
Abstract
Surface degassing is an important volatile discharge process for an active volcanic crater lake. The compositions of volcanic gases discharged from a lake surface (lake gas) were quantified by volcanic plume measurements using a Multi-GAS and alkaline-filter techniques at the Yudamari crater lake, Aso volcano, Japan. Compositions of the lake gases were quite variable and are clearly different from the gases from adjacent fumaroles. Differentiation processes of the lake gas, the lake water, and the fumarolic gases are evaluated based on their compositions. Concentrations of HCl in the lake gas and the lake water indicate that the lake gas composition is controlled by the equilibrium evaporation of the lake water at the lake temperature. Contrasting compositions of the lake gas and the lake water indicate that sulfate and elemental sulfur formation controls chemical differentiation in the lake. The composition of the hydrothermal fluids supplied to the lake is estimated based on mass balance of the lake gas and the lake water. The hydrothermal fluids have similar H2O/S and H2O/Cl ratios but lower CO2/S ratios than the fumarolic gases. This composition contrast indicates that the fumarolic gases are a mixture of magmatic gases and a vapor phase separated from the hydrothermal fluids supplied to the lake.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Lake gas
- Volcanic gas composition
- Multi-GAS
- Aso volcano
- Degassing
- Evaporation
- Gas solubility
- Hydrothermal system
1 Introduction
Volcanic lakes are efficient traps of volcanic volatiles supplied from depth, and a lake’s water composition is considered as an indicator of the flux and composition of supplied volcanic fluids (Delmelle and Bernard 2000). Chloride and sulfates are the most abundant dissolved constituents in volcanic lake waters, and their contents positively correlate with the acidity of the lake water, indicating that these components are supplied as acid volcanic gases to the lake (Varekamp et al. 2000). Volcanic gases are mixtures of various gas species—not only Cl and S species, but also of CO2, H2, and other inert gases. The concentration of inert gas species in lake waters have been rarely measured, but are likely very low because of their low solubility in water. The inert gases supplied to a lake are likely to escape from the lake surface immediately. In order to estimate the composition of the volcanic gas supplied to the lake, it is necessary to measure the composition of the gas emitted from the lake surface.
Evaporation and bubbling are major degassing processes of volcanic lakes. The stability of a volcanic lake is controlled by the energy and water budget between input by meteoric water and deep high-temperature fluids (e.g., volcanic gases) and output by lake water seepage, evaporation, radiation, and heat loss by conduction (Brown et al. 1989; Varekamp et al. 2000). Evaporation is a major energy and water sink in hot lakes that are heated by a large volcanic fluid input (Varekamp et al. 2000; Terada et al. 2012). For example, 50–90 % of the energy lost from Poás crater lake, Costa Rica, in 1985–1988 was by means of evaporation (Brown et al. 1991). Inert gas species such as CO2 may be discharged from the lake not only by evaporation but also by bubbling through the lake; these emission rates need to be quantified separately from the energy budget estimate. Recent airborne measurements of volcanic plumes from crater lakes of Ruapehu and White Island, New Zealand, demonstrated that the crater lakes are significant sources of CO2 degassing, similar to actively degassing volcanoes (Werner et al. 2006, 2008).
Gas emission from a lake often occurs by diffuse degassing from a large area of the lake surface. The composition of inert gas species discharged through lake degassing can be estimated by measuring the composition of bubbling gases or dissolved gases in the lake water (Inguaggiato and Rizzo 2004; Mazot et al. 2011). The composition determined by these methods, however, does not always represent the bulk composition of lake degassing, in particular when degassing activity is not homogeneous, such as when indicated by bubbling. Recently a new instrument, the Multi component gas analyzer system (Multi-GAS), was developed to estimate volcanic gas composition based on the measurements of an air-diluted volcanic gas plume composition (Shinohara 2005; Aiuppa et al. 2005). This method is also useful for measuring the composition of gases discharged from the surface of volcanic lakes. We applied this technique to estimate the composition of lake gases emitted from the Yudamari crater lake, Aso volcano, Japan. In this paper, we report the methods used for volcanic plume measurements and the results obtained and discuss the origin of the lake gases, by considering the differentiation processes in the lake as well as an underlying hydrothermal system.
2 Methods
A volcanic plume is a mixture of volcanic gas and air, and the original volcanic gas composition can be estimated by subtracting the atmospheric contribution from the plume composition. By combining the Multi-GAS and alkali-filter pack techniques, we can measure most of the major components in volcanic gases, including H2O, CO2, SO2, H2S, H2, HCl, and HF (Shinohara et al. 2011a, b). This method was developed to estimate the composition of volcanic gases emitted from an open vent or inaccessible vents, and is also applicable to plumes originating from lake degassing.
2.1 Multi-GAS
The Multi-GAS is an instrument comprising various gas analyzers and sensors to measure concentrations of volcanic gas species in volcanic plumes, and thus to estimate the original volcanic gas composition (Aiuppa et al. 2005; Shinohara 2005). Plume measurements with Multi-GAS were begun at Aso volcano in 2003. Measurements were made in 2003 and 2004 using the Multi-GAS instrument described by Shinohara (2005), whilst an upgraded system has been used from 2005 onwards (Shinohara et al. 2011b). The new Multi-GAS instrument has a non-dispersive infrared CO2–H2O analyzer (LI-840, LI-COR, Inc., Lincoln, USA), SO2 and H2S electrochemical sensors (KTS-512 and KHS-5TA, respectively, Komyo Rikagaku K.K., Kawasaki, Japan), and a H2 semiconductor sensor (Model GM12s, Sensor Tech K.K., Rittou, Japan). The electrochemical SO2 and H2S sensors have significant cross sensitivity, and filter-type scrubbers for H2S and SO2 were placed in front of the KTS-512 and KHS-5TA sensors, respectively, to reduce the cross-sensitivity (Shinohara et al. 2011b). Cross-sensitivity of the H2S sensor to SO2 is about 0.1 %, but is variable, ranging from 0.02 to 0.2 %, depending on the condition of the scrubber (Type KP-SO2J, Komyo Rikagaku K.K., Kawasaki, Japan). Cross-sensitivity of the SO2 sensor to H2S is about 1 %, which is insignificant for the measurements at Aso volcano since the measured SO2/H2S ratios are always larger than unity. The semiconductor H2 sensor is sensitive only to H2, but its sensitivity is affected by H2O concentrations. This effect is corrected for on the basis of the actual H2O concentration measured in the plume (Shinohara et al. 2011b). The H2S sensor was not used in 2003 and 2004, and the SO2/H2S concentration ratio in the plume was measured using Gas Detector Tubes (5La for SO2 in the range 4–60 ppm and 4LT for H2S in the range 0.2–2 ppm) manufactured by GASTECH Co (Ayase, Japan).
2.2 Alkali-filter
Relative concentrations of S, Cl, and F were measured with the alkali-filter technique (Pennisi and Le Cloarec 1998; Shinohara and Witter 2005). Each filter pack consisted of one 0.2-µm particle filter followed by two filter holders, each containing two filters impregnated with 0.2 ml of 1N KOH + 20 % glycerol solution. Sampling was performed at a flow rate of 1–2 l/min for 30–60 min. After sampling, filters in each holder were rinsed with pure water, and concentrations of F, Cl, and S were measured by ion-chromatography after oxidation with H2O2. Chloride and fluoride on the alkaline filters are considered as representative of HCl and HF gases trapped on the filter. S content on the filter is considered as representative of SO2, whereas the volcanic gases contain both SO2 and H2S. Laboratory tests revealed that the absorption efficiencies of SO2 and H2S by each holder are about 100 % and 0–30 %, respectively (Shinohara et al. 2011b), and the S trapped by the second filter holder is mostly derived from H2S, unless the first holder is saturated. For a large SO2/H2S ratio (e.g., >3), S content on the second filter holder is negligible; however, for a small SO2/H2S ratio, a significant amount of S is trapped on the second filter holder, suggesting the significant contribution of H2S. In order to reduce the H2S contribution, the S content is reported after correction by subtracting the S content on the second filter holder from that of the first filter holder (Shinohara et al. 2011b).
3 Yudamari Crater Lake, Aso Volcano
Yudamari crater lake, located within the summit crater of Nakadake stratocone, Aso volcano, Japan (Figs. 1 and 2), is one of the hottest and most acidic crater lakes in the world, with water temperature ranging 40–80 °C and acidity levels of pH = −1 to +1 (Ohsawa et al. 2003, 2010; Miyabuchi and Terada 2009). The high temperature and acidity indicate a large volcanic gas flux into the crater lake. Energy and material budget modeling of the Yudamari crater lake revealed that more than half of H2O in the lake is supplied as high-temperature volcanic gases and more than half of H2O in the lake is lost by evaporation (Saito et al. 2008; Terada et al. 2012).

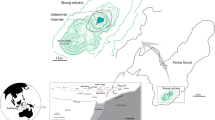
Location maps. a The location of Aso volcano in Japan; b Distribution of the Yudamari crater lake and the fumarolic area at the southern wall of the summit crater of Nakadake central cone, Aso volcano. Contours are in 10-m intervals. The numbers in circles and squares indicate the locations of plume measurement targeting plumes from the fumaroles and from the lake surface, respectively

A photograph of the Yudamari crater lake and the fumarolic area, taken on August 19, 2010
The lake is inaccessible because of the steep crater wall surrounding the crater lake, which is about 300 m in diameter and 120 m deep as measured from the rim (Fig. 1). In order to study the lake, various efforts have been made to conduct measurement using a rope stretched across the lake (Terada and Yoshikawa 2009). Water and lake sediments were sampled several times by bringing sampling devices to the lake surface with the rope (Ohsawa et al. 2003, 2010; Miyabuchi and Terada 2009). The instruments for continuous lake water temperature monitoring were also installed with the stretched rope (Terada et al. 2012).
The Nakadake cone is a presently active basalt to basaltic-andesitic stratocone of the Aso volcano. The activity of the Yudamari crater lake significantly changes with time and is closely correlated with the eruptive activity of the volcano (Ono et al. 1995; Sudo et al. 2006). Lake surface area and water temperature are frequently measured to monitor volcanic activity (Japan Metrological Agency Monthly Report; Terada et al. 2008; Fig. 3). The last significant eruptive activity occurred from 1988 to 1995 and was characterized by Strombolian and phreatomagmatic eruptions, and continuous ash venting (Ono and Watanabe 1985; Ono et al. 1995; Ikebe et al. 2008). After a quiet period during 1995–2003, the lake water volume decreased and the lake dried up, exposing the lake bottom. Several minor ash emissions occurred from 2003 to 2005 (Miyabuchi et al. 2008). The lake water level recovered to occupy most of the original lake area in 2006 and maintained a high level during most of the period of this study (Fig. 3). The lake water volume ranged from 2 to 8 × 105 m3 during the high water level period (Saito et al. 2008; Terada et al. 2008). Magnetotelluric surveys suggest that a hydrothermal fluid reservoir is located a few hundred meters beneath the crater lake (Kanda et al. 2008).

Variation in lake temperature, lake area, and fumarolic area temperature (Japan Meteorological Agency, Monthly Report; http://www.seisvol.kishou.go.jp/tokyo/STOCK/monthly_v-act_doc/monthly_vact.htm). As an approximate measure, the lake area is expressed in percentage relative to the maximum size, which is defined by the steep wall of the crater. Lake water volume or water level data are available for some of the periods (Saito et al. 2008; Terada et al. 2008, 2012). Temperature of the lake surface and the fumarolic area are measured with an infrared thermometer from the western rim of the crater at about 150 m from the target
The Nakadake cone is a persistently degassing volcano with SO2 emission rates ranging from 200 to 400 ton/d (Japan Meteorological Agency, Monthly Report). Volcanic gases are emitted from the crater lake surface and a fumarolic area located at the southern wall of the summit crater (Figs. 1 and 2). These two gases create volcanic plumes with different appearances, particularly under dry conditions. Volcanic plumes from the lake surface are white and dense, whereas the plumes from the fumaroles are transparent with a pale-blue color (Fig. 2). This contrast is likely caused by differences in water condensation of the plume reflecting temperature difference of the source gases (Matsushima and Shinohara 2006). Low-temperature gases are emitted from the lake surface and high-temperature gases from the fumaroles.
We attempted separate quantification of the composition of the gases emitted from the Yudamari crater lake (lake gas) and the fumarolic area at the southern wall (fumarolic gas). Since both the crater lake and the fumaroles are inaccessible, plume measurement is the only way to measure their compositions. Under certain wind conditions, the plumes derived from the two sources reach different locations on the crater rim, and each plume can be measured by selecting an appropriate measurement site (Fig. 1). Cross-contamination of these gases, however, is inevitable because of the adjacent location of the sources. The effect of contamination was minimized by removing sporadic peaks with different compositions in the Multi-GAS data. The effect of contamination on the alkaline-filter data, however, could not be reduced because these data are average compositions during sampling periods of 30–60 min.
4 Results
Gas compositions were obtained as concentration ratios, which commonly have an uncertainty of about 10 % attributed to the instrumental error. The measured ratios, however, have larger and more variable errors depending on the measurement conditions, such as the plume concentrations and atmospheric conditions. Since the gas source is not a single vent both for the lake gases and the fumarolic gases, there can be some heterogeneity in composition as well as composition fluctuation. Therefore, it is quite difficult to estimate the real error in composition determination. In some cases, multiple data sets were obtained within a day or two. Each data set was obtained by 30–60 min measurements. The multiple data agree well in many cases but also show fairly large scatters—up to 50 % in some cases (Table 1, Fig. 4). We consider these scatters as the maximum of the possible errors associated with the composition measurement.

Variation in gas concentration molal ratios estimated by the plume measurements. a CO2/SO2, b SO2/HCl, c HCl/.HF, d SO2/H2S and e H2/CO2. Circles and squares are the lake gas and the fumarolic gas compositions, respectively, and a diamond represents the data from September 18, 2007, considered as a mixture of the two gas types. Open and closed triangles and closed diamonds indicate the compositions estimated by Fourier transform infrared spectroscopy (FT-IR) measurements in previous studies. Shadowed areas indicate the low lake volume periods. Arrows on d indicate the data obtained when the SO2-filter was saturated, implying that the data are minimum estimates of the ratios
The compositions of lake gases and fumarolic gases show large variations with time but are within distinct ranges (Fig. 4). The magnitude of the temporal variation and the compositional contrast between the two are variable for different concentration ratios. The variation of CO2/SO2 ratios is relatively small and the difference between the ratio between the lake gas and the fumarolic gas is also relatively small compared with other ratios (Fig. 4a). Lake gas CO2/SO2 ratios are always smaller than the ratios for the fumarolic gases and averages of the two gas compositions are relatively constant at about two. The CO2/SO2 ratios of the lake and fumarolic gases vary inversely; small lake gas ratios are commonly associated with large fumarolic gas ratios. When the two gases have similar CO2/SO2 ratios, such as for October 4, 2004, February 28, 2008, and October 28, 2008, the ratio is close to two, which is a median value of the variation.
Fumarolic gas SO2/HCl ratios are relatively constant around 10, in particular excluding two data points obtained at site 2 and 3, which are likely affected by lake gas contamination (Table 1). In contrast, lake gas SO2/HCl ratios show a large variation and are larger than that for the fumarolic gases, with two exceptions obtained during the low lake volume periods in 2003 and 2005 (Fig. 4b). HCl/HF ratios show similar variation as SO2/HCl ratios; the HCl/HF ratios are relatively constant at ~5 for fumarolic gases, and are quite variable in lake gases (Fig. 4c). Large ratios (>20) were observed during the low lake volume periods, and very low ratios (<2) were observed during the period when the lake volume was increasing. The SO2/H2S ratios also show a similar level of variation as the SO2/HCl ratios; lake gas ratios are almost always larger than for fumarolic gases, and a low ratio is observed during the low lake volume period (Fig. 4d). A very large SO2/H2S ratio of 2,000 was measured for lake gases in 2009. Since cross-sensitivity of the H2S-sensor to SO2 is about 0.1 %, which corresponds to an SO2/H2S ratio of 1,000, this large value indicates that H2S content in this gas was below the detection limit. Lake and fumarolic gases show similar H2/CO2 ratios (Fig. 4e).
Compositions of volcanic gases were estimated based on volcanic plume measurement by Fourier transform infrared spectroscopy (FT-IR) at Aso volcano (Ono et al. 1999; Hirao et al. 2001; Mori and Notsu 2008). The measurements were conducted at the south-western rim of the crater overlooking the fumarolic area at the southern crater wall, which was used as an infrared light source. These studies intended to measure fumarolic gas composition but did not explicitly try to minimize contribution of the lake gases, and the variable contributions of lake gases might have affected the measurements. Compositions reported by Mori and Notsu (2008) agree with the range for the fumarolic gas as obtained in this study, whereas the compositions reported by Hirao et al. (2001), Ono et al. (1999) are similar to those of the lake gases, suggesting that the former study measured plumes mainly from the fumarolic area, whilst he latter studies measured a mixed plume with a large contribution from the lake gases (Fig. 4). The FT-IR results obtained by Mori and Notsu (2008) on the same day of our measurement (October 15, 2003) agree well with those for the fumarolic gas obtained by this study, in particular for the CO2/SO2 ratio.
5 Discussion
The lake gases are separated from the lake water at the surface, and both fluids are derived from a single hydrothermal fluid. The fumarolic area is located just by the lakeshore (100 m from the center), and gases from a common magmatic source are likely supplied to the lake and the fumarole. In the following sections, we will discuss conditions of differentiation processes to explain the compositional contrast between the lake gases, the lake waters and the fumarolic gases. First, we will discuss the distribution of volcanic gas species between the lake gases and the lake waters that occurs at the surface of the lake. Then, we will estimate compositions of the hydrothermal fluids supplied to the lake. Finally, we will compare compositions of the hydrothermal fluids and the fumarolic gases to evaluate differentiation processes of these fluids.
5.1 Differentiation in the Lake
Lake gases are separated from lake waters at the lake surface. Assuming equilibrium, the distribution of gas species between the lake gas and the water can be evaluated based on solubility data. Distribution of an inert gas component i between the vapor and liquid phases can be expressed by the Henry’s solubility constant (K i ), which is given as a function of temperature (Schulte et al. 2001; Fig. 5);
where C is molality (mol/kg) of a component indicated by the subscript in a phase indicated by the superscript; P, pressure in bars; X, mole fraction; and N w , a constant to convert mole fraction to molality (≈1,000/18.01 = 55.5). The partial pressure of each gas component can be calculated from the lake gas composition and saturated water vapor pressure at lake water temperature. Solubility of SO2, H2S, CO2, and H2 is low, and dissolved contents of these species in the lake water will be quite low. Among these species, SO2 is the most soluble gas component, but the SO2 mol fraction in the lake gas is 500 times that in the lake water at 60 °C, where K i = 0.6 and \(P_{{H_{2} O}} = 0. 2\). Other gas species, whose solubility is more than an order of magnitude less than that of SO2 in the crater lake temperature range, are predominant in the lake gas.

Henry’s law for solubility of volcanic gas species as a function of temperature (Schulte et al. 2001)
HCl is a soluble gas species, and Cl is the most dominant component of the lake water (Ohsawa et al. 2003, 2010; Miyabuchi and Terada 2009; Terada pers. com; Table 2). As HCl dissociates in an aqueous solution, its solubility does not obey the law of Henry’s solubility, but is expressed by the variation in vapor pressure as a function of temperature and concentration in the aqueous solution (Washburn 2003). By assuming the lake water as a simple HCl–H2O system, the HCl concentration in the lake water can be calculated from the lake gas HCl/H2O ratio at a given temperature based on a vapor pressure data set of HCl acid solution (Washburn 2003; Fig. 6). The lake water HCl concentrations calculated from the gas compositions agree well with the measured Cl concentrations in the lake water for the July–September 2007 and July 2008 datasets. This agreement indicates that the HCl and H2O contents in the lake gases are controlled by the equilibrium evaporation of the lake waters and that the evaporation can be modeled with a simple HCl–H2O solution in spite of the high concentration of sulfates in the lake water (Table 2). The negligible effect of the sulfate may be attributed to its smaller ionic dissociation constant compared to that of HCl.

Variations in HCl concentration in the lake water. Open squares and an open diamond represent HCl concentrations in the lake water calculated from the measured gas composition of the lake gas and of the mixed plume, respectively (Table 1). Closed diamonds show the measured HCl concentration in the lake water (Table 2; Ohsawa et al. 2003, 2010; Miyabuchi and Terada 2009; Terada pers. com.)
A large difference was observed between the calculated and measured HCl concentrations in March 2009 (Fig. 6). This difference can be due to contamination of plumes coming from the fumarolic area. Since HCl/H2O ratios of the fumarolic gases are one to two orders of magnitude larger than they are for the lake gases, even a small amount of contamination, such as a few %, can cause a significant deviation. The composition measured by the alkaline filter is an average plume composition during the 30–60 min sampling period and a small amount of cross contamination is inevitable in some cases, such as unstable wind conditions.
The lake gas compositions are controlled not only by simple gas dissolution in the lake water but also by various chemical reactions in the gas and water phases. The lake gases have high SO2/H2S ratios, commonly one to two orders of magnitude larger than the ratios for the fumarolic gases, except during the low lake volume period (Fig. 4d). Since SO2 is more soluble in an aqueous phase than H2S, these high ratios cannot be due to selective dissolution of these species into the lake water. High-temperature fumarolic gases commonly have high SO2/H2S ratios, and low-temperature fumarolic gases often have low SO2/H2S ratios (Mizutani and Sugiura 1966; Giggenbach 1987). However, very high SO2/H2S ratios, such as those obtained for the lake gases in this study, can also be found at low-temperature fumaroles because of S deposition by the following reaction (Mizutani and Sugiura 1966; Giggenbach 1987):
where Se is elemental S. When the initial ratio of SO2/H2S is higher than 0.5, the ratio increases by the reaction (2). Based on the temperature dependence of the equilibrium constant of reaction (2), S saturation temperatures are calculated (T-Se in Table 1). The S saturation temperatures of the lake gases are almost always higher than the lake temperatures, implying that the lake gases are supersaturated with Se and the high SO2/H2S ratios of the lake gases are likely the results of reaction (2). The higher S saturation temperatures indicate that reaction (2) does not achieve equilibrium, likely because of the short residence time of the gas species in the lake water (either as dissolved species or as bubbles). Formation of Se through reaction (2) agrees with the S-rich lake sediments (Miyabuchi and Terada 2009). A low SO2/H2S ratio of the lake gas was observed in October 2003 during the low lake volume period (Fig. 4d). The low ratio might be due to poor interaction of the gas species in the lake water.
Sulfate is the predominant S species in the lake water and is likely formed by disproportionation of SO2 through the following reactions (Giggenbach et al. 2003; Werner et al. 2008);
or
The sulfate formed by reaction (3) is also considered as a minor component of the lake water, which is supersaturated with H2S in terms of reaction (2). Therefore, reaction (4) is likely the major source of sulfate in the lake.
5.2 Hydrothermal Fluids Supplied to the Lake
The composition of the hydrothermal fluids supplied to the lake can be estimated based on mass balance of the lake gases and the lake water. In recent times, the energy and material budget of the Yudamari crater lake were studied extensively (Saito et al. 2008; Terada et al. 2008, 2012; Terada and Yoshikawa 2009). Energy and water budget modeling revealed that the water discharge rate by evaporation is 2–7 times the lake water seepage (Saito et al. 2008; Terada et al. 2012). Based on these water flux ratios and compositions of the lake gases and waters, compositions of their mixture are calculated (Fig. 7). Since both lake gases and waters have variable compositions, two end member compositions are assumed both for the lake water and the lake gas (open and closed small circles; Fig. 7), and a range of compositions is calculated to cover the possible composition range of the hydrothermal fluids (shaded area; Fig. 7). The lake sediments are rich in Se (Miyabuchi and Terada 2009). The lake water and the lake gas are not the only outputs of the lake; the Se deposition on the lake bottom too should be considered as an output of the lake in the geochemical model since the Se will react with neither the water nor the gases. Therefore, the Se deposition rate should be also considered for the mass balance calculations to estimate the hydrothermal fluids composition.

Relative concentrations of a H2O-S–Cl and b CO2-S–Cl. Open circles, open squares and closed diamonds are measured compositions of the fumarolic gases, the lake gases, and the lake waters. Dashed lines represent the mixing line of the lake gas and the lake water end member compositions shown by the small open and closed circles, respectively. The shadowed areas indicate the estimated ranges of the lake gas and water mixture compositions with the water mixing proportion ranging from 7:1 to 2:1. The area surrounded by the grey thick lines shows compositions of the hydrothermal fluids estimated with an assumption that half of the sulfur in the lake fluids was deposited as elemental sulfur (see text for details). The area surrounded by the dashed circle line shows compositions of the magmatic gases estimated as the average of the fumarolic gases and the hydrothermal fluids. The H2O content of the magmatic gases is estimated with an assumption that half of the H2O in the hydrothermal fluids is derived from the meteoric water (see text for details)
The S output fluxes as the Se, the lake gas SO2 and the lake water sulfate can be summarized as follows. The accumulation rate of the Se on the lake bottom was estimated based on S content in the lake sediments and a deposition rate of 250 t/d, which is comparable with the SO2 gas flux (100–200 t/d as S) from the volcano (Miyabuchi and Terada 2009). Although both the lake gas and the fumarolic gas contribute the SO2 emission, there is no quantitative estimate on their relative contribution. For simplicity, we assume that the lake gas is the major source of SO2 with the flux of 200 t/d S. The water output flux by the lake gas (evaporation) is two to seven times larger than that by the lake water (seepage; Terada et al. 2012). Since the H2O/S ratio of the lake water and gases are similar (Fig. 7a), the S output flux by the lake gas is also two to seven times larger than the flux by the lake water. As the lake gas S flux is 200 t/d, the S output flux by the lake water sulfate ranges from 30 to 100 t/d. The results show that the S output flux by the Se deposition is about half (250/550–250/480) of the total S output from the lake, and S content of the hydrothermal fluids supplied to the lake is about double that of the lake gas + water mixture (the areas surrounded by the grey lines; Fig. 7). This is a crude estimate because we do not have enough data to evaluate the relative SO2 flux of the lake gas and the fumarolic gas, and the correlation of compositions and fluxes of the lake water and lake gases. This estimate, however, indicates that the hydrothermal fluids are more S-rich than the lake gas + water mixture, and have a similar composition to the fumarolic gases on the H2O–S–Cl plot (Fig. 7a).
The deposition of half of the S in the hydrothermal system as Se can be an overestimate, because the reactions (2) and (4) convert only a small proportion of S species to Se. Reaction (4) can produce Se with only half the amount of sulfate. Efficiency of Se formation by reaction (2) depends on the initial SO2/H2S ratio. If the initial SO2/H2S ratio is similar to that of the fumarolic gases (ten), only 15 % of the total S can be converted to Se. In contrast, if the original SO2/H2S ratio of the fumarolic gases was small, such as around 0.5, a large amount of S can be formed by reaction (2) and the high ratio of the fumarolic gases can be considered as the result of reaction (2). This idea is consistent with the model that the fumarolic gases are a mixture of magmatic gases and a vapor phase formed under hydrothermal conditions as will be discussed in the next section. We need better constraints on the Se deposition rate not only to estimate the S budget in the lake but also to model the geochemical differentiation process of the hydrothermal system.
5.3 Differentiation in a Hydrothermal System
The estimated compositions of the hydrothermal fluids are plotted near the fumarolic gases with some overlap (Fig. 7). Both compositions have wide ranges with similar H2O/S and H2O/Cl ratios, but the hydrothermal fluids have significantly lower CO2/S and S/Cl ratios than do the fumarolic gases. The close occurrence of these two fluids suggests that a common magmatic gas is the source of the both fluids. The fumarolic gases can be either the source magmatic gas itself or the gas phase separated from the hydrothermal fluids in a hydrothermal system formed as a mixture of the magmatic gas and meteoric water.
The fumarolic gases are likely derived from high-temperature gases. Very high-temperatures of >500 °C were recorded by infrared thermometer measurements from the crater rim on some occasions (Fig. 3). Equilibrium temperatures calculated based on chemical and isotopic compositions also indicate that the fumarolic gases have a high-temperature origin. Apparent equilibrium temperatures (AET; Matsuo 1960; Ohba et al. 1994) for the following reaction were calculated for the fumarolic gas compositions (Table 1);
The calculated AETs range from 718 to 974 °C; an exception was observed on October 26, 2006, for which an AET of 498 °C was calculated (Table 1). The AETs agree with equilibrium temperatures estimated by other studies. High temperatures, ranging from 700 to 1050 °C, were estimated with the method of Mori and Notsu (1997) based on the CO/CO2 ratio measured by the FT-IR in 1996–2003 (Hirao et al. 2001; Mori and Notsu 2008). Tsunogai et al. (2011) measured the H2 isotope compositions of the Aso volcanic plumes in November 2010 and estimated an isotopic equilibrium temperature of 870 °C for the H2–H2O isotopic exchange reaction. The similar temperature estimates, regardless of the method used or the periods, indicate that the maximum temperature of the fumarolic gases are constant around 800–900 °C.
High-temperature emission of the fumarolic gases suggests that the fumarolic gas is a direct discharge of a magmatic gas, implying that the hydrothermal fluids are formed by differentiation of the fumarolic gas and meteoric water mixture. The composition contrast between the fumarolic gases and the hydrothermal fluids, however, disagrees with such a differentiation processes. Under acidic and high-temperature conditions, CO2 behaves as a conservative component. In order to create the CO2-poor hydrothermal fluids from the fumarolic gases, a CO2-rich fluid needs to be created as the counterpart. Emission of such CO2-rich fluids, however, is not observed around the Nakadake crater. Although diffuse emission of CO2-rich soil gases is a candidate of such a CO2-rich gas emission, a detailed soil gas survey concluded that soil CO2 flux in the crater area is only 0.12 t/d (Saito et al. 2007). Therefore, the fumarolic gas needs to be the differentiation counterpart of the hydrothermal fluids. This idea is supported by the inversely correlated variation in CO2/SO2 ratios of the fumarolic gas and lake gas with a constant median value of about two (Fig. 4a).
The fumarolic gases have higher CO2/S and S/Cl ratios than the hydrothermal fluids (Fig. 7b). This composition contrast is similar to the composition contrast between the lake gas and the lake water, and as that contrast is caused by vapor-liquid separation, this suggests that the composition contrast between fumarolic gases and hydrothermal fluids is also the result of vapor-liquid separation under hydrothermal conditions. Direct input of magmatic gases to the fumarolic gases, however, is also necessary because of the quite high measured and calculated equilibrium temperatures of the fumarolic gases. This idea is also supported by the fact that deviation in CO2/SO2 ratios of fumarolic gas and lake gas from the median value is not symmetric. Therefore, the fumarolic gases are likely a mixture of magmatic gases and a vapor phase separated from the hydrothermal fluids (Fig. 8). Because of the vapor loss to the fumarolic gases, the composition of the hydrothermal fluids also differs from the original composition of a magmatic gas-meteoric water mixture. The wide range of the fumarolic gas and the hydrothermal fluids compositions might be a result of the variable degrees and conditions of the vapor transfer to the fumarolic gases (Fig. 8).

A schematic diagram of a magmatic-hydrothermal system at Yudamari crater lake, Aso volcano. A hydrothermal system formed beneath the crater lake through mixing of magmatic gas and meteoric water. The hydrothermal fluids are supplied to the lake and are separated into the lake gas, the lake water and the elemental sulfur. The fumarolic gases are a mixture of the magmatic gas and a vapor phase in the hydrothermal system. The vapor phase loss to the fumarolic gases causes the hydrothermal fluids to be deficient in insoluble gas components such as CO2
Input of a vapor phase separated from the hydrothermal fluids results in composition shifts similar to those caused by magmatic gas scrubbing (Symonds et al. 2001). Since a crater lake and an associated hydrothermal system are effective sites for magmatic scrubbing, gas emission from a crater lake can be largely affected by this process. Werner et al. (2008) quantified CO2/SO2 ratios of the plumes discharged from White Island, New Zealand, by airborne measurements and observed inverse correlation with the crater lake growth; increases in the CO2/SO2 ratios were observed at the beginning of the crater lake formation, indicating significant magmatic gas scrubbing during this period. The simple magmatic gas scrubbing, however, will remove HCl more efficiently than SO2, and the high HCl contents in the fumarolic gases indicate that the fumarolic gases cannot be formed simply by magmatic scrubbing.
The magmatic gases should have intermediate compositions between the fumarolic gases and the hydrothermal fluids. If the discharge rate ratio of the fumarolic gases and the hydrothermal fluids is known, we can calculate the magmatic gas composition by mass balance. Emission rates of SO2 of the Aso volcano are regularly measured by the Japan Meteorological Agency and commonly range between 200–400 t/d, but contributions of the fumarolic gas and the lake gas are not separately quantified. Based on the energy and water budget of the lake, Terada et al. (2012) estimated that H2O flux by the fumarolic gas is 50–100 % of the flux from the lake surface. This estimate implies that the magmatic gas compositions are similar to the average compositions of the fumarolic gases and the hydrothermal fluids (Fig. 7). Since the hydrothermal system is formed by mixing of the magmatic gases and meteoric water (Fig. 8), the magmatic gas composition is more H2O-poor composition than the average compositions of the fumarolic gases and the hydrothermal fluids. Terada et al. (2012) estimated that the meteoric water contribution to the lake is similar to but commonly less than that by the high temperature steam. Therefore the H2O/S ratio of the magmatic gas is likely larger than a half of the ratio for the average composition of the hydrothermal fluids and fumarolic gases (Fig. 7).
Wide ranges of compositions are estimated for the hydrothermal fluids and the source magmatic gases because of poor constraints on the material budget such as correlation of compositions and fluxes, and flux ratios of the lake gas, lake water, and fumarolic gases. Further quantification of these correlations will provide better constraints for more precise geochemical modeling. In some occasions, gas plumes from the lake surface and the fumaroles ascend as separate plumes (Fig. 2), and DOAS or SO2-camera measurements (Mori and Burton 2006) in such occasions may enable separate quantification of discharge rates from the two sources. Such quantification of the discharge rates and compositions of the lake gases and the fumarolic gases is necessary for better estimating the magmatic gas composition and their differentiation processes.
6 Conclusions
Compositions of the lake gases and the fumarolic gases are quantified by plume measurements with Multi-GAS and alkaline-filter techniques. Compositions of both gases show large temporal variations and have distinct features; the fumarolic gases have larger CO2/SO2 ratios and smaller SO2/HCl and SO2/H2S ratios than those of the lake gases. The HCl/H2O ratio of the lake gas is close to that of a vapor phase in evaporation equilibrium with the lake water, implying that the lake gas and the lake water are differentiated by vapor-liquid equilibrium at lake surface conditions. Solubility of lake gas species indicates negligible gas contents in the lake water under the equilibrium conditions. Although dissolved gas contents are quite low, reactions of the gas species in the lake also control the lake gas and the lake water compositions. The large SO2/H2S ratios of the lake gases are attributed to Se formation at lake temperature. A disproportionation reaction of SO2 is the source of sulfate and Se in the lake and likely controls distribution of S species in the crater lake system.
Compositions of hydrothermal fluids supplied to the lake are estimated based on the compositions of the lake gases and the lake waters, and water discharge rates by lake gas evaporation and lake water seepage. The hydrothermal fluid compositions are also estimated considering formation of Se, which contributes about 50 % of the total S in the lake fluids. The estimated hydrothermal fluids compositions have H2O/S and H2O/Cl ratios similar to the fumarolic gases but have slightly smaller S/Cl ratios and significantly larger CO2/S ratios. The composition contrast between the fumarolic gases and the hydrothermal fluids and their occurrences suggest that these fluids are derived from vapor and liquid phases formed under hydrothermal conditions. Direct contribution of magmatic gases to these fluids is also suggested based on the high-temperature features of the fumarolic gases and gas-rich compositions of the lake gases. The large temporal variation in the lake gas and the fumarolic gas compositions is likely the results of changes in the mixing ratios of the magmatic gases and conditions of the hydrothermal phase separation.
References
Aiuppa A, Federico C, Giudice G, Gurrieri S (2005) Chemical mapping of a fumarolic field: La Fossa Crater, Vulcano Island (Aeolian Islands, Italy). Geophys Res Lett 32:L13309. doi:10.1029/2005GL023207
Brown H, Rymer H, Dowden J, Kapadia P, Stevenson D, Barquero J, Morales LD (1989) Energy budget analysis for crater lakes: implication for predicting volcanic activity. Nature 339:370–373
Brown GC, Rymer H, Stevenson D (1991) Volcano monitoring by microgravity and energy budget analysis. J Geol Soc London 148:585–593
Delmelle P, Bernard A (2000) Volcanic lakes. In: Sigurdson H (ed) Encyclopedia of volcanoes. Academic Press, San Diego, pp 877–895
Giggenbach WF (1987) Redox processes governing the chemistry of fumarolic gas discharges from White Island, New Zealand. Appl Geochem 2:143–161
Giggenbach WF, Shinohara H, Kusakabe M, Ohba T (2003) Formation of acid volcanic brines through interaction of magmatic gases, seawater, and rock within the White Island volcanic-hydrothermal system, New Zealand. Soc Econ Geol Sp Publ 10:19–40
Hirao T, Fujimitsu Y, Nishijima J, Ehara S (2001) Remote ovservation of volcanic gases by FT-IR at Aso Volcano. Geoth Volcanol Res Rep Kyushu Univ 10:116–121
Inguaggiato S, Rizzo A (2004) Dissolved helium isotope ratios in ground-waters: a new technique based on gas-water re-equilibration and its application to Stromboli volcanic system. Appl Geochem 19:665–673
Ikebe S, Watanabe K, Miyabuchi Y (2008) The sequence and style of the 1988–1995 eruptions of Nakadake, Aso Volcano, Kyushu, Japan. Bull Volcanol Soc Jpn 53:15–33 (in Japanese with English abstract)
Japan Metrological Agency. Monthly Report (in Japanese). http://www.seisvol.kishou.go.jp/tokyo/STOCK/monthly_v-act_doc/monthly_vact.htm
Kanda W, Tanaka Y, Utsugi M, Takakura S, Hashimoto T, Inoue H (2008) A preparation zone for volcanic explosions beneath Nakadake crater, Aso volcano as inferred from electrical resistivity surveys. J Volcanol Geotherm Res 178:32–45
Matsuo S (1960) On the origin of volcanic gases. J Earth Sci Nagoya Univ 8:222–245
Matsushima N, Shinohara H (2006) Visible and invisible volcanic plumes. Geophys Res Lett 33. doi:10.1029/2006GL026506
Mazot A, Rouwet D, Taran Y, Inguaggiato S, Varley N (2011) CO2 and He degassing at El Chichón volcano, Chiapas, Mexico: gas flux, origin and relationship with local and regional tectonics. Bull Volcanol 73:423–441
Miyabuchi Y, Terada A (2009) Subaqueous geothermal activity of acidic crater lake revealed by lacustrine sediments, Aso Volcano, Japan. J Volcanol Geotherm Res 187:140–145
Miyabuchi Y, Ikebe S, Watanabe K (2008) The July 10, 2003 and the January 14, 2004 ash emissions from a hot water pool of the Nakadake crater, Aso volcano, Japan. Bull Volcanol Soc Jpn 50:227–241 (in Japanese with English abstract)
Mizutani T, Sugiura T (1982) Variations in chemical and isotopic compositions of fumarolic gases from Showashinzan volcano, Hokkaido, Japan. Geochem J 16:63–71
Mizutani Y, Sugiura T (1966) The chemical equilibrium of the 2H2S + SO2 = 3S + 2H2O reaction of solfataras of the Nasudake Volcano. Bull Chem Soc Japan 39:2411–2414
Mori T, Burton M (2006) The SO2 camera: a simple, fast and cheap method for ground-based imaging of SO2 in volcanic plumes. Geophys Res Lett 33:L24804. doi:10.1029/2006GL027916
Mori T, Notsu K (1997) Remote CO, COS, CO2, SO2, HCl detection and temperature estimation of volcanic gas. Geophys Res Lett 24:2047–2050
Mori T, Notsu K (2008) Temporal variation in chemical composition of the volcanic plume from Aso volcano, Japan, measured by remote FT-IR spectroscopy. Geochem J 42:133–140
Ohba T, Hirabayashi J, Nogami K (1994) Water, heat and chloride budgets of the crater lake Yugama at Kusatsu-Shirane Volcano, Japan. Geochem J 28:217–231
Ohsawa S, Sudo Y, Mawatari H, Shimoda G, Utsugi M, Amita K, Yoshikawa S, Yamada M, Iwakura K, Onda Y (2003) Some geochemical features of Yudamari Crater Lake, Aso volcano, Japan. Geotherm Res Rep Kyushu Univ 12:62–65 (in Japanese with English abstract)
Ohsawa S, Saito T, Yoshikawa S, Mawatari H, Yamada M, Amita K, Takamatsu N, Sudo Y, Kagiyama T (2010) Color change of lake water at the active crater lake of Aso volcano, Yudamari, Japan: is it in response to change in water quality induced by volcanic activity? Limnology 11:207–215
Ono A, Koya M, Fujimitsu Y, Ehara S (1999) Remote observation of volcanic gases by Fourier Transform Infrared Spectroscopy (FT-IR) at Aso Volcano. Bull Volcanol Soc Jpn 44:123–130 (in Japanese with English abstract)
Ono K, Watanabe K (1985) Geological map of volcanoes 4; geological map of Aso Volcano 1:50,000 (in Japanese). Geological Survey of Japan, Tsukuba
Ono K, Watanabe K, Hoshizumi H, Ikebe S (1995) Ash eruption of the Naka-dake crater, Aso volcano, southwestern Japan. J Volcanol Geotherm Res 66:137–148
Pennisi M, Le Cloarec MF (1998) Variation of Cl, F, and S in Mount Etna’s plume, Italy, between 1992–1995. Geophys Res Lett 103:5061–5066
Saito M, Matsushima T, Matsuwo N, Shimizu H (2007) Observation of SO2 and CO2 fluxes in and around the active crater of Aso Nakadake Volcano. Sci Rep Dept Earth Planet Sci Kyushu Univ 22:51–52 (in Japanese with English abstract)
Saito T, Ohsawa S, Hashimoto T, Terada A, Yoshikawa S, Ohkura T (2008) Water, heat and chloride balances of the crater lake at Aso Volcano, Japan. J Geotherm Res Soc Jpn 30:107–120 (in Japanese with English abstract)
Schulte MD, Shock EL, Wood RH (2001) The temperature dependence of the standard-state thermodynamic properties of aqueous nonelectrolytes. Geochim Cosmochim Acta 65:3919–3930
Shinohara H (2005) A new technique to estimate volcanic gas composition: plume measurements with a portable multi-sensor system. J Volcanol Geotherm Res 143:319–333
Shinohara H, Witter J (2005) Volcanic gases emitted during mild Strombolian activity of Villarrica volcano. Chile. Geophys Res Lett 32:L20308. doi:10.1029/2005GL024131
Shinohara H, Hirabayashi J, Nogami K, Iguchi M (2011a) Evolution of volcanic gas composition during repeated culmination of volcanic activity at Kuchinoerabujima volcano, Japan. J Volcanol Geotherm Res 202:107–116. doi:10.1016/j.jvolgeores.2011.01.011
Shinohara H, Matsushima N, Kazahaya K, Ohwada M (2011b) Magma-hydrothermal system interaction inferred from volcanic gas measurements obtained during 2003–2008 at Meakandake volcano, Hokkaido, Japan. Bull Volcanol 73:409–421
Sudo Y, Tsuitsui T, Nakaboh M, Yoshikawa M, Yoshikawa S, Inoue H (2006) Ground deformation and magma reservoir at Aso Volcano: Location of deflation source derived from long-term geodetic surveys. Bull Volcanol Soc Jpn 51:291–309 (in Japanese with English abstract)
Symonds RB, Gerlach TM, Reed MH (2001) Magmatic gas scrubbing: implications for volcano monitoring. J Volcanol Geotherm Res 108:303–341
Terada A, Hashimoto T, Kagiyama T, Sasaki H (2008) Precise remote-monitoring technique of water volume and temperature of a crater lake in Aso volcano, Japan: implications for a sensitive window of a volcanic hydrothermal system. Earth Planet Space 60:705–710
Terada A, Yoshikawa S (2009) Development of observation techniques for inaccessible and extremely acidic crater lakes: installation of temperature telemetry buoys, dredging of lake sediments, and sampling of lake waters. J Geotherm Res Soc Japan 31:117–128 (in Japanese with English abstract)
Terada A, Hashimoto T, Kagiyama T (2012) Water flow model of active crater lake at Aso volcano, Japan: fluctuation of magmatic gas and groundwater fluxes from underlying hydrothermal systems. Bull Volcanol 74:641–655
Tsunogai U, Kamimura K, Anzai S, Nakagawa F, Komatsu DD (2011) Hydrogen isotopes in volcanic plumes: tracers for remote temperature sensing of fumaroles. Geochim Cosmochim Acta 75:4531–4546
Varekamp JC, Pasternack GB, Rowe GL Jr (2000) Volcanic lake systematics II. Chemical constraints. J Volcanol Geotherm Res 97:161–179
Washburn EW (2003) International critical tables of numerical data, physics, chemistry and technology, 1st electronic edn. Knovel, Norwich, NY, p 3414
Werner C, Christenson BW, Hagerty M, Britten K (2006) Variability of volcanic gas emissions during a crater lake heating cycle at Ruapehu Volcano, New Zealand. J Volcanol Geotherm Res 154:291–302
Werner C, Hurst T, Scott B, Sherburn S, Christenson BW, Britten K, Cole-Baker J, Mullan B (2008) Variability of passive gas emissions, seismicity, and deformation during crater lake growth at White Island Volcano, New Zealand, 2002–2006. J Geophys Res 113:B01204. doi:10.1029/2007JB005094
Acknowledgments
We wish to thank Dr. A. Terada for the unpublished lake water composition data, Drs. S. Onizawa, Toshiya Mori, Takehiko Mori, A. Namiki, Y. Sudo, N. Geshi and A. Yokoo for their discussion and assistance during the field works, Ms. M. Someya for assistance during chemical analyses, Drs. A. Aiuppa and C. Werner, for their detailed and constructive reviews, and Dr. B. Christenson for the careful editing. This work is partially supported by JSPS KAKENHI 22340130 to SH.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Shinohara, H., Yoshikawa, S., Miyabuchi, Y. (2015). Degassing Activity of a Volcanic Crater Lake: Volcanic Plume Measurements at the Yudamari Crater Lake, Aso Volcano, Japan. In: Rouwet, D., Christenson, B., Tassi, F., Vandemeulebrouck, J. (eds) Volcanic Lakes. Advances in Volcanology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-36833-2_8
Download citation
DOI: https://doi.org/10.1007/978-3-642-36833-2_8
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-36832-5
Online ISBN: 978-3-642-36833-2
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)