glutamate levels may be extremely different in the CNS and in the periphery under physiological and pathological conditions (see Sect. 4.4 below). Furthermore, excess glutamate causes massive cell death in the nervous system by a mechanism called excitotoxicity, which plays a cardinal role in numerous neurological diseases and injuries.

Glutamate and glutamate receptors. The amino acid glutamate is the major excitatory neurotransmitter in the mammalian CNS and is involved in most aspects of physiological brain function and development. Glutamate has a further signaling role in peripheral organs and tissues and in endocrine cells. In contrast, excessive exposure to elevated levels of glutamate that leads to neuronal death (a process termed excitotoxicity) plays a key role in numerous pathological conditions. Glutamate has two major classes of receptors: ionotropic glutamate receptors (iGluRs) that are ion-channels gated by glutamate, and metabotropic glutamate receptors (mGluRs) that are coupled to G-proteins. Each of these two receptor classes is further subdivided to specific receptor subtypes or groups, which contain multiple subunits
4.3 Glutamate Receptors
The vast majority of neuronal and glial cells express glutamate receptors (GluRs) of several types on their plasma membranes (Danbolt 2001). The GluRs (shown schematically in Fig. 4.1) are divided into two main groups: (1) The ionotropic glutamate receptors (iGluRs), which are ion channels opened/gated by glutamate; (2) The metabotropic glutamate receptors (mGluRs), which are G protein-coupled receptors (GPCRs) activated/gated by glutamate. The mGluRs belong to the large superfamily of GPCRs that activate intracellular signal transduction pathways (Tanabe et al. 1992; Masu et al. 1991; Monaghan et al. 1989; Hollmann and Heinemann 1994; Kew and Kemp 2005). Glutamate can activate all types of iGluRs and mGluRs. Yet, there are many glutamate agonists and antagonists that bind selectively to particular types of GluRs and activate or block their activity, respectively.
4.3.1 The Ionotropic Glutamate Receptors
The iGluRs are subdivided into three groups according to their pharmacology, structural similarities, and the type of synthetic agonist that activates them: (1) The N-methyl-D-aspartate (NMDA) receptors; (2) The alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors; and (3) The 2-carboxy-3-carboxymethyl-4-isopropenylpyrrolidine (Kainate; KA) receptors (Hollmann et al. 1989). iGluRs are found mainly in the CNS, where they have extremely important roles in many brain functions. On top of that, numerous iGluR subunits are expressed also in peripheral tissues outside the CNS. Their role in these tissues is out of the scope of the current review, and the reader may refer to Nedergaard et al. (2002), Hinoi et al. (2004), Gill and Pulido (2001) for further details on this important topic. Finally, as cited and discussed below, various types of immune cells, primarily T cells, clearly express high levels of iGluRs on their cell surface. The iGluRs in immune cells are functional receptors that upon direct binding of glutamate or its agonists or antagonists exert potent immune effects.
4.3.1.1 The AMPA Glutamate Receptors
The AMPA iGluRs (Fig. 4.1) are found in many parts of the brain, and are the most commonly expressed receptors in the nervous system. AMPA receptors are also expressed outside the CNS, for example in human T cells, and in other immune cells, as discussed later herein.
The AMPA iGluRs are both glutamate receptors and cation channels that are integral to the plasticity and synaptic transmission at many postsynaptic membranes. As to their structure, the AMPA iGluRs are homo- or hetero-oligomers composed of the GluR1-GluR4 subunits (Keinanen et al. 1990), and assemble as functional tetramers (Rosenmund et al. 1998). Thus, each AMPA receptor has four sites to which glutamate or AMPA iGluR agonists can bind, one for each subunit (Mayer 2005b). The binding site is believed to be formed by the N-tail and the extracellular loop between transmembrane domains three and four (Armstrong et al. 1998). When glutamate or AMPA agonists bind, these two loops move towards each other, opening the channel pore. Thus, the channel opens when two sites are occupied (Platt 2007), and increases its current as more binding sites are occupied (Rosenmund et al. 1998). The AMPA iGluR permeability to Ca2+ and other cations, such as Na+ and K+, is governed by the GluR2 subunit. If an AMPA iGluR lacks a GluR2 subunit, it will be permeable to Na+, K+ and Ca2+. The presence of a GluR2 subunit will almost certainly render the channel impermeable to Ca2+. This is determined by post-transcriptional modification – RNA editing – of the Q/R editing site of the GluR2 mRNA. Native AMPA iGluRs contain GluR2 and are therefore impermeable to Ca2+ ions (Kew and Kemp 2005).
4.3.1.2 The NMDA Glutamate Receptors
The NMDA iGluRs (Fig. 4.1) are the predominant molecular devices for controlling synaptic plasticity and memory function (Li and Tsien 2009). Alike the AMPA receptors, the NMDA iGluRs are expressed broadly in the CNS, but also outside it, for example in T cells, as discussed later in this review.
The NMDA iGluRs are hetero-oligomers composed of two obligatory NR1 subunits and two or three of the four regulatory subunits NR2A-D, which co-assemble to form a tetramer (Rosenmund et al. 1998) or pentamer (Hawkins et al. 1999). The NR1 subunit is necessary for Ca2+ conductivity of the receptor ion channel, while the NR2 subunits determine the electrophysiological and pharmacological properties of the receptor. A third subunit NR3A/B is a regulatory subunit that decreases the receptor’s channel activity (Das et al. 1998), and assembly of NR3 with NR1 subunits creates a functional glycine receptor that is not activated by glutamate (Chatterton et al. 2002). Activation of NMDA iGluRs requires binding of two agonists, glutamate and glycine, at their bindings sites on NR2 and NR1 subunits, respectively (Kew and Kemp 2005). In addition, membrane depolarization is needed to abrogate the blockade of these receptors by Mg2+, to render the receptors highly permeable to Ca2+ and Na+ ions (Moriyoshi et al. 1991; Collingridge and Singer 1990). Thus, a unique property of the NMDA iGluRs is their voltage-dependent activation, a result of the ion channel block by extracellular Mg2+ ions. This allows the flow of Na+ ions and small amounts of Ca2+ ions into the cell, and K+ ions out of the cell to be voltage-dependent. The Ca2+ flux through NMDA iGluRs is thought to play a critical role in synaptic plasticity, a cellular mechanism for learning and memory. The NMDA iGluRs are hence distinct in two ways: first, they are both ligand-gated and voltage-dependent; second, they require co-activation by two ligands – glutamate and glycine. Antagonists of the NMDA iGluRs are used as anesthetics for animals and sometimes humans, and are often used as recreational drugs due to their hallucinogenic properties, in addition to their unique effects at elevated dosages, such as dissociation. Common NMDA iGluRs antagonists include: Amantadine, Ketamine, Phencyclidine (PCP), Nitrous oxide, Dextromethorphan and Dextrorphan, Memantine, Ethanol, Riluzole, Xenon, HU-211 (also a cannabinoid), Lead and others.
4.3.1.3 The KA Glutamate Receptors
The KA iGluRs (Fig. 4.1) have a somewhat more limited distribution in the brain compared to the AMPA and NMDA iGluRs, and their function is still not well defined. The KA receptors play a role in both pre and postsynaptic neurons (Huettner 2003). The KA postsynaptic receptors are involved in excitatory neurotransmission, while the presynaptic KA receptors have been implicated in inhibitory neurotransmission by modulating release of the inhibitory neurotransmitter gamma-amino-butyric acid (GABA). Unlike AMPA iGluRs, KA iGluRs play only a minor role in signaling at synapses (Song and Huganir 2002). Rather, KA iGluRs may have a restrained role in synaptic plasticity, affecting the likelihood that the postsynaptic cell will fire in response to future stimulation (Mayer 2005a). Activating KA iGluRs in the presynaptic cell can affect the amount of neurotransmitters that are released (Mayer 2005a; Schmitz et al. 2001). This effect may occur quickly and last for a long time, and the effects of repetitive stimulation of KA iGluRs can be additive over time (Mayer 2005a; Schmitz et al. 2001).
The ion channel formed by KA iGluRs is permeable to Na+ and K+ ions, but impermeable to Ca2+. The KA iGluRs are composed of tetrameric assemblies of GluR5-7 and KA1/2 subunits. GluR5-7 subunits can form homomeric functional receptors, as well as combine with KA1 and KA2 to form heteromeric receptors with distinct pharmacological properties. KA1 and KA2 subunits do not form homomeric functional receptors (Keinanen et al. 1990; Lerma 2006).
4.3.2 The Metabotropic Glutamate Receptors
The mGluRs (Fig. 4.1) perform a variety of functions in the central and peripheral nervous systems. For example, they are involved in learning, memory, anxiety, and the perception of pain (Ohashi et al. 2002). The mGluRs are found in pre and postsynaptic neurons in synapses of the hippocampus, cerebellum and the cerebral cortex (Hinoi et al. 2001), as well as in other parts of the brain and in peripheral tissues (Chu and Hablitz 2000). Again, as discussed in this chapter, immune cells, primarily T cells, express mGluRs which are functional receptors in these cells, and upon their activation by glutamate or its mGluR agonists induce immune effects.
Like other types of metabotropic receptors, mGluRs have seven transmembrane domains that span the cell membrane (Platt 2007). Unlike iGluRs, mGluRs are not ion channels. They activate biochemical cascades, leading to the modification of other proteins. This can lead to changes in the synapse’s excitability, for example by presynaptic inhibition of neurotransmission (Sladeczek et al. 1993) or modulation and even induction of postsynaptic responses (Bonsi et al. 2005; Endoh 2004; Platt 2007). The mGluRs are also subdivided into three groups, termed group I, II and III (See Fig. 4.1), based on sequence similarity, pharmacology and intracellular signaling mechanisms. Group I mGluRs (mGluR1 and 5) are associated with Gq protein and coupled to phospholipase C (PLC), while group II (mGluR2 and 3) and III (mGluR4, 6, 7 and 8) are associated with Gi and G0 proteins and negatively coupled to adenylate cyclase. These eight mGluRs are products of different genes (Pin and Duvoisin 1995; Masu et al. 1991; Tanabe et al. 1992). mGluRs function as homodimers, with two glutamate molecules being required for full receptor activation (Kew and Kemp 2005).
4.4 Glutamate Receptors Expressed in Immune Cells
The vast majority of studies that investigated expression of GluRs in immune cells focused on lymphocytes of the T lineage, i.e. T cells. Hence, we will first describe GluR expression in T cells, and then describe the few studies that documented the expression of GluRs in other immune cells.
4.4.1 T Cells Express Ionotropic and Metabotropic Glutamate Receptors
4.4.1.1 Glutamate Binds to Normal Human T Cells with High Affinity
The first indirect evidence for the possible expression of GluRs in T cells was provided by a study testing the interaction of radiolabeled glutamate with naïve/resting normal human T cells (Kostanyan et al. 1997). [3H]glutamate was found to bind specifically to such T cells with a very high affinity (Kd = 2.36 × 10−7 M), and the binding was inhibited by glutamate-containing dipeptides. Moreover, binding of [3H]glutamate conjugated to dextran demonstrated that GluRs are expressed on the outer membrane of the T cells (Kostanyan et al. 1997). Yet, the exact identification of the specific GluR subtypes expressed in these T cells was not reported. Following this pioneering binding study, subsequent reports provided direct evidence for the expression of a plethora of iGluRs and mGluRs in human and rodent T cells (Table 4.1).
4.4.1.2 Naïve/Resting Normal Human T Cells Express on Their Cell Surface High Levels of AMPA Ionotropic Glutamate Receptors That Contain the GluR3 Subunit. Upon T Cell Receptor Activation, GluR3 Is Transiently Eliminated from the T Cell Surface by Granzyme B That Is Released by the Activated T Cells Themselves
The first demonstration of high expression and function of iGluR, of the AMPA subtype containing the GluR3 subunit, on the cell surface of normal human T cells (purified from blood of healthy individuals) was made in fact in our own studies (Ganor et al. 2003). By using several different methodologies, among them GluR3 specific RT-PCR, Western blotting, flow cytometry and immunofluorescent microscopy, we demonstrated for the first time that normal resting human T cells express high levels of iGluR AMPA GluR3 mRNA, as well as the GluR3 protein on their cell surface (Ganor et al. 2003). Furthermore, sequencing showed that the T cell-expressed GluR3 is identical with the brain GluR3. Interestingly, in a subsequent study we revealed that upon activation of the normal human peripheral T cells via their T cell receptor (TCR) with anti-CD3/CD28 antibodies (an in-vitro experimental approach that mimics T cell activation by an antigen presented to T cells by antigen presenting cells), GluR3 was eliminated from the surface of these T cells (Ganor et al. 2007). This process was mediated by the proteolytic enzyme granzyme B that was secreted by the activated T cells themselves, and that cleaved GluR3 from the T cell surface in an autocrine/paracrine manner (Ganor et al. 2007). Expression of GluR3 on the T cell surface was restored few days after TCR activation, when the cells reverted to their naïve/resting phenotype. This process of eliminating GluR3 transiently from the T cell surface by granzyme B may have an important regulatory role, and seems to operate also in neurons, as the neuronal GluR3 was also reported to serve as a substrate for granzyme B-mediated cleavage (Gahring et al. 2001). On top of all the above, we postulate that this proteolytic process may also have pathological consequences, since it may be relevant to the generation of a GluR3-derived autoantigenic peptide and later of GluR3 Abs found in ‘Autoimmune Epilepsy’ (Levite 2002). This topic is discussed in further details in Sect. 4.7 below.
Other studies further demonstrated the expression of iGluR of the NMDA family in human T cells. By using specific RT-PCR, sequencing and flow cytometry, normal resting human T cells were found to express the NR1 (Mashkina et al. 2007; Miglio et al. 2005b) and the NR2B subunits (Miglio et al. 2005b). Upon activation of the T cells with the mitogen phytohaemagglutinin (PHA), NR1 and NR2B levels increased and the NR2A and NR2D subunits were also expressed (Miglio et al. 2005b). These results show that the subunit composition of NMDA iGluRs in normal human T cells is altered following activation: while resting T cells express NR1 and NR2B-containing NMDA receptors, activated T cells express NR1 and NR2A/B/D-containing NMDA receptors. Interestingly, as discussed above, the AMPA GluRs are also altered following T cell activation, as shown by the disappearance of AMPA GluR3 from the cell surface of activated T cells (Ganor et al. 2007).
4.4.1.3 Normal Human T Cells Express Metabotropic Glutamate Receptors, and the Expression of Some mGluRs Is Modified by Activation of the Cells
The first mGluR subtypes identified in normal human T cells were mGluR5 and mGluR1 (Pacheco et al. 2004), which belong to the mGluRs group I. Several methodologies, including RT-PCR, flow cytometry and immunofluorescent microscopy, demonstrated the expression of these mGluRs at the RNA and protein levels. Interestingly, mGluR5 was expressed constitutively in naïve/resting human T cells, and in T cells activated by either PHA, the anti-CD3/TCR antibody OKT3, or antigen-pulsed dendritic cells (Pacheco et al. 2004, 2006). In contrast, mGluR1 was expressed only in activated, but not in naïve/resting normal human T cells (Pacheco et al. 2004, 2006). This is another example for the modification of GluRs composition after TCR activation, and hence for the differences in GluRs expression in resting vs. activated T cells. Another study confirmed at the RT-PCR level the constitutive expression of mGluR5 in both resting and activated human T cells, but showed the presence of mGluR1 transcripts also in resting T cells (Chiocchetti et al. 2006). Finally, RT-PCR was used to show that peripheral T cells from healthy individuals expressed transcripts for mGluR1, 2, 3 and 8, while T cells from patients with Amyotrophic Lateral Sclerosis (ALS) – a progressive and fatal neurodegenerative disease – showed reduced expression of mGluR2, but not the other mGluRs tested (Poulopoulou et al. 2005a). The possible relevance of this modified mGluR2 expression in T cells to the pathology of ALS is still unknown.
4.4.1.4 Mouse and Rat T Cells Express Ionotropic and Metabotropic Glutamate Receptors
Various GluR subtypes are expressed in T cells of non-human origin. In rat lymphocytes, RT-PCR was used to demonstrate the expression of the iGluR NMDA NR1 subunit, as well as group III mGluRs. Other GluRs were not expressed in these lymphocytes (Boldyrev et al. 2004). In rat thymus, a wider variety of mGluRs was expressed: immunohistochemical analysis and Western blotting revealed that T cells residing in the medulla express high levels of mGluR5 (group I) and moderate levels of mGluR2/3 (group II) and mGluR4 (group III), all of which showed decreased expression in T cells residing in the cortex (Rezzani et al. 2003). mGluRs were also shown to be differentially expressed in mouse thymocytes: RT-PCR, Western blotting and flow cytometry revealed the expression of group I mGluR5 in mature CD4+CD8+ and CD4+CD8−, but not in immature CD4−CD8− thymocytes; Group I mGluR1 showed an opposite pattern of expression; and group II mGluR2/3 were similarly expressed in all subsets (Storto et al. 2000). Finally, a recent study provided evidence that mouse thymocytes might also express iGluRs: PCR, confocal microscopy and flow cytometry were used to show the expression of NMDA iGluR subunits NR1, NR2A and NR2B. The NR1 subunit was expressed at higher levels on CD4+ or CD8+ single-positive thymocytes, compared to CD4+CD8+ double-positive cells (Affaticati et al. 2011).
Taken together, the above studies discussed in parts 4.4.1.1–4.4.1.4, show that normal human T cells, as well as mouse and rat T cells, definitely express iGluRs and mGluRs, and that T cell activation and/or maturation is an important mechanism able to either down- or up-regulate expression of specific iGluRs and mGluRs in normal T cells. As a result, resting and activated T cells express different GluRs, or different levels of these receptors.
4.4.1.5 Cancerous T Cells and Autoimmune T Cells, of Human and Mouse Origin, Express Ionotropic and Metabotropic Glutamate Receptors
Not only normal T cells express various types of GluRs. High levels of iGluRs and/or mGluRs are also expressed in cancer T cells: T-leukemia and T-lymphoma, as well as in autoimmune T cells (Table 4.1), such as encephalitogenic anti-myelin basic protein T cells that induce Experimental Autoimmune Encephalitis (EAE) - an animal model for Multiple Sclerosis (MS). A detailed description of GluRs expression in such autoimmune cells, and their relevance to cancer and MS, are discussed in Sect. 4.7 below.
4.4.2 Glutamate Receptors in Immune Cells Other Than T Cells: B Cells, Macrophages and Dendritic Cells
Few studies provided evidence for the expression of different GluRs in immune cells that do not belong to the T cell lineage. The key ones are cited below.
-
Study (1) A recent study demonstrated for the first time the expression of KA iGluRs in human B cells, and showed that human tonsilar B cells, CD19+ peripheral B cells, and the B cell line 8866 express transcripts and proteins for the GluR6, GluR7, KA1 and KA2 subunits (Sturgill et al. 2011). Interestingly, the authors further demonstrated that activation of such KA iGluRs by glutamate and KA increased IgE and IgG synthesis and cell proliferation (Sturgill et al.).
-
Study (2) Human monocytes-derived macrophages express both mGluR5 and mGluR1 (Chiocchetti et al. 2006), and rat alveolar macrophages (NR8383 cell line) express the NMDA subunits NR1 and NR2B (Dickman et al. 2004).
-
Study (3) In rat thymus, medullary dendritic cells (DCs) express high levels of mGluR5 and moderate levels of mGluR2/3 and 4, all of which are absent in cortical DCs (Rezzani et al. 2003).
Together, the studies cited here in part 4.4 show that GluRs are expressed in T cells and B cells, the two major types of lymphocytes that constitute the adoptive immune system, as well as in DCs and macrophages, two extremely important types of immune cells that belong to the innate immune system, but also bridge between the innate and the adoptive immune systems. Yet, while a great deal has been learned already about the effects of glutamate on T cells, the outcome of glutamate binding to other types of immune cells is to a large extent unknown. Accordingly, further studies are now needed to unveil the biological relevance of the interaction between glutamate and its receptors in such immune cells.
4.5 The Direct Effects of Glutamate on T Cell Function
4.5.1 Glutamate Binds Directly to Its Receptors in T Cells and Induces Potent Effects on T Cell Function. Glutamate’s Concentration and the Activation State of T Cells Are Major Factors in Dictating the Response to Glutamate. Thus, Resting and Activated T Cells Respond Differently to Glutamate
Collectively, several studies provide now clear evidence that glutamate on its own, directly and potently affects different T cell features and functions. This is in line with the direct effects of other neurotransmitters/neuropeptides on T cells (Levite 2008).
Glutamate-induced effects on T cells can be either activation or inhibition of a given T cell feature or function, depending on the context, and mainly on glutamate’s concentration and the activation state of the T cell. Thus, one can expect a different response to glutamate in the following three conditions:
Condition 1: The T cells being exposed to glutamate are in naïve/resting state, and not stimulated by any other stimuli;
Condition 2: Before encountering glutamate, the T cells have already been activated by other stimuli, such as antigen, mitogen, CD3 and CD28 antibodies, cytokines, growth factors or others;
Condition 3: The T cells are being simultaneously activated by glutamate and any other stimuli.
Concerning glutamate’s dose, this is indeed a very important factor which dictates the functional outcome of the interaction between glutamate and T cells, since glutamate induces different immune effects at low nanomolar (~10−9–10−8 M), mid micromolar (~105 M), or high millimolar (~10−3 M) concentrations (see Fig. 4.2). These different concentrations actually reflect the in-vivo levels of glutamate under normal and healthy physiological conditions on the one hand, and excess glutamate levels in numerous pathological conditions, on the other.

The direct effects of glutamate on human T cell function. (Left) Naïve/resting T cells express the GluR3 subunit of AMPA iGluRs. Whether AMPA receptors in resting T cells are homomeric tetramers containing only GluR3, or heteromeric tetramers containing also other AMPA iGluR subunits is still unknown. Low levels of glutamate at the nanomolar range act via GluR3 to increase T cell adhesion and chemotactic migration (Ganor et al. 2003). Such interactions may take place in the normal CNS and brain fluids, and may serve to assist T cells in their survey or exit of the CNS. Resting T cells express also the NMDA iGluR subunits NR1 and NR2B, as well as mGluR2, 3, 5 and 8, but their role in mediating resting T cell function is still elusive. (Right) Activated T cells lose GluR3 expression, due to potent enzymatic cleavage mediated by granzyme B, which is released by activated T cells and cleaves GluR3 of the cell surface (Ganor et al. 2007). GluR3 appears again once the cells return to the resting phase (Ganor et al. 2003, 2007). In parallel to losing GluR3, the activated T cells are no longer responsive to glutamate’s pro-adhesive and pro-migratory effect, evident at low nanomolar glutamate levels, and when the T cells are naïve/resting. Yet, such cells may express other AMPA iGluR subunits (that still needs to be formally demonstrated), as well as NMDA iGluRs composed of NR1 and NR2A/B/D and mGluR1 and 5. Mid levels of glutamate at the micromolar range act via both iGluRs and mGluRs to promote cell proliferation, by increasing iCa2+ via iGluRs and decreasing apoptosis via mGluRs. Such interactions may take place in blood under normal physiological conditions, and may be part of the normal process of T cell proliferation following antigen binding. In addition, excess levels of glutamate at the millimolar range, present in blood and/or CNS in a variety of pathological conditions, may activate mGluR5 to decrease proliferation and mGluR1 to increase cytokine secretion. Such interactions may control the expansion of the activated cells and help combating the disease
In the healthy cerebrospinal fluid (CSF) and brain extracellular fluid, glutamate is present at ≤10−6 M (Meldrum 2000). In the plasma of healthy individuals glutamate is present at a concentration of 10−5–10−4 M (Divino Filho et al. 1998; Graham et al. 2000; Meldrum 2000; Reynolds et al. 2002). Yet, glutamate levels are highest within the synaptic cleft and may reach a concentration as high as 10−3 M (Meldrum 2000). While these are the values for a normal healthy body, glutamate’s concentrations increase substantially, both in the plasma and in the brain, in numerous pathological conditions. Thus, glutamate’s plasma levels may increase substantially well above 10−4 M, either in a kaleidoscope of pathologies conditions, such as immune deficiency (Droge et al. 1993; Eck et al. 1989b; Ferrarese et al. 2001) and cancer (Droge et al. 1988; Eck et al. 1989a; Ollenschlager et al. 1989) (see Sect. 4.7 below), and glutamate’s brain levels increase in a variety of disorders that display a neuroinflammatory component, such as MS (see Sect. 4.7 below), traumatic brain injury, acute brain anoxia/ischemia, epilepsy, glaucoma, meningitis, ALS, and Alzheimer’s, Huntington’s disease and Parkinson’s disease (see Meldrum 2000; Sattler and Tymianski 2001), and (Pacheco et al. 2007) for specific numerical examples of elevation in glutamate levels). Importantly, excess glutamate is highly detrimental to neuronal cells, as it mediates over-activation of GluRs leading to neuronal death by a mechanism termed excitotoxicity (Sattler and Tymianski 2001).
As already mentioned above, another factor that crucially affects the responsiveness of T cell to glutamate is their activation state, i.e. whether they are naïve/resting or rather already activated, primarily since different GluRs are expressed in naïve/resting T cells and in activated T cells (see Fig. 4.2).
In the paragraphs that follow, we summarize the reported glutamate-mediated effects on various T cell functions. Each functional outcome is described while taking into account the concentrations of glutamate inducing the effect, the activation state of the cells, and the GluRs involved (Table 4.2). Finally, we suggest a model that incorporates all these findings into a unifying presentation of the effects of glutamate on T cells in health and disease (Fig. 4.2).
4.5.2 The Effects of Glutamate on Proliferation, Intracellular Ca2+ (iCa2+) and Apoptosis
4.5.2.1 Glutamate at a Very High Millimolar Concentration Range (10−3–10−2 M) Suppresses the Proliferation of Activated T Cells, Most Probably via Metabotropic Glutamate Receptors
An early observation suggested that elevated plasma glutamate levels correlate with a reduction in the mitogenic response of blood lymphocytes against pokeweed mitogen (Droge et al. 1988). Such an inverse correlation was reported for plasma glutamate ranging from 5 × 10−5 to 2 × 10−4 M (a range we refer to as mid-high), corresponding to the levels of glutamate measured in the plasma of either healthy individuals or patients with carcinoma, respectively (Droge et al. 1988). Later studies confirmed this initial observation, and provided direct evidence that glutamate hinders T cell proliferation. Glutamate at a high millimolar concentration range (10−3–10−2 M) inhibited the proliferation of T cells induced by PHA or anti-CD3 ± CD28 antibodies. Importantly, even at these high concentrations, glutamate did not affect the proliferation of normal naïve/resting human T cells (Lombardi et al. 2001, 2004; Pacheco et al. 2004), showing the marked differences between glutamate-induced effects on resting vs. activated T cells. The iGluRs agonists (S)-AMPA and KA, tested at the same concentration range, did not affect the proliferation of PHA-activated T-cells (Lombardi et al. 2004). Interestingly, while the iGluR agonist NMDA was similarly ineffective as AMPA and KA (Lombardi et al. 2004), the NMDA GluR antagonists D-AP5 and (+)-MK801 both inhibited PHA-induced (but not IL-2-induced) T cell proliferation (Miglio et al. 2005b). Together, these results suggest that AMPA and KA iGluRs do not mediate the anti-proliferative effect of glutamate on T cells. The exact contribution of the NMDA iGluRs to this effect requires further studies, which should better test the effects of several types of NMDA agonists on their own, as well as the effect of glutamate at mid-high and low concentrations, in the absence or presence of NMDA antagonists, on the proliferation (and hopefully also other responses) of resting vs. activated T cells.
In fact, the inhibitory effect of glutamate on the proliferation of activated T cells might be mediated by mGluRs, as the selective mGluR5 agonist CHPG (5 × 10−4 M) inhibited proliferation of anti-CD3-activated T cells (Pacheco et al. 2004). As such inhibitory effect was abrogated by the non-selective mGluR1/5 agonist (S)-3,5-DHPG (10−4 M), and coupled to the demonstration that both mGluR5 and mGluR1 are expressed in activated T cells (Table 4.1), these results suggest that glutamate might play a dual role in T cell function: on the one hand inhibiting T cell proliferation via mGluR5, and on the other hand reverting this effect via mGluR1 (Pacheco et al. 2004). Similar observations were indeed reported later, showing that glutamate released by antigen-pulsed DCs (see Sect. 4.6 below) acted initially via mGluR5 to impair T cell proliferation, but later stimulation of mGluR1 abrogated the anti-proliferative effect mediated by mGluR5 to allow robust T cell proliferation (Pacheco et al. 2006).
4.5.2.2 Glutamate at Low-Mid Nanomolar to Micromolar Concentrations Increases iCa2+ in Activated T Cells via Ionotropic Receptors. The Effect Is Not Seen in Resting T Cells or at Higher Glutamate Concentrations
Ca2+ signaling in response to antigenic stimulation is essential for proliferation of T cells (Guse 1998). Several studies reported that glutamate can clearly affect Ca2+ signaling in T cells, and that the effect is once again dependent first on glutamate’s concentration, and second on the activation state of the T cells. Thus, at low-mid nanomolar to micromolar concentrations, glutamate increased iCa2+ in activated (but not in resting) T cells (Lombardi et al. 2001). This iCa2+ potentiating effect induced by glutamate showed a bell-shape concentration-dependent relationship and was effective at 10−7–10−5 M with a maximal effect observed at 10−6 M. In contrast, glutamate at higher concentrations of 10−4–10−3 M was ineffective (Lombardi et al. 2001). The reported iCa2+ increase by glutamate in activated T cells was mediated by iGluRs, as the effect was mimicked by the iGluRs agonists NMDA, (S)-AMPA and KA at a similar concentration range, and several iGluRs antagonist and blockers (i.e. D-AP5, (+)-MK801, NBQX and KYNA) inhibited glutamate- and iGluR agonists-mediated iCa2+ rise (Lombardi et al. 2001). Interestingly, the prototype mGluR agonist (1S,3R)-ACPD was ineffective at the above concentration range (Lombardi et al. 2001), as well as the non-selective mGluR1/5 agonist (S)-3,5-DHPG, tested at concentrations up to 10−4 M (Pacheco et al. 2004).
Taken together with the observations discussed above in part 4.5.2.1, it seems that glutamate induces two different effects on activated T cells, at different concentration ranges and via different GluRs: (1) glutamate at low-mid nanomolar to micromolar concentrations increases iCa2+ in activated T cells via iGluRs; (2) glutamate at a very high millimolar concentration range inhibits the proliferation of activated T cells via mGluR5.
Interestingly, neither of these effects is induced by glutamate in naïve/resting human T cells.
4.5.2.3 Glutamate Affects Kv1.3 Voltage-Gated Potassium Currents in Resting T Cells, and Exerts Opposite Effects at Different Concentrations: Micromolar Glutamate Potentiates, While Higher, up to Millimolar Glutamate, Suppresses T Cell Kv1.3 Currents
How does glutamate increase Ca2+ signaling (the effect described above in part 4.5.2.2)? Ca2+ influx requires a negative membrane potential as its driving force, which in T cells is provided by voltage-gated K+ (Kv) channels (Lin et al. 1993), and especially the Kv1.3 channel (Cahalan et al. 2001). Interestingly, micromolar concentrations of glutamate (10−6–10−5 M) potentiated Kv1.3 currents in normal resting human T cells, while higher concentrations (up to 10−3 M), suppressed such currents (Poulopoulou et al. 2005b). These observations provide a plausible mechanism for the distinct effects of glutamate on Ca2+ signaling and proliferation. Unfortunately, the ability of glutamate to modulate Kv1.3 currents was tested only in resting human T cells, and therefore additional studies are necessary to test the effects of glutamate at different concentrations on voltage-gated K+ currents in activated T cells, and to find out if these correlate with the glutamate-induced effects on iCa2+ on the one hand, and on proliferation on the other, in such activated T cells.
4.5.2.4 Glutamate Protects T Cells from Activation-Induced Cell Death (AICD) via Metabotropic Glutamate Receptors, and Also Inhibits FasL Expression Involved in AICD
It is very well known that antigen stimulation and TCR triggering of T cells induce robust proliferation, cytokine secretion, and the upregulation of many other T cell features and functions. In contrast, chronic TCR stimulation, or re-stimulation of T cells within a short period after the first antigenic stimulation, induces apoptosis of the cells via a mechanism termed Activation-Induced Cell Death (AICD), which is crucial for maintenance of peripheral tolerance and for limiting an ongoing immune response (Green et al. 2003). As glutamate at high concentrations clearly suppresses the proliferation of activated T cells (discussed above in part 4.5.2.1), one may speculate that glutamate may also induce T cell apoptosis and promote AICD. Yet, the opposite was found to be true: glutamate at a broad concentration range of 10−8–10−4 M suppressed AICD (Chiocchetti et al. 2006). Thus, glutamate in fact seems to contribute to improved/prolonged T cell survival by protecting the cells from apoptosis. Glutamate exerted its maximal effect at 10−6 M, and this effect was mimicked by the prototype mGluR agonist (1S,3R)-ACPD, as well as by the non-selective group I mGluR agonists quisqualate and (S)-3,5-DHPG and the selective mGluR5 agonist CHPG (Chiocchetti et al. 2006). In line with that, several group I mGluRs antagonists and blockers (i.e. AIDA, LY367385 and MPEP) antagonized glutamate- and mGluR agonists-mediated inhibition of AICD in activated T cells (Chiocchetti et al. 2006). The protection of activated T cells from AICD by glutamate was further shown to result from inhibition of FasL expression (Chiocchetti et al. 2006), which is known to be involved in AICD (Green et al. 2003).
Together, the above-discussed data show that:
-
1.
At low-mid concentration, glutamate induces the following effects in activated T cells via different GluRs expressed in these cells: (a) Increase in iCa2+ via iGluRs; (b) Improved/prolonged survival in conditions of AICD via mGluRs.
-
2.
In contrast, high concentrations of glutamate have an anti-proliferative effect on mitogen- or CD3/CD28 Ab-activated T cells, probably via mGluRs.
Such unique ability of glutamate to induce somewhat opposing effects on activated T cells at different dose ranges may be important for either limiting or maintaining ongoing immune responses, respectively. Yet, additional studies are needed for exploring whether glutamate indeed exerts similar effects in-vivo in physiological and/or pathological conditions.
4.5.3 Glutamate at Low Nanomolar Concentrations Induces Adhesion to Fibronectin and Laminin, as well as Chemotactic Migration to CXCL12/SDF-1 of Naïve/Resting Human T Cells, via AMPA Ionotropic Glutamate Receptors
Following our discovery that normal resting human T cells express high levels of iGluR AMPA GluR3 on their cell surface (Ganor et al. 2003), we investigated whether glutamate, operating via such AMPA receptors, can on its own trigger T cell function in naïve/resting T cells. We found that glutamate at low-mid concentrations of 10−9–10−6 M induces adhesion of normal resting human T cells to two major glycoproteins of the extracellular matrix (ECM): fibronectin and laminin (Ganor et al. 2003). T-cell adhesion to ECM is a very important and required T cell function, needed for migration, extravasation and homing of T cells into tissues under physiological and pathological conditions. This pro-adhesive effect of glutamate showed a bell-shape concentration-dependent relationship, effective at a very broad low-mid concentration range of 10−12–10−6 M, with a maximal effect observed at low 10−9 M. Glutamate-induced T cell adhesion to fibronectin and laminin was mediated via AMPA iGluRs, since it was mimicked by the AMPA iGluRs agonists AMPA and KA, and blocked by several AMPA iGluRs antagonist such as CNQX and NBQX. Glutamate induced the adhesion of normal human T cells to the ECM glycoproteins via activating specific adhesion receptors expressed on the T cell surface, namely the α5β1 and α6β1 integrins. We proved this by demonstrating that anti-VLA-5 monoclonal antibody, which blocks the α5 integrin chain that binds fibronectin, blocked glutamate-induced T cells adhesion to fibronectin, and likewise the anti-VLA-6 monoclonal antibody, which blocks the α6 integrin chain that binds laminin, blocked glutamate-induced adhesion to laminin (Ganor et al. 2003). Interestingly, upon TCR activation and granzyme B cleavage of GluR3 from the cell surface, human T cells ‘lost’ their glutamate-induced adhesion to laminin (Ganor et al. 2007), suggesting a possible mechanism by which activated T cells shut off the pro-adhesive effect of glutamate via shedding their surface GluR3.
Glutamate on its own (10−8 M again) induced an additional very important function: T cell chemotactic migration. Thus, glutamate increased the CXCR4-mediated chemotactic migration of naïve/resting normal human T cells towards the key chemokine CXCL12/SDF-1 (Ganor et al. 2003), which is a key player in cell migration in health and disease. Glutamate-induced T cell chemotaxis was mediated by activation of CXCR4, the specific receptor for CXCL12/SDF-1, expressed on the cell surface of the T cells since anti-CXCR4 monoclonal antibody blocked the effect. These results demonstrate that glutamate, at very low nanomolar concentrations and via acting on AMPA iGluRs highly expressed in normal naïve/resting human T cells, has both a pro-adhesive and a pro-migratory effect on such cells.
How does glutamate affect T-cell integrin function? The answer is still unknown but we speculate that this process may be linked to the depolarization and opening of voltage-gated Kv1.3 channels expressed in naïve/resting T cells, since we previously reported that β1 integrins are physically associated with such channels, and that T cell adhesion can be induced by depolarization (Levite et al. 2000). Hence, a possible scenario might be that glutamate at low concentrations induces depolarization of resting T cells, thereby opening Kv1.3 channels and inducing outward K+ currents in such cells (Poulopoulou et al. 2005b), and this in turn leads also to integrin activation and to the subsequent T cell adhesion to ECM glycoproteins.
4.5.4 Glutamate Affects the Secretion of Several Cytokines by T Cells
All immune responses absolutely require, and are completely dependent on, autocrine and paracrine cytokine signaling, i.e. the secretion of specific cytokines at the right time and place by immune cells, and the binding of these cytokines to their cognate receptors expressed by other immune cells, as well as on those that secreted them. T cells secrete and respond to a variety of cytokines, and different T cell subpopulations secrete and respond preferentially to different cytokines. CD4+ T helper (Th) cells are crucial for orchestrating the adoptive immune response by recruiting and activating other cells of the immune system, and these activities are also dependent on specific cytokines.
Traditionally, Th cells were classified as either Th1 or Th2 cells based on cytokine secretion, signaling pathways and lineage-specific transcription factors. Th1 cells mainly secrete IFNγ and IL-2 and promote immunity against intracellular pathogens, while Th2 cells secrete IL-4, IL-5, IL-10 and IL-13 and promote humoral responses and the defense against extracellular parasites. More recently, several additional T cell subsets have been identified, among them Th9 and Th17 cells (Nakayama and Yamashita 2010; Zhou et al. 2009). As T cell effector function critically depends on cytokine secretion, any stimuli able to stimulate, modulate or suppress the cytokines secreted by T cells may have important outcomes on the type, efficiency and control of the T cell-mediated immune reactions in response to viruses, bacteria, cancer or any other stimuli.
Can glutamate affect cytokine secretion by T cells? Several studies have provided evidence that glutamate can indeed do so. At high concentrations of 10−3 M glutamate increased IFNγ and IL-10 secretion by anti-CD3 activated T cells, but at five times higher concentration glutamate decreased IFNγ, IL-10 and IL-5 secretion by these T cells. Once again, glutamate had different effects on naïve/resting vs. activated T cells, since glutamate had no effect on cytokine secretion by naïve/resting cells (Lombardi et al. 2004). Glutamate-induced suppression of IFNγ secretion by activated T cells may involve iGluRs, as the effect on IL-2-stimulated T cells was mimicked by NMDA at 5 × 10−4 M (Mashkina et al. 2007).
In contrast to these glutamate-induced effects exerted at very high concentrations of 1 − 5 × 10−3 M, glutamate at ~1,000-fold lower concentration of 10−6 M may operate via mGluRs to modulate IL-6 production and enhance the secretion of TNFα, IFNγ, IL-2 and IL-10 (Pacheco et al. 2006). Glutamate released by antigen-pulsed DCs (see part 4.6.3 below) acted on mGluR5 expressed in T cells, in the context of a DC-T cell co-culture (i.e. the immunological synapse), to impair early IL-6 production. At later time points, when antigen-pulsed DCs induced T cell activation and expression of mGluR1, glutamate operated via mGluR1 to counteract the suppressive effect on IL-6 production and also enhanced the secretion of TNFα, IFNγ, IL-2 and IL-10 (Pacheco et al. 2006). Of note, although the levels of glutamate secreted by DCs to the co-culture media were estimated at the micromolar range, the actual concentration of glutamate within the immunological synapse might in fact be higher. Taken together, the studies cited above show clearly that glutamate has the ability to increase or suppress the secretion of several key T cells cytokines. Yet, further studies are needed to elucidate the exact effects exerted by glutamate at different concentrations on the secretion of specific cytokines by various T cell subpopulations, among them Th1, Th2, Teffs, Tregs, Th17, CD4+, CD8+ and others. Exploring these effects can be very rewarding scientifically and even clinically.
4.5.5 Proposed Summary for the Effects of Glutamate on T Cell Function
Based on the multitude of studies discussed above, we propose the following model to describe the functional dialogues between glutamate, its receptors – the different types of GluRs – and human T cells (Fig. 4.2), taking place in three different conditions:
-
Condition 1: Activated T cells encountering physiological mid micromolar levels of glutamate
Activated T cells express on their cell surface: (1) NMDA iGluR subunits NR1 and NR2A/B/D; (2) AMPA iGluRs that do not contain the GluR3 subunit; (3) mGluR5 and mGluR1. The expression of these GluRs allows the activated T cells to respond to the physiological levels of glutamate at a mid concentration range (10−6–10−4 M) present in blood. Such interactions lead to increased iCa2+ (via iGluRs) and decreased AICD (via mGluRs) of the activated T cells.
All these glutamate-induced effects may contribute to an improved T cell survival and function following the encounter of T cells with various antigens derived either from invading microbial and/or viral threats, or from tumor antigens, when the cancer is attacked by T cells. One may envision that glutamate-induced protection of activated T cells from AICD may be especially important in conditions of chronic/repeated T cell exposure to such antigens.
-
Condition 2: Activated T cells encountering pathological excess millimolar levels of glutamate
Activated T cells express mGluRs that allow the cells to respond to elevated levels of glutamate (10−3 M concentration range), present in the plasma and/or in the CNS in various pathologies. Activated T cells are expected to encounter such excess glutamate in these two very different body locations, i.e. blood and CNS, since they are constantly migrating in the circulation, and routinely cross the blood–brain barrier (BBB) and enter the CNS for immunosurveillance and neuroprotection in physiological and pathological conditions. The interaction of mGluRs expressed by activated T cells with excess glutamate present in many pathological conditions may have functional outcomes, leading to decreased T cell proliferation and increased cytokine production. These could be important for preventing or controlling the expansion of the activated cells on the one hand, and for improved combat of diseases on the other.
-
Condition 3: Naïve/resting T cells encountering physiological low levels of glutamate
At normal conditions, most of the T cells in the body are in a naïve/resting a state. Furthermore, few days after TCR activation, T cells revert to a naïve/resting phenotype. We found that in parallel to that reversion, T cells regain their expression of the AMPA GluR3 that was cleaved of their cell surface by granzyme B after their TCR activation. Yet, the cells lose now their expression of mGluR1. So some GluRs are regained, while others are lost upon entering the resting phase. The exposure of naïve/resting T cells to the low glutamate levels present in the healthy brain fluids triggers their adhesion to fibronectin and laminin, and induces their chemotactic migration towards key chemokines present in the CNS. This could assist T cells in their patrol and survey of the CNS, and maybe also in their exit back to the periphery. As the levels of glutamate in blood are higher under normal physiological conditions (at the micromolar range), the interactions between such high glutamate and resting T cells in the periphery may not necessarily be productive.
4.6 Production of Glutamate by Immune Cells
Neuronal cells produce glutamate, and this is most probably the major source for glutamate under physiological conditions. Upon release, glutamate exerts various effects on neuronal cells, as well as on other target cells among them immune cells, as discussed above. In addition, published evidences point now to the ability of various immune cells to release glutamate from their own sources. The main evidences supporting this notion are summarized below.
4.6.1 Neutrophils Secrete Glutamate, Which in Turn Decreases Endothelial Cell Permeability
Neutrophils are the most common type of white blood cells, comprising about 50–70% of all white blood cells. Neutrophils are phagocytic cells that can ingest other cells, though they do not survive the act. Neutrophils are the first immune cells to arrive at a site of infection through chemotaxis. Activated neutrophils were shown to secret glutamate, which subsequently acted on mGluR1, 4 and 5 expressed by human brain and dermal microvascular endothelial cells, resulting in decreased cell permeability (Collard et al. 2002). These findings suggest that neutrophil-derived glutamate may be an important factor that alters endothelial cell permeability during injury and/or inflammation. Whether this process contributes, for instance, to the in vivo transmigration and entry of activated T cells across blood vessels into the brain, is an important yet still open question that surely deserves more investigation.
4.6.2 Monocytes/Macrophages and Activated Microglia Cells Secrete Glutamate, Which in Turn Leads to Neuronal Death
Early observations suggested that activated microglia (featuring a phenotype similar to brain macrophages) could secrete glutamate, which acts on NMDA iGluRs to induce neuronal cell death (Piani et al. 1991, 1992). These results were later confirmed by many studies, demonstrating that the activation of both monocytes/macrophages and microglia may lead to the secretion of NMDA iGluR-activating substances, including glutamate itself, which in turn are able to cause excitotoxic neuronal death. Importantly, this process, by which macrophages/microglia-derived glutamate leads to neuronal death, is accepted as an important process contributing to neuronal damage in human immunodeficiency virus type 1 infection (for review see Kaul et al. 2005).
4.6.3 Immature Dendritic Cells Release Low Levels of Glutamate, While Activated Mature Dendritic Cells Release Much Higher Levels of Glutamate
A recent study reported that immature DCs were able to release low levels of glutamate. Interestingly, DC maturation (induced by either lipopolysaccharide [LPS], TNFα, or the superantigen staphylococcal enterotoxin A [SEA]) markedly enhanced the capacity of DCs to release glutamate (Pacheco et al. 2006). Moreover, glutamate was also released by antigen-pulsed DCs that were co-cultured with T cells, suggesting that glutamate release from DCs is a general feature of antigen presentation across the immunological synapse formed between DCs and T cells. More recently, these results were confirmed by showing that mouse DCs also secret glutamate, which in turn acted on mouse thymocytes in the context of the immunological synapse (Affaticati et al. 2011).
4.7 Involvement of Glutamate and Its Receptors in Cancer, Autoimmune Diseases and Human Immunodeficiency Virus Type 1 Infection
4.7.1 Cancer
4.7.1.1 Glutamate Antagonists Block the Growth of Solid (Non Immune) Tumors: Glioma, Breast and Lung Carcinoma, Colon Adenocarcinoma and Neuroblastoma
Clear evidence has accumulated to support a role of glutamate and its receptors, both iGluRs and mGluRs, in promoting tumor growth, while glutamate antagonists block it. Few examples include the following: (1) Blocking Ca2+-permeable AMPA iGluRs in human glioblastoma cells inhibited cell locomotion and induced apoptosis (Ishiuchi et al. 2002); (2) Treatment of gliomas with the NMDA iGluR antagonists MK801 or memantine slowed tumor growth (Takano et al. 2001); (3) The NMDA iGluR antagonist dizocilpine exerted an anti-proliferative effect preferably on colon adenocarcinoma, astrocytoma, and breast and lung carcinoma cells. In addition, breast and lung carcinoma, colon adenocarcinoma and neuroblastoma cells responded most favorably to the AMPA iGluR antagonist GYKI52466. Such anti-proliferative effects of iGluR antagonists were Ca2+-dependent and resulted from decreased cell division and increased cell death (Rzeski et al. 2001; Stepulak et al. 2005); (4) Blocking group II mGluRs reduced the growth of glioma cells in-vivo (Arcella et al. 2005), and mGluR3 and mGluR4 controlled the proliferation of brain tumor cells (Nicoletti et al. 2007). Importantly, many studies have also shown that GluRs are expressed in a variety of neuronal and non-neuronal cancer cell lines and tumors (Kalariti et al. 2005; Stepulak et al. 2009).
4.7.1.2 T-leukemia and T-lymphoma Express Ionotropic and Metabotropic Glutamate Receptors, and Glutamate, via AMPA Receptors, Promotes These Cancerous T Cells by Augmenting Their in vivo Extravasation and Their Expression of Matrix Metalloproteinase 9 (MMP-9) and CD147. Glutamate, via Metabotropic Receptors, Also Augments iCa2+ and the Expression of the Early Ca2+-Inducible Genes c-fos and c-jun in T-leukemia
Various cancerous immune cells express GluRs, as shown in Table 4.1. Moreover, we showed in our own studies that human T-leukemia (Jurkat) and T-lymphoma (HuT-78) cell lines express on their cell surface high levels of the specific AMPA iGluR GluR3 subunit (Ganor et al. 2003, 2009), the very same iGluR subunit we found in high levels in resting normal human T cells (Ganor et al. 2003, 2009).
Human T-leukemia cells (Jurkat) express also the AMPA iGluR GluR2 and GluR4 subunits (Stepulak et al. 2009); the KA iGluR GluR6, GluR7, KA1 and KA2 subunits (Stepulak et al. 2009); and the NMDA iGluR NR1, NR2A-D and NR3A,B subunits (Miglio et al. 2007, 2005b; Stepulak et al. 2009). Group I mGluRs are also expressed in several human T-cancerous cell lines: mGluR5 in T-leukemia (Jurkat, FRO) and T-lymphoma (H9, HuT-78), and mGluR1 in T-leukemia (Jurkat, FRO, SUP-T1) and T-lymphoma (HuT-78) cell lines (Pacheco et al. 2004; Chiocchetti et al. 2006). Another study confirmed the expression of group I mGluRs in T-leukemia (Jurkat) cells, and further demonstrated that these cells also express group I and II mGluRs (Stepulak et al. 2009). In addition, mGluRs are expressed by other types of cancerous immune cells, which are not of T cell origin: both group I mGluR5 and mGluR1 were shown to be expressed in human monocytic leukemia cells (THP-1) (Chiocchetti et al. 2006), and mGluR5, but not mGluR1, was reported in B lymphoblast SKW6.4 cells (Pacheco et al. 2004).
Coupled with the early studies reporting on elevated plasma glutamate levels in patients with malignancies (Droge et al. 1988; Eck et al. 1989a; Ollenschlager et al. 1989), the above observations suggest an important role for glutamate/GluRs in tumor biology, including T cell cancers. Indeed, several studies have clearly demonstrated that glutamate affects directly the behavior of T-leukemia and T-lymphoma cells (illustrated schematically in Fig. 4.3).

Glutamate promotes T leukemia and T lymphoma via iGluRs and mGluRs expressed in these cells. Glutamate, in nanomolar range, such as that found primarily in the CSF and brain extracellular fluids (Meldrum 2000), activates its AMPA iGluRs expressed in T leukemia and T lymphoma and induces the following: (1) Elevated extravasation in-vivo into solid organs; (2) Elevated expression of CD147, a cancer-associated MMP inducer and MMP9, also associated with cancer metastasis (Ganor et al. 2009). Also, glutamate, via mGluRs expressed in T leukemia and T lymphoma induces the following: (1) Elevated intracellular Ca2+; (2) Elevated expression of the early Ca2+ inducible genes c-fos and c-jun (Miglio et al. 2005a)
Investigating the effect of glutamate on iCa2+ rise in human T-leukemia Jurkat cells, Miglio et al. showed that group I mGluRs, i.e. mGluR5 and mGluR1 are expressed in Jurkat cells (see above), and evoke iCa2+ increase. Thus, at a broad concentration range of 10−7–10−3 M, the prototype mGluR agonist (1S,3R)-ACPD, the non-selective mGluR1/5 agonist (S)-3,5-DHPG, and the selective mGluR5 agonist CHPG produced such effect (Miglio et al. 2005a). Moreover, several selective group I mGluR antagonists (i.e. AIDA, LY367385, MPEP) antagonized the effect of DHPG (Miglio et al. 2005a). The reported rise in iCa2+ resulted also in up-regulation of the early Ca2+-inducible genes c-fos and c-jun (Miglio et al. 2005a). As the protein products of these genes play important roles in the regulation of the cell cycle (Tay et al. 1996), these findings suggest that glutamate might modulate Jurkat cell function by activating multiple downstream signaling events that regulate cell proliferation and cytokine mRNA transcription.
Miglio et al. further showed that the NMDA iGluR antagonists (+)-MK801 and D-AP5 at concentrations of 1 − 5 × 10−4 M, inhibited the growth of Jurkat T-leukemia cells by promoting their apoptosis (Miglio et al. 2007). Interestingly, direct treatment with either glutamate or NMDA induced adhesion of the T-leukemia cells to fibronectin. This pro-adhesive effect showed a bell-shape concentration-dependent relationship, was effective at 10−8–10−5 M with a maximal effect observed at 10−6 M glutamate and 10−5 M NMDA, and was blocked by the NMDA iGluRs antagonists (+)-MK801 and D-AP5 (Miglio et al. 2007). As we previously reported that glutamate induces such pro-adhesive effect also in normal resting non-cancer human T cells (Ganor et al. 2003), these observations suggest that activation by glutamate of iGluRs in T cells, and possibly other integrin-expressing cells, is a common mechanism by which glutamate controls T cell adhesion.
Finally, our own studies revealed several other cancer-promoting functional outcomes of the interaction between glutamate and its AMPA iGluRs expressed in human T cell cancers. Thus, ex vivo treatment of Jurkat human T-leukemia cells with glutamate at low concentration of 10−8 M enhanced their subsequent in-vivo engraftment into chick embryo liver and chorioallantoic membrane (Ganor et al. 2009). In correlation with this pro-metastatic in-vivo effect, glutamate also induced in-vitro a significant elevation of the cancer-associated matrix metalloproteinase (MMP) inducer CD147, as well as increased the secretion of the cancer-associated MMP-9 in the human T-cancerous cells (Ganor et al. 2009). These multiple and potent glutamate-induced effects were mediated by iGluR AMPA receptors expressed in these cancerous T cells, since the effect was mimicked by AMPA, and blocked by the AMPA antagonist CNQX (Ganor et al. 2009). These findings suggest that glutamate may facilitate the spread of T-leukemia and T-lymphoma in-vivo and their penetration from the circulation into solid organs.
Importantly, the reported pro-adhesive and pro-metastaic effects of glutamate on T cell cancers seem to be operational at low concentrations of 10−9 M, such as those found primarily in the CSF and brain extracellular fluids (Meldrum 2000). This argues that increased engraftment of T cell cancers would reach its optimum within the CNS, after such immune cancer cells cross the BBB, enter the nervous system, and encounter glutamate that will operate via iGluRs. Importantly, in the periphery, the interaction of T cell cancers with glutamate would rather increase their proliferation, in line with all the above-mentioned findings demonstrating that GluR antagonists limit tumor growth. Such increased glutamate-mediated cancer cell proliferation would probably be operational at higher glutamate levels, since a multitude of early studies clearly indicated that glutamate levels might increase in the plasma of patients with malignancies (Droge et al. 1988; Eck et al. 1989a; Ollenschlager et al. 1989).
4.7.2 Multiple Sclerosis
Multiple Sclerosis (MS) and its animal model Experimental Autoimmune Encephalomyelitis (EAE) are demyelinating diseases caused primarily by autoreactive T cells, which enter the CNS and attack the nerve enwrapping myelin sheath (Hemmer et al. 2002; Merrill and Benveniste 1996). In addition, myelin-producing cells of the CNS (i.e. oligodendrocytes) and some axons are lost as a result of the inflammatory attack on the CNS.
4.7.2.1 AMPA GluR3 Is Highly Expressed in Mouse EAE-Inducing Anti-myelin Basic Protein T Cell Mouse Clones; Treatment of EAE-Afflicted Mice and Rats with Antagonists of AMPA Ionotropic Glutamate Receptors Results in Substantial Amelioration of the Disease
Interestingly, two previous studies reported that treatment of mice (Pitt et al. 2000) or rats (Smith et al. 2000) sensitized for EAE, with NBQX, the AMPA iGluR antagonist, resulted in substantial amelioration of disease. The authors concluded that NBQX was beneficial for EAE since it blocked AMPA iGluRs expressed in neuronal or glia cells.
In addition to this interpretation and conclusion, we proposed that NBQX suppressed EAE in these studies because on top of inhibiting GluRs in neurons and glia, it blocked AMPA iGluRs expressed in the autoagressive encephalitogenic T cells that cause the disease. Hence, we believe that by blocking the T cell expressed AMPA iGluRs, NBQX could have prevented the activation of the autoagressive T-cells by glutamate released from nerve endings at the sites of inflammation/damage in the CNS, thereby reducing their pathogenic potential and conferring EAE suppression (Ganor et al. 2003). Our proposal is based on: (1) our demonstration that iGluR AMPA GluR3 is highly expressed not only in normal human T cells, but also in mouse encephalitogenic EAE-inducing anti-myelin basic protein T cell clones (Ganor et al. 2003); (2) the evidences discussed above in parts 4.5.2.2, 4.5.2.4, and 4.5.4, showing the multiple beneficial effects of glutamate at low-mid concentration on activated T cells. For reminder, glutamate-induced effects on activated T cells, mediated by various types of GluRs, include increase in iCa2+ currents and the improvement/prolongation of T cell survival in conditions of AICD by protection from apoptosis and inhibition of FasL expression. In view of all these, one can envision that a glutamate antagonists able to prevent the beneficial effects of glutamate on activated T cells would also be able to inhibit activated autoimmune cells in EAE/MS.
4.7.2.2 Lymphocytes of MS Patients Express iGluR AMPA GluR3, Which Is Upregulated During Relapse and in Patients with Neurological Evidence of Disease Activity. Furthermore, Glutamate Augments the Proliferation and Chemotactic Migration of the Autoreactive T Cells
In line and confirmation with the observations described above in part 4.7.2.1, a later important study by Sarchielli et al. demonstrated that iGluR AMPA GluR3 is expressed in lymphocytes of MS patients, and that its expression is upregulated during relapse and in patients with neurological evidence of disease activity (Sarchielli et al. 2007). An equally exciting additional finding made in this study was that either glutamate or AMPA (10−8–10−5 M) enhanced the proliferation of the autoreactive T cells in response to myelin-derived proteins, and also augmented the chemotactic migration of these autoimmune T cells towards CXCL12/SDF-1 (Sarchielli et al. 2007). Another study also reported that the inhibition of PHA-induced cell proliferation caused by 10−3 M glutamate (see above) was lower in T cells of MS patients (Lombardi et al. 2003).
Interestingly, adhesion to laminin in the CNS plays a crucial role in the recruitment, transmigration and penetration of myelin autoreactive T cells: the parenchymal basement membranes containing certain laminin isoforms were found to be permissive for encephalitogenic T cell transmigration, while those containing others were restrictive (Sixt et al. 2001). Based on our previous findings, showing that glutamate on its own induces adhesion of naïve/resting human T cells to laminin, as well as to fibronectin (Ganor et al. 2003), we speculate that encounter of resting T cells with glutamate could cause their adhesion to laminin-containing brain parenchyma, and could further promote their directional migration towards chemokines secreted in specific sites within the CNS in MS.
Together, these findings support the idea that glutamate-induced activation of MS-associated T cells may indeed play a vital role in MS (illustrated schematically in Fig. 4.4), and if this is indeed true, blocking glutamate signaling in MS may turn to be beneficial.

Glutamate and its receptors may play an important role in Multiple Sclerosis (MS) and Experimental Autoimmune Encephalomyelitis (EAE). The main evidences supporting an important role played by glutamate and its AMPA iGluRs in MS and EAE are the following: (1) The iGluR AMPA GluR3 subunit is expressed in lymphocytes of MS patients, and its expression is upregulated during relapse and in patients with neurological evidence of disease activity (Sarchielli et al. 2007); (2) Glutamate or AMPA (10−8–10−5 M) enhances the proliferation of the autoreactive T cells in response to myelin-derived proteins, and also augments the chemotactic migration of these autoreactive Tcells towards CXCL12/SDF-1 (Sarchielli et al. 2007); (3) AMPA GluR3 is expressed in mouse encephalitogenic anti-myelin basic protein T cell clones (Ganor et al. 2003); (4) There is an abnormal response to glutamate of T lymphocytes from MS patients (Lombardi et al. 2003); (5) Glutamate/AMPA receptor antagonists block EAE in mice and rats (Pitt et al. 2000; Smith et al. 2000)
4.7.3 Rheumatoid Arthritis
Rheumatoid Arthritis (RA) is a chronic systemic inflammatory disorder that may affect many tissues and organs, but principally attacks synovial joints. The process produces an inflammatory response of the synovium (synovitis) secondary to hyperplasia of synovial cells, excess synovial fluid, and the development of pannus in the synovium. Although the cause of RA is unknown, autoimmunity plays a pivotal role in both its chronicity and progression, and RA is considered a systemic autoimmune disease. A recent review discusses a possible involvement of glutamatergic signaling machineries in the pathophysiology of RA (Hinoi and Yoneda 2011). The authors cover recent molecular/biological analyses, including their own, which propose a novel function for glutamate as an extracellular signal mediator operating in an autocrine and/or paracrine manner in several peripheral and non-neuronal tissues, including the bone and cartilage. In particular, a drastic increase was demonstrated in the endogenous levels of both glutamate and aspartate in the synovial fluid with intimate relevance to increased edema and sensitization to thermal hyperalgesia in experimental arthritis models. In their review, the authors outline the role of glutamate in synovial fibroblasts in addition to the possible involvement of glutamatergic signaling machineries in the pathogenesis of joint diseases such as RA.
4.7.4 Systemic Lupus Erythematosus
Systemic Lupus Erythematosus (SLE) is a systemic autoimmune disease (or autoimmune connective tissue disease) that can affect any part of the body. SLE most often harms the heart, joints, skin, lungs, blood vessels, liver, kidneys, and nervous system. As occurs in other autoimmune diseases, the immune system attacks the body’s cells and tissues, resulting in inflammation and tissue damage. It is a type III hypersensitivity reaction caused by antibody-immune complex formation. In a recent study, high-performance liquid chromatography was used for glutamate measurements in freshly isolated, non-cultured peripheral T cells of patients with SLE and healthy controls. The authors found that the mean ± SD serum concentrations of glutamate were lower in patients with either clinically quiescent SLE (77 ± 27 μM [n = 18]) or active SLE (61 ± 36 μM [n = 16]) than in healthy controls (166 ± 64 μM [n = 24]) (Poulopoulou et al. 2008).
4.7.5 ‘Autoimmune Epilepsy’ and ‘Autoimmune Encephalitis’ Mediated by Glutamate Receptor Antibodies
It is known and widely accepted that excess glutamate contributes to epilepsy. In fact, epilepsy is conceived as a disease caused by either too much glutamate-mediated excitation or too low gamma-amino-butyric acid (GABA)-mediated inhibition. In line with this notion, excess glutamate or its agonists are highly neurotoxic and causes seizures in animal models. For decades, epilepsy was considered a brain disorder caused solely by various neurological factors, and not by any immunological factors like antibodies (Abs). Breaking this dogma, a series of studies and publications by several groups including ours over the last ~15 years provided plentiful evidence in support of ‘Autoimmune Epilepsy’, a term coined in 2002 by M. Levite (Levite 2002) and used in subsequent studies (Ganor et al. 2004, 2005a), referring to human epilepsies in which deleterious Abs, primarily iGluR Abs, are found in serum and/or CSF of the epilepsy patients, and where such Abs are suspected to contribute to the initiation and/or worsening of the seizures themselves, and/or to any of the neuropsychobehavioral impairments that often accompany the seizures (Levite and Ganor 2008). For review of the vast majority of relevant publications on the topic of ‘Autoimmune Epilepsy’ and GluR Abs in human diseases the reader is refereed to (Levite and Ganor 2008; Vincent et al. 2011) and for some original papers to (Andrews et al. 1996; Andrews and McNamara 1996; Antozzi et al. 1998; Baranzini et al. 2002; Ganor et al. 2004, 2005a; Levite et al. 1999; Levite and Hermelin 1999; Mantegazza et al. 2002; Rogers et al. 1994; Twyman et al. 1995; Tziperman et al. 2007; Wiendl et al. 2001).
In summary, three types of GluR Abs were found thus far in patients with epilepsy, encephalitis and/or SLE, and most if not all of them can cause brain damage. The main features of these three types of GluR Abs are summarized below, as well as shown in schematically Fig. 4.5 and its detailed legend.

Glutamate receptor autoantibodies (Abs) of several types are found in patients with Epilepsy, Encephalitis and Systemic Lupus Erythematosus (SLE), and can cause brain damage. Three types of glutamate receptor Abs were found thus far in patients with epilepsy, encephalitis and/or (SLE), and they are all detrimental to the CNS. The three glutamate receptor Abs types are: (1) AMPA GluR3 Abs. GluR3 Abs, especially those directed against the B peptide of GluR3, termed GluR3B Abs, are found mainly in ~30% of epilepsy patients. These AMPA GluR3 Abs bind, activate and kill neurons, and cause brain pathology in animal models (for review see Levite and Ganor 2008). In addition, we recently found in GluR3B-immunized mice that GluR3B Abs lower seizure threshold and cause neuropsychiatric and behavioral impairments, and are also frequent among epilepsy patients with neuro/psycho/cognitive impairments (Goldberg et al., paper in preparation). In addition, by electrophysiological recordings we showed that affinity purified GluR3B Abs activate GluR3-containing homomeric and heteromeric AMPA receptor complexes and induce the characteristic ion currents, without the requirement of neuronal, glial or blood ancillary molecules. Furthermore, the ion currents induced by the GluR3B Abs were completely blocked by CNQX, a selective AMPA agonist (Cohen-Kashi Malina et al. 2006) (insert). (2) NMDA R1 (NR1) Abs. These Abs are directed against the NR1 subunit of the NMDA iGluRs. According to several recent studies, the NMDA NR1 Abs cause ‘Anti-NMDA receptor encephalitis’ – a severe, treatable and potentially reversible disorder presenting with memory deficits, psychiatric symptoms, autonomic instability including hypoventilation and seizures (Dalmau et al. 2011; Florance et al. 2009; Irani and Vincent 2011). (3) NMDA NR2A/B Abs. These Abs are directed against the NR2 subunit of the NMDA iGluRs and were found thus far in ~30% of SLE patients (with our without neuropsychiatric problems), ~18% of epilepsy patients, and in some patients with encephalitis. Once they reach the CNS, the NMDA NR2A/B Abs can bind and subsequently kill neurons in the hippocamous or amygdala via induction of excitotoxicity and apoptosis (Huerta et al. 2006; Kowal et al. 2004; Levite and Ganor 2008)
-
1.
Anti-AMPA-GluR3 (GluR3) Abs. These Abs are directed primarily against amino acids (aa) 372–395 of GluR3, termed GluR3B peptide, or against aa 245–274 of GluR3 termed GluR3A peptide. The GluR3 Abs, mainly GluR3B Abs, were found thus far in ~30% of patients with different types of epilepsy. Furthermore, studies of our own group, as well as of others, showed that these GluR3 or GluR3B Abs can often activate GluRs and damage neurons and glia in-vitro and in-vivo. Thus, GluR3B Abs bind to neurons, possess a unique ability to activate their glutamate-receptor antigen, kill neurons and glia in-vitro and in-vivo (either by excitotoxicity or by complement fixation independent of receptor activation), and cause multiple brain damage. In animal models (mice, rats or rabbits), GluR3 Abs may cause seizures, augment their severity or modulate their threshold (for review of all these effects see (Levite and Ganor 2008) and the studies cited therein).
Interestingly, in one of our studies we showed by electrophysiological recordings that affinity purified GluR3B Abs on their own activate GluR3-containing homomeric and heteromeric AMPA receptor complexes and induce the characteristic ion currents, without the requirement of neuronal, glial or blood ancillary molecules. Furthermore, the ion currents induced by GluR3B Abs were completely blocked by CNQX, a selective AMPA agonist (Cohen-Kashi Malina et al. 2006). Thus, GluR3B Abs can partially mimic glutamate.
In other studies, we showed that GluR3B-immunized mice and rats exhibited significant brain pathology in-vivo (Ganor et al. 2005b; Levite and Hermelin 1999). The neuropathological findings included, for example, thickening of cerebral meninges with lymphocyte infiltrates, cerebellar cortical abiotrophy with loss of Purkinje cells, occasional reactive gliosis, and loss of mature neurons compensated by increased neurogenesis. In a recent still unpublished study, we also observed that GluR3B-immunized mice exhibit lower threshold to chemoconvulsant-induced seizures, and suffer from several neuropsychiatric and behavioral impairments compared to several control groups (Goldberg H, Ganor Y and Levite M, paper in preparation). Finally, in this study we also found a striking, surprising and significant correlation in epilepsy patients between the presence of GluR3B Abs in their serum and the presence of their neuro/psycho/cognitive impairments. For remider: various neuropsychiatric impairment sometimes accompany the seizures.
-
2.
Anti-NMDA-R1 (NR1) Abs. These Abs are directed against the NR1 subunit of NMDA iGluRs. According to several recent studies, NMDA NR1 Abs cause ‘Anti-NMDA receptor encephalitis’ - a severe, treatable and potentially reversible disorder presenting with memory deficits, psychiatric symptoms, autonomic instability including hypoventilation and seizures (Dalmau et al. 2011; Florance et al. 2009; Irani and Vincent 2011). Importantly, Hughes et al. demonstrated recently that patient’s NMDA receptor Abs cause a selective and reversible decrease in NMDA surface receptors and synaptic localization that correlate with patient’s Abs titers (Hughes et al. 2011). The authors further found that the mechanism of this decrease is selective Ab-mediated capping and internalization of surface NMDA receptors (since Fab fragments from patient’s Abs did not decrease NMDA receptor density). This is in line with another study of this group (Moscato et al. 2011). Furthermore, in another study, Manto et al. showed that CSF of patient with NMDA Abs and encephalitis increased the concentrations of glutamate in the extracellular space. The increase was dose-dependent and was dramatic with purified IgG. Patient’s CSF also impaired both the NMDA- and the AMPA-mediated synaptic regulation of glutamate (Manto et al. 2011). Taken together, all these findings show that NR1 Abs can clearly impair glutamate signaling and cause neurological abnormalities.
-
3.
Anti-NMDA-NR2A/B (NR2) Abs. These Abs are directed against the NR2 subunit of the NMDA iGluRs. They were found in 18% of epilepsy patients tested thus far, ~30% of SLE patients (with our without neuropsychiatric problems), and in some patients with encephalitis. Once the NR2A/B Abs reach the CNS, they can bind and subsequently kill neurons in the hippocamous or amygdala via induction of excitotoxicity and apoptosis (Huerta et al. 2006; Kowal et al. 2004; Levite and Ganor 2008).
In summary, the three types of GluR Abs, directed against AMPA GluR3, NMDA NR1 or NMDA NR2A/B, are found in human patients, mainly suffering from epilepsy and encephalitis, and are detrimental to the nervous system.
What triggers the autoimmune production of all these GluR Abs is still unknown. Yet, at least for the AMPA GluR3 Abs, we proposed that the AMPA GluR3 expressed in peripheral T cells may be the primary source of the GluR3-derived antigens for these Abs, which later damage neurons. We also proposed that the cleavage of the AMPA GluR3 from the surface of activated T cells by granzyme B that is produced and released by such activated cells, and the subsequent shedding of GluR3B-containing fragments into the extracellular milieu is a key immunogenic event, which at certain inflammatory conditions leads to the production of the detrimental GluR3B Abs (Ganor et al. 2007). We further speculate that the genetic background of the individual may play a role too in dictating whether autoimmunity to GluR3 Abs will develop or not. Our suggested models/scenarios for the production of GluR3 Abs are drawn schematically and discussed in detail in our recent review (Levite and Ganor 2008). All the above suggestions call for further studies.
4.7.6 Human Immunodeficiency Virus Type 1 (HIV-1) Infection
Patients who are infected with the human immunodeficiency virus type 1 (HIV-1) were found to have, on average, markedly elevated plasma glutamate levels (Eck et al. 1989b). Moreover, increased glutamate levels were reported in CSF of patients with HIV-1 dementia. Importantly, the levels of glutamate in the CSF, but not in the plasma, were related to the degree of dementia and brain atrophy. Of remider: HIV-1 reaches the brain following entry of HIV-1-infected monocytes/macrophages across the BBB. Although neurons are not infected with HIV-1, the predominant pathways to neuronal injury in HIV-1 dementia include indirect effects through release of macrophage, microglial and astrocyte toxins, as well as direct injury by viral proteins. Among the released toxins is glutamate, which is produced by macrophages themselves (see Sect. 4.6 above) and overstimulates neuronal GluRs, resulting in neuronal death by excitotoxicity, similar to the events taking place in other neurodegenerative diseases (for review see Kaul et al. 2005). These observations suggest that increased CSF glutamate, most probably produced locally by infected macrophages (and other cell types) plays a pathogenic role in HIV-1 dementia, thus supporting the treatment of these patients with GluR antagonists.
References
Affaticati P, Mignen O, Jambou F, Potier MC, Klingel-Schmitt I, Degrouard J, Peineau S, Gouadon E, Collingridge GL, Liblau R, Capiod T, Cohen-Kaminsky S (2011) Sustained calcium signalling and caspase-3 activation involve NMDA receptors in thymocytes in contact with dendritic cells. Cell Death Differ 18(1):99–108
Andrews PI, McNamara JO (1996) Rasmussen’s encephalitis: an autoimmune disorder? Curr Opin Neurobiol 6(5):673–678
Andrews PI, Dichter MA, Berkovic SF, Newton MR, McNamara JO (1996) Plasmapheresis in Rasmussen’s encephalitis. Neurology 46(1):242–246
Antozzi C, Granata T, Aurisano N, Zardini G, Confalonieri P, Airaghi G, Mantegazza R, Spreafico R (1998) Long-term selective IgG immuno-adsorption improves Rasmussen’s encephalitis. Neurology 51(1):302–305
Arcella A, Carpinelli G, Battaglia G, D’Onofrio M, Santoro F, Ngomba RT, Bruno V, Casolini P, Giangaspero F, Nicoletti F (2005) Pharmacological blockade of group II metabotropic glutamate receptors reduces the growth of glioma cells in vivo. Neuro Oncol 7(3):236–245
Armstrong N, Sun Y, Chen GQ, Gouaux E (1998) Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature 395(6705):913–917
Baranzini SE, Laxer K, Saketkhoo R, Elkins MK, Parent JM, Mantegazza R, Oksenberg JR (2002) Analysis of antibody gene rearrangement, usage, and specificity in chronic focal encephalitis. Neurology 58(5):709–716
Boldyrev AA, Kazey VI, Leinsoo TA, Mashkina AP, Tyulina OV, Johnson P, Tuneva JO, Chittur S, Carpenter DO (2004) Rodent lymphocytes express functionally active glutamate receptors. Biochem Biophys Res Commun 324(1):133–139
Bonsi P, Cuomo D, De Persis C, Centonze D, Bernardi G, Calabresi P, Pisani A (2005) Modulatory action of metabotropic glutamate receptor (mGluR) 5 on mGluR1 function in striatal cholinergic interneurons. Neuropharmacology 49(Suppl 1):104–113
Cahalan MD, Wulff H, Chandy KG (2001) Molecular properties and physiological roles of ion channels in the immune system. J Clin Immunol 21(4):235–252
Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cui J, Tu S, Sevarino KA, Nakanishi N, Tong G, Lipton SA, Zhang D (2002) Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415(6873):793–798
Chiocchetti A, Miglio G, Mesturini R, Varsaldi F, Mocellin M, Orilieri E, Dianzani C, Fantozzi R, Dianzani U, Lombardi G (2006) Group I mGlu receptor stimulation inhibits activation-induced cell death of human T lymphocytes. Br J Pharmacol 148(6):760–768
Chu Z, Hablitz JJ (2000) Quisqualate induces an inward current via mGluR activation in neocortical pyramidal neurons. Brain Res 879(1–2):88–92
Cohen-Kashi Malina K, Ganor Y, Levite M, Teichberg VI (2006) Autoantibodies against an extracellular peptide of the GluR3 subtype of AMPA receptors activate both homomeric and heteromeric AMPA receptor channels. Neurochem Res 31(10):1181–1190
Collard CD, Park KA, Montalto MC, Alapati S, Buras JA, Stahl GL, Colgan SP (2002) Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem 277(17):14801–14811
Collingridge GL, Singer W (1990) Excitatory amino acid receptors and synaptic plasticity. Trends Pharmacol Sci 11(7):290–296
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R (2011) Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 10(1):63–74
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65(1):1–105
Das S, Sasaki YF, Rothe T, Premkumar LS, Takasu M, Crandall JE, Dikkes P, Conner DA, Rayudu PV, Cheung W, Chen HS, Lipton SA, Nakanishi N (1998) Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature 393(6683):377–381
Dickman KG, Youssef JG, Mathew SM, Said SI (2004) Ionotropic glutamate receptors in lungs and airways: molecular basis for glutamate toxicity. Am J Respir Cell Mol Biol 30(2):139–144
Divino Filho JC, Hazel SJ, Furst P, Bergstrom J, Hall K (1998) Glutamate concentration in plasma, erythrocyte and muscle in relation to plasma levels of insulin-like growth factor (IGF)-I, IGF binding protein-1 and insulin in patients on haemodialysis. J Endocrinol 156(3):519–527
Droge W, Eck HP, Betzler M, Schlag P, Drings P, Ebert W (1988) Plasma glutamate concentration and lymphocyte activity. J Cancer Res Clin Oncol 114(2):124–128
Droge W, Murthy KK, Stahl-Hennig C, Hartung S, Plesker R, Rouse S, Peterhans E, Kinscherf R, Fischbach T, Eck HP (1993) Plasma amino acid dysregulation after lentiviral infection. AIDS Res Hum Retroviruses 9(9):807–809
Eck HP, Drings P, Droge W (1989a) Plasma glutamate levels, lymphocyte reactivity and death rate in patients with bronchial carcinoma. J Cancer Res Clin Oncol 115(6):571–574
Eck HP, Frey H, Droge W (1989b) Elevated plasma glutamate concentrations in HIV-1-infected patients may contribute to loss of macrophage and lymphocyte functions. Int Immunol 1(4):367–372
Endoh T (2004) Characterization of modulatory effects of postsynaptic metabotropic glutamate receptors on calcium currents in rat nucleus tractus solitarius. Brain Res 1024(1–2):212–224
Ferrarese C, Aliprandi A, Tremolizzo L, Stanzani L, De Micheli A, Dolara A, Frattola L (2001) Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 57(4):671–675
Florance NR, Davis RL, Lam C, Szperka C, Zhou L, Ahmad S, Campen CJ, Moss H, Peter N, Gleichman AJ, Glaser CA, Lynch DR, Rosenfeld MR, Dalmau J (2009) Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol 66(1):11–18
Foster AC, Fagg GE (1984) Acidic amino acid binding sites in mammalian neuronal membranes: their characteristics and relationship to synaptic receptors. Brain Res 319(2):103–164
Gahring L, Carlson NG, Meyer EL, Rogers SW (2001) Granzyme B proteolysis of a neuronal glutamate receptor generates an autoantigen and is modulated by glycosylation. J Immunol 166(3):1433–1438
Ganor Y, Besser M, Ben-Zakay N, Unger T, Levite M (2003) Human T cells express a functional ionotropic glutamate receptor GluR3, and glutamate by itself triggers integrin-mediated adhesion to laminin and fibronectin and chemotactic migration. J Immunol 170(8):4362–4372
Ganor Y, Goldberg-Stern H, Amrom D, Lerman-Sagie T, Teichberg VI, Pelled D, Futerman AH, Zeev BB, Freilinger M, Verheulpen D, Van Bogaert P, Levite M (2004) Autoimmune epilepsy: some epilepsy patients harbor autoantibodies to glutamate receptors and dsDNA on both sides of the blood–brain barrier, which may kill neurons and decrease in brain fluids after hemispherotomy. Clin Dev Immunol 11(3–4):241–252
Ganor Y, Goldberg-Stern H, Lerman-Sagie T, Teichberg VI, Levite M (2005a) Autoimmune epilepsy: distinct subpopulations of epilepsy patients harbor serum autoantibodies to either glutamate/AMPA receptor GluR3, glutamate/NMDA receptor subunit NR2A or double-stranded DNA. Epilepsy Res 65(1–2):11–22
Ganor Y, Gottlieb M, Eilam R, Otmy H, Teichberg VI, Levite M (2005b) Immunization with the glutamate receptor-derived peptide GluR3B induces neuronal death and reactive gliosis, but confers partial protection from pentylenetetrazole-induced seizures. Exp Neurol 195(1):92–102
Ganor Y, Teichberg VI, Levite M (2007) TCR activation eliminates glutamate receptor GluR3 from the cell surface of normal human T cells, via an autocrine/paracrine granzyme B-mediated proteolytic cleavage. J Immunol 178(2):683–692
Ganor Y, Grinberg I, Reis A, Cooper I, Goldstein RS, Levite M (2009) Human T-leukemia and T-lymphoma express glutamate receptor AMPA GluR3, and the neurotransmitter glutamate elevates the cancer-related matrix-metalloproteinases inducer CD147/EMMPRIN, MMP-9 secretion and engraftment of T-leukemia in vivo. Leuk Lymphoma 50(6):985–997
Gill SS, Pulido OM (2001) Glutamate receptors in peripheral tissues: current knowledge, future research, and implications for toxicology. Toxicol Pathol 29(2):208–223
Graham TE, Sgro V, Friars D, Gibala MJ (2000) Glutamate ingestion: the plasma and muscle free amino acid pools of resting humans. Am J Physiol Endocrinol Metab 278(1):E83–89
Green DR, Droin N, Pinkoski M (2003) Activation-induced cell death in T cells. Immunol Rev 193:70–81
Guse AH (1998) Ca2+ signaling in T-lymphocytes. Crit Rev Immunol 18(5):419–448
Hawkins LM, Chazot PL, Stephenson FA (1999) Biochemical evidence for the co-association of three N-methyl-D-aspartate (NMDA) R2 subunits in recombinant NMDA receptors. J Biol Chem 274(38):27211–27218
Hemmer B, Cepok S, Nessler S, Sommer N (2002) Pathogenesis of multiple sclerosis: an update on immunology. Curr Opin Neurol 15(3):227–231
Hinoi E, Yoneda Y (2011) Possible involvement of glutamatergic signaling machineries in pathophysiology of rheumatoid arthritis. J Pharmacol Sci 116(3):248–256
Hinoi E, Ogita K, Takeuchi Y, Ohashi H, Maruyama T, Yoneda Y (2001) Characterization with [3H]quisqualate of group I metabotropic glutamate receptor subtype in rat central and peripheral excitable tissues. Neurochem Int 38(3):277–285
Hinoi E, Takarada T, Ueshima T, Tsuchihashi Y, Yoneda Y (2004) Glutamate signaling in peripheral tissues. Eur J Biochem 271(1):1–13
Hollmann M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17:31–108
Hollmann M, O’Shea-Greenfield A, Rogers SW, Heinemann S (1989) Cloning by functional expression of a member of the glutamate receptor family. Nature 342(6250):643–648
Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B (2006) Immunity and behavior: antibodies alter emotion. Proc Natl Acad Sci USA 103(3):678–683
Huettner JE (2003) Kainate receptors and synaptic transmission. Prog Neurobiol 70(5):387–407
Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, Parsons TD, Lynch DR, Dalmau J, Balice-Gordon RJ (2011) Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci 30(17):5866–5875
Irani SR, Vincent A (2011) NMDA receptor antibody encephalitis. Curr Neurol Neurosci Rep 11(3):298–304
Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, Miwa A, Kurihara H, Nakazato Y, Tamura M, Sasaki T, Ozawa S (2002) Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med 8(9):971–978
Kalariti N, Pissimissis N, Koutsilieris M (2005) The glutamatergic system outside the CNS and in cancer biology. Expert Opin Investig Drugs 14(12):1487–1496
Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA (2005) HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ 12(Suppl 1):878–892
Keinanen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B, Seeburg PH (1990) A family of AMPA-selective glutamate receptors. Science 249(4968):556–560
Kew JN, Kemp JA (2005) Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berl) 179(1):4–29
Komuro H, Rakic P (1993) Modulation of neuronal migration by NMDA receptors. Science 260(5104):95–97
Kostanyan IA, Merkulova MI, Navolotskaya EV, Nurieva RI (1997) Study of interaction between L-glutamate and human blood lymphocytes. Immunol Lett 58(3):177–180
Kowal C, DeGiorgio LA, Nakaoka T, Hetherington H, Huerta PT, Diamond B, Volpe BT (2004) Cognition and immunity; antibody impairs memory. Immunity 21(2):179–188
Lerma J (2006) Kainate receptor physiology. Curr Opin Pharmacol 6(1):89–97
Levite M (2002) Autoimmune epilepsy. Nat Immunol 3(6):500
Levite M (2008) Neurotransmitters activate T-cells and elicit crucial functions via neurotransmitter receptors. Curr Opin Pharmacol 8(4):460–471
Levite M, Ganor Y (2008) Autoantibodies to glutamate receptors can damage the brain in epilepsy, systemic lupus erythematosus and encephalitis. Expert Rev Neurother 8(7):1141–1160
Levite M, Hermelin A (1999) Autoimmunity to the glutamate receptor in mice–a model for Rasmussen’s encephalitis? J Autoimmun 13(1):73–82
Levite M, Fleidervish IA, Schwarz A, Pelled D, Futerman AH (1999) Autoantibodies to the glutamate receptor kill neurons via activation of the receptor ion channel. J Autoimmun 13(1):61–72
Levite M, Cahalon L, Peretz A, Hershkoviz R, Sobko A, Ariel A, Desai R, Attali B, Lider O (2000) Extracellular K(+) and opening of voltage-gated potassium channels activate T cell integrin function: physical and functional association between Kv1.3 channels and beta1 integrins. J Exp Med 191(7):1167–1176
Li F, Tsien JZ (2009) Memory and the NMDA receptors. N Engl J Med 361(3):302–303
Lin CS, Boltz RC, Blake JT, Nguyen M, Talento A, Fischer PA, Springer MS, Sigal NH, Slaughter RS, Garcia ML et al (1993) Voltage-gated potassium channels regulate calcium-dependent pathways involved in human T lymphocyte activation. J Exp Med 177(3):637–645
Lombardi G, Dianzani C, Miglio G, Canonico PL, Fantozzi R (2001) Characterization of ionotropic glutamate receptors in human lymphocytes. Br J Pharmacol 133(6):936–944
Lombardi G, Miglio G, Canonico PL, Naldi P, Comi C, Monaco F (2003) Abnormal response to glutamate of T lymphocytes from multiple sclerosis patients. Neurosci Lett 340(1):5–8
Lombardi G, Miglio G, Dianzani C, Mesturini R, Varsaldi F, Chiocchetti A, Dianzani U, Fantozzi R (2004) Glutamate modulation of human lymphocyte growth: in vitro studies. Biochem Biophys Res Commun 318(2):496–502
Mantegazza R, Bernasconi P, Baggi F, Spreafico R, Ragona F, Antozzi C, Bernardi G, Granata T (2002) Antibodies against GluR3 peptides are not specific for Rasmussen’s encephalitis but are also present in epilepsy patients with severe, early onset disease and intractable seizures. J Neuroimmunol 131(1–2):179–185
Manto M, Dalmau J, Didelot A, Rogemond V, Honnorat J (2011) In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J Rare Dis 5:31
Mashkina AP, Tyulina OV, Solovyova TI, Kovalenko EI, Kanevski LM, Johnson P, Boldyrev AA (2007) The excitotoxic effect of NMDA on human lymphocyte immune function. Neurochem Int 51(6–7):356–360
Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S (1991) Sequence and expression of a metabotropic glutamate receptor. Nature 349(6312):760–765
Mayer ML (2005a) Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron 45(4):539–552
Mayer ML (2005b) Glutamate receptor ion channels. Curr Opin Neurobiol 15(3):282–288
Mayer ML, Westbrook GL (1987) The physiology of excitatory amino acids in the vertebrate central nervous system. Prog Neurobiol 28(3):197–276
Meldrum BS (2000) Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 130(4S Suppl):1007S–1015S
Merrill JE, Benveniste EN (1996) Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci 19(8):331–338
Miglio G, Varsaldi F, Dianzani C, Fantozzi R, Lombardi G (2005a) Stimulation of group I metabotropic glutamate receptors evokes calcium signals and c-jun and c-fos gene expression in human T cells. Biochem Pharmacol 70(2):189–199
Miglio G, Varsaldi F, Lombardi G (2005b) Human T lymphocytes express N-methyl-D-aspartate receptors functionally active in controlling T cell activation. Biochem Biophys Res Commun 338(4):1875–1883
Miglio G, Dianzani C, Fallarini S, Fantozzi R, Lombardi G (2007) Stimulation of N-methyl-D-aspartate receptors modulates Jurkat T cell growth and adhesion to fibronectin. Biochem Biophys Res Commun 361(2):404–409
Monaghan DT, Bridges RJ, Cotman CW (1989) The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol 29:365–402
Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S (1991) Molecular cloning and characterization of the rat NMDA receptor. Nature 354(6348):31–37
Moscato EH, Jain A, Peng X, Hughes EG, Dalmau J, Balice-Gordon RJ (2011) Mechanisms underlying autoimmune synaptic encephalitis leading to disorders of memory, behavior and cognition: insights from molecular, cellular and synaptic studies. Eur J Neurosci 32(2):298–309
Nakayama T, Yamashita M (2010) The TCR-mediated signaling pathways that control the direction of helper T cell differentiation. Semin Immunol 22(5):303–309
Nedergaard M, Takano T, Hansen AJ (2002) Beyond the role of glutamate as a neurotransmitter. Nat Rev Neurosci 3(9):748–755
Nicoletti F, Arcella A, Iacovelli L, Battaglia G, Giangaspero F, Melchiorri D (2007) Metabotropic glutamate receptors: new targets for the control of tumor growth? Trends Pharmacol Sci 28(5):206–213
Ohashi H, Maruyama T, Higashi-Matsumoto H, Nomoto T, Nishimura S, Takeuchi Y (2002) A novel binding assay for metabotropic glutamate receptors using [3H] L-quisqualic acid and recombinant receptors. Z Naturforsch C 57(3–4):348–355
Ollenschlager G, Karner J, Karner-Hanusch J, Jansen S, Schindler J, Roth E (1989) Plasma glutamate–a prognostic marker of cancer and of other immunodeficiency syndromes? Scand J Clin Lab Invest 49(8):773–777
Pacheco R, Ciruela F, Casado V, Mallol J, Gallart T, Lluis C, Franco R (2004) Group I metabotropic glutamate receptors mediate a dual role of glutamate in T cell activation. J Biol Chem 279(32):33352–33358
Pacheco R, Oliva H, Martinez-Navio JM, Climent N, Ciruela F, Gatell JM, Gallart T, Mallol J, Lluis C, Franco R (2006) Glutamate released by dendritic cells as a novel modulator of T cell activation. J Immunol 177(10):6695–6704
Pacheco R, Gallart T, Lluis C, Franco R (2007) Role of glutamate on T-cell mediated immunity. J Neuroimmunol 185(1–2):9–19
Piani D, Frei K, Do KQ, Cuenod M, Fontana A (1991) Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett 133(2):159–162
Piani D, Spranger M, Frei K, Schaffner A, Fontana A (1992) Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol 22(9):2429–2436
Pin JP, Duvoisin R (1995) The metabotropic glutamate receptors: structure and functions. Neuropharmacology 34(1):1–26
Pitt D, Werner P, Raine CS (2000) Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 6(1):67–70
Platt SR (2007) The role of glutamate in central nervous system health and disease–a review. Vet J 173(2):278–286
Poulopoulou C, Davaki P, Koliaraki V, Kolovou D, Markakis I, Vassilopoulos D (2005a) Reduced expression of metabotropic glutamate receptor 2 mRNA in T cells of ALS patients. Ann Neurol 58(6):946–949
Poulopoulou C, Markakis I, Davaki P, Nikolaou C, Poulopoulos A, Raptis E, Vassilopoulos D (2005b) Modulation of voltage-gated potassium channels in human T lymphocytes by extracellular glutamate. Mol Pharmacol 67(3):856–867
Poulopoulou C, Papadopoulou-Daifoti Z, Hatzimanolis A, Fragiadaki K, Polissidis A, Anderzanova E, Davaki P, Katsiari CG, Sfikakis PP (2008) Glutamate levels and activity of the T cell voltage-gated potassium Kv1.3 channel in patients with systemic lupus erythematosus. Arthritis Rheum 58(5):1445–1450
Reynolds JD, Amory DW, Grocott HP, White WD, Newman MF (2002) Change in plasma glutamate concentration during cardiac surgery is a poor predictor of cognitive outcome. J Cardiothorac Vasc Anesth 16(4):431–436
Rezzani R, Corsetti G, Rodella L, Angoscini P, Lonati C, Bianchi R (2003) Cyclosporine-A treatment inhibits the expression of metabotropic glutamate receptors in rat thymus. Acta Histochem 105(1):81–87
Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, Hughes TE, Heinemann SF, McNamara JO (1994) Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science 265(5172):648–651
Rosenmund C, Stern-Bach Y, Stevens CF (1998) The tetrameric structure of a glutamate receptor channel. Science 280(5369):1596–1599
Rzeski W, Turski L, Ikonomidou C (2001) Glutamate antagonists limit tumor growth. Proc Natl Acad Sci USA 98(11):6372–6377
Sarchielli P, Di Filippo M, Candeliere A, Chiasserini D, Mattioni A, Tenaglia S, Bonucci M, Calabresi P (2007) Expression of ionotropic glutamate receptor GLUR3 and effects of glutamate on MBP- and MOG-specific lymphocyte activation and chemotactic migration in multiple sclerosis patients. J Neuroimmunol 188(1–2):146–158
Sattler R, Tymianski M (2001) Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol 24(1–3):107–129
Schmitz D, Mellor J, Nicoll RA (2001) Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science 291(5510):1972–1976
Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM (2001) Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood–brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol 153(5):933–946
Sladeczek F, Momiyama A, Takahashi T (1993) Presynaptic inhibitory action of a metabotropic glutamate receptor agonist on excitatory transmission in visual cortical neurons. Proc Biol Sci 253(1338):297–303
Smith T, Groom A, Zhu B, Turski L (2000) Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med 6(1):62–66
Song I, Huganir RL (2002) Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci 25(11):578–588
Stepulak A, Sifringer M, Rzeski W, Endesfelder S, Gratopp A, Pohl EE, Bittigau P, Felderhoff-Mueser U, Kaindl AM, Buhrer C, Hansen HH, Stryjecka-Zimmer M, Turski L, Ikonomidou C (2005) NMDA antagonist inhibits the extracellular signal-regulated kinase pathway and suppresses cancer growth. Proc Natl Acad Sci USA 102(43):15605–15610
Stepulak A, Luksch H, Gebhardt C, Uckermann O, Marzahn J, Sifringer M, Rzeski W, Staufner C, Brocke KS, Turski L, Ikonomidou C (2009) Expression of glutamate receptor subunits in human cancers. Histochem Cell Biol 132(4):435–445
Storto M, de Grazia U, Battaglia G, Felli MP, Maroder M, Gulino A, Ragona G, Nicoletti F, Screpanti I, Frati L, Calogero A (2000) Expression of metabotropic glutamate receptors in murine thymocytes and thymic stromal cells. J Neuroimmunol 109(2):112–120
Sturgill JL, Mathews J, Scherle P, Conrad DH (2011) Glutamate signaling through the kainate receptor enhances human immunoglobulin production. J Neuroimmunol 233(1–2):80–89
Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M (2001) Glutamate release promotes growth of malignant gliomas. Nat Med 7(9):1010–1015
Tanabe Y, Masu M, Ishii T, Shigemoto R, Nakanishi S (1992) A family of metabotropic glutamate receptors. Neuron 8(1):169–179
Tay DL, Hoffbrand AV, Wickremasinghe GR (1996) Expression of c-fos and c-jun proteins and AP-1 binding activity during cell cycle progression of HL60 cells and phytohemagglutinin-stimulated lymphocytes. Exp Hematol 24(2):277–284
Twyman RE, Gahring LC, Spiess J, Rogers SW (1995) Glutamate receptor antibodies activate a subset of receptors and reveal an agonist binding site. Neuron 14(4):755–762
Tziperman B, Garty BZ, Schoenfeld N, Hoffer V, Watemberg N, Lev D, Ganor Y, Levite M, Lerman-Sagie T (2007) Acute intermittent porphyria, Rasmussen encephalitis, or both? J Child Neurol 22(1):99–105
Vincent A, Bien CG, Irani SR, Waters P (2011) Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 10(8):759–772
Wiendl H, Bien CG, Bernasconi P, Fleckenstein B, Elger CE, Dichgans J, Mantegazza R, Melms A (2001) GluR3 antibodies: prevalence in focal epilepsy but no specificity for Rasmussen’s encephalitis. Neurology 57(8):1511–1514
Zhou L, Chong MM, Littman DR (2009) Plasticity of CD4+ T cell lineage differentiation. Immunity 30(5):646–655
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag/Wien
About this chapter
Cite this chapter
Ganor, Y., Levite, M. (2012). Glutamate in the Immune System: Glutamate Receptors in Immune Cells, Potent Effects, Endogenous Production and Involvement in Disease. In: Levite, M. (eds) Nerve-Driven Immunity. Springer, Vienna. https://doi.org/10.1007/978-3-7091-0888-8_4
Download citation
DOI: https://doi.org/10.1007/978-3-7091-0888-8_4
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-0887-1
Online ISBN: 978-3-7091-0888-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)