
Abstract
Large genomes in plants are composed primarily of long terminal repeat (LTR) retrotransposons, which replicate and propagate by a “copy-and-paste” mechanism dependent on enzymes encoded by the retrotransposons themselves. The enzymes direct a life cycle involving transcription, translation, packaging, reverse transcription, and integration. Loss of any coding capacity will render a retrotransposon incapable of completing its life cycle autonomously. Nevertheless, retrotransposons lacking complete open reading frames for one or more of their proteins are abundant in the genome. These nonautonomous retrotransposons can, however, be complemented in trans by proteins expressed by another retrotransposon, restoring mobility. It is sufficient for a nonautonomous LTR retrotransposon to retain the signals needed for recognition by the transcription machinery and the proteins of autonomous elements. The degree to which nonautonomous retrotransposons interfere with the propagation of autonomous elements has major evolutionary consequences for the genome, affecting the relative rate of gain versus loss of retrotransposons and thereby genome size.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Retrotransposons
5.1.1 Retrotransposons, Drivers of Genome Evolution
As described in elsewhere in this volume (Chap. 1), transposable elements (TEs) can be grouped into 2 major Classes, 9 Orders and 29 Superfamilies (Wicker et al. 2007). Class I, the retrotransposons, is composed of TEs that replicate via an RNA intermediate by a “copy-and-paste” mechanism. Class II elements move generally by “cut-and-paste” as DNA segments. However, Subclass 2 of Class II includes as well the Helitron (Kapitonov and Jurka 2007) and Maverick/Polinton elements that propagate by what could be called “cut and copy” (Fischer and Suttle 2011). This chapter will be focused on retrotransposons.
The most abundant TEs in plant genomes are the long terminal repeat (LTR) retrotransposons, the structures of which are described below. Most plant genomes contain hundreds of LTR retrotransposon families, each in low or moderate copy numbers. However, the large plant genomes contain a few very abundant and replicatively successful retrotransposon families. In the Triticeae (barley, wheat, and relatives), the BARE1, WIS, and Angela elements account for more than 10 % of the genome (Vicient et al. 1999a; Kalendar et al. 2000; Soleimani et al. 2006; Wicker et al. 2009). A whole-genome survey of barley showed that 50 % of the genome is comprised of only 14 TE families, 12 being LTR retrotransposons (Wicker et al. 2009). Why certain LTR retrotransposon families have been able to expand to large numbers while others have not is unknown, though of great interest. Some abundant LTR retrotransposon families are activated by stresses such as drought (Kalendar et al. 2000) or UV light (Ramallo et al. 2008), but so are other retrotransposons that are nevertheless rare in the genome (Grandbastien et al. 2005). Moreover, it is also a reasonable conjecture that selective forces act to drive copy numbers down for some families because of their propensity, for example, to insert into genes.
As a consequence of their overall abundance, LTR retrotransposons are responsible for major variations in genome size other than those explained by genome duplication and polyploidization. For example, Arabidopsis thaliana and sorghum, respectively, having 120 Mbp and 700 Mbp genomes, contain a similar amount of Class II transposons, with the difference in their genome size explained mainly by the differential abundance of LTR retrotransposons (Arabidopsis Genome Initiative 2000; Paterson et al. 2009). In barley, a whole-genome survey showed that less than a dozen LTR retrotransposon families account for almost half of the genome, while Class II elements contribute about 5 % (Wicker et al. 2009). Earlier, we showed that the difference in genome size between two particular Hordeum species can be explained primarily by the difference in BARE1 abundance (Vicient et al. 1999b).
5.1.2 Replication of Autonomous Retrotransposons
The Class I transposable elements all employ a replication cycle in which transcribed RNA is copied into dsDNA by reverse transcriptase. The two largest orders of Class I TEs are named by the presence or absence of an LTR at either end of the retrotransposon (Fig. 5.1). The LINEs (Long Interspersed Nuclear Elements; Goodier and Kazazian 2008) are generally seen as the canonical non-LTR retrotransposons, though the DIRS (Dictyostelium Intermediate Repeat Sequence), PLE (Penelope-like element), and SINE (Short Interspersed Nuclear Elements) retrotransposons also lack LTRs (Wicker et al. 2007). The non-LTR retrotransposons are found throughout the clades of eukaryotes. While they predominate in the genomes of vertebrates and some fungi (Spanu et al. 2010), they are generally much less abundant in plants.

Main groups of autonomous and nonautonomous retrotransposons. (a) Autonomous LTR retrotransposons. Above, the basic structure of an LTR retrotransposon, comprising: the long terminal repeats (LTRs); the primer binding site (PBS), which is the (−)-strand priming site for reverse transcription; the polypurine tract (PPT), which is the (+)-strand priming site for reverse transcription; the PBS and PPT are part of the internal domain, which in autonomous elements includes the protein-coding open reading frame(s). Below, the major superfamilies of LTR retrotransposons, Gypsy and Copia. The open reading frame(s) of the internal domain are gag, encoding the capsid protein Gag; ap, aspartic proteinase; rt–rh, reverse transcriptase–RNase H; int, integrase. The position of the env domain encoding the envelope protein in those Gypsy and Copia clades that contain it is shown. (b) Nonautonomous retrotransposons. BARE2 is an example of a major conserved group having a specific deletion that generates a nonautonomous subfamily. Elements like Morgane have a degenerate or truncated, but still recognizable open reading frame. LARD elements have a long internal domain with conserved structure but lacking coding capacity. TRIM elements have virtually no internal domain except for the PBS and PPT signals. (c) Autonomous and nonautonomous non-LTR retrotransposons. Shown are the autonomous order LINE of the L1 superfamily (ape = apurinic endonuclease) and the nonautonomous order SINE. A gray bar indicates a noncoding domain
The LINEs are considered to be the primordial Class I elements due to their simple structure, specifying only reverse transcriptase and endonuclease activities in the basic forms. Not only lacking LTRs, the non-LTR retrotransposons also function without an integrase gene (Figs. 5.1 and 5.2). Instead, the reverse transcriptase primes DNA synthesis from the poly-A tail of the element’s transcript (Fig. 5.2), later ligating the end of the newly synthesized DNA into the insertion point.

Replication mechanism of a non-LTR retrotransposon. Replication of a LINE of superfamily L1 is shown. The element contains ORF1, specifying an RNA-binding protein, and an open reading frame encoding an apurinic endonuclease (ape) and reverse transcriptase–RNase H (rt–rh). During replication, the LINE is transcribed (Step 1), the open reading frames translated (Step 2; for simplicity only the RT is shown), assembled into a ribonucleoprotein particle (Step 3), and transported into the nucleus (step not shown). The APE nicks the target site, at which point the RNA anneals (Step 4). The free 3′ hydroxyl group of the nicked target is used to prime reverse transcription by a process called target-primed reverse transcription (Step 5). The other strand of the target DNA is also nicked, and the second strand of the LINE is synthesized by the RT (Step 6). The process is completed and the new copy is now inserted at the target site (Step 7). The process is reviewed by Han and Boeke (2005)
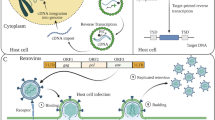
The first step of replication of an LTR retrotransposon (Fig. 5.3) is transcription of an integrated element. The LTRs both drive transcription, by providing a promoter at the 5′ end of the retrotransposon, and specify RNA termination and polyadenylation, using signals in the LTR that are operational at the 3′ end of the inserted element. Transcription by pol II thus begins within the 5′ LTR and terminates within the 3′ LTR before its 3′ end. The RNA transcripts meet two fates: they are translated to form the protein products needed for the retrotransposon life cycle; they are packaged into virus-like particles (VLPs) and later reverse transcribed into cDNA. If the same RNA serves in both pathways, translation must precede reverse transcription for two reasons. First, packaging removes the RNA from access to the translation machinery. Second, during reverse transcription the RNA is hydrolyzed by the action of RNaseH.

Lifecycle of LTR retrotransposons. An element of superfamily Copia with a single open reading frame (ORF) is depicted diagrammatically, integrated into the genome, within the nucleus (gray curve). The plasma membrane is represented as a black curve. The major steps of the life cycle are shown in green circles. If the step depends on the proteins encoded by the retrotransposon and is therefore potentially blocked in a nonautonomous retrotransposon in the absence of complementation, it is shown in a red hexagon. The steps are (1) transcription of a copy integrated into the genome, from the promoter in the long terminal repeat (LTR); (2) nuclear export; (3) alternative translation or buckling of two transcripts destined for packaging and reverse transcription; (4) translation either of separate gag and pol ORFs or of one common ORF to produce the capsid protein Gag and a polyprotein containing aspartic proteinase (AP), reverse transcriptase (RT), RNAseH, and integrase (INT), the order of the protein units being shown being as for elements of superfamily Gypsy; (5) assembly of a virus-like particle (VLP) from Gag containing RNA transcripts, integrase, reverse transcriptase–RnaseH; (6) reverse transcription by RT; (7) localization of the VLP to the nucleus; (8) passage of the cDNA–integrase complex into the nucleus and integration of the cDNA into the genome
Packaging into VLPs is mediated by two signals present in the untranslated leader (UTL) between the PBS and the beginning of gag. These are the PSI (Packaging SIgnal) and DIS (DImerization Signal) motifs, which form conserved secondary structures in the RNA as stem–loops. In retroviruses, and by extension in retrotransposons, PSI mediates packaging of the transcript into its specific particle (Lu et al. 2011; Miyazaki et al. 2011). The DIS directs so-called kissing-loop interactions leading to dimerization of the transcripts during, or just before, packaging (Paillart et al. 2004). Such signals are highly important for propagation of retroviruses, because any change in their structures may severely weaken both the replication and the infection processes.
Translation of the RNA produces the capsid protein Gag, sometimes in a separate reading frame from the enzymes reverse transcriptase and integrase. The proteins are derived from the polyprotein by the endoproteolytic action of aspartic proteinase, also part of the polyprotein. The Gag is assembled into the VLP capsids, into which the RNA template for reverse transcription is packaged as well as reverse transcriptase and integrase. Because the promoter and terminator are internal to the LTRs, the transcripts lack the 5′ end of the 5′ LTR and the 3′ end of the 3′ LTR (Fig. 5.4); these are restored by the complex reverse transcription mechanism of LTR retrotransposons. The mechanism (Fig. 5.4) achieves this through two template switches by reverse transcriptase. The overall replication pathway is fully distinct from that of the LINES. Reverse transcriptase initiates first-strand synthesis from a tRNA primer at the primer binding site (PBS) adjacent to the 5′ LTR. The second strand is primed at the polypurine tract (PPT) adjacent to the 3′ LTR.

Reverse transcription of LTR retrotransposons. Diagrammatically represented are the major steps. (a) Attachment of a tRNA primer at the primer-binding site (PBS) of the retrotransposon transcript (black line), adjacent to the 3′ end of the 5′ LTR regions R and U5, to initiate reverse transcription. (b) Extension of the minus-strand cDNA (shown as a gray line) to the end of the transcript to form minus-strand strong-stop DNA (−sssDNA); (c) Degradation of the RNA from the RNA/DNA hybrid by RNaseH, exposing the repeat (R) domain that is present at both ends of the transcript. (d) Transfer of the exposed −sssDNA to the 3′ end of the transcript by hybridization of the R domain. (e) Extension of the minus-strand and concomitant degradation of the hybridized regions of the transcript by RNase H until the polypurine tract (PPT) of the cDNA is exposed, whereupon plus-strand cDNA (dotted line) synthesis is initiated from RNA fragments (short black lines) as primers. The plus strand is extended to the 5′ end of the minus-strand cDNA, and generating a complementary copy of the PBS, and forms plus-strand strong-stop DNA (+sssDNA). (f) The RNA primers are removed by RNAseH, exposing the PBS on the +sssDNA. (g) Transfer of the +sssDNA, mediated by hybridization of the PBS domain, and continuation of cDNA synthesis requiring strand displacement, each strand serving as a template for the other. (h) Completion of cDNA synthesis to generate a double-stranded linear molecular with intact LTRs at either end. The details and representation are essentially as presented earlier (Telesnitsky and Goff 1997)
Following reverse transcription, the VLP is targeted to the nucleus, the cDNA enters the nucleus, and integration takes place (Fig. 5.3). In contrast to non-LTR retrotransposons (Fig. 5.2), the DNA copy is inserted by integrase (INT), an enzyme specialized for this job (Fig. 5.5). Integrase creates staggered cuts at the target site, trims extra nucleotides from the 3′ termini of the LTRs, and then joins the 3′ termini to the free 5′ ends at the staggered cut (Fig. 5.5). In addition, some retrotransposons contain an open reading frame for an envelope protein (see below).

Integration mechanism of an LTR retrotransposon. The retrotransposon is represented as a loop bounded by two LTRs. Each LTR is flanked by an extra dinucleotide basepair (in this case AA/TT, as found in retrotransposon BARE of barley), which is copied by RT from the dinucleotide found between the PBS and the 3′ end of the 5′ LTR during reverse transcription. The integrase is represented, bound to the LTRs, as a tetramer (Dolan et al. 2009; Cherepanov et al. 2011), forming a pre-integration complex together with the retrotransposon. The genomic DNA target is shown as a pair of gray lines beneath the preintegration complex. (a) The pre-integration complex and target site. (b) The integrase makes a 4- to 6-bp staggered cut in the genomic DNA and trims the dinucleotide from the 3′ end of each LTR, generating 5′ overhangs on both the retrotransposon and at the target site (shown as “P” for 5′ phosphate). (c) Integration of the LTR retrotransposon. The 3′ ends of the LTR are joined to the 5′ overhangs of the target. The trans-esterification reaction, in which the target is cleaved and retrotransposon joined, proceeds as a single-step. (d) Following the integration reaction and removal of the remaining dinucleotide from the 5′ end of each LTR, the gaps generated by the staggered cut remain. The repair of these gaps generates the target-site duplication (TSD) flanking the retrotransposon
The LTR retrotransposons are divided into two main superfamilies, Gypsy and Copia, which differ diagnostically in the order of their encoded protein domains (Fig. 5.1). The groups are each found in almost all eukaryotic lineages and most likely originated from two independent gene fusion events predating the radiation of the eukaryotes. Sequence and structural similarities indicate that the retroviruses evolved from Gypsy elements through the acquisition of the env gene that encodes an envelope protein with transmembrane domains. The protein mediates the formation of an envelope, derived from the plasma membrane, around retroviruses, which consequently can bud from the plasma membrane, leave the host cell, and go on to infect other cells. The gypsy family of Drosophila, the type element of the superfamily, has retroviral-like properties because it can be infectious under laboratory conditions (Kim et al. 1994).
In fact, the env domain is not restricted to animal retroviruses; an env-bearing clade of Gypsy elements is widespread in plants (Vicient et al. 2001). Moreover, env domains can be found in a clade of Copia retrotransposons (Laten et al. 2005; see also a review on this topic, Chap. 6). The likely early division of the Copia and Gypsy lineages and the distinct position of env in the clades of the two superfamilies argues for independent gain of function in both cases and begs a function in the organisms where an extracellular segment of the life cycle has not been demonstrated.
5.2 Nonautonomous Transposable Elements
Retrotransposons play a major role in genome size variation over evolutionary time (discussed above) and are dynamic in their induction both by biotic and abiotic stresses (Wessler 1996; Kalendar et al. 2000; Grandbastien et al. 2005; Ramallo et al. 2008) as well as by “genome stress” (McClintock 1984; Kashkush et al. 2003; Belyayev et al. 2010). Nevertheless, most copies of retrotransposons encountered in a random segment of the genome contain deletions or mutations affecting their open reading frames (ORFs), if they have them at all. These elements, which appear at first glance to be incapable of replicating, can form the majority of the retrotransposon population. This observation may lead the casual onlooker to conclude that Ohno was correct when he referred to the nongenic component of the genome as “junk” (Ohno 1972). However, while the genome may contain “fossils,” or no-longer active transposable elements, these are no more junk, an anthropomorphic term, than are pseudogenes or dinosaur bones.
Many of the apparently fossilized TEs, in fact, can be brought back to life when mobilized by another element; it takes more than a few mutations to kill a TE. This was recognized early on when McClintock observed both autonomous and nonautonomous controlling elements, respectively, Ac and Ds (McClintock 1948; Jones 2005). The canonical autonomous elements contain intact open reading frames and promoters, as well as the structural motifs that are recognized by the TE enzymes and processing signals recognized by the enzymes of general cellular DNA and RNA metabolism.
The nonautonomous but active mobile elements can still be transcribed and mobilized in trans by proteins from autonomous elements; others may have lost the motifs required for trans activation and are both nonautonomous and nonmobile. Among the Class II transposons such as those studied by McClintock, the term “nonautonomous element” has referred to those that cannot express transposase and catalyze their own transposition. They form binary systems with the autonomous elements able to drive their transposition. The classical examples of these include the Ac–Ds (McClintock 1948; Fedoroff et al. 1983; Jones 2005) and Suppressor–Mutator (Spm; Fedoroff 1999) systems, although similar ones are widespread (Hartl et al. 1992).
5.2.1 Nonautonomous Retrotransposons
For Class I elements, the phenomenon of non-autonomy has several additional facets because of the complexity of their replicative life cycle (Figs. 5.2, 5.3, 5.4, and 5.5; Sabot and Schulman 2006). In Class II transposons, a nonautonomous element can be mobilized as long as its termini are recognized by transposase. The LTR retrotransposons must be transcribed and translated, then transcripts packaged, together with integrase and reverse transcriptase, into VLPs formed from self-encoded Gag (Fig. 5.3). Reverse transcription, targeting and entering of the nucleus, and finally integration must occur. While any of these steps may be blocked by lack of a self-encoded protein (Fig. 5.3), all potentially can be complemented in trans if a translationally or enzymatically defective LTR retrotransposon nevertheless possesses the correct recognition signals for proteins encoded by an autonomous and competent element.
5.2.2 Types of Nonautonomous Retrotransposons
The many nonautonomous TEs fall into several categories. The first group, referred to here as Type 1, is comprised of previously autonomous elements that have been variously mutated or deleted so that one or more of their motifs or encoded proteins are no longer functional. In many cases, parts of their protein coding domains may still be recognizable even if they are rendered nonfunctional by substitutions, stop codons, or both. Because retrotransposons encode a polyprotein, any upstream mutation generating a frameshift or stop codon will have polar effects, knocking out expression of the downstream proteins until an efficient start codon is reached. Therefore, nonautonomous elements encompass not only those where some or all of their coding capacity has been deleted but also otherwise autonomous elements with a point mutation leading to polar truncation of translation. The diverse Type 1 are, therefore, expected to be very widespread among the retrotransposons and could still be activated in trans by autonomous elements. A particular nonautonomous copy may have been integrated as a fully functional, autonomous copy and accumulated mutations thereafter, or may have been propagated from a genomic copy that was already nonautonomous.
A second category, Type 2, more interesting than the first because it sheds light on what is minimally required for transposition, consists of groups of nonautonomous mobile elements that have conserved structures or deletions in which one, several, or all protein-coding domains are missing. Type 2 elements have made a successful “lifestyle” of being nonautonomous. Members of this category likely arose from among the variety of mutated forms in the first category. Effective, repeated replication and propagation of particular individual elements gave rise to families or subfamilies of elements with conserved deletions. Further, stepwise deletions and cycles of replication and propagation may lead to conserved groups of elements lacking all protein-coding domains.
Type 3, like Type 2, contains nonautonomous elements of conserved structure, but these are not derived from transposable elements. Instead, they coincidentally possess the signals required for replication due to their role in other or earlier cellular functions. Classic examples of this category are the SINE elements, which will be discussed in more detail below.
Type 4 contains many elements that can no longer be mobilized in trans without restoring mutations. These are both nonautonomous and inactive and may be derived from members of either of the first two categories. These are the true fossils of the genome. Further insertions, deletions, and point mutations may render them unrecognizable as derivatives of transposable elements.
5.2.3 Examples of Type 2 Nonautonomous Retrotransposons
A good example of a Type 2 nonautonomous element is the BARE2 retrotransposon of barley (Tanskanen et al. 2007), a member of the Copia superfamily. BARE2 is a conserved, abundant, and insertionally polymorphic subfamily of the BARE family of retrotransposons and has most of its protein-coding domains intact. However, it has a small, conserved deletion that removes the gag start codon, so that it cannot produce this protein. Instead, the capsid protein is supplied to it by BARE1 for packaging (Tanskanen et al. 2007). Further along the pathway of ORF loss are the Morgane elements of wheat and its relatives (Sabot et al. 2006). Morgane lacks the Gag entirely; the degenerate polyprotein is, however, still recognizable as belonging to the Gypsy superfamily, though it is riddled with stop codons. Nevertheless, Morgane possesses the PBS and PPT motifs needed for reverse transcription.
An endpoint of ORF degeneration, on a continuum from BARE2 through Morgane and onward to complete loss of coding capacity, is represented by the Large Retrotransposon Derivative (LARD) elements. LARDs code for no protein, but possess a long internal domain with a predicted well-conserved RNA structure (Kalendar et al. 2004). The LARDs were found to be abundant (estimated 1.3 × 103 full-length copies and 1.16 × 104 solo LTRs in barley), polymorphic in their insertion sites, and widespread within the grass tribe Triticeae, possessing 4.4-kb LTRs and ∼3.5-kb internal domains flanked by the PBS and PPT priming sites for reverse transcriptase. The conserved RNA structure and priming sites suggests that LARDs have evolved to be reverse transcribed and packaged by the proteins of another retrotransposon, apparently of the Gypsy superfamily.
If a retrotransposon can replicate without encoding proteins, the internal domain may be dispensed with as well, providing that the RNA template for cDNA still can be packaged. This requires retention of the PSI and DIS motifs, described above. Such reduced elements, where the signals for replication have been retained but the rest of the internal domain virtually completely deleted, are exemplified by the Terminal Repeat retrotransposon In Miniature (TRIM; Witte et al. 2001; Kalendar et al. 2008). These lack protein-coding capacity and have only very short internal domains, but nevertheless are abundant and conserved in plants.
Among the TRIM retrotransposons, Cassandra is a particularly interesting family (Kalendar et al. 2008). These elements are 565–860 bp overall, comprising 240–350 bp LTRs flanking a PBS, PPT, and as little as 34 bp in between these signals. Their LTRs all contain conserved 5S RNA sequences and associated RNA polymerase (pol) III promoters and terminators. These resemble the 5S RNA components of ribosomes. The predicted Cassandra RNA 5S secondary structures resemble those of cellular 5S rRNA, with high information content specifically in the pol III promoter region. Cassandra thus appears both to have adapted a ubiquitous cellular gene for ribosomal RNA for use as a promoter and to co-opt an as-yet-unidentified group of retrotransposons for the proteins needed in its lifecycle. The occurrence of Cassandra in the ferns, tree ferns, and in all the angiosperms that have been investigated to date places their origin at least in the Permian, 250 MYA, and suggests that their means of replication as nonautonomous elements has been highly successful for a very long time.
5.2.4 Examples of Type 3 Nonautonomous Retrotransposons
Similar to the TRIMs in their degree of reduction are the short interspersed elements (SINEs), nonautonomous Class I elements that are mobilized by non-LTR retrotransposons. Rather than being derived from LINEs by reduction or mutation, SINEs comprise a diverse group of sequences, sharing the ability to be recognized by the enzymatic machinery of the LINEs (Goodier and Kazazian 2008). They are highly abundant in mammalian genomes, with numbers ranging from 104 to 106 (Kramerov and Vassetzky 2005), but are also found in plants and elsewhere (Deragon and Zhang 2006). Although sharing a mechanism of propagation and a classification as a Order of Class I elements (Wicker et al. 2007), SINEs are polyphyletic in origin and are derived variously from tRNA, rRNA, and other pol III transcripts (Kramerov and Vassetzky 2005). They are generally 150–200 bp; those originating from tRNA possess the tRNA sequence at their 5′ ends and homology at their 3′ ends to a LINE from the same genome, which is thought to provide binding sites for LINE-encoded proteins. The 3′ tails are generally AT rich, betraying origins as reverse-transcribed gene transcripts. Although the enzymology of SINE retroposition is not fully understood, at least for the Alu SINE element of humans, one of the LINE L1 proteins, ORF2p, is needed while the other, ORF1p, may aid the movement (Kroutter et al. 2009).
5.2.5 Classification of Nonautonomous Retrotransposons
Classification of nonautonomous retrotransposons, and nonautonomous transposable elements in general, can be problematic. The current consensus classification (Wicker et al. 2007; see also a review on this topic, Chap. 1) hierarchically divides TEs, respectively, by the presence of an RNA transposition intermediate (Class), mobility during reverse transcription and the number of DNA strands cut at the TE donor site (Subclass), major differences in insertion mechanism (Order), large-scale features such as the structure of protein or noncoding domains (Superfamily), and DNA sequence conservation (Families and Subfamilies). Type 1 nonautonomous elements are relatively easy to fully classify down to the family level. Type 2 elements such as BARE2, if their internal domains retain coding capacity, can generally be placed as subfamilies within TE families. Highly reduced elements, such as the TRIMs discussed below, may be impossible to define below the level of subclass on the basis of sequence analysis and may require experimental data such as evidence for packaging or interactions with the gene products of autonomous elements for more precise phylogenetic placement.
Type 3 elements present a special problem for classification because they can be polyphyletic in origin. Moreover, while some SINEs, for example, may rely on a particular partner for mobilization, others are relatively nonspecific (Kajikawa and Okada 2002). The same may be the case for highly reduced nonautonomous LTR retrotransposons such as TRIMs. For such elements, association to the level of order based on mechanistic considerations may be the limit to what is possible. Depending on their origin or degree of degeneracy, Type 4 nonautonomous elements may or may not be possible to classify. The scheme of Wicker et al. (2007) allows for an “X” to denote ambiguity in the classification of a TE by the three-letter code defining its phylogenetic position.
5.3 Population Structure of Nonautonomous Elements
A thought-provoking feature of the highly reduced, nonautonomous TEs, such as SINEs and TRIMs among the retrotransposons and MITEs among the DNA transposons, is their exceptional abundance. One can view the great abundance of small nonautonomous elements and the comparative rarity of large autonomous elements metaphorically, as abundant but small parasites carried by individual large organisms. While the relative numbers of organismal hosts and parasites reflect an ecosystem’s carrying capacity as related to size and niche, the meaning of this model for replicating entities within a genome is far from clear. The mechanisms behind the differences in abundance between autonomous TEs and their small, nonautonomous derivatives or partners are likewise opaque. However, the high probability of formation and the low cost or the selective advantage of the symbiotic lifestyle of nonautonomous elements may be the factors affecting their prevalence.
5.4 Evolution of Autonomous and Nonautonomous Retrotransposons
The minimalist SINEs and TRIMs illustrate the principal that so long as processing and recognition signals such as, for TRIMs, the PBS, PPT, PSI, and DIS remain present in cis, all of the proteins needed for propagation can be supplied in trans. Hence, the nonautonomous TEs provide a model for the de novo evolution of mobile elements. Today, the proteins for replication and packaging are supplied in trans to nonautonomous elements. In the deep past, the proteins ancestral to those of modern TEs could have acted in trans to mobilize nascent Class I or Class II elements. The various coding domains and replication signals need not have been assembled simultaneously but could have been captured or added sequentially. The respective likelihoods of TEs arising de novo and nonautonomous derivatives appearing are not equal, however. The abundance of nonautonomous elements in the genome demonstrates that the loss of coding capacity occurs often. Independent evolution of new types of TEs, based on the presence of relatively few (two classes, nine orders; Wicker et al. 2007) types of transposable elements in the eukaryotes, appears to happen rarely.
One can nevertheless begin to model the evolution of TEs based on the nonautonomous elements as the minimal functional unit needing to be assembled in cis. Focusing on the retrotransposons, mobility requires propagation of a copy, which requires an integrase enzymatic function to break the genomic DNA and integrate a mobile DNA segment into the chromosome. The LTR retrotransposon integrases are part of a large range of DNA-active enzymes that share the DDD or DDE motif at the active site, including the V(D)J recombinases and the bacterial transposases (Keith et al. 2008). This implies a common origin; recent structural studies of the enzymes strongly support this view (Hickman et al. 2010; Montaño and Rice 2011). Early on, it was noticed that retrotransposons, retroviruses, and bacteriophage Mu all share the terminal TG…CA ends that are found within LTRs (Temin 1980). The formation of terminal inverted repeats (TIRs) flanking a promoter within the ancestral retrotransposon provided recognition and binding sites for the primordial integrase, allowing its propagation. Research to identify the amino acid residues of integrase that interact with the LTR (Dolan et al. 2009) should eventually allow a clear picture to emerge of the coevolution of integrases and their recognition sites.
An LTR is, in essence, a pair of TIRs flanking a promoter, terminator, and polyadenylation signal, the whole of which is then repeated twice. The short TIRs recognized by the integrase almost universally share the 5′ TG…CA 3′ termini that form the outer nucleotides of the TIRs. Promoters are plentiful in the genome, and terminators, polyadenylation signals, and 5- or 6-bp repeats are short enough to occur with high frequency. In between the two LTRs, one needs the PBS and PPT signals as a minimum for reverse transcription. Although it seems at first glance to be unlikely that two LTR repeat units would occur close to one another in the genome by sheer chance, the process of replication by reverse transcriptase, involving two strand jumps, homogenizes the two ends of the final double-stranded cDNA, creating the LTRs. It is not so implausible to imagine that the acquisition of a tRNA gene near a promoter and of a purine-rich tract near a terminator, together with the presence of a stretch of a few 10s of bases of similar nucleotides at either end, would have permitted reverse transcription to create two LTRs, each possessing the promoter and terminator flanking the genes.
The reverse transcriptase itself appears to be derived from an ancient family of enzymes involved in nucleic acid metabolism, in this case polymerization. This view is supported by the presence in plants, animals, fungi, protists, and bacteria of a conserved family of genes, rvt, which encode polymerases able to incorporate both ribonucleotides and deoxyribonucleotides (Gladyshev and Arkhipova 2011). All retrotransposon reverse transcriptases have in their catalytic center a highly conserved motif, generally YVDD, which is surrounded by several small hydrophobic amino acids, together referred to as the reverse transcriptase signature.
The eukaryotic telomerase enzyme, which adds telomeres to the ends of chromosomes through reverse transcription of an RNA template, contains a similar motif in its catalytic center (Autexier and Lue 2006; Lue et al. 2005; Lingner et al. 1997). Structure-based alignments indicate that the rvt enzymes most closely resemble modern LINE reverse transcriptases and belong with them in a larger family including the reverse transcriptases of LTR retrotransposons, retroviruses, pararetroviruses, telomerases, and the PLE order of Class I elements. Thus, Class I reverse transcriptase and telomerase are descendants of a common ancestral enzyme. The earliest retrotransposon reverse transcriptase probably then fused with an RNaseH gene. Subsequent acquisition of regulatory sequences gave rise to the structurally simplest known Class I elements, the non-LTR retrotransposons. Once a template is primed, reverse transcriptases are generally nonspecific. Hence, reverse transcription of a primordial retrotransposon could well have been carried out in trans by an enzyme not encoded by the TE itself.
Autonomous LTR retrotransposons appear to have arisen as a fusion of a reverse transcriptase and an integrase. Such a fusion event appears to have occurred at least twice, each leading to the formation of the two main LTR retrotransposon superfamilies, Gypsy and Copia (Fig. 5.1). The LTRs of Gypsy and Copia elements are very similar in their overall structure and function and in the presence of TG....CA ends. The similarities are unsurprising, considering both the similarity in the integrases that recognize the LTRs and the reliance of all LTRs on conserved transcriptional machinery. As argued above, LTRs may arise relatively easily over evolutionary time. Hence, if the primeval Gypsy and Copia elements evolved independently, they could have acquired LTRs independently. Alternatively, both have evolved from an ancestral LTR-containing intermediate.
5.5 Conclusions
The life cycle of retrotransposons involves stages of transcription, translation, packaging, reverse transcription, and integration. Loss of any of the functions will render a retrotransposon incapable of completing its life cycle autonomously. However, complementation in trans by proteins expressed by another retrotransposon can restore the ability of nonautonomous elements to transpose. Nonautonomous elements may be unable to express one or more proteins, or they may lack coding capacity entirely. It appears that all that needs to be retained are the signals required in cis, respectively, within the element residing in the genome, for transcription, termination, and polyadenylation, within the transcript for dimerization, packaging, and reverse transcription, and within the cDNA copy for integration. The signals are enough for transcripts of nonautonomous elements to hitch a ride in the VLPs of an autonomous retrotransposon and be carried as cDNA to elsewhere in the genome.
Because of the many ways in which full function can be lost from an autonomous retrotransposon, the nonautonomous elements probably form the majority of all TEs. Moreover, major groups of nonautonomous elements have highly conserved, but deleted internal domains where the open reading frame normally resides; these tend to be abundant. These groups have become specialized as effectively propagating nonautonomous elements. Besides clarifying how much of the genomic DNA that does not code for long ORFs may nevertheless be mobile, the trans-complementation model helps explain how autonomous retrotransposons may have evolved through sequential gain of function.
An important question which remains unanswered is the effect of nonautonomous retrotransposons on their autonomous partners: are they propagating at the expense of the partners providing proteins in trans? For example, if nonautonomous elements are freer to optimize very efficient packaging structures in the absence of constraints to maintain open reading frames, will they block replication of the autonomous partners, leading to their ultimate demise? While scenarios can be modeled, the question will need to be addressed by finding and studying the partnerships experimentally. The answer has major evolutionary consequences for the genome, affecting the relative rates of gain versus loss of retrotransposons and thereby genome size (Hawkins et al. 2009).
A related question is to what extent a nonautonomous retrotransposon group is dependent on a particular autonomous family for replication, and to what extent the nonautonomous elements are generalists and can be complemented by many or all autonomous elements. A specialist group will disappear if its autonomous partners in the genome should all become nonautonomous or inactive. A third alternative over evolutionary time is, like a surfing sailboat moving from wave to wave, to develop specificity for a new, active group as the older one declines. This is conceivable given the high mutation rates of retrotransposon replication. Despite the importance of retrotransposons to genome dynamics and gene activity (e.g., through epigenetic effects), our understanding of their biology is still in a primitive state.
References
Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815
Autexier C, Lue NF (2006) The structure and function of telomerase reverse transcriptase. Annu Rev Biochem 75:493–517
Belyayev A, Kalendar R, Brodsky L, Nevo E, Schulman AH, Raskina O (2010) Transposable elements in a marginal plant population: temporal fluctuations provide new insights into genome evolution of wild diploid wheat. Mob DNA 1:6
Cherepanov P, Maertens GN, Hare S (2011) Structural insights into the retroviral DNA integration apparatus. Curr Opin Struct Biol 2:249–256
Deragon J, Zhang X (2006) Short interspersed elements (SINEs) in plants: origin classification and use as phylogenetic markers. Syst Biol 55:949–956
Dolan J, Chen A, Weber IT, Harrison RW, Leis J (2009) Defining the DNA substrate binding sites on HIV-1 integrase. J Mol Biol 385:568–579
Fedoroff NV (1999) The supressor-mutator element and the evolutionary riddle of transposons. Genes Cells 4:11–19
Fedoroff N, Wessler S, Shure M (1983) Isolation of the transposable maize controlling elements Ac and Ds. Cell 35:235–242
Fischer MG, Suttle CA (2011) A virophage at the origin of large DNA transposons. Science 332:231–234
Gladyshev EA, Arkhipova IR (2011) A widespread class of reverse transcriptase-related cellular genes. Proc Natl Acad Sci USA 108:20311–20316
Goodier JL, Kazazian HHJ (2008) Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135:23–35
Grandbastien MA, Audeon C, Bonnivard E, Casacuberta JM, Chalhoub B, Costa AP, Le QH, Melayah D, Petit M, Poncet C, Tam SM, Van Sluys MA, Mhiri C (2005) Stress activation and genomic impact of Tnt1 retrotransposons in Solanaceae. Cytogenet Genome Res 110:229–241
Han JS, Boeke JD (2005) LINE-1 retrotransposons: modulators of quantity and quality of mammalian gene expression? Bioessays 27:775–784
Hartl DL, Lozovskaya ER, Lawrence JG (1992) Nonautonomous transposable elements in prokaryotes and eukaryotes. Genetica 86:47–53
Hawkins JS, Proulx SR, Rapp RA, Wendel JF (2009) Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc Natl Acad Sci USA 106:17811–17816
Hickman AB, Chandler M, Dyda F (2010) Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit Rev Biochem Mol Biol 45:50–69
Jones RN (2005) McClintock’s controlling elements: the full story. Cytogenet Genome Res 109:90–103
Kajikawa M, Okada N (2002) LINEs mobilize SINEs in the eel through a shared 3′ sequence. Cell 111:433–444
Kalendar R, Tanskanen J, Immonen S, Nevo E, Schulman AH (2000) Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc Natl Acad Sci USA 97:6603–6607
Kalendar R, Vicient CM, Peleg O, Anamthawat-Jonsson K, Bolshoy A, Schulman AH (2004) LARD retroelements: novel non-autonomous components of barley and related genomes. Genetics 166:1437–1450
Kalendar R, Tanskanen JA, Chang W, Antonius K, Sela H, Peleg P, Schulman AH (2008) Cassandra retrotransposons carry independently transcribed 5S RNA. Proc Natl Acad Sci USA 105:5833–5838
Kapitonov VV, Jurka J (2007) Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet 23:521–529
Kashkush K, Feldman M, Levy AA (2003) Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat Genet 32:102–106
Keith JH, Schaeper CA, Fraser TS, Fraser MJ Jr (2008) Mutational analysis of highly conserved aspartate residues essential to the catalytic core of the piggyBac transposase. BMC Mol Biol 9:73
Kim A, Terzian C, Santamaria P, Pélisson A, Prud’homme N, Bucheton A (1994) Retroviruses in invertebrates: the gypsy retrotransposon is apparently an infectious retrovirus of Drosophila melanogaster. Proc Natl Acad Sci USA 91:1285–1289
Kramerov D, Vassetzky N (2005) Short retroposons in eukaryotic genomes. Int Rev Cytol 247:165–221
Kroutter EN, Belancio VP, Wagstaff BJ, Roy-Engel AM (2009) The RNA polymerase dictates ORF1 requirement and timing of LINE and SINE retrotransposition. PLoS Genet 5:e1000458
Laten HM, Havecker ER, Farmer LM, Voytas DF (2005) SIRE1, an endogenous retrovirus family from Glycine max, is highly homogeneous and evolutionarily young. Mol Biol Evol 20:1222–1230
Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276:561–567
Lu K, Heng X, Summers MF (2011) Structural determinants and mechanism of HIV-1 genome packaging. J Mol Biol 410:609–633
Lue NF, Bosoy D, Moriarty TJ, Autexier C, Altman B, Leng S (2005) Telomerase can act as a template- and RNA-independent terminal transferase. Proc Natl Acad Sci USA 102:9778–9783
McClintock B (1948) Mutable loci in maize. Year B Carnegie Inst Wash 47:155–169
McClintock B (1984) The significance of responses of the genome to challenge. Science 226:792–801
Miyazaki Y, Miyake A, Nomaguchi M, Adachi A (2011) Structural dynamics of retroviral genome and the packaging. Front Microbiol 2:264
Montaño SP, Rice PA (2011) Moving DNA around: DNA transposition and retroviral integration. Curr Opin Struct Biol 21:370–378
Ohno S (1972) So much ‘junk’ in our genome. Brookhaven Symp Biol 23:366–370
Paillart JC, Shehu-Xhilaga M, Marquet R, Mak J (2004) Dimerization of retroviral RNA genomes: an inseparable pair. Nat Rev Microbiol 2:461–472
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Mehboob ur R, Ware D, Westhoff P, Mayer KFX, Messing J, Rokhsar DS (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Ramallo E, Kalendar R, Schulman AH, Martínez-Izquierdo JA (2008) Reme1: a Copia retrotransposon in melon is transcriptionally induced by UV light. Plant Mol Biol 66:137–150
Sabot F, Schulman AH (2006) Parasitism and the retrotransposon life cycle in plants: a hitchhiker’s guide to the genome. Heredity 97:381–388
Sabot F, Sourdille P, Chantret N, Bernard M (2006) Morgane, a new LTR retrotransposon group, and its subfamilies in wheats. Genetica 128:439–447
Soleimani VD, Baum BR, Johnson DA (2006) Quantification of the retrotransposon BARE-1 reveals the dynamic nature of the barley genome. Genome 49:389–396
Spanu PD, Abbott JC, Amselem J, Burgis TA, Soanes DM, Stüber K, Loren V, van Themaat E, Brown JK, Butcher SA, Gurr SJ, Lebrun MH, Ridout CJ, Schulze-Lefert P, Talbot NJ, Ahmadinejad N, Ametz C, Barton GR, Benjdia M, Bidzinski P, Bindschedler LV, Both M, Brewer MT, Cadle-Davidson L, Cadle-Davidson MM, Collemare J, Cramer R, Frenkel O, Godfrey D, Harriman J, Hoede C, King BC, Klages S, Kleemann J, Knoll D, Koti PS, Kreplak J, López-Ruiz FJ, Lu X, Maekawa T, Mahanil S, Micali C, Milgroom MG, Montana G, Noir S, O’Connell RJ, Oberhaensli S, Parlange F, Pedersen C, Quesneville H, Reinhardt R, Rott M, Sacristán S, Schmidt SM, Schön M, Skamnioti P, Sommer H, Stephens A, Takahara H, Thordal-Christensen H, Vigouroux M, Wessling R, Wicker T, Panstruga R (2010) Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 330:1543–1546
Tanskanen JA, Sabot F, Vicient C, Schulman AH (2007) Life without GAG: The BARE-2 retrotransposon as a parasite's parasite. Gene 390:166–174
Telesnitsky A, Goff SP (1997) Reverse transcriptase and the generation of retroviral DNA in retroviruses. In: Coffin JM, Hughes SH, Varmus HE (eds) Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp 121–160
Temin HM (1980) Origin of retroviruses from cellular moveable genetic elements. Cell 21:599–600
Vicient CM, Kalendar R, Anamthawat-Jonsson K, Schulman AH (1999a) Structure functionality and evolution of the BARE-1 retrotransposon of barley. Genetica 107:53–63
Vicient CM, Suoniemi A, Anamthawat-Jónsson K, Tanskanen J, Beharav A, Nevo E, Schulman AH (1999b) Retrotransposon BARE-1 and its role in genome evolution in the genus Hordeum. Plant Cell 11:1769–1784
Vicient CM, Kalendar R, Schulman AH (2001) Envelope-containing retrovirus-like elements are widespread transcribed and spliced and insertionally polymorphic in plants. Genome Res 11:2041–2049
Wessler SR (1996) Turned on by stress: plant retrotransposons. Curr Biol 6:959–961
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Wicker T, Taudien S, Houben A, Keller B, Graner A, Platzer M, Stein N (2009) A whole-genome snapshot of 454 sequences exposes the composition of the barley genome and provides evidence for parallel evolution of genome size in wheat and barley. Plant J 59:712–722
Witte CP, Le QH, Bureau T, Kumar A (2001) Terminal-repeat retrotransposons in miniature (TRIM) are involved in restructuring plant genomes. Proc Natl Acad Sci USA 98:13778–13783
Acknowledgment
Research on which this review is based was carried out under a grant from the Academy of Finland, Decision 123074.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Schulman, A.H. (2012). Hitching a Ride: Nonautonomous Retrotransposons and Parasitism as a Lifestyle. In: Grandbastien, MA., Casacuberta, J. (eds) Plant Transposable Elements. Topics in Current Genetics, vol 24. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-31842-9_5
Download citation
DOI: https://doi.org/10.1007/978-3-642-31842-9_5
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-31841-2
Online ISBN: 978-3-642-31842-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)