
Abstract
The p75 neurotrophin receptor (p75NTR) regulates a wide range of cellular functions, including programmed cell death, axonal growth and degeneration, cell proliferation, myelination, and synaptic plasticity. The multiplicity of cellular functions governed by the receptor arises from the variety of ligands and co-receptors which associate with p75NTR and regulate its signaling. P75NTR promotes survival through interactions with Trk receptors, inhibits axonal regeneration via partnerships with Nogo receptor (Nogo-R) and Lingo-1, and promotes apoptosis through association with Sortilin. Signals downstream of these interactions are further modulated through regulated intramembrane proteolysis (RIP) of p75NTR and by interactions with numerous cytosolic partners. In this chapter, we discuss the intricate signaling mechanisms of p75NTR, emphasizing how these signals are differentially regulated to mediate these diverse cellular functions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The discovery of nerve growth factor (NGF) as the factor released by the target to promote survival and differentiation quickly led to the hunt for the receptor involved in mediating its actions. Early studies characterizing radiolabeled NGF binding to peripheral neurons revealed that NGF bound its receptors in a complex manner that likely involved multiple sites (Frazier et al. 1974a, b). Subsequently, two distinct NGF binding sites, one with high affinity and one with low affinity, were demonstrated on sensory neurons (Sutter et al. 1979). Cross-linking studies confirmed two receptor components in sympathetic neurons (Massague et al. 1981) and PC12 cells (Grob et al. 1983; Massague et al. 1982) of approximately 140–200 kDa and 70–100 kDa, with the lower molecular weight species being most abundant. These binding studies set the stage for the expression cloning of the NGF receptor by Chao et al. (1986) and, independently, by the Shooter lab (Radeke et al. 1987). Both groups identified the cDNA for the lower molecular weight species; it was termed the p75 receptor, which proved to be the low affinity binding site. Eventually, the proto-oncogene tropomyosin-related kinase A, or TrkA, was recognized as another NGF receptor accounting for the higher molecular weight component (Kaplan et al. 1991; Klein et al. 1991), and a complex of both p75 and TrkA was shown to comprise the high affinity binding site (Hempstead et al. 1991).
The cloning of the p75 receptor was a major achievement in the field; however, it opened a Pandora’s box of puzzles and paradoxes. Prior to the cloning of p75, it had been suggested that only the high-affinity binding component was able to mediate the neurite growth-promoting effects of NGF (Sonnenfeld and Ishii 1982; Stach and Wagner 1982), raising the question of the role of the low affinity component. Once TrkA was identified as another receptor for NGF, it was quickly established as the primary mediator of NGF’s survival and differentiation effects, being a tyrosine kinase with potent signaling capability (Ibanez et al. 1992; Loeb et al. 1991). For several years the p75 receptor was thought to simply function as a binding partner for TrkA or acting to increase the local concentration of NGF to facilitate activation of TrkA (Chao and Hempstead 1995). However, there were a number of observations that piqued the interest of researchers in the field, causing them to further explore the role of p75; for example, the receptor is expressed widely in the developing nervous system, with expression in peripheral neurons, within the spinal cord, and throughout the brain (Ernfors et al. 1991). It is expressed by many neuronal cell types, as well as neural stem cells, some astrocytes, oligodendrocyte precursors, Schwann cells, and olfactory ensheathing glia (Cragnolini and Friedman 2008). Several nonneural tissues also express the receptor during some stage of development, such as kidney and muscle (Ernfors et al. 1991; Wheeler and Bothwell 1992). In contrast, the Trk receptors exhibit a much more restricted expression pattern. In addition, the p75 receptor is strongly upregulated in many neurons and glial cells following injury, suggesting that it has a functional role in such conditions (discussed below). Finally, there are portions of the intracellular domain, where signaling would initiate, that are highly conserved across species, from chicken to human. These findings prompted further study of the p75 receptor, and in the 25 years since its initial cloning, it has been shown to regulate an amazing array of cellular responses, including cell survival, cell cycle, neurite outgrowth, synaptic function, and myelination. This chapter will discuss our current understanding of the molecular mechanisms by which the p75 receptor mediates these diverse signals.
2 Structure
After the cloning of p75 it was quickly recognized that it not only bound NGF but also brain-derived neurotrophic factor (BDNF) (Rodriguez-Tebar et al. 1990), neurotrophin-3 (NT3) (Rodriguez-Tebar et al. 1992), and neurotrophin-4 (NT4) (Ryden et al. 1995), with similar affinity, although with somewhat different kinetics (Rodriguez-Tebar et al. 1992). The ability of the receptor to bind all neurotrophins led to its designation as the p75 neurotrophin receptor (p75NTR) as opposed to the p75 NGF receptor. The p75NTR interacts with the neurotrophins through the four cysteine-rich domains in its extracellular domain (Baldwin and Shooter 1995). The initial X-ray crystallography structural analysis of the extracellular domain of p75NTR bound to NGF indicated that the receptor monomer binds NGF in an asymmetrical fashion, resulting in a 1:2 ratio (He and Garcia 2004). However, considerable biochemical data indicated that p75NTR associates with neurotrophins in a 2:2 ratio. Binding analyses using cross-linkers to attach neurotrophins to the receptor indicated a dimer of p75NTR bound to a neurotrophin dimer (Grob et al. 1985). Further crystallographic analyses support a 2:2 complex between neurotrophins and p75NTR (Feng et al. 2010; Gong et al. 2008), and it has been suggested that the 1:2 asymmetrical binding may represent an intermediate in the formation of the 2:2 complex (Feng et al. 2010).
At least a fraction of p75NTR has been shown to preexist as a disulfide-linked dimer (Grob et al. 1985; Ross et al. 1984), and a highly conserved cysteine (257) in the transmembrane domain responsible for linking the monomers was recently identified, although non-covalent dimerization still occurred even when cysteine 257 was mutated (Vilar et al. 2009). Further analysis revealed that a conserved AxxxG266 sequence in the transmembrane region, which is often found in self-associating transmembrane proteins, is required for the formation of dimers. Through their studies, the authors elucidated an interesting aspect of the receptor’s structural dynamics that provided a mechanism by which p75NTR transduces its signal upon ligand binding: the disulfide in the transmembrane domain acts as a pivot point, such that when the extracellular domain clamps down on a neurotrophin, the intracellular domains separate. The parting of the dimerized intracellular portions of the receptor facilitates binding of signaling molecules necessary for p75NTR-mediated cell death (Vilar et al. 2009).
The intracellular domain (ICD) of p75NTR contains a region similar to the Tumor necrosis factor receptor (TNFR) and the Fas antigen (Chapman 1995; Chapman and Kuntz 1995; Feinstein et al. 1995). Since TNFR and Fas mediate apoptotic signals, this portion of their ICD was termed the “death domain.” The 3-D structure of p75NTR’s death domain was determined by NMR and was similar to the structure of the Fas death domain, although there were a few differences. In particular, the death domain of Fas and TNFR self-assemble, while that of p75NTR does not (Liepinsh et al. 1997). This result is in agreement with the ability of TNFR and Fas to signal by recruiting other death domain-containing proteins while the intracellular interactors of p75NTR so far identified do not contain a death domain.
In addition to the full length form of p75NTR, a splice variant was reported lacking exon III, which encodes the cysteine-rich domains 2, 3, and 4 that are required for neurotrophin binding (von Schack et al. 2001). The original p75NTR knockout mouse was created by deleting exon III (Lee et al. 1992); thus the short form of p75NTR (s-p75NTR) could still be detected in these mice, in principle. The existence of s-p75NTR, however, remains rather controversial, and its function is not known. Nevertheless, an alternative mutant mouse was created lacking exon IV, such that both splice isoforms of p75NTR are deleted (von Schack et al. 2001). These mice exhibit a number of neurological and vascular defects similar to the exon III knockout mice, but with a more severe phenotype. However, understanding the phenotype of the exon IV mutants is complicated by the fact that the targeting strategy created a cryptic truncated protein encoding an extracellular stalk with the entire transmembrane and intracellular domains of the receptor (Paul et al. 2004). Since expression of the intracellular domain of the receptor can initiate signaling independent of ligand (Majdan et al. 1997), some phenotypic characteristics of this mouse may be due to the expression of this fragment. Clearly, results from using either of these genetically altered mice need to be interpreted with caution, and further study is needed to understand the role of s-p75NTR.
3 Apoptotic Signaling
Although p75NTR was first discovered for its ability to bind NGF, which promotes neuronal survival, the most investigated function of the receptor is, ironically, its ability to induce programmed cell death. One of the earliest indications of this function was revealed in a study by Bredesen’s group demonstrating that ectopic expression of p75NTR in an immortalized neural cell line increased apoptosis after serum withdrawal (Rabizadeh et al. 1993). These results proved challenging to reproduce in primary cells with the endogenous receptor; however, the groups of Barde and Chao found that activation of endogenous p75NTR by NGF could induce apoptosis in early retinal neurons in the chick (Frade et al. 1996) and oligodendrocytes in rat (Casaccia-Bonnefil et al. 1996), respectively. The ability of p75NTR to induce programmed cell death in response to ligand binding has now been observed in a wide variety of neuronal and non-neuronal cell types, including sympathetic (Bamji et al. 1998; Linggi et al. 2005; Teng et al. 2005), motor (Sedel et al. 1999), and hippocampal neurons (Volosin et al. 2008); photoreceptor cells (Srinivasan et al. 2004); oligodendrocytes (Casaccia-Bonnefil et al. 1996); Schwann cells (Khursigara et al. 2001; Syroid et al. 2000); and other cells (Bunone et al. 1997; Volosin et al. 2006; Wang et al. 2000). These in vitro studies together with the analysis of p75 NTR −/− mice have established this receptor as a critical regulator of developmental apoptosis, promoting the naturally occurring elimination of neurons within the developing basal forebrain (Naumann et al. 2002), trigeminal ganglia (Agerman et al. 2000), retina (Frade et al. 1996), superior cervical ganglion (Bamji et al. 1998), and spinal cord (Frade and Barde 1999). This developmental role of p75NTR has been particularly well characterized in sympathetic neurons. These neurons express TrkA and p75NTR, which together mediate a survival signal in response to NGF (discussed below); however, Miller and colleagues demonstrated that selective activation of p75NTR by BDNF led to apoptosis (Bamji et al. 1998). Furthermore, deletion of the receptor resulted in an increase in the number of neurons during the development of the superior cervical ganglia, suggesting that p75NTR mediates normal developmental death in this population. Ginty’s group later demonstrated that these neurons produce BDNF in response to NGF and suggested a model in which neurons receiving robust trophic support through NGF-induced activation of TrkA produce BDNF, thereby promoting p75NTR-dependent death of neighboring neurons receiving insufficient NGF signal (Deppmann et al. 2008). Their computer simulations based on this model quite accurately predicted the normal developmental kinetics of cell death in the superior cervical ganglia.
3.1 Activation of the Mitochondrial Cascade
Over the past decade, significant progress has been made in understanding the cellular mechanisms through which p75NTR promotes apoptosis, although many facets of the receptor’s signaling remain enigmatic. Members of the TNF receptor superfamily can activate two pathways that regulate cell survival. Through their death domain, they recruit other death domain-containing adaptors, such as TRADD and FADD, leading to caspase-8 activation and induction of a terminal caspase cascade (Dempsey et al. 2003). Despite attempts to detect activation of caspase-8 (Gu et al. 1999; Troy et al. 2002), no evidence supports p75NTR utilizing this pathway, which agrees with the structural divergence of p75NTR’s death domain from that of TNFR and Fas. The second pathway initiated by many members of the TNF receptor family involves stimulation of the stress kinase c-Jun N-terminal kinase (JNK) and of the transcription factor NF-κB (Dempsey et al. 2003). JNK activation causes cell death by inducing phosphorylation of the transcription factor c-Jun and the tumor suppressors p53 and p73 (Dhanasekaran and Reddy 2008), resulting in transcriptional upregulation of an array of pro-apoptotic genes, including Bax (Miyashita and Reed 1995), PUMA (Nakano and Vousden 2001), Bak (Bogoyevitch and Kobe 2006), and Caspase-6 (MacLachlan and El-Deiry 2002), among others (Wu 2004). In addition, JNK directly phosphorylates several Bcl-2 family proteins, causing inhibition of pro-survival members such as Bcl-2 (Yamamoto et al. 1999) and activation of pro-death members such as Bim (Lei and Davis 2003) and Bad (Donovan et al. 2002). These events ultimately lead to the release of cytochrome c from mitochondria and caspase-dependent apoptosis (Bogoyevitch and Kobe 2006).
An accumulation of evidence has indicated that p75NTR-induced apoptosis occurs via this mitochondrial cascade. Activation of JNK in response to ligand binding to endogenous p75NTR has been demonstrated in oligodendocytes (Casaccia-Bonnefil et al. 1996), sympathetic neurons (Bamji et al. 1998), and hippocampal neurons (Friedman 2000), and inhibition of the kinase prevented the induction of apoptosis (Friedman 2000; Harrington et al. 2002; Kenchappa et al. 2010; Yeiser et al. 2004; Yoon et al. 1998). Overexpressing p75NTR in cortical neurons also resulted in activation of JNK (Bhakar et al. 2003). In mammals, there are three genes encoding the JNK family, JNK1–3. While JNK1 and JNK2 are ubiquitously expressed, JNK3 is selectively expressed in the nervous system and heart (Gupta et al. 1996; Kuan et al. 2003; Mohit et al. 1995) and has been suggested to be the primary isoform mediating neuronal death in response to a variety of ligands and insults (Dhanasekaran and Reddy 2008). Of these three JNK isoforms, JNK3 was selectively activated following ligand binding to p75NTR in oligodendrocytes (Harrington et al. 2002) and sympathetic neurons (Kenchappa et al. 2010), and gene deletion of JNK3 prevented receptor-mediated apoptosis both in vitro and in vivo (Kenchappa et al. 2010; Li et al. 2007).
Further support for p75NTR activating a JNK-p53 apoptotic pathway comes from the fact that cell death mediated by the receptor is associated with upregulation of p53 (Aloyz et al. 1998; Linggi et al. 2005). Induction of apoptosis by p75NTR has also been linked to phosphorylation of Bim (Becker et al. 2004) and Bad (Bhakar et al. 2003), cytochrome c release (Bhakar et al. 2003), and cleavage of procaspase-3, -6, -7, or -9 (Bhakar et al. 2003; Tabassum et al. 2003). Curiously, however, the receptor does not require c-Jun for killing sympathetic neurons (Palmada et al. 2002).
3.2 Cytosolic Factors Linking p75NTR to JNK
Like many other receptors of the tumor necrosis factor (TNF) receptor superfamily, p75NTR promotes downstream signaling via association with a number of cytosolic interactors (Fig. 1). One group of p75NTR interactors that contributes to activation of JNK is the family of TNF receptor-associated factors (TRAFs). TRAF family proteins are distinguished by a conserved C-terminal domain that is responsible for their oligomerization and interactions with the cytoplasmic domains of TNF receptor family members (Zotti et al. 2012). With the exception of TRAF1, all TRAF family members also feature an N-terminal domain containing RING and zinc finger structures that are critical for their signaling function. The RING finger domain in the TRAFs acts as an E3 ubiquitin ligase, but instead of targeting proteins for proteasomal degradation, the TRAFs form a ubiquitin chain through Lys 63 linkages, which serve as protein–protein interaction motifs (Ha et al. 2009; Hacker et al. 2011). TRAF1–6 have been reported to associate with p75NTR, with TRAF2, 4, and 6 shown to modulate p75NTR-induced cell death via interactions with the ICD of the receptor (Khursigara et al. 1999; Ye et al. 1999). However, the role of TRAF6 in p75NTR signaling has been the most thoroughly studied. TRAF6 associates with p75NTR in a ligand-dependent manner (Khursigara et al. 1999) and mediates signaling from the receptor to both JNK and NF-κB (Khursigara et al. 1999; Yeiser et al. 2004). Sympathetic neurons from traf6−/− mice fail to activate JNK in response to BDNF binding to p75NTR and fail to undergo apoptosis (Yeiser et al. 2004). Furthermore, there is reduced developmental cell death in the superior cervical ganglia in traf6−/− mice relative to the wild type, indicating that TRAF6 is essential for p75NTR-mediated apoptotic signaling in vivo.

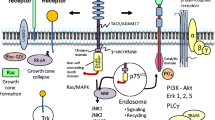
Signaling pathways mediated by p75NTR that regulate cell survival and apoptosis. In response to neurotrophin binding, p75NTR promotes JNK activation via interactions with NRAGE, TRAF6, and NRIF, thus leading to apoptosis. Activation of JNK by p75NTR also occurs through induction of sphingomyelinases. The chopper domain of p75NTR promotes apoptosis by facilitating depletion of internal K+ through GIRK channels. Other cytosolic interactors contribute to p75NTR-mediated cell death, including NADE, MAGE-G1, and Necdin. In response to pro-neurotrophins, p75NTR inhibits Trk-mediated survival signaling via induction of PTEN and the resultant inhibition of PI3K-Akt survival signaling. Promotion of cell survival by p75NTR is facilitated by its interactions with Trk receptors which enhance Trk-mediated PI3K-Akt survival signaling, as well as other Trk-mediated survival pathways. P75NTR may also promote survival via activation of NFκB, possibly through associations between RIP2 and TRAF6 (abbreviations: DD p75NTR death domain, C p75NTR chopper domain, K Trk receptor tyrosine kinase domain)
TRAF6 also associates with the neurotrophin receptor-interacting factor (NRIF) to promote JNK activation (Gentry et al. 2004; Linggi et al. 2005). NRIF is a zinc-finger protein that was first identified in a yeast 2-hybrid screen for proteins interacting with the ICD of p75NTR (Casademunt et al. 1999). NRIF and TRAF6 can directly interact, and overexpression of NRIF together with TRAF6 enhanced TRAF6-mediated JNK activation (Gentry et al. 2004). Furthermore, BDNF-induced JNK activation and cell death were significantly attenuated in nrif−/− sympathetic neurons (Linggi et al. 2005). Gene deletion revealed that NRIF was required for developmental apoptosis in the retina (Casademunt et al. 1999), which is a p75NTR-dependent process (Frade et al. 1996). Thus, interaction of NRIF with TRAF6 and p75NTR appears to be critical for p75NTR-mediated JNK activation and apoptosis. However, expression of NRIF alone in mouse embryonic fibroblasts was not sufficient to activate the kinase, although it did induce cell death (Linggi et al. 2005). Exactly how NRIF contributes to the activation of JNK is not clear, but it may facilitate oligomerization of TRAF6, which is necessary for it to mediate its biological actions (Yin et al. 2009).
Another intracellular binding partner of p75NTR that is linked to JNK activation is the neurotrophin receptor-interacting MAGE homolog, NRAGE (also known as Maged1 and dlxin) (Salehi et al. 2002). NRAGE contains a melanoma-associated antigen (MAGE) domain, which is a region of homology defining the MAGE family of proteins. The function of the MAGE proteins is poorly understood, but many have been implicated in the regulation of cell cycle and apoptosis (Sang et al. 2011). Ectopic expression of NRAGE along with p75NTR in a sympathetic precursor cell line enabled NGF-dependent cell death, thereby implicating this interactor in the apoptotic pathway activated by p75NTR (Salehi et al. 2000). Overexpression of NRAGE in PC12 cells led to potent activation of JNK, release of cytochrome c from mitochondria, and the induction of caspases -3, -6, and -9, ultimately resulting in cell death (Salehi et al. 2002). These results suggested that NRAGE could be involved in p75NTR-mediated stimulation of JNK. Corroborating evidence came from analysis of nrage−/− mice: p75NTR-induced JNK activation in nrage−/− sympathetic neurons was significantly reduced compared to wild-type neurons (Bertrand et al. 2008). Furthermore, the null animals have an increased number of neurons in their superior cervical ganglia, like p75 NTR −/− mice, and sympathetic neurons isolated from nrage−/− mice were resistant to p75NTR-mediated apoptosis (Bertrand et al. 2008). These results suggest a function for NRAGE as an adaptor protein, linking the receptor to JNK activation and apoptosis. Whether NRAGE, TRAF6, and NRIF form a complex or function independently to regulate the kinase remains an open question; however, they may function at different stages of the cascade to affect the kinetics of JNK activity (discussed below). It should be noted that sequestering the anti-apoptotic factor XIAP (Jordan et al. 2001; Kendall et al. 2005) and promoting degradation of the anti-apoptotic transcription factor Che1 (Di Certo et al. 2007) have also been suggested as mechanisms through which NRAGE affects cell survival, though these interactions have not been studied in the context of p75NTR signaling.
Another mechanism through which p75NTR has been suggested to regulate JNK involves production of the lipid signaling molecule ceramide (Fig. 1). When the field was searching for evidence of signaling by p75NTR, a NGF-mediated increase in ceramide levels through activation of neutral sphingomyelinase in T9 glioma cells was one of the first signals detected (Dobrowsky et al. 1994). Multiple reports have since confirmed the ability of p75NTR to stimulate ceramide production in other cell types, including in oligodendrocytes (Casaccia-Bonnefil et al. 1996), hippocampal neurons (Brann et al. 2002), Schwann cells (Hirata et al. 2001), and mesencephalic neurons (Blochl and Sirrenberg 1996). One known downstream effect of elevated ceramide is activation of JNK (Westwick et al. 1995), and thus ceramide may couple p75NTR to JNK phosphorylation. Indeed, in cultured hippocampal neurons activation of p75NTR resulted in upregulation of ceramide, stimulation of JNK, and cell death (Brann et al. 2002). Furthermore, inhibition of sphingomyelinase in these neurons prevented ceramide accumulation, JNK activation, and the induction of apoptosis. However, increasing ceramide levels does not always result in cell death. In fact, p75NTR-mediated ceramide production has also been linked to promotion of cell survival (DeFreitas et al. 2001; McCollum and Estus 2004). Understanding this lipid signaling pathway is complicated by the fact that ceramide is a central intermediate in sphingolipid metabolism and can have a variety of effects depending on the specific fatty acid chain attached and its cellular concentration and localization (Horres and Hannun 2012). Further studies are needed to elucidate the mechanisms by which p75NTR activates sphingomyelinase and to reveal how ceramide elicits its effects in various cellular contexts.
3.3 Other Factors Involved in p75NTR Mediated Apoptosis
Apart from TRAF6, NRIF, and NRAGE, several other cytosolic proteins have been shown to associate with p75NTR and suggested to regulate its apoptotic signaling. For example, p75NTR-associated cell death executor (NADE), a novel protein isolated in a two-hybrid screening for proteins binding to the ICD of the receptor, was reported to associate with endogenous p75NTR in PC12 cells (Mukai et al. 2000). Overexpression of NADE together with p75NTR in HEK 293 cells induced apoptosis (Mukai et al. 2000) and expression of a fragment of NADE lacking the region identified as necessary for promoting apoptosis blocked receptor-mediated cell death in oligodendrocytes (Mukai et al. 2002). Currently, though, how NADE contributes to p75NTR-mediated apoptotic signaling is unknown. In addition, MAGE-G1, MAGE-H1 and the MAGE-related protein, Necdin, have also been shown to interact with p75NTR (Kuwako et al. 2004; Tcherpakov et al. 2002). Both Necdin and MAGE-G1 associate with E2F1, a transcription factor that is important for G1/S transition in the cell cycle and that can induce apoptosis in postmitotic cells (Ginsberg 2002). When the ICD of p75NTR was overexpressed in a neuroblastoma cell line, Necdin and MAGE-G1 bound to the receptor ICD, thereby releasing E2F1 and triggering apoptosis (Kuwako et al. 2004; Lopez-Sanchez et al. 2007). Additional studies are needed to determine whether Necdin and MAGE-G1 regulate ligand-mediated cell death in primary cells. The p75NTR has also been reported to promote apoptosis through upregulation of the sugar binding protein Galectin-1 (Plachta et al. 2007). Embryonic stem (ES) cells engineered to express p75NTR degenerated when they were induced to differentiate into neurons. This degeneration correlated with expression of Galectin-1, which promoted death of the ES cells as well as cortical neurons (Plachta et al. 2007). Furthermore, mice lacking Galectin-1 were resistant to neuronal apoptosis caused by pilocarpine-induced seizures (Bischoff et al. 2012), which was demonstrated to be a p75NTR-dependent process (Roux et al. 1999; Volosin et al. 2008). The mechanisms by which this lectin causes cell death remain to be determined.
3.4 Regulated Intramembrane Proteolysis of p75NTR
In a manner similar to Notch and amyloid precursor protein (APP), p75NTR undergoes regulated intramembrane proteolysis (RIP). Proteolysis of p75NTR was first described as a response to phorbol esters in HEK293 cells transfected with the receptor (Jung et al. 2003; Kanning et al. 2003). The extracellular region of p75NTR is first cleaved by the metalloproteinase TNFα-converting enzyme (TACE, also known as ADAM17), thereby producing a 24 kDa membrane-bound C-terminal fragment (p75NTR-CTF) (Weskamp et al. 2004). This cleavage event appears to be quite promiscuous in terms of the amino acid sequence; however, deletion analysis revealed that at least 15 residues extracellular to the transmembrane domain are required (Zampieri et al. 2005). Following release of the soluble ectodomain, the p75NTR-CTF is then further cleaved within its transmembrane region by the γ-secretase complex, thereby releasing the 19 kDa intracellular domain of the receptor (p75NTR-ICD). Similar to proteolysis by TACE, cleavage by γ-secretase is quite permissive for various amino acids; nevertheless, there must be some sequence specificity for both enzymes since substituting the transmembrane domain of Fas for that of p75NTR blocked cutting by γ-secretase and replacing the 15 amino acid juxtamembrane sequence with the Fas sequence blocked p75NTR proteolysis by TACE (Zampieri et al. 2005). The order of the two cleavage reactions is also invariant, with TACE acting on the receptor prior to γ-secretase. This was determined by studies in which cleavage of p75NTR by γ-secretase was prevented by TACE inhibition, but inhibition of γ-secretase did not affect TACE activity, thus indicating that release of the extracellular domain is required for further proteolysis of the receptor within the transmembrane domain (Kenchappa et al. 2010; Zampieri et al. 2005). Since the initial finding of RIP of p75NTR in response to phorbol esters, a number of reports have demonstrated that proteolysis of p75NTR occurs through a ligand-dependent mechanism; for example, treatment of sympathetic neurons with BDNF (Kenchappa et al. 2006, 2010), Schwann cells with NGF (Frade 2005), and cerebellar neurons with myelin-associated glycoprotein (Domeniconi et al. 2005) (this ligand is discussed below) resulted in RIP. It is unclear, however, whether ligand activated p75NTR always results in RIP.
One functional role of p75NTR cleavage, like for many γ-secretase substrates, is to facilitate signaling to the nucleus. Release of the p75NTR-ICD may facilitate nuclear translocation of associated factors such as NRIF. Although NRIF was shown to be required for p75NTR-mediated apoptotic signaling based on analyses of nrif−/− mice (Casademunt et al. 1999; Linggi et al. 2005), exactly how it contributed to the cell death was not clear. NRIF contains a classic C2H2 zinc-finger motif (Casademunt et al. 1999), which is typically found among DNA binding transcription factors (Wolfe et al. 2000), suggesting that in addition to facilitating JNK activation, NRIF could bind DNA and regulate transcription. The recognition of p75NTR proteolysis by γ-secretase revealed a possible mechanism by which NRIF could be translocated from the surface-bound ICD of the receptor to the nucleus. Indeed, it was demonstrated that BDNF-induced cleavage of the receptor in sympathetic neurons facilitated nuclear localization of NRIF and, subsequently, apoptosis (Kenchappa et al. 2006). Blocking receptor cleavage prevented both localization of NRIF to the nucleus and cell death. A similar signaling cascade has been detected in hippocampal neurons, where neuronal death due to pilocarpine-induced seizures was associated with p75NTR proteolysis and NRIF nuclear translocation. Moreover, the number of apoptotic neurons after seizure was significantly reduced in p75 NTR −/− (Troy et al. 2002) and in nrif−/− mice (Volosin et al. 2008).
The mechanism of NRIF nuclear translocation also depends on TRAF6-mediated ubiquitylation. TRAF6 was shown to ubiquitylate NRIF following ligand binding to p75NTR, and blocking this event by mutating the ubiquitin-attachment site of NRIF prevented its nuclear translocation and inhibited p75NTR-mediated apoptosis (Geetha et al. 2005). The ubiquitylation of NRIF required p75NTR cleavage (Kenchappa et al. 2006), suggesting that receptor proteolysis facilitates an interaction between NRIF and TRAF6, enabling ubiquitylation of NRIF, which is needed for it to enter the nucleus, and oligomerization of TRAF6, which promotes the activation of JNK (Fig. 2).
The cleavage of p75NTR and the activation of JNK were recently shown to occur through interdependent pathways. In sympathetic neurons, JNK activation was required for ligand-induced proteolysis of the receptor by both TACE and γ-secretase (Kenchappa et al. 2010), as blocking JNK activity or deleting JNK3 prevented receptor cleavage by both proteases. The activation of JNK facilitated the transcriptional upregulation of TACE and, through an unknown mechanism, stimulated both TACE and γ-secretase, thereby inducing p75NTR processing. Interestingly, the release of the receptor’s ICD, along with NRIF and TRAF6, was necessary for prolonged JNK stimulation by the receptor. Expression of a non-cleavable mutant p75NTR prevented JNK activation at 24 h, yet the kinase was still activated for the first hour after ligand binding (Kenchappa et al. 2010). Hence, there appears to be a biphasic activation of JNK by p75NTR, with an early signal, perhaps initiated through NRAGE, inducing proteolytic processing of the receptor, which allows NRIF and TRAF6 to promote long-term stimulation of the kinase as well as nuclear signaling, ultimately resulting in cell death (Fig. 2).
In contrast to the evidence that proteolytic processing of p75NTR induces apoptosis by releasing the p75NTR-ICD, in certain cellular contexts programmed cell death may be activated by the p75NTR-CTF alone. Coulson et al. found that overexpression of the p75NTR-CTF was sufficient to promote the apoptosis of dorsal root ganglion (DRG) neurons and that the death domain was not necessary (Coulson et al. 2000; Underwood et al. 2008). This function of the p75NTR-CTF required a 29 amino acid sequence in the cytoplasmic juxtamembrane region of the receptor termed the “chopper domain” (Coulson et al. 2000). Coulson and colleagues demonstrated that ectopic expression of membrane-associated fragments of p75NTR containing the chopper domain promoted apoptosis by inducing a Rac-dependent increase in phosphatidylinositol 4,5-bisphosphate (PIP2). In turn, PIP2 stimulated G-protein-coupled inwardly rectifying potassium (GIRK) channels, causing a depletion of internal potassium that ultimately activated an apoptotic protease activating factor 1 (APAF-1)-dependent cell death pathway (Coulson et al. 2004, 2008; Skeldal et al. 2011). It should be cautioned, however, that these studies relied on overexpression of the CTF; thus, further studies are needed to determine how the various fragments of the receptor regulate cell death under different physiological conditions.

Cell death signaling initiated by regulated intramembrane proteolysis (RIP) of p75NTR. Stimulation of p75NTR by neurotrophins promotes an early phase of JNK activation, occurring within 30 min of ligand binding. Through a mechanism currently unknown, JNK induces sequential proteolytic cleavage of p75NTR by TACE and γ-secretase. Release of the p75NTR intracellular domain promotes TRAF6-dependent ubiquitylation and nuclear translocation of NRIF, as well as persistent JNK activation, ultimately leading to induction of programmed cell death (abbreviations: DD death domain, C chopper domain, U ubiquitin)
3.5 Proneurotrophins and Sortilin
The initial discovery that p75NTR can induce programmed cell death was somewhat puzzling, as in vitro studies indicated that relatively high concentrations of neurotrophins were needed to induce apoptosis, and in certain cell types, cross-reactivity of neurotrophins with Trk receptors could potentially promote an opposing, pro-survival signal. An answer was found, at least in part, by Hempstead’s group, who discovered that precursor forms of neurotrophins are biologically active, selective ligands for p75NTR. Like most secreted proteins, neurotrophins are initially synthesized as larger precursors, which are enzymatically cleaved to generate the mature form of the protein (Edwards et al. 1988; Suter et al. 1991). Proneurotrophins have an amino-terminal pro-domain that assists in their proper folding and dimerization (Heymach and Shooter 1995; Rattenholl et al. 2001a, b). The pro-domain can be proteolytically removed by furin and pro-protein convertases in the endoplasmic reticulum and Golgi apparatus (Seidah et al. 1996). Alternatively, the cleavage of the pro-domain can also be mediated by plasmin and matrix metalloproteases following secretion of the proneurotrophin into the extracellular milieu (Lee et al. 2001). While it was originally thought that mature neurotrophins are the only physiologically active ligands for p75NTR, it is now well established that endogenous proneurotrophins can be secreted to function as potent activators of p75NTR signaling (Beattie et al. 2002; Harrington et al. 2004; Lebrun-Julien et al. 2010; Lee et al. 2001; Teng et al. 2005).
Proneurotrophins do not activate Trk receptors (Boutilier et al. 2008; Lee et al. 2001) and have been demonstrated to induce significant p75NTR-mediated cell death at sub-nanomolar concentrations (Lee et al. 2001). Thus, proteolytic processing determines the functional fate of nascent neurotrophins, with uncleaved forms selectively triggering p75NTR-mediated cell death and mature forms activating either p75NTR or Trk receptors, depending upon the cellular context. Proneurotrophins induce programmed cell death by binding to a high affinity protein complex containing p75NTR and its co-receptor Sortilin, a member of the Vps10p-domain receptor family (Nykjaer et al. 2004; Teng et al. 2005). Mammalian members of the Vps10p family, which consists of Sortilin, SorLA, and SorCS-1, -2, and -3, are type I transmembrane receptors with multifunctional roles that include the modulation of protein sorting and trafficking, as well as regulation of signal transduction (Willnow et al. 2008). Proneurotrophins bind to Sortilin via their pro-domain and to p75NTR by their mature domain, thus facilitating the association of these two receptors to initiate programmed cell death (Nykjaer et al. 2004, 2005; Teng et al. 2005). Following initial reports that Sortilin mediates neurotrophin-induced cell death in vitro (Nykjaer et al. 2004; Teng et al. 2005), studies have indicated that Sortilin is required for developmental p75NTR-mediated cell death in vivo. For example, mice lacking Sortilin have a reduction in the developmental apoptosis of retinal ganglion cells that is indistinguishable from that of p75NTR-deficient mice (Jansen et al. 2007). However, Sortilin may not be required for all p75NTR-mediated cell death, as these mice did not have defects in the apoptosis of sympathetic neurons during the developmental time period in which p75NTR-mediated death is known to occur (Jansen et al. 2007). Loss of Sortilin did, however, impair age-related degeneration of these neurons, suggesting that proneurotrophins may not have been involved in the early development of the sympathetic neurons, but do have a role in their loss during aging.
3.6 Apoptotic Role of p75NTR in Pathology
In addition to its critical role during neurodevelopment, p75NTR is a stress-activated receptor that stimulates the death of cells within injured tissue. Though the receptor is downregulated in most regions of the nervous system after early postnatal development, reexpression of p75NTR occurs in response to many forms of cellular damage. For example, increases in p75NTR expression have been reported following neuronal axotomy (Ernfors et al. 1989; Giehl et al. 2001; Harrington et al. 2004; Koliatsos et al. 1991; Taniuchi et al. 1986), mechanical damage (Beattie et al. 2002; Brunello et al. 1990; Rende et al. 1993), elevated intraocular pressure (Wei et al. 2007), seizures (Roux et al. 1999; Volosin et al. 2008), and focal ischemia (Kokaia et al. 1998). Beyond measuring increases in expression of the receptor, multiple studies have more definitively demonstrated that p75NTR signaling is responsible for injury-induced cell death in vivo. In one such study, unilateral administration of kainic acid to the basal forebrain resulted in reexpression of p75NTRin the degenerating cholinergic neurons, which correlated with their apoptosis. Administration of a function-blocking p75NTR antibody prevented this cell death, thereby indicating that p75NTR signaling contributes to excitotoxin-induced death of basal forebrain neurons (Oh et al. 2000). Similarly, expression of p75NTR was induced and associated with programmed cell death caused by axotomy of corticospinal neurons, and antibodies to p75NTR prevented this apoptosis (Giehl et al. 2001). Although these two studies indicated that p75NTR promotes neuronal death after injury, whether proneurotrophins contribute to the death caused by these injuries was not known. In a later report, injury to the spinal cord was found to induce production of proNGF and to stimulate p75NTR-dependent apoptosis of spinal cord oligodendrocytes (Beattie et al. 2002). ProNGF extracted from the injured region elicited apoptosis of cultured oligodendrocytes expressing p75NTR but not of p75 NTR −/− oligodendrocytes. Thus, this work suggested that proNGF functions to promote the elimination of damaged cells by activating p75NTR after spinal cord injury (Beattie et al. 2002). A subsequent study by Yoon and colleagues demonstrated that axotomy of corticospinal neurons also resulted in apoptosis of the neurons through a proNGF–p75NTR-dependent mechanism (Harrington et al. 2004). Following lesion of the internal capsule, proNGF was detected in cerebral spinal fluid, indicating that proNGF is produced and secreted in vivo after brain injury. In the cortex of lesioned animals, an interaction between proNGF and p75NTR was detected in vivo, and disruption of this interaction by infusion of an antibody specific for proNGF prevented the apoptosis caused by the injury (Harrington et al. 2004). These experiments provided the first conclusive evidence that proNGF is a pathophysiological ligand that induces apoptosis in response to neuronal damage. Since then, a growing body of evidence has linked proneurotrophins to cell death induced by various types of injury. For example, hippocampal seizures stimulated the upregulation and secretion of proNGF in vivo, and antibodies specific for proNGF prevented seizure-induced apoptosis of neurons within the dentate gyrus (Volosin et al. 2008). In another study, increases in proNGF, along with p75NTR and Sortilin, were reported in the retina after exposure of albino mice to intense light, and blockade of Sortilin with the pro-domain of proNGF attenuated light-induced retinal cell death (Santos et al. 2012). The ability of proneurotrophins to induce cell death in response to cellular damage is likely not specific to proNGF, as proBDNF has also been demonstrated to promote apoptosis in a number of cell culture models (Fan et al. 2008; Taylor et al. 2012; Teng et al. 2005), and upregulation of proBDNF has been detected in in vivo injury models, such as in an animal model of cochlear damage (Tan and Shepherd 2006). ProBDNF has also been implicated in apoptosis occurring due to neuronal axotomy, as infusion of a proBDNF antibody prevented the death of sensory neurons induced by lesion of the sciatic nerve in vivo (Fan et al. 2008).
While the signaling mechanisms responsible for the induction of p75NTR and the proneurotrophins after injury are not well understood, several studies have provided some clues. One possibility is that cellular damage prevents the proteolytic processing of neurotrophins, thus increasing the release of death inducing proneurotrophins. A recent study by Friedman and colleagues has revealed that following kainic acid-induced seizures, the proneurotrophin-processing enzyme matrix metalloproteinase-7 (MMP-7) and its inhibitor tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) were regulated in a manner that would hinder cleavage of proneurotrophins and lead to increased release of proNGF (Le and Friedman 2012). Decreased MMP-7 production has also been observed in samples from human patients and animal models with diabetic retinopathy (Ali et al. 2011). These findings suggest that regulation of proteolytic processing of proneurotrophins is one mechanism by which the levels of these factors are modulated, though a greater understanding of the pathways regulating their release in the unprocessed versus the mature form is needed. Along with increases in the levels of proneurotrophins, upregulation of p75NTR after injury may occur due to inflammatory signals released in response to tissue damage. The inflammatory cytokines interleukin-12 (IL-12), tumor necrosis factor alpha (TNFα), and interleukin-1β have been demonstrated to increase p75NTR expression in a variety of in vitro systems, such as in cultured hippocampal neurons (Choi and Friedman 2009), natural killer cells (Rogers et al. 2010), or astrocytes (Choi and Friedman 2009). Interestingly, a recent report indicated that trauma-induced upregulation of p75NTR could also result from calcium influx. Within axotomized hippocampal neurons, cellular responses to GABA change from hyperpolarizing to depolarizing, leading to increased intracellular calcium and the subsequent activation of Rho kinase (ROCK). The activation of ROCK resulted in upregulation of p75NTR, ultimately leading to neuronal death (Shulga et al. 2012). Thus, multiple signals may contribute to trauma-induced upregulation of the receptor. However, the mechanisms by which these signals increase p75NTR transcription are still poorly understood. The ubiquitous transcription factor Sp1 has been linked to p75NTR basal expression (Poukka et al. 1996) and upregulation following hypo-osmotic stress (Kommaddi et al. 2011a; Ramos et al. 2007), but whether this factor is involved in other forms of injury is not known. It should also be added that long-term treatment of SH-SY5Y cell lines with IGF1 resulted in a significant upregulation of p75NTR levels (Costantini et al. 2006), suggesting that a factor that responds to IGF1 signaling may also be involved.
In addition to regulating cell death following injury, p75NTR signaling has been suggested to contribute to neurodegeneration caused by a number of diseases. Among these disorders, the link between p75NTR and Alzheimer’s disease (AD) has been most studied. Besides Purkinje neurons in the cerebellum, p75NTR is expressed at high levels in cholinergic neurons of the adult basal forebrain, a population of neurons that undergoes severe degeneration early in the progression of AD pathology. Additionally, several in vitro studies have indicated that amyloid beta 1–42 (Aβ), the main component of plaques commonly found within brains of AD patients, is a pro-apoptotic ligand for p75NTR (Costantini et al. 2005; Hashimoto et al. 2004; Yaar et al. 1997). These findings have led to the hypothesis that activation of p75NTR by Aβ contributes to neurodegeneration caused by AD. This idea has remained controversial, however, due to other reports indicating that expression of p75NTR is protective against Aβ-induced toxicity (Bengoechea et al. 2009; Zhang et al. 2003). Nonetheless, a role for p75NTR in Aβ-induced neurotoxicity was recently strengthened by an in vivo finding that deletion of p75NTR prevented the degeneration of cholinergic basal forebrain neurons in vivo following Aβ injection into the hippocampus (Sotthibundhu et al. 2008). Furthermore, when p75 NTR −/− mice were crossed with the Thy1-hAPPLond/Swe mouse model of AD, the degeneration of hippocampal and forebrain cholinergic fibers was dramatically rescued (Knowles et al. 2009). Just as for the in vitro studies, however, these in vivo studies were also challenged by a recent study by Wang et al, which indicated that p75NTR signaling induces production of Aβ, since deletion of the p75 NTR gene in the APPswe/PS1dE mouse model of AD resulted in decreased production of Aβ within cortical neurons (Wang et al. 2011). Despite some differences, these findings together suggest that p75NTR signaling by Aβ peptides contributes to overall AD pathology. Apart from Aβ-induced apoptosis, studies have also implicated proNGF in AD pathology. Increased expression of proNGF has been detected in human brains affected by AD (Pedraza et al. 2005; Peng et al. 2004), and proNGF isolated from these brain samples induced p75NTR-mediated death of cultured sympathetic neurons (Pedraza et al. 2005; Podlesniy et al. 2006). Thus, in addition to activation of p75NTR by Aβ, enhanced production of proNGF may contribute to neurodegeneration within the AD brain. While these studies provide multiple links between p75NTR signaling and AD-induced neurodegeneration, collective evidence suggests that the degeneration of neurons in AD occurs near the end-stages of the disease (Jack et al. 2010). Hence, understanding whether p75NTR plays a critical role in the onset and early progression of AD remains essential.
While the majority of studies related to p75NTR and neurodegenerative disease have focused on the contributions of the receptor to AD, it is perhaps not surprising that p75NTR has been linked to a number of other disorders. For example, p75NTR may contribute to degeneration of motor neurons during the progression of amyotrophic lateral sclerosis (ALS). Though p75NTR is downregulated in motor neurons of the spinal cord during the perinatal period, reexpression of the receptor was detected in spinal motorneurons of an ALS mouse model (Copray et al. 2003; Lowry et al. 2001), as well as in spinal cord samples from human patients with ALS (Lowry et al. 2001; Seeburger et al. 1993). Furthermore, the receptor was implicated in ALS-associated motoneuron death by a study in which knockdown of p75NTR delayed locomotor impairment and mortality in the SOD1G93A mouse model of ALS (Turner et al. 2003). However, when the SOD1G93A mice were crossed with the p75 NTR −/− mice, prolonged survival was only detected in the female mice, and this improvement did not correlate with increased motorneuron survival, but with reduced astrocytosis (Kust et al. 2003). Nevertheless, the SOD mutation represents a very small fraction of ALS patients, thus further study into the role of the receptor in this disease is warranted.
Degeneration of dopaminergic neurons in Parkinson’s disease (PD) could also involve p75NTR. A study by Simon and colleagues demonstrated that loss of the Engrailed transcription factors results in increased expression of p75NTR in the ventral midbrain (Alavian et al. 2009). This finding has implications for Parkinson’s disease because mice deficient in Engrailed-1 and Engrailed-2 exhibit progressive loss of mesencephalic dopaminergic neurons and have PD-like motor deficiencies (Alavian et al. 2009). Importantly, knocking down p75NTR or addition of a receptor-blocking antibody prevented the apoptosis of mesencephalic dopaminergic neurons in cultures from the engrailed 1, 2 double knockout mice. Upregulation of p75NTR in dopaminergic nigro-striatal neurons has also been reported following kainic-acid treatment (Wang et al. 2008). However, direct evidence for p75NTR expression in nigral dopaminergic neurons in PD and causal evidence linking expression of p75NTR to PD-associated nigral neurodegeneration in vivo is still missing.
In addition to these neurodegenerative conditions, evidence continues to grow implicating p75NTR in the pathology of other neurological diseases. For example, p75NTR has been suggested as having a role in spongiform encephalomyelopathy (Stoica et al. 2008), diabetes-related impairment of neovascularization (Caporali et al. 2008), and psoriasis (Truzzi et al. 2011), among others. The abundance of links between p75NTR and such a variety of diseases indicates that the receptor may function in a broader sense as a stress-induced apoptotic signal that is activated by a mechanism common to all of these pathological conditions. Thus, further elucidation of the mechanisms by which p75NTR is upregulated and activated during these pathological conditions and of the contributions of the receptor to the resulting neurodegeneration may be of critical therapeutic importance.
4 Promotion of Cell Survival
Despite its currently known role in eliciting programmed cell death, early studies of p75NTR demonstrated that in a variety of cellular contexts the receptor has the opposite function: to promote cell survival. Though p75NTR-induced apoptosis has been more widely studied, the receptor has been demonstrated to promote cell survival in a wide variety of cell types. One of the first indications that the receptor can promote neuronal survival came from analysis of p75 NTR −/− mice, which revealed significant loss of sensory innervation of limbs (Lee et al. 1992). Subsequently, the number of neurons in the dorsal root ganglia (DRG) was reported to be reduced by 50–75 % in the knockout mice (Murray et al. 1999). Although the DRG is a very heterogeneous population of neurons, a decrease in virtually all types of neurons was detected, based on morphological criteria (Bergmann et al. 1997; Gjerstad et al. 2002) or expression of various markers (Jiang et al. 2004). Since then, numerous reports have suggested that p75NTR promotes survival in a wide range of cell types, with the majority suggesting that this is through cooperation with the Trk family, leading to a high affinity receptor complex or enhanced Trk signaling.
4.1 P75NTR Interactions with the Trks
Shortly after TrkA was identified as a receptor for NGF, Chao and colleagues demonstrated that p75NTR interacts with TrkA to form a high-affinity binding complex (Hempstead et al. 1991). While TrkA alone was found to bind NGF with nanomolar affinity, co-expression with p75NTR was discovered to increase this interaction by 100-fold (Esposito et al. 2001; Hempstead et al. 1991). Thus, p75NTR can augment Trk-mediated survival by increasing its interaction with neurotrophins. Given that neurotrophins are typically present in limiting amounts in the target tissues, the presence of high-affinity receptors is an obvious advantage. The requirement for p75NTR in forming the high-affinity complex was initially offered as an explanation for the sensory neuron loss in the animals lacking the receptor. Indeed, neurotrophin dose–response curves revealed that higher doses of NGF were needed to promote survival of sensory and sympathetic neurons from p75 NTR −/− mice (Davies et al. 1993; Lee et al. 1992). However, a critical element unanswered by this interpretation of the data relates to the fact that, unlike the loss of neurons in the DRG, p75 NTR −/− mice actually have excess sympathetic neurons (Bamji et al. 1998; Deppmann et al. 2008; Jansen et al. 2007). As discussed above, p75NTR contributes to normal, developmental apoptosis of sympathetic neurons, which could explain the increased neuronal number in the knockout mice, yet why the receptor functions differently in sensory neurons has yet to be resolved.
Although functional interaction between p75NTR and Trk receptors is clear, the molecular details are not fully understood. Surprisingly, the transmembrane and intracellular domains of p75NTR, but not the neurotrophin-binding portion of the extracellular domain, are required for the high-affinity complex (Esposito et al. 2001). Furthermore, structure analysis by X-ray crystallography and complementation assays (using fragments of beta-galactosidase) indicated that complexes of each receptor bind NGF independently and that there is no direct interaction between p75NTR and TrkA (Wehrman et al. 2007). The structural analysis disagrees with many early cross-linking experiments (discussed above) and co-immunoprecipitation studies in HEK293 cells (e.g., Bibel et al. 1999) that indicate the presence of a complex of both receptors. Clearly, further study is required to resolve the nature of the high affinity complex.
It is also important to note that a p75NTR homolog, neurotrophin receptor homolog 2 (NRH2), was recently identified (Kanning et al. 2003). Like p75NTR, NRH2 can also undergo cleavage by TACE and γ-secretase (Kanning et al. 2003), associate with Sortilin (Kim and Hempstead 2009), and form a high-affinity NGF receptor with TrkA (Murray et al. 2004). NRH2 is co-expressed with p75NTR in multiple neuronal subtypes. Thus, understanding how NRH2 and p75NTR function together with their co-receptors is necessary to interpret the phenotype of p75 NTR −/− mice.
Remarkably, Trk–p75NTR interactions not only facilitate the formation of a high-affinity receptor complex but also regulate the neurotrophin selectivity of the tyrosine kinase receptor. For example, in the absence of p75NTR, TrkA can respond to both NT3 and NGF; however, the Trk–p75NTR complex is highly selective for NGF (Benedetti et al. 1993; Clary and Reichardt 1994). During the development of sympathetic neurons, NT3–TrkA interaction is necessary for neuronal survival (Ernfors et al. 1994; Farinas et al. 1994; Francis et al. 1999). Ginty and colleagues demonstrated that intermediate targets, such as blood vessels, produce NT3 and promote axon growth, but not survival, through TrkA. However, as the neurons innervate their NGF-secreting targets, p75NTR is upregulated, causing TrkA to become selective for NGF over NT3. The NGF–Trk–p75NTR complex is then retrogradely transported to promote survival (Kuruvilla et al. 2004). Hence, p75NTR can function as a switch factor, allowing differential TrkA responses. Similar selectivity is observed with TrkB ligands; co-expression of p75NTR with TrkB increased its selectivity for BDNF over NT3 and NT4 (Bibel et al. 1999).
Beyond regulating the affinity and selectivity of Trks for neurotrophins, p75NTR also potentiates Trk survival signaling. Prevention of neurotrophin binding to p75NTR attenuated TrkA signaling in several in vitro systems (Barker and Shooter 1994; Lachance et al. 1997; Ryden et al. 1997; Verdi et al. 1994). The mechanism by which p75NTR enhances Trk signaling remains poorly understood. Barker and colleagues demonstrated that co-expression of p75NTR with TrkA attenuated TrkA ubiquitylation and delayed the NGF-dependent internalization and degradation of the receptor (Makkerh et al. 2005). Therefore, one mechanism utilized by p75NTR to augment Trk-mediated survival signaling is prolonging cell surface expression of the Trk receptor. Chao and colleagues identified a large transmembrane protein, Ankyrin repeat-rich membrane spanning (ARMS/Kidins220), that interacts with both p75NTR and Trk (Kong et al. 2001). ARMS is tyrosine phosphorylated following neurotrophin treatment and is expressed in many of the neuronal populations that receive neurotrophin stimulation (Kong et al. 2001). While these data suggest that ARMS may serve as a link between p75NTR and Trk receptors, expression of ARMS was discovered to decrease association of TrkA with p75NTR (Chang et al. 2004), and the functional role of the protein has yet to be determined. Recently, however, analysis of mice lacking ARMS revealed substantial apoptosis of sensory neurons (Cesca et al. 2011), a phenotype similar to that observed in p75 NTR −/− animals (Murray et al. 1999), thus further highlighting the potential importance of this interactor in regulation of cell survival through p75NTR–Trk interaction.
In addition to p75NTR regulating Trk function at the cell surface, there is evidence that intracellular signaling pathways are modulated by co-activation of the receptors. One such signaling event particularly affected by this interaction is activation of the pro-survival kinase Akt. Treatment of PC12 cells with NGF in the presence of an antibody that blocked binding to p75NTR inhibited the activation of Akt (Bui et al. 2002). Similarly, silencing of P75NTR in PC12 cells or cerebellar granule neurons reduced neurotrophin-induced activation of the kinase (Ceni et al. 2010). These authors also reported that activation of Akt required proteolysis of p75NTR. In contrast to what was observed for p75NTR in sympathetic neurons (Kenchappa et al. 2006), Ceni et al. reported that the cleavage of p75NTR was induced by Trk activation. They have since extended these findings, demonstrating that Trk activation promoted phosphorylation of TACE, which activated the protease and lead to cleavage of p75NTR, which was necessary for potentiation of neurotrophin-induced survival signaling (Kommaddi et al. 2011b).
Although p75NTR can function in cooperation with Trks to promote survival signals, Friedman and colleagues found that in basal forebrain neurons, p75NTR reduced Akt signaling by increasing the levels of active PTEN (phosphatase and tensin homolog deleted on chromosome 10), an inhibitor of the PI3 kinase–Akt pathway (Song et al. 2010). ProNGF binding to p75NTR upregulated PTEN, which resulted in apoptosis of the neurons, even if TrkB was activated by BDNF. Since proNGF signals through binding a complex of p75NTR and Sortilin, the effects of p75NTR on Akt activity appears to depend on its co-receptor.
4.2 P75NTR Activation of NFκB
Apart from enhancing Trk survival signaling, there is evidence that p75NTR can activate an independent pro-survival signal; for example, selective activation of p75NTR prevented the death of certain neuroblastoma (Cortazzo et al. 1996) and breast cancer cells (Verbeke et al. 2010), of hippocampal neurons treated with NMDA (Bui et al. 2002), and of both sensory neurons (Longo et al. 1997) and cortical subplate neurons deprived of trophic support (DeFreitas et al. 2001). In addition, the receptor appears to play a protective role after certain injuries; e.g., p75 NTR −/− mice have increased death of primary auditory neurons following acoustic trauma (Tan et al. 2010). The pro-survival effects of p75NTR appear to be more of a modulatory signal, as they are not as potent as the effects of tyrosine kinases like the Trks. The molecular mechanisms by which p75NTR promotes survival independent of the Trk receptors are not fully understood; however, one downstream pathway that has been identified involves the transcription factor nuclear factor kappa B (NFκB). NFκB is best characterized for its role in the immune system, where it is activated by many cytokine and Toll-like receptors, leading to upregulation of other cytokines and pro-survival genes (Baldwin 2012). The activation of NFκB by p75NTR was first reported in Schwann cells (Carter et al. 1996) and has since been demonstrated in a variety of cell types, including Schwannoma cells (Gentry et al. 2000), primary Schwann cells (Khursigara et al. 2001), trigeminal neurons (Hamanoue et al. 1999), and hippocampal neurons (Culmsee et al. 2002). NFκB exists as a dimer, held in the cytosol through binding to its inhibitor IκB. The transcription factor is activated through phosphorylation of IκB by the IκB kinase (IKK) complex, leading to proteasomal degradation of the inhibitor and release of the NFκB dimer to translocate into the nucleus (Baldwin 2012). As mentioned above, neurotrophin binding to p75NTR can recruit members of the TRAF family, which activate the IKK complex (Ha et al. 2009; Hacker et al. 2011); specifically, TRAF6 was shown to mediate activation of NFκB, as Schwann cells from traf6−/− mice did not respond to p75NTR activation (Yeiser et al. 2004). Since TRAF6 promotes both NFκB and JNK activation, it was recognized as a potential nodal point for determining survival vs. apoptotic signaling. How TRAF6 selectively promotes one pathway over the other remains to be fully elucidated; however, the finding that the adaptor protein receptor-interacting protein 2 (RIP2) directly associates with the death domain of p75NTR provided an important clue. Chao and colleagues demonstrated that expression of RIP2 in Schwann cells conferred NGF-dependent activation of NFκB through interaction with TRAF6. Expression of RIP2 in these cells also reduced JNK activation and the subsequent apoptosis (Khursigara et al. 2001). Thus, RIP2 expression may serve as the key toggle, switching TRAF6 signaling to NFκB from JNK (Fig. 1).
Another well-established activator of NFκB is Akt. Although evidence suggests that p75NTR enhances Trk activation of Akt, as discussed above, p75NTR has also been reported to promote Akt activation in a manner that was independent of Trk signaling in hippocampal neurons (Arevalo and Rodriguez-Tebar 2006), melanoma cells, and mutant PC12 cells lacking TrkA (Roux et al. 2001). Therefore, NFκB may be among the downstream pro-survival signals activated by p75NTR in some contexts.
It is also notable that the p75NTR was recently shown to regulate the stability of HIF1a, a transcription factor induced by oxidative stress that controls the expression of a wide variety of genes involved in protection from reactive oxygen species and, importantly, promoting cell survival (Hu et al. 2003). Le Moan et al. (2011) reported that the ICD of the receptor can bind the E3 ubiquitin ligase Siah2, which targets HIF1a for degradation. The interaction between p75NTR-ICD and Siah2 lead to upregulation of HIF1a and increased expression of vascular endothelial growth factor, which promoted angiogenesis after retinal hypoxia. While the authors did not address a potential role in regulating survival, given that many target genes of HIF1a are pro-survival, it will be interesting to determine whether this pathway has a role in promoting neuronal survival in response to activation of the receptor.
Because of the ability of p75NTR to augment Trk-mediated survival signaling and ligand selectivity, the overall effect of p75NTR on cell survival is quite variable by cell type and highly dependent upon the presence or absence of the Trk receptor. In general, though not always, simultaneous activation of both Trk receptors and p75NTR by mature neurotrophins results in cell survival. However, selective activation of p75NTR by neurotrophins in the absence of Trk-receptor activation more often promotes cell death than survival. For example, NGF treatment of sympathetic neurons, which express both p75NTR and TrkA, promotes neuronal survival. Stimulation of these neurons with BDNF, however, results in apoptosis, as these neurons do not express TrkB (Bamji et al. 1998). The proliferative state of the cell also may influence the effects of p75NTR signaling, as the majority of reports describing p75NTR mediated cell death have involved post-mitotic neurons, while studies of p75NTR in proliferative cells have revealed more variable survival outcomes (Skeldal et al. 2011).
5 Regulation of the Cell Cycle
One of the first effects described for selective activation of p75NTR in the absence of Trks was cell cycle arrest in a glioma cell line treated with NGF (Dobrowsky et al. 1994). The effects of p75NTR on cell cycle have since been linked to several receptor-interacting proteins, including SC-1 (Chittka et al. 2004), NRIF (Benzel et al. 2001), and NRAGE (Salehi et al. 2000). Like NRIF, SC-1 is a C2H2 zinc finger-containing protein, and translocation of SC-1 to the nucleus was demonstrated in response to NGF binding to p75NTR in transfected COS cells (Chittka and Chao 1999). Expression of SC-1 in these cells blocked cell proliferation through a mechanism involving repression of cyclin E transcription (Chittka et al. 2004). Chao and colleagues have also demonstrated that expression of p75NTR-ICD inhibited cyclin E mRNA production in HeLa cells, and the endogenous p75NTR-ICD in PC12 cells could be localized to the cyclin E promoter by chromatin immunoprecipitation following NGF treatment (Parkhurst et al. 2010). These results suggest that the ICD of p75NTR translocates to the nucleus along with SC-1 to modulate genes involved in the cell cycle. Whether SC-1 has a role in the receptor’s apoptotic signal remains an open question.
NRIF (Benzel et al. 2001) and NRAGE (Salehi et al. 2000) have also been implicated in the regulation of cell cycle based on the observation that ectopic expression of either factor in HEK 293 cells induced cell cycle arrest. It is interesting that these two proteins inhibit proliferation in immortalized fibroblasts but cause apoptosis when expressed in neurons, Schwann cells (Linggi et al. 2005), or neural precursors (Salehi et al. 2000, 2002). Perhaps the differential effects relate to the presence of the tumor suppressor p53, which is mutated or inhibited in many immortalized cells, including HEK 293s. Cell death induced by ectopic expression of NRIF is dependent on p53 (Linggi et al. 2005) and over-expression of NRAGE in breast cancer cells upregulated p53 (Du et al. 2009). One interpretation of these findings is that in response to p75NTR activation, NRIF and NRAGE may alter expression of key cell cycle genes, thereby triggering an increase in p53 expression, ultimately resulting in cell death, but in cells with p53 mutated or blocked, the effects are limited to inhibiting proliferation.
6 Regulation of Synaptic Plasticity
In addition to regulating neuronal survival, the balance of signaling between the Trks and p75NTR plays an important role in modulating synaptic efficacy. BDNF, acting through TrkB, is essential for the strengthening of synaptic function, referred to as long-term potentiation (LTP) (Figurov et al. 1996; Kang et al. 1997; Korte et al. 1998; Patterson et al. 2001). In contrast, p75NTR has a critical role in the weakening of synaptic connections, a process called long-term depression (LTD). Analysis of p75NTR knockout mice revealed a deficiency in the ability to induce LTD in hippocampal slices, although LTP was not impaired (Rosch et al. 2005; Woo et al. 2005). The mechanism by which p75NTR regulates synaptic function has yet to be fully resolved; however, glutamate receptor expression appears to be altered in the null animals. Lu and colleagues found reduced levels of the NMDA receptor subunit NR2B in p75 NTR −/− hippocampal lysates, and NMDA currents measured in slices from the null mice were not affected by the NR2B antagonist ifenprodil, while in wild types they were blocked (Woo et al. 2005). In addition, Korte’s group found impaired AMPA receptor function and decreased levels of the AMPA receptor subunits GluR2 and 3 in the hippocampus of p75 NTR −/− mice (Rosch et al. 2005). Glutamatergic signaling is essential for the development of LTD, and both NMDA and AMPA receptors have been implicated as contributing to the synaptic changes, depending on the mechanism of induction (Hunt and Castillo 2012). Therefore, the altered expression of these receptors in p75 NTR −/− mice likely contributes to their inability to induce LTD. Furthermore, these changes in glutamatergic signaling may underlie the impairments in learning, inhibitory avoidance, and habituation that have been observed in the p75NTR null mice (Peterson et al. 1999).
P75NTR may also regulate synaptic plasticity through modulating dendritic structure. No gross morphological changes in the structure of the hippocampus have been detected in the p75 NTR −/− animals; however, careful measurements of dendritic spines revealed an increase in their density and complexity (Zagrebelsky et al. 2005). Moreover, immunoelectron microscopy revealed that p75NTR is expressed on dendritic shafts and spines in the hippocampus (Woo et al. 2005). These results suggest that the receptor may be important for normal pruning and refining of these postsynaptic structures. Correspondingly, overexpression of p75NTR in hippocampal slices resulted in reduced spine density and complexity. Decreases in spine size and number are associated with LTD (Okamoto et al. 2004; Zhou et al. 2004) and increases in size and number correlate with LTP (Desmond and Levy 1986; Fifkova and Anderson 1981; Van Harreveld and Fifkova 1975); therefore, it is interesting to speculate that during LTD induction, p75NTR may cause retraction of dendritic spines. As described below, the receptor can activate the Rho family of GTPases, which regulate the actin-cytoskeleton (Yamashita et al. 1999). Thus, local dendritic activation of the receptor may result in collapse or shrinkage of a spine through activation of Rho or inhibition of Rac, ultimately resulting in reduced synaptic efficacy. However, how morphological changes in dendritic spines affect synaptic plasticity remains an open question.
The ligand responsible for activating p75NTR during LTD has been suggested to be proBDNF; however, this remains somewhat controversial. Pang et al. (2004) reported that proBDNF cleavage by the extracellular protease plasmin to produce mature BDNF was required for the induction of LTP. Previous studies demonstrated that tissue plasmingen activator (tPA), which activates plasmin from plasminogen, is secreted from axon terminals (Krystosek and Seeds 1981) and is required for LTP (Baranes et al. 1998; Frey et al. 1996; Huang et al. 1996), but what role the protease played in the process was not clear. The finding that tPA targets proBDNF provided a relevant substrate. Moreover, the fact that tPA/plasmin is acting extracellularly suggested that proBDNF is released into the synaptic cleft, where it could activate p75NTR if it’s not cleaved. Indeed, Woo et al. (2005) found that perfusion of hippocampal slices with proBDNF enhanced LTD, but slices from p75 NTR −/− mice were insensitive. More recently, this group also demonstrated that synaptic competition is regulated by a balance between pro- and mature-BDNF. Using cocultures of Xenopus myocytes and motor neurons, they demonstrated that when two neurons innervate one myocyte, the active terminal promotes cleavage of proBDNF to mature BDNF, which stabilizes the synapse through activation of TrkB. In contrast, at the less active terminal proBDNF is not cleaved and causes axon retraction through binding to p75NTR (Je et al. 2012). Such activity-dependent synaptic competition is a common principle in many areas of the developing nervous system, although the underlying molecular mechanisms are not known. The finding that pro-/mature BDNF levels can be dynamically regulated to act as the punishment and reward signal provides some important clues as to the mechanisms that could underlie this competition. Nevertheless, despite these elegant studies, results from the Barde group cast doubt on a role for proBDNF in synaptic plasticity. They reported that proBDNF was not secreted by hippocampal neurons in culture and that the induction of LTD was unaffected by conditional deletion of BDNF (both the pro- and mature-form of the neurotrophin) in neurons (Matsumoto et al. 2008). Clearly, additional studies are needed to fully understand the role of p75NTR in synaptic plasticity.
7 Promotion of Peripheral Myelination
All cells of the neural crest lineage express p75NTR during development, including those that become Schwann cells in the sciatic nerve. Schwann cells continue to express p75NTR until myelination, at which time it is downregulated. This reduction in its expression was attributed to axonal signals, whose identity still remains unknown (Lemke and Chao 1988). Due to its regulated expression in the sciatic nerve, questions have persisted as to whether p75NTR plays a role in some aspects of the myelination process by Schwann cells. Anton et al. (1994) first reported that p75NTR is involved in migration of Schwann cells: Schwann cell migration out of DRGs onto sciatic nerve explants was enhanced by NGF treatment, but REX, a p75NTR antibody, blocked its effect. The effect was, however, observed only with explants from denervated sciatic nerves and not with intact sciatic nerves, suggesting that p75NTR may play a role only after injury. When Schwann cell migration was examined during embryogenesis in trigeminal ganglia, however, p75NTR did have an effect: the extent of Schwann cell migration to the axon tips was significantly reduced in p75NTR knockout mice compared to littermates (Bentley and Lee 2000). Since trigeminal ganglion neurons also express p75NTR, Bentley and Lee examined Schwann cell migration in vitro using sciatic nerve explants. The extent of Schwann cell migration was reduced in cultures from p75NTR knockout mice compared to that in the wild-type counterparts, although NGF addition had no effect. These results suggest that p75NTR expression specifically in Schwann cells regulates their migration both during development and after injury.
Schwann cells have to migrate along the axon before they form myelin sheaths around axons. The fact that p75NTR plays a role in Schwann cell migration suggests that the receptor could regulate the myelination process. This question was first addressed by Eric Shooter’s group in 2002. Cosgaya et al. (2002) reported that blocking p75NTR signaling resulted in inhibition of myelin formation both in DRG-Schwann cell cocultures as well as in developing sciatic nerves. In particular, EM analysis of sciatic nerves that were injected with REX antibody at P0, before the onset of myelination, revealed that the myelin sheath became thinner 4 days later, without affecting the number of myelinated axons, suggesting that p75NTR promotes Schwann cell myelination in vivo. As for the ligand that activates p75NTR in Schwann cells, BDNF, which the group had previously reported to promote Schwann cell myelination (Chan et al. 2001), was shown to be responsible. Attributing the effect of BDNF to p75NTR in Schwann cells was indirect, however, because p75NTR is expressed both in axons and Schwann cells at the neonate stage in the sciatic nerve. For instance, Cosgaya et al. (2002) illustrated that while the full-length TrkB levels were barely detectable, p75NTR levels were very high in Schwann cells. While modulating BDNF signaling by either injecting excess BDNF or scavenging the neurotrophin with TrkB-Fc affected myelination in wild type mice, no effect on myelination was observed in p75NTR knockout mice. Since p75NTR is the only receptor in Schwann cells that could elicit downstream signaling to promote myelination, the lack of effect was attributed to BDNF acting through p75NTR in Schwann cells.
The idea that BDNF promotes myelination through p75NTR in Schwann cells was challenged by the Murray group (Xiao et al. 2009). In their study, a DRG-Schwann cell coculture system was utilized, wherein p75NTR expression was regulated both in DRG and Schwann cells independently. To delete p75NTR from DRG neurons, DRG neurons were isolated from p75NTR knockout mice, maintained in NGF, and subsequently seeded with rat Schwann cells. To knockdown p75NTR in Schwann cells, they transduced rat Schwann cells with p75NTR-shRNA prior to seeding them onto NGF-dependent DRG neurons. Surprisingly, adding BDNF to the NGF-dependent DRG neurons from p75NTR knockout mice failed to promote myelination, while in wild-type counterparts BDNF increased myelin protein levels by 1.5–2-fold. When p75NTR expression was knocked down in Schwann cells before they were seeded onto NGF-dependent DRG neurons, however, there was little effect, regardless of whether BDNF was present or not. This study thus demonstrated that it is p75NTR in DRG neurons and not in Schwann cells that influences Schwann cell myelination.
Tep et al., on the other hand, reported opposite results by using the same DRG-Schwann cell coculture system, wherein p75NTR expression was knocked down only in Schwann cells before they were seeded onto NGF-dependent DRG neurons (Tep et al. 2012): the number of myelinated nerves were reduced by ≥50 %. As for the reason why the two studies differ, Tep et al. stated that while Xiao et al. isolated DRG neurons at P2 and cultured them in the presence of NGF for 2–3 weeks, Tep et al. cultured DRG neurons from embryonic day 15 animals and cultured them for a week before Schwann cells were seeded. It is not clear whether the age of DRG neurons subjected to myelination in culture is responsible for the opposite outcome, since the studies were conducted independently. Since p75NTR was shown to affect the extent of remyelination after injury (Song et al. 2006), it is possible that under injury conditions in the adult PNS, p75NTR expressed in DRG neurons controls aspects of remyelination. What now awaits is the analysis of conditional p75NTR knockout mice in Schwann cells and neurons, individually, to resolve the issue.
Regardless of where p75NTR exerts its effect, the question remained as to what signaling pathway p75NTR employs to promote myelination in Schwann cells. P75NTR was shown to regulate small GTPases that are critical for cytoskeletal reorganization, such as RhoA (Domeniconi et al. 2005; Harrington et al. 2008; Passino et al. 2007; Wang et al. 2002; Yamashita et al. 1999, 2002; Yamashita and Tohyama 2003; Yamauchi et al. 2004) and Rac1 (Deinhardt et al. 2011; Harrington et al. 2002; Tep et al. 2012) in a variety of tissues both in and outside of the nervous system. During Schwann cell myelination, p75NTR-mediated Rac1 activation appears to be important, as partitioning-defective 3 (Par3) was shown to bind p75NTR through its PDZ-binding tri-peptide motifs in Schwann cells (Chan et al. 2006). Par3 is a member of the polarization complex that includes Par6 and protein kinase C, regulating activation of Rac1 and Cdc42 (Goldstein and Macara 2007). In Schwann cells, Par3 was colocalized with p75NTR at axon–glial interfaces in response to BDNF (Chan et al. 2006) and was shown to mediate Rac1 activation by BDNF (Tep et al. 2012). Rac1 activation was also significantly reduced in neonate sciatic nerves from p75NTR knockout mice, supporting the notion that p75NTR activates Rac1 as it forms a complex with Par3 at the axon–glial interface (Tep et al. 2012). Thus, Rac1 activation appears to be a pathway induced by p75NTR that regulates Schwann cell myelination.
8 Regulation of Neurite Growth and Axonal Degeneration
Although p75NTR was shown to activate members of the Rac/Rho family in Schwann cells, thereby affecting myelination, the role of p75NTR-dependent RhoA activation has been best studied in the context of neurite outgrowth. The regulation of RhoA by p75NTR was first demonstrated by Yamashita et al, who identified RhoA as a receptor-interacting factor in a yeast two-hybrid screen. They found that unliganded p75NTR promoted RhoA activation and neurotrophin binding prevented this activation, thereby promoting neurite outgrowth (Yamashita et al. 1999). Soon after this finding, p75NTR was shown to regulate RhoA in response to a group of myelin proteins, including myelin associated glycoprotein (MAG), Oligodendrocyte myelin glycoprotein (OMgp), and Nogo (Wang et al. 2002; Wong et al. 2002; Yamashita et al. 2002). These myelin proteins are expressed by oligodendrocytes and are known to inhibit neurite outgrowth, which has made them a major focus for studies aimed at axonal regeneration after spinal cord injury (Akbik et al. 2012). All three of these inhibitors bind to a glycosylphosphatidylinositol (gpi)-linked receptor, the Nogo receptor (NgR). The NgR lacks an intracellular domain, but it can associate with p75NTR and another transmembrane protein, Lingo-1, to regulate RhoA (Mi et al. 2004) (Fig. 3).

Regulation of axonal growth by p75NTR. A complex comprised of p75NTR, NogoR, and Lingo-1 is formed in response to myelin-derived inhibitors such as MAG, OMgp, and Nogo. In response to stimulation by these ligands, Rho-GDI is recruited to the intracellular domain of p75NTR. Concurrently, association of Kalirin-9 with the p75NTR intracellular domain is decreased, thus resulting in enhanced activation of Rho family members by Kalirin-9 and inhibition of axonal growth
The ability of p75NTR to regulate Rho family members depends on a number of intracellular interacting factors. The activation of RhoA by myelin proteins occurs through the recruitment of the Rho inhibitor RhoGDI to p75NTR, thereby releasing RhoA (Yamashita and Tohyama 2003). RhoGDI competes with the guanine nucleotide exchange factor (GEF), Kalirin 9, for p75NTR binding (Harrington et al. 2008), such that upon receptor activation, Kalirin 9 is released, allowing binding of RhoGDI. Once RhoA is freed from RhoGDI, it is then activated by Kalirin 9 (Fig. 3). It should be added that RhoA activation by MAG in cerebellar granule neurons required cleavage of p75NTR (Domeniconi et al. 2005). It is not known how receptor cleavage facilitates the stimulation of Rho or whether other myelin inhibitory molecules that bind the NgR–p75NTR–Lingo-1 complex also induce p75NTR cleavage. In addition to regulating RhoA, Hempstead’s group recently demonstrated that proneurotrophin binding to p75NTR caused a decrease in Rac activity. Rac typically counterbalances Rho, such that decreasing Rac has a similar effect to activating Rho. The reduction in Rac activation after proneurotrophin binding was linked to dissociation of the Rac GEF Trio from a complex of p75NTR with another Sortilin family member, SorCS2, suggesting that the release of Trio reduces basal Rac activity (Deinhardt et al. 2011). Hence, there may be multiple mechanisms by which p75NTR regulates members of the Rho family, depending on the ligand and the co-receptors present.
As might be expected, evidence suggests that p75NTR can regulate axonal growth and retraction at the tip of the growing axon in the growth cone. Treatment of Xenopus spinal neurons with MAG caused p75NTR-dependent repulsion of the growth cone (Wong et al. 2002). Similarly, proneurotrophins were recently reported to induce growth cone collapse in cortical neurons (Sun et al. 2012) and in hippocampal neurons (Deinhardt et al. 2011). In contrast to the inhibitory effects of proneurotrophins and myelin proteins on growth cones, mature neurotrophin binding to p75NTR increased filopodial length in the growth cone of subplate neurons (McQuillen et al. 2002), as well as in retinal ganglion and dorsal root ganglion cells, where it was demonstrated that the receptor promoted filopodia growth through inhibition of RhoA (Gehler et al. 2004).
The recognition of p75NTR as a signal transducer for MAG, OMgp and Nogo created a considerable amount of interest in targeting the receptor as a means of promoting regeneration following CNS injury; however, the regulation of axonal regeneration is very complex, and the role of p75NTR is not well understood. Despite the fact that p75 NTR null neurons failed to activate RhoA and respond to myelin inhibitors in culture (Wang et al. 2002; Yamashita et al. 2002; Yamashita and Tohyama 2003; Zheng et al. 2005) and after spinal cord injury (Dubreuil et al. 2003), there was no effect on regeneration of corticospinal tract after spinal cord injury in the p75 NTR knockout mice (Song et al. 2004; Zheng et al. 2005). The reason why p75NTR does not appear to regulate regeneration after spinal cord injury is not clear, but this may be due to the fact that p75NTR expression is upregulated in oligodendrocytes as well as denervated axons following spinal cord injury (Beattie et al. 2002; Tep et al. 2013), potentially confounding its effect on regeneration. It is also possible that the NgR can partner with other receptors in addition to p75NTR; for example, another member of the TNF receptor superfamily, Troy, was shown to function as a signal transducer for the NgR (Park et al. 2005; Shao et al. 2005). In addition, another receptor, PirB, was identified that also binds myelin inhibitory molecules (Atwal et al. 2008), and Fujita et al. (2011b) reported that myelin inhibitory molecules induced interaction between the PirB and p75NTR in cerebellar granule neurons. In that study, the authors demonstrated that optic nerve regeneration was significantly improved after injury in p75NTR knockout mice, suggesting that p75NTR does play a role in regeneration at least in the optic system, which is less complex than spinal cord injury in terms of affected cell types and the cellular responses to injury. The question remains, however, whether this effect is mediated through the interaction of p75NTR with PirB, since PirB knockout mice had no effect on optic nerve regeneration (Fujita et al. 2011a). The ultimate outcome of whether p75NTR promotes axon regrowth or retraction after an injury may depend, in part, on the type of injury and the different ligands produced that bind to p75NTR and its signaling partners: p75NTR interacts with Lingo-1 and NgR or PirB in response to myelin inhibitory molecules (Mi et al. 2004; Wang et al. 2002), Sortilin in response to proNGF (Nykjaer et al. 2004), EphA in response to Ephrin-As (Lim et al. 2008), in addition to TrkA, TrkB, and TrkC in response to NGF, BDNF, and NT3.
Does myelin play a similarly inhibitory role in intact brains in the absence of any injury? Freda Miller’s group addressed this question by examining axonal sprouting of p75NTR-expressing cholinergic septal neurons into the corpus callosum in the adult brain (Park et al. 2010). Surprisingly, there were many more cholinergic fibers entering the corpus callosum in p75 NTR knockout than in the wild type mice, suggesting that p75NTR normally prevents misrouting of these fibers in vivo. P75NTR appeared to have induced degeneration of the fibers that entered the white matter tract, since cholinergic axons degenerated when plated on myelin. Similar axonal breakdown was observed in cultured sympathetic neurons when BDNF was applied selectively to axons while the soma was maintained in NGF (Singh et al. 2008). The axonal degeneration induced by p75NTR in vitro and in vivo extended beyond the collapse of growth cones, as there was overt breakdown of axonal fibers; however, there was no apoptosis of the neurons. The signaling mechanisms involved in the degeneration appear to involve both Rho and caspase-6, as activation of both enzymes was detected in axons following stimulation of p75NTR, and inhibition of either Rho or caspase-6 blocked the axonal breakdown (Park et al. 2010). Local activation of caspases in distal axons during axonal degeneration has been increasingly reported; for example, degeneration due to withdrawal of NGF selectively from distal axons of DRG neurons grown in compartmentalized chambers was dependent on both caspase-3 and -6 (Simon et al. 2012). These caspases were not activated in the soma; thus the neurons did not undergo apoptosis. How such degenerative/apoptotic signaling is restricted to the axon is not known. The caspase inhibitor XIAP is also upregulated in mature sympathetic neurons, making them resistant to injection of cytochrome c (Potts et al. 2003). Thus, NGF acting at the soma may prevent the spread of caspase activation through upregulation of such inhibitors. Additional studies are needed to understand the spatial control of such signaling pathways.
Conclusion
Since the initial cloning of p75NTR, the field has undergone a remarkable transformation in its understanding of this puzzling receptor. Initially, p75NTR was viewed as a non-signaling, auxiliary binding component for members of the Trk family that simply enabled neurotrophins to bind with high affinity to the Trks. Now, p75NTR is considered a key signaling component for multiple ligands through the formation of a variety of receptor complexes that regulate a wide array of biological responses, from cell survival to neurite growth, axon degeneration, or myelin formation. Many questions remain to be answered regarding the mechanisms utilized by p75NTR to mediate its responses, such as how the receptor can selectively activate one pathway over another, some of which can be in direct opposition (e.g., survival vs. apoptosis). The genes regulated by receptor proteolysis and the ICD-binding proteins that translocate to the nucleus are also yet to be identified. What role Sortilin and other Vps10p family members play in p75NTR signaling beyond binding to the pro-domain of proneurotrophins remains to be determined. Even the molecular nature of the high affinity complex between p75NTR and the Trks is still poorly understood. Beyond these questions regarding receptor signaling, it will be equally important to explore the role of the receptor in the many injuries and diseases where it is upregulated. Given that p75NTR has the potential to signal both survival and degeneration/apoptosis, it is enticing to consider the possibility of developing novel therapeutics that switch the receptor’s degeneration signal to one promoting survival. Certainly, further understanding of how the receptor functions will provide additional insights for developing novel therapeutics for many neuropathologies.
In this chapter, we attempted to cover the many aspects of signaling through the p75NTR receptor and the biological outcomes; nevertheless, we could not fully encompass the many contributions made by the large number of researchers who have studied this complex system. We apologize for those omissions.
References
Agerman K, Baudet C, Fundin B, Willson C, Ernfors P (2000) Attenuation of a caspase-3 dependent cell death in NT4- and p75-deficient embryonic sensory neurons. Mol Cell Neurosci 16:258–268
Akbik F, Cafferty WB, Strittmatter SM (2012) Myelin associated inhibitors: a link between injury-induced and experience-dependent plasticity. Exp Neurol 235:43–52
Alavian KN, Sgado P, Alberi L, Subramaniam S, Simon HH (2009) Elevated P75NTR expression causes death of engrailed-deficient midbrain dopaminergic neurons by Erk1/2 suppression. Neural Dev 4:11
Ali TK, Al-Gayyar MM, Matragoon S, Pillai BA, Abdelsaid MA et al (2011) Diabetes-induced peroxynitrite impairs the balance of pro-nerve growth factor and nerve growth factor, and causes neurovascular injury. Diabetologia 54:657–668
Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J et al (1998) p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol 143:1691–1703
Anton ES, Weskamp G, Reichardt LF, Matthew WD (1994) Nerve growth factor and its low-affinity receptor promote Schwann cell migration. Proc Natl Acad Sci U S A 91:2795–2799
Arevalo MA, Rodriguez-Tebar A (2006) Activation of casein kinase II and inhibition of phosphatase and tensin homologue deleted on chromosome 10 phosphatase by nerve growth factor/p75NTR inhibit glycogen synthase kinase-3beta and stimulate axonal growth. Mol Biol Cell 17:3369–3377
Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y et al (2008) PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322:967–970
Baldwin AS (2012) Regulation of cell death and autophagy by IKK and NF-kB: critical mechanisms in immune function and cancer. Immunol Rev 246:327–345
Baldwin AN, Shooter EM (1995) Zone mapping of the binding domain of the rat low affinity nerve growth factor receptor by the introduction of novel N-glycosylation sites. J Biol Chem 270:4594–4602
Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R et al (1998) The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140:911–923
Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER (1998) Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron 21:813–825
Barker PA, Shooter EM (1994) Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to TrkA on PC12 cells. Neuron 13:203–215
Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL et al (2002) ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36:375–386
Becker EB, Howell J, Kodama Y, Barker PA, Bonni A (2004) Characterization of the c-Jun N-terminal kinase-BimEL signaling pathway in neuronal apoptosis. J Neurosci 24:8762–8770
Benedetti M, Levi A, Chao MV (1993) Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci U S A 90:7859–7863
Bengoechea TG, Chen Z, O’Leary DA, Masliah E, Lee KF (2009) p75 reduces beta-amyloid-induced sympathetic innervation deficits in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A 106:7870–7875
Bentley CA, Lee KF (2000) p75 is important for axon growth and schwann cell migration during development. J Neurosci 20:7706–7715
Benzel I, Barde YA, Casademunt E (2001) Strain-specific complementation between NRIF1 and NRIF2, two zinc finger proteins sharing structural and biochemical properties. Gene 281:19–30
Bergmann I, Priestley JV, McMahon SB, Brocker EB, Toyka KV, Koltzenburg M (1997) Analysis of cutaneous sensory neurons in transgenic mice lacking the low affinity neurotrophin receptor p75. Eur J Neurosci 9:18–28
Bertrand MJ, Kenchappa RS, Andrieu D, Leclercq-Smekens M, Nguyen HN et al (2008) NRAGE, a p75NTR adaptor protein, is required for developmental apoptosis in vivo. Cell Death Differ 15:1921–1929
Bhakar AL, Howell JL, Paul CE, Salehi AH, Becker EB et al (2003) Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of Bad. J Neurosci 23:11373–11381
Bibel M, Hoppe E, Barde YA (1999) Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J 18:616–622
Bischoff V, Deogracias R, Poirier F, Barde YA (2012) Seizure-induced neuronal death is suppressed in the absence of the endogenous lectin Galectin-1. J Neurosci 32:15590–15600
Blochl A, Sirrenberg C (1996) Neurotrophins stimulate the release of dopamine from rat mesencephalic neurons via Trk and p75Lntr receptors. J Biol Chem 271:21100–21107
Bogoyevitch MA, Kobe B (2006) Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev 70:1061–1095
Boutilier J, Ceni C, Pagdala PC, Forgie A, Neet KE, Barker PA (2008) Proneurotrophins require endocytosis and intracellular proteolysis to induce TrkA activation. J Biol Chem 283:12709–12716
Brann AB, Tcherpakov M, Williams IM, Futerman AH, Fainzilber M (2002) Nerve growth factor-induced p75-mediated death of cultured hippocampal neurons is age-dependent and transduced through ceramide generated by neutral sphingomyelinase. J Biol Chem 277:9812–9818
Brunello N, Reynolds M, Wrathall JR, Mocchetti I (1990) Increased nerve growth factor receptor mRNA in contused rat spinal cord. Neurosci Lett 118:238–240
Bui NT, Konig HG, Culmsee C, Bauerbach E, Poppe M et al (2002) p75 neurotrophin receptor is required for constitutive and NGF-induced survival signalling in PC12 cells and rat hippocampal neurones. J Neurochem 81:594–605
Bunone G, Mariotti A, Compagni A, Morandi E, Della Valle G (1997) Induction of apoptosis by p75 neurotrophin receptor in human neuroblastoma cells. Oncogene 14:1463–1470
Caporali A, Pani E, Horrevoets AJ, Kraenkel N, Oikawa A et al (2008) Neurotrophin p75 receptor (p75NTR) promotes endothelial cell apoptosis and inhibits angiogenesis: implications for diabetes-induced impaired neovascularization in ischemic limb muscles. Circ Res 103:e15–e26
Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R et al (1996) Selective activation of NF-kappa B by nerve growth factor through the neurotrophin receptor p75. Science 272:542–545
Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV (1996) Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 383:716–719
Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G, Barde YA (1999) The zinc finger protein NRIF interacts with the neurotrophin receptor p75(NTR) and participates in programmed cell death. EMBO J 18:6050–6061
Ceni C, Kommaddi RP, Thomas R, Vereker E, Liu X et al (2010) The p75NTR intracellular domain generated by neurotrophin-induced receptor cleavage potentiates Trk signaling. J Cell Sci 123:2299–2307
Cesca F, Yabe A, Spencer-Dene B, Arrigoni A, Al-Qatari M et al (2011) Kidins220/ARMS is an essential modulator of cardiovascular and nervous system development. Cell Death Dis 2:e226
Chan JR, Cosgaya JM, Wu YJ, Shooter EM (2001) Neurotrophins are key mediators of the myelination program in the peripheral nervous system. Proc Natl Acad Sci U S A 98:14661–14668
Chan JR, Jolicoeur C, Yamauchi J, Elliott J, Fawcett JP et al (2006) The polarity protein Par-3 directly interacts with p75NTR to regulate myelination. Science 314:832–836
Chang MS, Arevalo JC, Chao MV (2004) Ternary complex with Trk, p75, and an ankyrin-rich membrane spanning protein. J Neurosci Res 78:186–192
Chao MV, Hempstead BL (1995) p75 and Trk: a two-receptor system. Trends Neurosci 18:321–326
Chao MV, Bothwell MA, Ross AH, Koprowski H, Lanahan AA et al (1986) Gene transfer and molecular cloning of the human NGF receptor. Science 232:518–521
Chapman BS (1995) A region of the 75 kDa neurotrophin receptor homologous to the death domains of TNFR-I and Fas. FEBS Lett 374:216–220
Chapman BS, Kuntz ID (1995) Modeled structure of the 75-kDa neurotrophin receptor. Protein Sci 4:1696–1707
Chittka A, Chao MV (1999) Identification of a zinc finger protein whose subcellular distribution is regulated by serum and nerve growth factor. Proc Natl Acad Sci U S A 96:10705–10710
Chittka A, Arevalo JC, Rodriguez-Guzman M, Perez P, Chao MV, Sendtner M (2004) The p75NTR-interacting protein SC1 inhibits cell cycle progression by transcriptional repression of cyclin E. J Cell Biol 164:985–996
Choi S, Friedman WJ (2009) Inflammatory cytokines IL-1beta and TNF-alpha regulate p75NTR expression in CNS neurons and astrocytes by distinct cell-type-specific signalling mechanisms. ASN Neuro 1(2):e00010
Clary DO, Reichardt LF (1994) An alternatively spliced form of the nerve growth factor receptor TrkA confers an enhanced response to neurotrophin 3. Proc Natl Acad Sci U S A 91:11133–11137
Copray JC, Jaarsma D, Kust BM, Bruggeman RW, Mantingh I et al (2003) Expression of the low affinity neurotrophin receptor p75 in spinal motoneurons in a transgenic mouse model for amyotrophic lateral sclerosis. Neuroscience 116:685–694
Cortazzo MH, Kassis ES, Sproul KA, Schor NF (1996) Nerve growth factor (NGF)-mediated protection of neural crest cells from antimitotic agent-induced apoptosis: the role of the low-affinity NGF receptor. J Neurosci 16:3895–3899
Cosgaya JM, Chan JR, Shooter EM (2002) The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 298:1245–1248
Costantini C, Rossi F, Formaggio E, Bernardoni R, Cecconi D, Della-Bianca V (2005) Characterization of the signaling pathway downstream p75 neurotrophin receptor involved in beta-amyloid peptide-dependent cell death. J Mol Neurosci 25:141–156
Costantini C, Scrable H, Puglielli L (2006) An aging pathway controls the TrkA to p75NTR receptor switch and amyloid beta-peptide generation. EMBO J 25:1997–2006
Coulson EJ, Reid K, Baca M, Shipham KA, Hulett SM et al (2000) Chopper, a new death domain of the p75 neurotrophin receptor that mediates rapid neuronal cell death. J Biol Chem 275:30537–30545
Coulson EJ, Reid K, Shipham KM, Morley S, Kilpatrick TJ, Bartlett PF (2004) The role of neurotransmission and the Chopper domain in p75 neurotrophin receptor death signaling. Prog Brain Res 146:41–62
Coulson EJ, May LM, Osborne SL, Reid K, Underwood CK et al (2008) p75 neurotrophin receptor mediates neuronal cell death by activating GIRK channels through phosphatidylinositol 4,5-bisphosphate. J Neurosci 28:315–324
Cragnolini AB, Friedman WJ (2008) The function of p75NTR in glia. Trends Neurosci 31:99–104
Culmsee C, Gerling N, Lehmann M, Nikolova-Karakashian M, Prehn JH et al (2002) Nerve growth factor survival signaling in cultured hippocampal neurons is mediated through TrkA and requires the common neurotrophin receptor P75. Neuroscience 115:1089–1108
Davies AM, Lee KF, Jaenisch R (1993) p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron 11:565–574
DeFreitas MF, McQuillen PS, Shatz CJ (2001) A novel p75NTR signaling pathway promotes survival, not death, of immunopurified neocortical subplate neurons. J Neurosci 21:5121–5129
Deinhardt K, Kim T, Spellman DS, Mains RE, Eipper BA et al (2011) Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci Signal 4:ra82
Dempsey PW, Doyle SE, He JQ, Cheng G (2003) The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev 14:193–209
Deppmann CD, Mihalas S, Sharma N, Lonze BE, Niebur E, Ginty DD (2008) A model for neuronal competition during development. Science 320:369–373
Desmond NL, Levy WB (1986) Changes in the postsynaptic density with long-term potentiation in the dentate gyrus. J Comp Neurol 253:476–482
Dhanasekaran DN, Reddy EP (2008) JNK signaling in apoptosis. Oncogene 27:6245–6251
Di Certo MG, Corbi N, Bruno T, Iezzi S, De Nicola F et al (2007) NRAGE associates with the anti-apoptotic factor Che-1 and regulates its degradation to induce cell death. J Cell Sci 120:1852–1858
Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA (1994) Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science 265:1596–1599
Domeniconi M, Zampieri N, Spencer T, Hilaire M, Mellado W et al (2005) MAG induces regulated intramembrane proteolysis of the p75 neurotrophin receptor to inhibit neurite outgrowth. Neuron 46:849–855
Donovan N, Becker EB, Konishi Y, Bonni A (2002) JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem 277:40944–40949
Du Q, Zhang Y, Tian XX, Li Y, Fang WG (2009) MAGE-D1 inhibits proliferation, migration and invasion of human breast cancer cells. Oncol Rep 22:659–665
Dubreuil CI, Winton MJ, McKerracher L (2003) Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol 162:233–243
Edwards RH, Selby MJ, Garcia PD, Rutter WJ (1988) Processing of the native nerve growth factor precursor to form biologically active nerve growth factor. J Biol Chem 263:6810–6815
Ernfors P, Henschen A, Olson L, Persson H (1989) Expression of nerve growth factor receptor mRNA is developmentally regulated and increased after axotomy in rat spinal cord motoneurons. Neuron 2:1605–1613
Ernfors P, Wetmore C, Eriksdotter-Nilsson M, Bygdeman M, Stromberg I et al (1991) The nerve growth factor receptor gene is expressed in both neuronal and non-neuronal tissues in the human fetus. Int J Dev Neurosci 9:57–66
Ernfors P, Lee KF, Kucera J, Jaenisch R (1994) Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell 77:503–512
Esposito D, Patel P, Stephens RM, Perez P, Chao MV et al (2001) The cytoplasmic and transmembrane domains of the p75 and Trk A receptors regulate high affinity binding to nerve growth factor. J Biol Chem 276:32687–32695
Fan YJ, Wu LL, Li HY, Wang YJ, Zhou XF (2008) Differential effects of pro-BDNF on sensory neurons after sciatic nerve transection in neonatal rats. Eur J Neurosci 27:2380–2390
Farinas I, Jones KR, Backus C, Wang XY, Reichardt LF (1994) Severe sensory and sympathetic deficits in mice lacking neurotrophin-3. Nature 369:658–661
Feinstein E, Kimchi A, Wallach D, Boldin M, Varfolomeev E (1995) The death domain: a module shared by proteins with diverse cellular functions. Trends Biochem Sci 20:342–344
Feng D, Kim T, Ozkan E, Light M, Torkin R et al (2010) Molecular and structural insight into proNGF engagement of p75NTR and sortilin. J Mol Biol 396:967–984
Fifkova E, Anderson CL (1981) Stimulation-induced changes in dimensions of stalks of dendritic spines in the dentate molecular layer. Exp Neurol 74:621–627
Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B (1996) Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381:706–709
Frade JM (2005) Nuclear translocation of the p75 neurotrophin receptor cytoplasmic domain in response to neurotrophin binding. J Neurosci 25:1407–1411
Frade JM, Barde YA (1999) Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development 126:683–690
Frade JM, Rodriguez-Tebar A, Barde YA (1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383:166–168
Francis N, Farinas I, Brennan C, Rivas-Plata K, Backus C et al (1999) NT-3, like NGF, is required for survival of sympathetic neurons, but not their precursors. Dev Biol 210:411–427
Frazier WA, Boyd LF, Bradshaw RA (1974a) Properties of the specific binding of 125I-nerve growth factor to responsive peripheral neurons. J Biol Chem 249:5513–5519
Frazier WA, Boyd LF, Szutowicz A, Pulliam MW, Bradshaw RA (1974b) Specific binding sites for 125I-nerve growth factor in peripheral tissues and brain. Biochem Biophys Res Commun 57:1096–1103
Frey U, Muller M, Kuhl D (1996) A different form of long-lasting potentiation revealed in tissue plasminogen activator mutant mice. J Neurosci 16:2057–2063
Friedman WJ (2000) Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci 20:6340–6346
Fujita Y, Endo S, Takai T, Yamashita T (2011a) Myelin suppresses axon regeneration by PIR-B/SHP-mediated inhibition of Trk activity. EMBO J 30:1389–1401
Fujita Y, Takashima R, Endo S, Takai T, Yamashita T (2011b) The p75 receptor mediates axon growth inhibition through an association with PIR-B. Cell Death Dis 2:e198
Geetha T, Kenchappa RS, Wooten MW, Carter BD (2005) TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor. EMBO J 24:3859–3868
Gehler S, Shaw AE, Sarmiere PD, Bamburg JR, Letourneau PC (2004) Brain-derived neurotrophic factor regulation of retinal growth cone filopodial dynamics is mediated through actin depolymerizing factor/cofilin. J Neurosci 24:10741–10749
Gentry JJ, Casaccia-Bonnefil P, Carter BD (2000) Nerve growth factor activation of nuclear factor kappaB through its p75 receptor is an anti-apoptotic signal in RN22 schwannoma cells. J Biol Chem 275:7558–7565
Gentry JJ, Rutkoski NJ, Burke TL, Carter BD (2004) A functional interaction between the p75 neurotrophin receptor interacting factors, TRAF6 and NRIF. J Biol Chem 279:16646–16656
Giehl KM, Rohrig S, Bonatz H, Gutjahr M, Leiner B et al (2001) Endogenous brain-derived neurotrophic factor and neurotrophin-3 antagonistically regulate survival of axotomized corticospinal neurons in vivo. J Neurosci 21:3492–3502
Ginsberg D (2002) E2F1 pathways to apoptosis. FEBS Lett 529:122–125
Gjerstad MD, Tandrup T, Koltzenburg M, Jakobsen J (2002) Predominant neuronal B-cell loss in L5 DRG of p75 receptor-deficient mice. J Anat 200:81–87
Goldstein B, Macara IG (2007) The PAR proteins: fundamental players in animal cell polarization. Dev Cell 13:609–622
Gong Y, Cao P, Yu HJ, Jiang T (2008) Crystal structure of the neurotrophin-3 and p75NTR symmetrical complex. Nature 454:789–793
Grob PM, Berlot CH, Bothwell MA (1983) Affinity labeling and partial purification of nerve growth factor receptors from rat pheochromocytoma and human melanoma cells. Proc Natl Acad Sci U S A 80:6819–6823
Grob PM, Ross AH, Koprowski H, Bothwell M (1985) Characterization of the human melanoma nerve growth factor receptor. J Biol Chem 260:8044–8049
Gu C, Casaccia-Bonnefil P, Srinivasan A, Chao MV (1999) Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci 19:3043–3049
Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK et al (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J 15:2760–2770
Ha H, Han D, Choi Y (2009) TRAF-mediated TNFR-family signaling. Curr Protoc Immunol Chapter 11:Unit11 9D
Hacker H, Tseng PH, Karin M (2011) Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol 11:457–468
Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM (1999) p75-mediated NF-kappaB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci 14:28–40
Harrington AW, Kim JY, Yoon SO (2002) Activation of Rac GTPase by p75 Is Necessary for c-jun N-Terminal Kinase-Mediated Apoptosis. J Neurosci 22:156–166
Harrington AW, Leiner B, Blechschmitt C, Arevalo JC, Lee R et al (2004) Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proc Natl Acad Sci U S A 101:6226–6230
Harrington AW, Li QM, Tep C, Park JB, He Z, Yoon SO (2008) The role of Kalirin9 in p75/nogo receptor-mediated RhoA activation in cerebellar granule neurons. J Biol Chem 283:24690–24697
Hashimoto Y, Kaneko Y, Tsukamoto E, Frankowski H, Kouyama K et al (2004) Molecular characterization of neurohybrid cell death induced by Alzheimer’s amyloid-beta peptides via p75NTR/PLAIDD. J Neurochem 90:549–558
He XL, Garcia KC (2004) Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science 304:870–875
Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV (1991) High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature 350:678–683
Heymach JV Jr, Shooter EM (1995) The biosynthesis of neurotrophin heterodimers by transfected mammalian cells. J Biol Chem 270:12297–12304
Hirata H, Hibasami H, Yoshida T, Ogawa M, Matsumoto M et al (2001) Nerve growth factor signaling of p75 induces differentiation and ceramide-mediated apoptosis in Schwann cells cultured from degenerating nerves. Glia 36:245–258
Horres CR, Hannun YA (2012) The roles of neutral sphingomyelinases in neurological pathologies. Neurochem Res 37:1137–1149
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC (2003) Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol 23:9361–9374
Huang YY, Bach ME, Lipp HP, Zhuo M, Wolfer DP et al (1996) Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proc Natl Acad Sci U S A 93:8699–8704
Hunt DL, Castillo PE (2012) Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol 22:496–508
Ibanez CF, Ebendal T, Barbany G, Murray-Rust J, Blundell TL, Persson H (1992) Disruption of the low affinity receptor-binding site in NGF allows neuronal survival and differentiation by binding to the trk gene product. Cell 69:329–341
Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS et al (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9:119–128
Jansen P, Giehl K, Nyengaard JR, Teng K, Lioubinski O et al (2007) Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat Neurosci 10:1449–1457
Je HS, Yang F, Ji Y, Nagappan G, Hempstead BL, Lu B (2012) Role of pro-brain-derived neurotrophic factor (proBDNF) to mature BDNF conversion in activity-dependent competition at developing neuromuscular synapses. Proc Natl Acad Sci U S A 109:15924–15929
Jiang Y, Nyengaard JR, Zhang JS, Jakobsen J (2004) Selective loss of calcitonin gene-related Peptide-expressing primary sensory neurons of the a-cell phenotype in early experimental diabetes. Diabetes 53:2669–2675
Jordan BW, Dinev D, LeMellay V, Troppmair J, Gotz R et al (2001) Neurotrophin receptor-interacting mage homologue is an inducible inhibitor of apoptosis protein-interacting protein that augments cell death. J Biol Chem 276:39985–39989
Jung KM, Tan S, Landman N, Petrova K, Murray S et al (2003) Regulated intramembrane proteolysis of the p75 neurotrophin receptor modulates its association with the TrkA receptor. J Biol Chem 278:42161–42169
Kang H, Welcher AA, Shelton D, Schuman EM (1997) Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron 19:653–664
Kanning KC, Hudson M, Amieux PS, Wiley JC, Bothwell M, Schecterson LC (2003) Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J Neurosci 23:5425–5436
Kaplan DR, Martin-Zanca D, Parada LF (1991) Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature 350:158–160
Kenchappa RS, Zampieri N, Chao MV, Barker PA, Teng HK et al (2006) Ligand-dependent cleavage of the P75 neurotrophin receptor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neurons. Neuron 50:219–232
Kenchappa RS, Tep C, Korade Z, Urra S, Bronfman FC et al (2010) p75 neurotrophin receptor-mediated apoptosis in sympathetic neurons involves a biphasic activation of JNK and up-regulation of tumor necrosis factor-alpha-converting enzyme/ADAM17. J Biol Chem 285:20358–20368
Kendall SE, Battelli C, Irwin S, Mitchell JG, Glackin CA, Verdi JM (2005) NRAGE mediates p38 activation and neural progenitor apoptosis via the bone morphogenetic protein signaling cascade. Mol Cell Biol 25:7711–7724
Khursigara G, Orlinick JR, Chao MV (1999) Association of the p75 neurotrophin receptor with TRAF6. J Biol Chem 274:2597–2600
Khursigara G, Bertin J, Yano H, Moffett H, DiStefano PS, Chao MV (2001) A prosurvival function for the p75 receptor death domain mediated via the caspase recruitment domain receptor-interacting protein 2. J Neurosci 21:5854–5863
Kim T, Hempstead BL (2009) NRH2 is a trafficking switch to regulate sortilin localization and permit proneurotrophin-induced cell death. EMBO J 28:1612–1623
Klein R, Jing SQ, Nanduri V, O’Rourke E, Barbacid M (1991) The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 65:189–197
Knowles JK, Rajadas J, Nguyen TV, Yang T, LeMieux MC et al (2009) The p75 neurotrophin receptor promotes amyloid-beta(1–42)-induced neuritic dystrophy in vitro and in vivo. J Neurosci 29:10627–10637
Kokaia Z, Andsberg G, Martinez-Serrano A, Lindvall O (1998) Focal cerebral ischemia in rats induces expression of P75 neurotrophin receptor in resistant striatal cholinergic neurons. Neuroscience 84:1113–1125
Koliatsos VE, Crawford TO, Price DL (1991) Axotomy induces nerve growth factor receptor immunoreactivity in spinal motor neurons. Brain Res 549:297–304
Kommaddi RP, Dickson KM, Barker PA (2011a) Stress-induced expression of the p75 neurotrophin receptor is regulated by O-GlcNAcylation of the Sp1 transcription factor. J Neurochem 116:396–405
Kommaddi RP, Thomas R, Ceni C, Daigneault K, Barker PA (2011b) Trk-dependent ADAM17 activation facilitates neurotrophin survival signaling. FASEB J 25:2061–2070
Kong H, Boulter J, Weber JL, Lai C, Chao MV (2001) An evolutionarily conserved transmembrane protein that is a novel downstream target of neurotrophin and ephrin receptors. J Neurosci 21:176–185
Korte M, Kang H, Bonhoeffer T, Schuman E (1998) A role for BDNF in the late-phase of hippocampal long-term potentiation. Neuropharmacology 37:553–559
Krystosek A, Seeds NW (1981) Plasminogen activator release at the neuronal growth cone. Science 213:1532–1534
Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ et al (2003) A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A 100:15184–15189
Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G et al (2004) A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell 118:243–255
Kust BM, Brouwer N, Mantingh IJ, Boddeke HW, Copray JC (2003) Reduced p75NTR expression delays disease onset only in female mice of a transgenic model of familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 4:100–105
Kuwako K, Taniura H, Yoshikawa K (2004) Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J Biol Chem 279:1703–1712
Lachance C, Belliveau DJ, Barker PA (1997) Blocking nerve growth factor binding to the p75 neurotrophin receptor on sympathetic neurons transiently reduces trkA activation but does not affect neuronal survival. Neuroscience 81:861–871
Le Moan N, Houslay DM, Christian F, Houslay MD, Akassoglou K (2011) Oxygen-dependent cleavage of the p75 neurotrophin receptor triggers stabilization of HIF-1alpha. Mol Cell 44:476–490
Le AP, Friedman WJ (2012) Matrix metalloproteinase-7 regulates cleavage of pro-nerve growth factor and is neuroprotective following kainic acid-induced seizures. J Neurosci 32:703–712
Lebrun-Julien F, Bertrand MJ, De Backer O, Stellwagen D, Morales CR et al (2010) ProNGF induces TNFalpha-dependent death of retinal ganglion cells through a p75NTR non-cell-autonomous signaling pathway. Proc Natl Acad Sci U S A 107:3817–3822
Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH et al (1992) Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69:737–749
Lee R, Kermani P, Teng KK, Hempstead BL (2001) Regulation of cell survival by secreted proneurotrophins. Science 294:1945–1948
Lei K, Davis RJ (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A 100:2432–2437
Lemke G, Chao M (1988) Axons regulate Schwann cell expression of the major myelin and NGF receptor genes. Development 102:499–504
Li QM, Tep C, Yune TY, Zhou XZ, Uchida T et al (2007) Opposite regulation of oligodendrocyte apoptosis by JNK3 and Pin1 after spinal cord injury. J Neurosci 27:8395–8404
Liepinsh E, Ilag LL, Otting G, Ibanez CF (1997) NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J 16:4999–5005
Lim YS, McLaughlin T, Sung TC, Santiago A, Lee KF, O’Leary DD (2008) p75(NTR) mediates ephrin-A reverse signaling required for axon repulsion and mapping. Neuron 59:746–758
Linggi MS, Burke TL, Williams BB, Harrington A, Kraemer R et al (2005) Neurotrophin receptor interacting factor (NRIF) is an essential mediator of apoptotic signaling by the p75 neurotrophin receptor. J Biol Chem 280:13801–13808
Loeb DM, Maragos J, Martin-Zanca D, Chao MV, Parada LF, Greene LA (1991) The trk proto-oncogene rescues NGF responsiveness in mutant NGF-nonresponsive PC12 cell lines. Cell 66:961–966
Longo FM, Manthorpe M, Xie YM, Varon S (1997) Synthetic NGF peptide derivatives prevent neuronal death via a p75 receptor-dependent mechanism. J Neurosci Res 48:1–17
Lopez-Sanchez N, Gonzalez-Fernandez Z, Niinobe M, Yoshikawa K, Frade JM (2007) Single mage gene in the chicken genome encodes CMage, a protein with functional similarities to mammalian type II Mage proteins. Physiol Genomics 30:156–171
Lowry KS, Murray SS, McLean CA, Talman P, Mathers S et al (2001) A potential role for the p75 low-affinity neurotrophin receptor in spinal motor neuron degeneration in murine and human amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2:127–134
MacLachlan TK, El-Deiry WS (2002) Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proc Natl Acad Sci U S A 99:9492–9497
Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C et al (1997) Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci 17:6988–6998
Makkerh JP, Ceni C, Auld DS, Vaillancourt F, Dorval G, Barker PA (2005) p75 neurotrophin receptor reduces ligand-induced Trk receptor ubiquitination and delays Trk receptor internalization and degradation. EMBO Rep 6:936–941
Massague J, Guillette BJ, Czech MP, Morgan CJ, Bradshaw RA (1981) Identification of a nerve growth factor receptor protein in sympathetic ganglia membranes by affinity labeling. J Biol Chem 256:9419–9424
Massague J, Buxser S, Johnson GL, Czech MP (1982) Affinity labeling of a nerve growth factor receptor component on rat pheochromocytoma (PC12) cells. Biochim Biophys Acta 693:205–212
Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R et al (2008) Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci 11:131–133
McCollum AT, Estus S (2004) NGF acts via p75 low-affinity neurotrophin receptor and calpain inhibition to reduce UV neurotoxicity. J Neurosci Res 77:552–564
McQuillen PS, DeFreitas MF, Zada G, Shatz CJ (2002) A novel role for p75NTR in subplate growth cone complexity and visual thalamocortical innervation. J Neurosci 22:3580–3593
Mi S, Lee X, Shao Z, Thill G, Ji B et al (2004) LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci 7:221–228
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293–299
Mohit AA, Martin JH, Miller CA (1995) p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 14:67–78
Mukai J, Hachiya T, Shoji-Hoshino S, Kimura MT, Nadano D et al (2000) NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J Biol Chem 275:17566–17570
Mukai J, Shoji S, Kimura MT, Okubo S, Sano H et al (2002) Structure-function analysis of NADE: identification of regions that mediate nerve growth factor-induced apoptosis. J Biol Chem 277:13973–13982
Murray SS, Bartlett PF, Cheema SS (1999) Differential loss of spinal sensory but not motor neurons in the p75NTR knockout mouse. Neurosci Lett 267:45–48
Murray SS, Perez P, Lee R, Hempstead BL, Chao MV (2004) A novel p75 neurotrophin receptor-related protein, NRH2, regulates nerve growth factor binding to the TrkA receptor. J Neurosci 24:2742–2749
Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7:683–694
Naumann T, Casademunt E, Hollerbach E, Hofmann J, Dechant G et al (2002) Complete deletion of the neurotrophin receptor p75NTR leads to long-lasting increases in the number of basal forebrain cholinergic neurons. J Neurosci 22:2409–2418
Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P et al (2004) Sortilin is essential for proNGF-induced neuronal cell death. Nature 427:843–848
Nykjaer A, Willnow TE, Petersen CM (2005) p75NTR – live or let die. Curr Opin Neurobiol 15:49–57
Oh JD, Chartisathian K, Chase TN, Butcher LL (2000) Overexpression of neurotrophin receptor p75 contributes to the excitotoxin-induced cholinergic neuronal death in rat basal forebrain. Brain Res 853:174–185
Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7:1104–1112
Palmada M, Kanwal S, Rutkoski NJ, Gustafson-Brown C, Johnson RS et al (2002) c-jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J Cell Biol 158:453–461
Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K et al (2004) Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306:487–491
Park JB, Yiu G, Kaneko S, Wang J, Chang J et al (2005) A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron 45:345–351
Park KJ, Grosso CA, Aubert I, Kaplan DR, Miller FD (2010) p75NTR-dependent, myelin-mediated axonal degeneration regulates neural connectivity in the adult brain. Nat Neurosci 13:559–566
Parkhurst CN, Zampieri N, Chao MV (2010) Nuclear localization of the p75 neurotrophin receptor intracellular domain. J Biol Chem 285:5361–5368
Passino MA, Adams RA, Sikorski SL, Akassoglou K (2007) Regulation of hepatic stellate cell differentiation by the neurotrophin receptor p75NTR. Science 315:1853–1856
Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H et al (2001) Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron 32:123–140
Paul CE, Vereker E, Dickson KM, Barker PA (2004) A pro-apoptotic fragment of the p75 neurotrophin receptor is expressed in p75NTRExonIV null mice. J Neurosci 24:1917–1923
Pedraza CE, Podlesniy P, Vidal N, Arevalo JC, Lee R et al (2005) Pro-NGF isolated from the human brain affected by Alzheimer’s disease induces neuronal apoptosis mediated by p75NTR. Am J Pathol 166:533–543
Peng S, Wuu J, Mufson EJ, Fahnestock M (2004) Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol 63:641–649
Peterson DA, Dickinson-Anson HA, Leppert JT, Lee KF, Gage FH (1999) Central neuronal loss and behavioral impairment in mice lacking neurotrophin receptor p75. J Comp Neurol 404:1–20
Plachta N, Annaheim C, Bissiere S, Lin S, Ruegg M et al (2007) Identification of a lectin causing the degeneration of neuronal processes using engineered embryonic stem cells. Nat Neurosci 10:712–719
Podlesniy P, Kichev A, Pedraza C, Saurat J, Encinas M et al (2006) Pro-NGF from Alzheimer’s disease and normal human brain displays distinctive abilities to induce processing and nuclear translocation of intracellular domain of p75NTR and apoptosis. Am J Pathol 169:119–131
Poukka H, Kallio PJ, Janne OA, Palvimo JJ (1996) Regulation of the rat p75 neurotrophin receptor promoter by GC element binding proteins. Biochem Biophys Res Commun 229:565–570
Potts PR, Singh S, Knezek M, Thompson CB, Deshmukh M (2003) Critical function of endogenous XIAP in regulating caspase activation during sympathetic neuronal apoptosis. J Cell Biol 163:789–799.
Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM et al (1993) Induction of apoptosis by the low-affinity NGF receptor. Science 261:345–348
Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM (1987) Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature 325:593–597
Ramos A, Ho WC, Forte S, Dickson K, Boutilier J et al (2007) Hypo-osmolar stress induces p75NTR expression by activating Sp1-dependent transcription. J Neurosci 27:1498–1506
Rattenholl A, Lilie H, Grossmann A, Stern A, Schwarz E, Rudolph R (2001a) The pro-sequence facilitates folding of human nerve growth factor from Escherichia coli inclusion bodies. Eur J Biochem 268:3296–3303
Rattenholl A, Ruoppolo M, Flagiello A, Monti M, Vinci F et al (2001b) Pro-sequence assisted folding and disulfide bond formation of human nerve growth factor. J Mol Biol 305:523–533
Rende M, Provenzano C, Tonali P (1993) Modulation of low-affinity nerve growth factor receptor in injured adult rat spinal cord motoneurons. J Comp Neurol 338:560–574
Rodriguez-Tebar A, Dechant G, Barde YA (1990) Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron 4:487–492
Rodriguez-Tebar A, Dechant G, Gotz R, Barde YA (1992) Binding of neurotrophin-3 to its neuronal receptors and interactions with nerve growth factor and brain-derived neurotrophic factor. EMBO J 11:917–922
Rogers ML, Bailey S, Matusica D, Nicholson I, Muyderman H et al (2010) ProNGF mediates death of Natural Killer cells through activation of the p75NTR-sortilin complex. J Neuroimmunol 226:93–103
Rosch H, Schweigreiter R, Bonhoeffer T, Barde YA, Korte M (2005) The neurotrophin receptor p75NTR modulates long-term depression and regulates the expression of AMPA receptor subunits in the hippocampus. Proc Natl Acad Sci U S A 102:7362–7367
Ross AH, Grob P, Bothwell M, Elder DE, Ernst CS et al (1984) Characterization of nerve growth factor receptor in neural crest tumors using monoclonal antibodies. Proc Natl Acad Sci U S A 81:6681–6685
Roux PP, Colicos MA, Barker PA, Kennedy TE (1999) p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J Neurosci 19:6887–6896
Roux PP, Bhakar AL, Kennedy TE, Barker PA (2001) The p75 neurotrophin receptor activates Akt (protein kinase B) through a phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem 276:23097–23104
Ryden M, Murray-Rust J, Glass D, Ilag LL, Trupp M et al (1995) Functional analysis of mutant neurotrophins deficient in low-affinity binding reveals a role for p75LNGFR in NT-4 signalling. EMBO J 14:1979–1990
Ryden M, Hempstead B, Ibanez CF (1997) Differential modulation of neuron survival during development by nerve growth factor binding to the p75 neurotrophin receptor. J Biol Chem 272:16322–16328
Salehi AH, Roux PP, Kubu CJ, Zeindler C, Bhakar A et al (2000) NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron 27:279–288
Salehi AH, Xanthoudakis S, Barker PA (2002) NRAGE, a p75 neurotrophin receptor-interacting protein, induces caspase activation and cell death through a JNK-dependent mitochondrial pathway. J Biol Chem 277:48043–48050
Sang M, Wang L, Ding C, Zhou X, Wang B et al (2011) Melanoma-associated antigen genes – an update. Cancer Lett 302:85–90
Santos AM, Lopez-Sanchez N, Martin-Oliva D, de la Villa P, Cuadros MA, Frade JM (2012) Sortilin participates in light-dependent photoreceptor degeneration in vivo. PLoS One 7:e36243
Sedel F, Bechade C, Triller A (1999) Nerve growth factor (NGF) induces motoneuron apoptosis in rat embryonic spinal cord in vitro. Eur J Neurosci 11:3904–3912
Seeburger JL, Tarras S, Natter H, Springer JE (1993) Spinal cord motoneurons express p75NGFR and p145trkB mRNA in amyotrophic lateral sclerosis. Brain Res 621:111–115
Seidah NG, Benjannet S, Pareek S, Savaria D, Hamelin J et al (1996) Cellular processing of the nerve growth factor precursor by the mammalian pro-protein convertases. Biochem J 314(Pt 3):951–960
Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S et al (2005) TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron 45:353–359
Shulga A, Magalhaes AC, Autio H, Plantman S, di Lieto A et al (2012) The loop diuretic bumetanide blocks posttraumatic p75NTR upregulation and rescues injured neurons. J Neurosci 32:1757–1770
Simon DJ, Weimer RM, McLaughlin T, Kallop D, Stanger K et al (2012) A caspase cascade regulating developmental axon degeneration. J Neurosci 32:17540–17553
Singh KK, Park KJ, Hong EJ, Kramer BM, Greenberg ME et al (2008) Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat Neurosci 11:649–658
Skeldal S, Matusica D, Nykjaer A, Coulson EJ (2011) Proteolytic processing of the p75 neurotrophin receptor: a prerequisite for signalling?: Neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75(NTR). Bioessays 33:614–625
Song XY, Zhong JH, Wang X, Zhou XF (2004) Suppression of p75NTR does not promote regeneration of injured spinal cord in mice. J Neurosci 24:542–546
Song XY, Zhou FH, Zhong JH, Wu LL, Zhou XF (2006) Knockout of p75(NTR) impairs re-myelination of injured sciatic nerve in mice. J Neurochem 96:833–842
Song W, Volosin M, Cragnolini AB, Hempstead BL, Friedman WJ (2010) ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. J Neurosci 30:15608–15615
Sonnenfeld KH, Ishii DN (1982) Nerve growth factor effects and receptors in cultured human neuroblastoma cell lines. J Neurosci Res 8:375–391
Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ (2008) Beta-amyloid(1–42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci 28:3941–3946
Srinivasan B, Roque CH, Hempstead BL, Al-Ubaidi MR, Roque RS (2004) Microglia-derived pronerve growth factor promotes photoreceptor cell death via p75 neurotrophin receptor. J Biol Chem 279:41839–41845
Stach RW, Wagner BJ (1982) Decrease in the number of lower affinity (type II) nerve growth factor receptors on embryonic sensory neurons does not affect fiber outgrowth. J Neurosci Res 7:103–110
Stoica G, Lungu G, Kim HT, Wong PK (2008) Up-regulation of pro-nerve growth factor, neurotrophin receptor p75, and sortilin is associated with retrovirus-induced spongiform encephalomyelopathy. Brain Res 1208:204–216
Sun Y, Lim Y, Li F, Liu S, Lu JJ et al (2012) ProBDNF collapses neurite outgrowth of primary neurons by activating RhoA. PLoS One [Electronic Resource] 7:e35883
Suter U, Heymach JV Jr, Shooter EM (1991) Two conserved domains in the NGF propeptide are necessary and sufficient for the biosynthesis of correctly processed and biologically active NGF. EMBO J 10:2395–2400
Sutter A, Riopelle RJ, Harris-Warrick RM, Shooter EM (1979) Nerve growth factor receptors. Characterization of two distinct classes of binding sites on chick embryo sensory ganglia cells. J Biol Chem 254:5972–5982
Syroid DE, Maycox PJ, Soilu-Hanninen M, Petratos S, Bucci T et al (2000) Induction of postnatal schwann cell death by the low-affinity neurotrophin receptor in vitro and after axotomy. J Neurosci 20:5741–5747
Tabassum A, Khwaja F, Djakiew D (2003) The p75(NTR) tumor suppressor induces caspase-mediated apoptosis in bladder tumor cells. Int J Cancer 105:47–52
Tan J, Shepherd RK (2006) Aminoglycoside-induced degeneration of adult spiral ganglion neurons involves differential modulation of tyrosine kinase B and p75 neurotrophin receptor signaling. Am J Pathol 169:528–543
Tan J, Clarke M, Barrett G, Millard R (2010) The p75 neurotrophin receptor protects primary auditory neurons against acoustic trauma in mice. Hear Res 268(1–2):46–59
Taniuchi M, Clark HB, Johnson EM Jr (1986) Induction of nerve growth factor receptor in Schwann cells after axotomy. Proc Natl Acad Sci U S A 83:4094–4098
Taylor AR, Gifondorwa DJ, Robinson MB, Strupe JL, Prevette D et al (2012) Motoneuron programmed cell death in response to proBDNF. Dev Neurobiol 72:699–712
Tcherpakov M, Bronfman FC, Conticello SG, Vaskovsky A, Levy Z et al (2002) The p75 neurotrophin receptor interacts with multiple MAGE proteins. J Biol Chem 277:49101–49104
Teng HK, Teng KK, Lee R, Wright S, Tevar S et al (2005) ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci 25:5455–5463
Tep C, Kim ML, Opincariu LI, Limpert AS, Chan JR et al (2012) Brain-derived neurotrophic factor (BDNF) induces polarized signaling of small GTPase (Rac1) protein at the onset of Schwann cell myelination through partitioning-defective 3 (Par3) protein. J Biol Chem 287:1600–1608
Tep C, Lim TH, Ko PO, Getahun S, Ryu JC et al (2013) Oral administration of a small molecule targeted to block proNGF binding to p75 promotes myelin sparing and functional recovery after spinal cord injury. J Neurosci 33:397–410
Troy CM, Friedman JE, Friedman WJ (2002) Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J Biol Chem 277:34295–34302
Truzzi F, Marconi A, Atzei P, Panza MC, Lotti R et al (2011) p75 neurotrophin receptor mediates apoptosis in transit-amplifying cells and its overexpression restores cell death in psoriatic keratinocytes. Cell Death Differ 18:948–958
Turner BJ, Cheah IK, Macfarlane KJ, Lopes EC, Petratos S et al (2003) Antisense peptide nucleic acid-mediated knockdown of the p75 neurotrophin receptor delays motor neuron disease in mutant SOD1 transgenic mice. J Neurochem 87:752–763
Underwood CK, Reid K, May LM, Bartlett PF, Coulson EJ (2008) Palmitoylation of the C-terminal fragment of p75(NTR) regulates death signaling and is required for subsequent cleavage by gamma-secretase. Mol Cell Neurosci 37:346–358
Van Harreveld A, Fifkova E (1975) Swelling of dendritic spines in the fascia dentata after stimulation of the perforant fibers as a mechanism of post-tetanic potentiation. Exp Neurol 49:736–749
Verbeke S, Meignan S, Lagadec C, Germain E, Hondermarck H et al (2010) Overexpression of p75(NTR) increases survival of breast cancer cells through p21(waf1). Cell Signal 22:1864–1873
Verdi JM, Birren SJ, Ibanez CF, Persson H, Kaplan DR et al (1994) p75LNGFR regulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron 12:733–745
Vilar M, Charalampopoulos I, Kenchappa RS, Simi A, Karaca E et al (2009) Activation of the p75 neurotrophin receptor through conformational rearrangement of disulphide-linked receptor dimers. Neuron 62:72–83
Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ (2006) Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci 26:7756–7766
Volosin M, Trotter C, Cragnolini A, Kenchappa RS, Light M et al (2008) Induction of proneurotrophins and activation of p75NTR-mediated apoptosis via neurotrophin receptor-interacting factor in hippocampal neurons after seizures. J Neurosci 28:9870–9879
von Schack D, Casademunt E, Schweigreiter R, Meyer M, Bibel M, Dechant G (2001) Complete ablation of the neurotrophin receptor p75NTR causes defects both in the nervous and the vascular system. Nat Neurosci 4:977–978
Wang S, Bray P, McCaffrey T, March K, Hempstead BL, Kraemer R (2000) p75(NTR) mediates neurotrophin-induced apoptosis of vascular smooth muscle cells. Am J Pathol 157:1247–1258
Wang KC, Kim JA, Sivasankaran R, Segal R, He Z (2002) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420:74–78
Wang YQ, Bian GL, Bai Y, Cao R, Chen LW (2008) Identification and kainic acid-induced up-regulation of low-affinity p75 neurotrophin receptor (p75NTR) in the nigral dopamine neurons of adult rats. Neurochem Int 53:56–62
Wang YJ, Wang X, Lu JJ, Li QX, Gao CY et al (2011) p75NTR regulates Abeta deposition by increasing Abeta production but inhibiting Abeta aggregation with its extracellular domain. J Neurosci 31:2292–2304
Wehrman T, He X, Raab B, Dukipatti A, Blau H, Garcia KC (2007) Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 53:25–38
Wei Y, Wang N, Lu Q, Zhang N, Zheng D, Li J (2007) Enhanced protein expressions of sortilin and p75NTR in retina of rat following elevated intraocular pressure-induced retinal ischemia. Neurosci Lett 429:169–174
Weskamp G, Schlondorff J, Lum L, Becherer JD, Kim TW et al (2004) Evidence for a critical role of the tumor necrosis factor alpha convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR). J Biol Chem 279:4241–4249
Westwick JK, Bielawska AE, Dbaibo G, Hannun YA, Brenner DA (1995) Ceramide activates the stress-activated protein kinases. J Biol Chem 270:22689–22692
Wheeler EF, Bothwell M (1992) Spatiotemporal patterns of expression of NGF and the low-affinity NGF receptor in rat embryos suggest functional roles in tissue morphogenesis and myogenesis. J Neurosci 12:930–945
Willnow TE, Petersen CM, Nykjaer A (2008) VPS10P-domain receptors – regulators of neuronal viability and function. Nat Rev Neurosci 9:899–909
Wolfe SA, Nekludova L, Pabo CO (2000) DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct 29:183–212
Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM (2002) A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 5:1302–1308
Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT et al (2005) Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci 8:1069–1077
Wu GS (2004) The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther 3:156–161
Xiao J, Wong AW, Willingham MM, Kaasinen SK, Hendry IA et al (2009) BDNF exerts contrasting effects on peripheral myelination of NGF-dependent and BDNF-dependent DRG neurons. J Neurosci 29:4016–4022
Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB et al (1997) Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer’s disease. J Clin Invest 100:2333–2340
Yamamoto K, Ichijo H, Korsmeyer SJ (1999) BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol 19:8469–8478
Yamashita T, Tohyama M (2003) The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci 6:461–467
Yamashita T, Tucker KL, Barde YA (1999) Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 24:585–593
Yamashita T, Higuchi H, Tohyama M (2002) The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. J Cell Biol 157:565–570
Yamauchi J, Chan JR, Shooter EM (2004) Neurotrophins regulate Schwann cell migration by activating divergent signaling pathways dependent on Rho GTPases. Proc Natl Acad Sci U S A 101:8774–8779
Ye X, Mehlen P, Rabizadeh S, VanArsdale T, Zhang H et al (1999) TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J Biol Chem 274:30202–30208
Yeiser EC, Rutkoski NJ, Naito A, Inoue J, Carter BD (2004) Neurotrophin signaling through the p75 receptor is deficient in traf6−/− mice. J Neurosci 24:10521–10529
Yin Q, Lin SC, Lamothe B, Lu M, Lo YC et al (2009) E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol 16:658–666
Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV (1998) Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci 18:3273–3281
Zagrebelsky M, Holz A, Dechant G, Barde YA, Bonhoeffer T, Korte M (2005) The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J Neurosci 25:9989–9999
Zampieri N, Xu CF, Neubert TA, Chao MV (2005) Cleavage of p75 neurotrophin receptor by alpha-secretase and gamma-secretase requires specific receptor domains. J Biol Chem 280:14563–14571
Zhang Y, Hong Y, Bounhar Y, Blacker M, Roucou X et al (2003) p75 neurotrophin receptor protects primary cultures of human neurons against extracellular amyloid beta peptide cytotoxicity. J Neurosci 23:7385–7394
Zheng B, Atwal J, Ho C, Case L, He XL et al (2005) Genetic deletion of the Nogo receptor does not reduce neurite inhibition in vitro or promote corticospinal tract regeneration in vivo. Proc Natl Acad Sci U S A 102:1205–1210
Zhou Q, Homma KJ, Poo MM (2004) Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44:749–757
Zotti T, Vito P, Stilo R (2012) The seventh ring: exploring TRAF7 functions. J Cell Physiol 227:1280–1284
Acknowledgments
This work was supported by grants NS038220 (B.D.C. and B.K.) and NS039472 and NS050585 (S.O.Y.) from the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg 2014
About this chapter
Cite this chapter
Kraemer, B.R., Yoon, S.O., Carter, B.D. (2014). The Biological Functions and Signaling Mechanisms of the p75 Neurotrophin Receptor. In: Lewin, G., Carter, B. (eds) Neurotrophic Factors. Handbook of Experimental Pharmacology, vol 220. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-45106-5_6
Download citation
DOI: https://doi.org/10.1007/978-3-642-45106-5_6
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-45105-8
Online ISBN: 978-3-642-45106-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)