
Abstract
The Notch signaling pathway plays a pivotal role in development, physiology and diseases such as cancer. In this chapter, we first give an overview of the different molecular mechanisms that regulate Notch signaling. Each subject is covered in more depth in the subsequent chapters of this book. Next, we will use the inflammatory system as an example to discuss the physiological function of Notch signaling. This is followed by a discussion of recent advances in the different pathophysiological roles of Notch signaling in leukemia as well as a wide range of solid cancers. Finally, we discuss how information about pathogenic mutations in Notch pathway components, combined with structural biological data, are beginning to provide important biological and mechanistic insights about the pathway.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- A:
-
Alanine
- ACC:
-
Adenoid cystic carcinoma
- ADAM:
-
A Disintegrin And Metalloprotease
- ANKs:
-
Ankyrin
- AML:
-
Acute Myeloid Leukemia
- AOS:
-
Adams-Oliver Syndrome
- ASCL1:
-
Achaete-scute homolog 1
- AVD:
-
Aortic valve disease
- AVS:
-
Aortic valve stenosis
- BAV:
-
Bicuspid aortic valve
- BCC:
-
Basal cell carcinoma
- BMDM:
-
Bone marrow derived macrophages
- BMI1:
-
B lymphoma Mo-MLV insertion region 1 homolog
- BRD4:
-
BromoDomain-containing 4
- CADASIL:
-
Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
- CDK9:
-
Cyclin-dependent kinase 9
- CLL:
-
Chronic lymphocytic leukemia
- COA:
-
Coarctation of the aorta
- CR:
-
Cysteine-rich
- CSCs:
-
Cancer stem cells
- cSCC:
-
Cutaneous squamous cell carcinoma
- DKO:
-
Double knockout
- DLL:
-
DELTA-LIKE
- DN-MAML:
-
Dominant-Negative Mastermind
- DSL:
-
DELTA, SERRATE, LAG-2
- E:
-
Glutamic acid
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-Mesenchymal Transition
- EP300:
-
E1A Binding Protein P300
- ER:
-
Endoplasmic reticulum or estrogen receptor
- ERK:
-
Extracellular signal-Regulated Kinase
- ETS1:
-
E26 avian leukemia oncogene 1
- F:
-
Phenylalanine
- FBXW7:
-
F-box and WD repeat domain-containing 7
- FOXA2:
-
Forkhead box A2
- G:
-
Glycine
- GABPA:
-
GA Binding Protein Transcription Factor Alpha Subunit
- GBM:
-
Glioblastoma
- GOF:
-
Gain-Of-Function
- GSI:
-
γ-Secretase Inhibitor
- HCC:
-
Hepatocellular carcinoma
- HCS:
-
Hadju-Cheney syndrome
- HD:
-
Heterodimerization Domain
- HDACs:
-
Histone deacetylases
- HER2:
-
Human epidermal growth factor receptor 2
- Hes1:
-
Hairy and Enhancer of Split 1
- HEY1:
-
Hairy/enhancer-of-split related with YRPW motif 1
- HLH:
-
Helix-loop-helix
- HLHS:
-
Hypoplastic left heart syndrome
- H&NHLHS:
-
head and neck
- I:
-
Isoleucine
- IκB:
-
Inhibitor of kappa B
- IKKα:
-
Inhibitor of Kappa-B Kinase subunit alpha
- IL1R:
-
Interleukin 1 receptor
- IL4:
-
Interleukin 4
- IL6:
-
Interleukin 6
- IL10:
-
Interleukin 10
- IL12:
-
Interleukin 12
- IL13:
-
Interleukin 13
- IFNγ:
-
Interferon γ
- IRAK2:
-
Interleukin 1 receptor-associated kinase-like 2
- IRF8:
-
Interferon-regulatory factor 8
- JAG:
-
JAGGED
- K:
-
Lysine
- KO:
-
Knockout
- KMT2D:
-
Lysine methyltransferase 2D
- L:
-
Leucine
- LNR:
-
Lin-12/Notch Repeat
- LOF:
-
Loss-Of-function
- LPS:
-
Lipopolysaccharide
- lSCC:
-
Lung squamous cell carcinoma
- MAML:
-
MASTERMIND-LIKE
- MAPK:
-
Mitogen-activated protein kinase
- MCL:
-
Mantle cell lymphoma
- MINT:
-
Msx2-interating protein
- miR:
-
microRNA
- MST:
-
Mammalian sterile 20-like kinase
- mTOR:
-
mammalian target of rapamycin
- Myc:
-
myelocytomatosis proto-oncogene
- nCC:
-
Noncutaneous carcinoma
- NCoR:
-
Nuclear receptor corepressor
- NECD:
-
Notch extracellular domain
- NEXT:
-
Notch EXtracellular Truncation
- NF-κB:
-
Nuclear Factor-κB
- NICD:
-
Notch intracellular domain
- NRR:
-
Negative regulatory region
- NSCLC:
-
Non-small cell lung cancer
- OLIG2:
-
Oligodendrocyte transcription factor
- P:
-
Proline
- PanIN:
-
Pancreatic intraepithelial neoplasia
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDZ:
-
PSD-95/Dlg/ZO-1
- PEST:
-
proline (P), glutamic acid (E), serine (S) and threonine (T)
- PI3K:
-
PhosphatidylInositol 4,5-bisphosphate 3-Kinase
- PIM:
-
Proto-Oncogene, Serine/Threonine Kinase
- PIN1:
-
Peptidylprolyl Cis/Trans Isomerase, NIMA-Interacting 1
- POFUT1:
-
Protein O-fucosyltransferase 1
- PR:
-
Progesterone receptor
- Ptcra:
-
invariant preTα chain of the pre-T cell receptor
- PTEN:
-
Phosphatase and tensin homolog
- PTM:
-
Post-translational modification
- R:
-
Arginine
- RAM:
-
RBPJ-associated module
- RAS:
-
Rat sarcoma virus oncogene
- RBPJ:
-
Recombination signal binding protein for immunoglobulin kappa J region
- RCC:
-
Renal cell cancer
- RUNX1:
-
Runt related transcription factor 1
- RUNX3:
-
Runt related transcription factor 3
- S:
-
Serine
- SCC:
-
Squamous cell carcinoma
- SCLC:
-
Small cell lung cancer
- SHARP:
-
SMRT/HDAC1 Associated Repressor Protein
- SLE:
-
Systemic lupus erythematosus
- SMRT:
-
Silencing mediator for retinoid or thyroid-hormone receptors
- SMZL:
-
Splenic marginal zone lymphoma
- SPEN:
-
Split ENds family transcriptional repressor
- SPOC:
-
Spen paralog and ortholog C-terminal domain
- T:
-
Threonine
- T-ALL:
-
T-cells acute lymphoblastc leukemia
- TAD:
-
Trans-activation domain
- TAV:
-
Tricuspid aortic valve
- TCR:
-
T-cell receptor
- TGFβ:
-
Transforming growth factor beta
- TKO:
-
Triple knockout
- TLR:
-
Toll-like receptor
- TLR4:
-
Toll-like receptor 4
- TM:
-
Transmembrane
- TMZ:
-
Temozolomide
- TNBC:
-
Triple-negative breast cancer
- TNFα:
-
Tumor necrosis factor alpha
- TORC1/2:
-
mTOR signaling complex 1/2
- V:
-
Valine
- Vegfr1:
-
Vascular endothelial growth factor receptor 1
- YAP/TAZ:
-
Yes-associated protein and WW domain containing transcription regulator 1
- ZNF143:
-
Zinc finger protein 143
1 Overview on Notch signaling
The Notch mutant phenotype was first described over a hundred years ago by John Dexter, who noticed the appearance of notches at the wing margins of fruit flies Drosophila melanogaster . Thomas Hunt Morgan identified the alleles of the corresponding genes (Morgan 1917). Several decades later, the Notch gene, encoding a transmembrane receptor controlling Drosophila neurogenesis , was identified (Artavanis-Tsakonas et al. 1983; Wharton et al. 1985; Kidd et al. 1986; del Amo et al. 1993). Soon after that, it became apparent that the Notch gene is evolutionary conserved and controls a plethora of developmental decisions, regulating homeostasis as well as development and differentiation of several different tissues and cell types during both embryonic and postnatal life . Thus, it is one of a few signaling pathways, like Wnt, transforming growth factor beta (TGFβ) and Hedgehog that is repeatedly used in multicellular organisms throughout embryonal and adult development. In Integration of Drosophila and Human Genetics to Understand Notch Signaling Related Diseases, Yamamoto and colleagues introduce how biological and genetic experiments in Drosophila contributed to the identification of key players in Notch signaling , and further discuss how mechanistic information obtained in flies can be translated to understand Notch signaling related genetic disorders in human. Notch signaling has also been implicated in carcinogenesis, of which we will highlight in this chapter and further dedicate several chapters in this book (The Notch3 Receptor and Its Intracellular Signaling-Dependent Oncogenic Mechanisms and Notch in Leukemia).
The mechanisms of how Notch signaling pathway regulates a wide range of functions can be grouped in three main categories: lateral inhibition , lateral induction and lineage decisions. During lateral inhibition, equipotent cells establish a hierarchy mediated by NOTCH receptors and ligands. During these signaling events, one cell “A” signals more to the adjacent ones preventing them to adopt the same “A” cell fate. In the lateral induction model, non-equipotent cells are involved. In particular, one group of cells signals to another group determining the acquisition of different cell fates. Finally, in the lineage decision model, asymmetrical cell division allows daughter cells to adopt different cell fates by the differential expression and/or segregation of NOTCH receptors or modulators of the Notch pathway. These models are described in depth in “Modeling the Notch Response”, “Integration of Drosophila and Human Genetics to Understand Notch Signaling Related Diseases”, “Notch and Stem Cells” and “Notch and Senescence” of this book. T-cell differentiation is a well-characterized example of the lineage decision model that was investigated in depth. In particular, loss-of-function (LOF) of Notch leads to a complete block in T-cell development (Radtke et al. 1999), whereas gain-of-function (GOF) of Notch, by introducing a constitutive-active form of Notch into hematopoietic progenitor cells, leads to T-cells acute lymphoblastic leukemia (T-ALL) in mice (Pear et al. 1996). In fact, the human NOTCH1 gene was identified in T-ALL patients as a hot spot of chromosomal translocations (Ellisen et al. 1991). The role of Notch in the early stages of T-cell development is discussed by Osborne and colleagues in “Notch and T Cell Function – A Complex Tale” of this book. Regarding pathogenesis, Chiang and colleagues discuss the aspects of Notch signaling in leukemogenesis (Notch in Leukemia) and Screpanti and colleagues focus on NOTCH3 related functions (The Notch3 Receptor and Its Intracellular Signaling-Dependent Oncogenic Mechanisms).
2 Molecular Mechanisms Controlling the Notch Signal Transduction Pathway
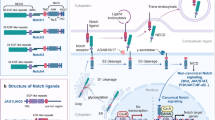
At the molecular level, the Notch signaling pathway is a seemingly simple pathway that does not involve any second messengers. Ligand-triggered activation of the NOTCH receptor leads to the release of the cleaved NOTCH intracellular domain (NICD) that drives the signaling response (Fig. 1). NOTCH receptors are single-pass transmembrane proteins that are synthesized in the endoplasmic reticulum (ER) and processed in the Golgi apparatus. During their maturation, NOTCH receptors are proteolytically processed by cleavage at the S1 site (Blaumueller et al. 1997; Logeat et al. 1998; Lake et al. 2009) and further post-translationally modified (discussed in detail in “Regulation of Notch Function by O-Glycosylation” of this book), producing the mature heterodimeric NOTCH receptor that is exposed on the plasma membrane. In mammals, four NOTCH receptors (NOTCH1-4) are expressed in a tissue- and cell-type specific manner. Mature NOTCH receptors consist of a NOTCH extracellular domain (NECD) and an intracellular portion (NICD) which are connected by a transmembrane (TM) domain. The NECD is characterized by epidermal growth factor (EGF)-like repeats that vary in number among the different isoforms, followed by three Lin-12/Notch repeats (LNR) and finally by a hydrophobic region required for the heterodimerization of the receptor. The LNR and heterodimerization (HD) domains form a negative regulatory region (NRR) that prevents ligand-independent cleavage of the receptor at the S2 cleavage site (Sanchez-Irizarry et al. 2004). The NOTCH-TM domain contains the S3 cleavage site which is the target of the γ-secretase complex that releases the NICD (Fortini 2002). The NICD is characterized by an N-terminal RBPJ (recombination signal binding protein for immunoglobulin kappa J region)-associated module (RAM) followed by ankyrin repeats (ANKs) that together form the RBPJ-interacting region (Tamura et al. 1995). These domains are followed by a transactivation domain (TAD) required for transcriptional activation and by a proline (P), glutamic acid (E), serine (S) and threonine (T) (PEST)-rich domain involved in regulating the turnover of the NICD protein. It must be noted that the TAD is not conserved in all NOTCH proteins but it is specifically found within NOTCH1 and NOTCH2, suggesting different mechanisms of transcriptional activation used by the different NOTCH proteins.

Overview of the Notch signaling cascade. Ligand binding to NOTCH receptor leads to proteolysis-dependent release of the NOTCH intracellular domain (NICD). Structural aspects of the ligand/receptor interaction are discussed in “Structural Insights into Notch Receptor-Ligand Interactions” whereas mechanotransduction of the signal and glycosylation of the NOTCH receptor are discussed in “The Molecular Mechanism of Notch Activation” and “Regulation of Notch Function by O-Glycosylation”, respectively. The first cleavage, mediated by ADAM metalloproteases, generates an intermediate proteolytic product called NEXT (Notch EXtracellular Truncation) which is substrate for a γ-secretase complex that releases the NICD. The NICD subqsequently translocates into the nucleus where it interacts with the transcription factor RBPJ and cofactor MAML leading to activation of Notch target genes (see “CSL-Associated Corepressor and Coactivator Complexes” in this book). Several Notch target genes are involved in feedback regulation of the Notch pathway, as in the case of HES1 which also regulates its own expression leading to an oscillatory control (see Oscillatory Control of Notch Signaling in Development” in this book). The Notch pathway is also regulated by endocytosis and vesicle trafficking of the NOTCH receptor (see “Endocytic Trafficking of the Notch Receptor” in this book) which can lead to degradation or ligand independent activation of the pathway (see “Mechanisms of Non-canonical Signaling in Health and Disease: Diversity to Take Therapy up a Notch?” in this book)
Similar to NOTCH receptors, the NOTCH ligands are single-pass transmembrane proteins. They are members of two different families: the DELTA/DELTA-LIKE and the SERRATE/JAGGED families. The Drosophila genome encodes one member of each family (Delta and Serrate) while mammalian ligands are more complex as three members of the DELTA family [DELTA-LIKE (DLL) 1, 3 and 4] and two members of the JAGGED family (JAG1 and 2) are encoded. All NOTCH ligands present with a DSL (DELTA, SERRATE, LAG-2) domain that contains the NOTCH receptor-interacting region followed by EGF repeats which vary in number among the different members of the families (Parks et al. 2006; D’Souza et al. 2008). Compared to the DELTA family, only the JAGGED family presents a cysteine-rich (CR) region proximal to the TM domain. Additionally, the intracellular domain of some Notch ligands is characterized by a PDZ (PSD-95/Dlg/ZO-1) domain that supports interactions with proteins of the adherens junctions (Mizuhara et al. 2005).
A major breakthrough in the Notch field was the recent elucidation of the molecular structure of the NOTCH/ligand complex (see “Structural Insights into Notch Receptor-Ligand Interactions” of this book). Genetic and biochemical studies already revealed that Notch receptor glycosylation is pivotal for its function. Reassuringly, the structures showed that sugars are in the midst of the receptor/ligand structure. This aspect and the complex regulation by NOTCH glycosylation are discussed in “Regulation of Notch Function by O-Glycosylation”. In addition to glycosylation, the exact molecular mechanisms of receptor/ligand interactions and the signal triggering mechanisms are discussed in “Structural Insights into Notch Receptor-Ligand Interactions” by Handford and colleagues considering the lipid environment and in “The Molecular Mechanism of Notch Activation” by Gordon and colleagues considering mechano-transduction and pulling-forces between two adjacent cells that express NOTCH ligand and NOTCH receptor.
The signaling cascade activated upon ligand binding is remarkably simple; in fact, two consecutive proteolytic cleavages of the NOTCH receptor release the NICD from the membrane. First, ADAM (a disintegrin and metalloprotease) metalloproteases (Brou et al. 2000; Mumm et al. 2000) cleave off the majority of the NECD; this is known as the S2 cleavage. Subsequently, the intracellular domain of the remaining Notch receptor (NICD) is liberated by an intramembrane cleavage mediated by the γ-secretase complex, a process known as S3 cleavage. The intricate regulation of receptor cleavage and endocytic trafficking as part of this process is discussed in detail by Klein and colleagues in “Endocytic Trafficking of the Notch Receptor” of this book. Upon activation, the NICD translocates into the nucleus, associates with transcription factor RBPJ and activates the expression of Notch target genes (Fig. 1). Pivotal cofactors within the RBPJ/NICD complex are MASTERMIND-LIKE (MAML) proteins which are required for the complex to be fully functional (Wu et al. 2000; Lin et al. 2002; Wu et al. 2002; Nam et al. 2003; Nam et al. 2006; Wilson and Kovall 2006); this trimeric complex recruits several additional coactivators such as acetyltransferase EP300 [E1A Binding Protein P300, (Oswald et al. 2001; Hansson et al. 2009; Jung et al. 2013)]. This is known as the canonical pathway of Notch activation and these nuclear events are discussed in “CSL-Associated Corepressor and Coactivator Complexes”. Regarding non-canonical Notch signaling, which is represented for example by RBPJ-independent events, Vaccari and colleagues elucidate these aspects of Notch signaling in “Mechanisms of Non-canonical Signaling in Health and Disease: Diversity to Take Therapy up a Notch?”. Interestingly, the protein half-life of the NICD is pivotal for amplitude and duration of the Notch response. Several post-translational modifications (PTMs) of the NICD, such as phosphorylation, acetylation and methylation are key in this process, and they culminate in the ubiquitin-dependent proteasomal degradation of the NICD, thereby terminating the Notch response (Fryer et al. 2002; Fryer et al. 2004; Palermo et al. 2012; Hein et al. 2015; Borggrefe et al. 2016). This is particularly relevant in pathophysiological conditions such as leukemogenesis. Here, stabilizing NOTCH mutations are found in several leukemias, such as T-ALL and chronic lymphocytic leukemia (CLL). Additionally, the NOTCH ubiquitin-ligase FBXW7 (F-box and WD repeat domain-containing 7) is frequently mutated in leukemia patients. The interested reader is referred to our recent review (Borggrefe et al. 2016) as well as “The Notch3 Receptor and Its Intracellular Signaling-Dependent Oncogenic Mechanisms” and “Notch in Leukemia” of this book.
In the absence of a Notch signal, the central transcription factor RBPJ remains in the nucleus bound to its target nucleotide sequence and recruits corepressors to prevent the expression of Notch target genes. In the last few years several groups including our have set out to characterize the composition of the RBPJ corepressor and coactivator complexes (Oswald et al. 2001; Hansson et al. 2009; Jung et al. 2013; Kao et al. 1998; Oswald et al. 2002; Oswald et al. 2005; Salat et al. 2008; Borggrefe and Oswald 2009; Moshkin et al. 2009; Liefke et al. 2010; Mulligan et al. 2011; Yatim et al. 2012; Oswald et al. 2016; Xu et al. 2017 and “The Notch Interactome: Complexity in Signaling Circuitry” and “Oscillatory Control of Notch Signaling in Development” of this book) and to unveil their structure (Nam et al. 2003; Nam et al. 2006; Wilson and Kovall 2006; Kovall and Hendrickson 2004; Kovall 2007; VanderWielen et al. 2011; Collins et al. 2014; Contreras et al. 2015; Yuan et al. 2016 and “Structural Insights into Notch Receptor-Ligand Interactions” of this book). These studies resulted in two important findings: First, the Notch signaling pathway is not based on a simple ON/OFF-state concerning Notch target gene expression; second, the individual RBPJ/NICD complex does not operate alone but functions as homodimer and may collaborate with other DNA binding proteins. The first observation is supported by the characterization of the protein interaction network of SHARP [SMRT (silencing mediator for retinoid or thyroid-hormone receptors)/HDAC1 (histone deacetylase)-associated repressor protein; also known as mouse MINT (Msx2-interating protein) or SPEN (Split ENds family transcriptional repressor)] which, focusing on its SPOC (Spen paralog and ortholog C-terminal) domain, unveiled an interesting and surprising scenario (Oswald et al. 2016). In fact, while previously SHARP was exclusively considered as a transcriptional repressor (Oswald et al. 2002; Oswald et al. 2005; Salat et al. 2008), proteomics studies revealed that SHARP does not exclusively interact with the corepressor NCoR (nuclear receptor corepressor) complex but also with the coactivator KMT2D (lysine methyltransferase 2D) complex (Oswald et al. 2016). These observations identified SHARP as a key regulator of the Notch pathway where NCoR and KMT2D compete for the same binding site of SHARP (Oswald et al. 2016). Thus, it is likely that SHARP is a central chromatin regulator tuning the output of the Notch response by balancing histone methylation and deacetylation.
The second observation is based on the identification of NICD homodimers that are required to specifically induce a subset of Notch target genes such as Hes1 (hairy and enhancer of split 1), Myc (myelocytomatosis proto-oncogene) and Ptcra [invariant preTα chain of the pre-TCR (T-cell receptor)] that are characterized by paired RBPJ binding sites oriented and spaced in a specific manner (Nam et al. 2007; Liu et al. 2010; Hass et al. 2015). Additionally, genome-wide studies unveiled that NOTCH1 and RBPJ binding occurs at sites that are also bound by additional transcription factors such as AML1 [acute myeloid leukemia 1, also known as RUNX1 (Runt related transcription factor 1)], ETS1 (E26 avian leukemia oncogene 1), GABPA (GA binding protein transcription factor alpha subunit) and ZNF143 [Zinc finger protein 143, (Wang et al. 2011a; Ngondo-Mbongo et al. 2013; Wang et al. 2014)], suggesting that several transcriptional factors synergize to fine-tune the expression of Notch target genes. Alternatively, competitive binding may have different transcriptional outputs in regard to the expression of Notch target genes.
Apart from chromatin regulation prior to the Notch response and combinatorial activities of several transcription factors, differential gene regulation is achieved by different promoter structures and feedback loops, which can result in oscillatory mechanisms that play key roles in development (Fig. 1). One particularly well-studied example is the basic helix-loop-helix (HLH) transcription factor HES1, encoded by a prototypic Notch target gene. Kageyama and colleagues discuss in depth the function of HES1 in “Oscillatory Control of Notch Signaling in Development” of this book.
3 Notch in Inflammation
Notch signaling has been shown to play important roles in both innate and adaptive immunity. In innate immunity, Notch signaling promotes the differentiation of specific cell types as well as supports the activation of specific cells. Macrophages are key mediators of innate immunity but are also involved in supporting specific aspects of the adaptive immunity. Based on the activating stimulus, macrophages polarize into so called M1 or M2 states: while M1 macrophages are involved in supporting inflammatory responses by producing inflammatory molecules such as interleukin 12 (IL12), IL6 or tumor necrosis factor alpha (TNFα), M2 macrophages regulate the resolution of inflammation by producing anti-inflammatory molecules such as IL10 or TGFβ (Porta et al. 2015; Kapellos and Iqbal 2016; Patel et al. 2017). Polarized macrophages can be further distinguished in M2a, M2b or M2c based on the different gene expression profile and the activating stimulus, for example IL4 and IL13 induce the M2a phenotype, the M2b is induced by exposure to immune complexes with Toll-like receptors (TLRs) or interleukin 1 receptor (IL1R) while M2c is induced by IL10 (Mantovani et al. 2004).
In bone marrow derived macrophages (BMDMs) from Rbpj conditional knockout (KO) mice (Rbpj flox/flox;Mx1-Cre) the expression of lipopolysaccharide (LPS)-induced genes is inhibited (Xu et al. 2012). RBPJ positively regulates LPS-mediated transcription via the canonical Notch signaling pathway as treatment with inhibitors of the γ-secretase complex (GSIs), that block the activation of the Notch pathway, Adam10 or Notch1 deficiencies impair gene expression of LPS targets (Xu et al. 2012). Mechanistically RBPJ controls the expression of IRAK2 (interleukin-1 receptor-associated kinase-like 2) protein that supports a cascade that culminates with the synthesis of IRF8 (interferon-regulatory factor 8) (Xu et al. 2012), a key transcription factor of the inflammatory gene expression program (Mancino et al. 2015). The control of this program in macrophages is more complex as LPS treatment also leads to upregulation of Notch target genes, such as HES1 and HEY1 (Hairy/enhancer-of-split related with YRPW motif 1), which are involved in a negative feedback loop that controls the expression of pro-inflammatory cytokines (Hu et al. 2008). Importantly, treatment with interferon γ (IFNγ) leads to downregulation of HES1 and HEY1 gene expression. This suggests a mechanism how IFNγ may augment the production of pro-inflammatory cytokines (Hu et al. 2008). As these studies pointed out to the RBPJ-dependent induction of Il12 gene upon LPS stimulation (Xu et al. 2012; Hu et al. 2008), another study could demonstrate that this effect does not involve the transcriptional activity of the NICD/RBPJ complex as overexpression of a dominant negative form of MAML (DN-MAML) does not influence the expression of Il12 in BMDMs (Boonyatecha et al. 2012). The reasons for this contrasting results are still not clear but it must be noted that another study could show that the pro-inflammatory cytokine IL6 is positively and directly regulated by the Notch signaling pathway upon treatment of BMDMs with LPS and IFNγ. In fact, Il6 expression is downregulated by GSIs and upregulated by overexpression of NICD1 upon LPS and IFNγ treatment and finally the Il6 locus is bound by NOTCH1 (Wongchana and Palaga 2012). Fung and colleagues observed that NOTCH3 expression increases during differentiation of human monocytes into macrophages in culture, while DLL4 expression increases upon pro-inflammatory stimulation of human macrophages (Fung et al. 2007). Of note, the LPS-mediated DLL4 induction is dependent on TLR4 (Toll-like receptor 4) and NF-κB (nuclear factor-κB) pathways and triggers the Notch signaling cascade that finally increases the pro-inflammatory properties of human macrophages (Fung et al. 2007). Similarly, also JAG1 is induced upon LPS stimulation of human macrophages in an NF-κB-dependent manner (Foldi et al. 2010) as well as Notch1 induction is observed upon macrophages activation and GSIs pretreatment leads to reduced expression of pro-inflammatory genes upon stimulation with LPS and IFNγ (Palaga et al. 2008), suggesting Notch signaling as an important determinant of macrophages-mediated inflammatory responses. Myeloid-specific LOF of Notch1, obtained from LysMCre;Notch1 flox/flox mice, leads to decreased macrophages recruitment at wounds as well as GSIs treatment results in failure of Vegfr1 (vascular endothelial growth factor receptor 1) induction upon macrophages stimulation with LPS and INFγ (Outtz et al. 2010).
In peritoneal macrophages, Notch signaling determines a switch from pro-inflammatory cytokines (TNFα and IL6) to anti-inflammatory cytokines (IL10) production upon stimulation with LPS in a way that is dependent on the PEST domain of NICD proteins (Zhang et al. 2012). This pro-inflammatory inhibitory effect of Notch signaling is based on the inhibition of the MAPK (mitogen-activated protein kinase) pathway leading to reduced transcriptional activity of NF-κB (Zhang et al. 2012). In contrast, another study observed that Notch signaling increases pro-inflammatory properties of macrophage derived Raw 264.7 cells upon LPS stimulation by promoting nuclear translocation of NF-κB (Monsalve et al. 2009). The reasons for the differences observed in these studies are not clear and more work is required to better dissect the role of Notch signaling upon LPS stimulation in these cells.
RBPJ controls also the M2 polarization of macrophages as RBPJ KO macrophages from Rbpj flox/flox;Lyz2-Cre mice treated with chitin, a major structural component of fungi and helminthes that induce the M2 polarization, present impaired expression of genes associated with the M2 phenotype (Foldi et al. 2016). It must also be noted that Rbpj KO results in M2 polarization of BMDM upon LPS stimulation (Wang et al. 2010a), suggesting that RBPJ may play different roles in the M1 vs M2 polarization based on the activating stimulus. Additionally, stimulation of macrophages with IL4, an interleukin that drives the M2 polarization, leads to upregulation of Jag1 (Outtz et al. 2010).
Interestingly, in a mouse model of systemic lupus erythematosus (SLE), Notch signaling is required to induce macrophage polarization versus the M2b phenotype through PI3K (phosphatidylInositol 4,5-bisphosphate 3-kinase)/AKT-ERK (Extracellular signal-regulated kinase)-1/2 and p38 MAPK signaling pathways (Zhang et al. 2010).
In summary, an important role for Notch in inflammation is evident, but further studies are required to differentiate between direct and indirect effects and to clarify how the Notch pathway orchestrates different polarization of macrophages.
4 Dysregulation of Notch Signaling in Diseases
Accurate regulation of the Notch signaling pathway is required for development, differentiation and homeostasis of a wide variety of tissues during both adult and embryonic life (see “The Notch3 Receptor and Its Intracellular Signaling-Dependent Oncogenic Mechanisms”, “Notch and Neurogenesis”, “Notch and Stem Cells”, “Notch and Senescence”, “Control of Blood Vessel Formation by Notch Signaling” and “Notch and T Cell Function – A Complex Tale” in this book) and dysregulation of Notch signaling is associated with many diseases (see “Integration of Drosophila and Human Genetics to Understand Notch Signaling Related Diseases”, “Mechanisms of Non-canonical Signaling in Health and Disease: Diversity to Take Therapy up a Notch?”, “The Notch3 Receptor and Its Intracellular Signaling-Dependent Oncogenic Mechanisms”, “Notch and Senescence”, “Control of Blood Vessel Formation by Notch Signaling” and “Notch in Leukemia” in this book).
Notch signaling has been associated with several congenital disorders, for example Notch LOF has been linked to Alagille and Adams-Oliver syndromes (AOS) whereas Notch GOF results in Hadju-Cheney syndrome [HCS, see “Integration of Drosophila and Human Genetics to Understand Notch Signaling Related Diseases” in this book and Masek and Andersson 2017]. In addition, missense mutations that affect the structure of NOTCH receptors have been found in genetic diseases. For example, NOTCH3 mutations that affect specific domains of the NECD have been linked to CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy). There are several instances where somatic mutations of NOTCH or Notch pathway components or modulators lead to cancer . We will briefly discuss the current knowledge about Notch signaling in cancer and the interested reader is referred to “The Notch3 Receptor and Its Intracellular Signaling-Dependent Oncogenic Mechanisms” and “Notch in Leukemia” of this book and other recent reviews (Aster et al. 2017).
4.1 Notch in Leukemia
In 1991, recurring mutations in the NOTCH1 gene were first described in patients with T-ALL , thus implicating Notch signaling directly in leukemogenesis (Weng et al. 2004). Those mutations lead to a C-terminal truncation of the intracellular NOTCH1 protein, thereby removing the destabilizing PEST domain and leading to increased NICD1 half-life (Weng et al. 2004). Other NOTCH1 activating mutations in T-ALL have also been identified in the NECD leading to constitutive cleavage of the NOTCH receptor (Weng et al. 2004). Similar activating NOTCH1 mutations were also identified in CLL (Puente et al. 2011), in line with previous data showing activation of the Notch pathway in CLL (Rosati et al. 2009). These data suggest Notch signaling as a potential therapeutic target in the treatment of T-ALL and CLL and lead to some clinical trials in the last years.
GSIs can be used to prevent the activation of the Notch pathway by blocking the release of the NICD from the membrane. However, this approach is unfortunately limited due to two reasons: 1) GSIs cause severe gastrointestinal side effects due to the important role of Notch signaling in differentiation of the highly proliferating gut epithelium; 2) Drug resistance to GSIs also fairly frequently arises and it is associated with mutational loss of PTEN [phosphatase and tensin homolog, (Palomero et al. 2007)] or FBXW7 (O’Neil et al. 2007) and dependent on BRD4 [bromodomain-containing protein 4, (Knoechel et al. 2014)] as well as on miR (microRNA)-223 (Kumar et al. 2014). The problems encountered with the clinical use of GSIs pointed out the need for a better dissection of the molecular mechanisms that define the Notch signaling response with the final goal to identify additional potential therapeutic targets to block Notch signaling or its oncogenic target genes. This will be of benefit not exclusively for T-ALL and CLL as aberrant Notch signaling is also observed in acute myeloid leukemia [AML, (Thiel et al. 2017)], mantle cell lymphoma [MCL, (Kridel et al. 2012)] and splenic marginal zone lymphoma [SMZL, (Rossi et al. 2012)].
4.2 Notch in Solid Tumors
NOTCH1 was originally identified as an oncogene in leukemia but surprisingly NOTCH genes have also been found to have tumor suppressive roles in other contexts (Table 1). In this section we will discuss the different functions of Notch signaling in different types of solid tumors.
4.2.1 Notch in Glioblastoma
Glioblastoma (GBM) represents one of the most aggressive forms of brain tumor and the Notch signaling pathway has been implicated in the molecular pathogenesis of gliomas. NOTCH1 receptor as well as JAG1 and DLL1 ligands are upregulated in GBM cell lines and in primary human gliomas and their knockdown results in decreased luciferase activity, using a Notch-dependent reporter assay (Purow et al. 2005). When human cell lines, transfected with NOTCH1 siRNAs, were intracranially injected into recipient mice, an increased survival was observed compared to controls (Purow et al. 2005). In line with these observations, GSIs treatment of GBM neurospheres reduces their proliferation while overexpression of an active form of NOTCH2 has the opposite effect (Fan et al. 2010). This phenotype is linked to cancer stem cells (CSCs), as GSIs treatment downregulates the expression of CSCs markers such as CD133, NESTIN, BMI1 (B lymphoma Mo-MLV insertion region 1 homolog) and OLIG2 (oligodendrocyte transcription factor 2). The most striking observation is that GSIs treatment reduces the mortality in mouse models (Fan et al. 2010), suggesting Notch signaling as a good candidate for therapeutic intervention. Even if GSIs lead to increased apoptosis of GBM neurosphere cells, as revealed by increased cleaved CASPASE-3 (Fan et al. 2010), the molecular mechanisms behind are poorly defined. Similarly, expression of DN-MAML reduces the proliferation of GBM cells but the same study pointed out to a cell type-specific dependence on different NOTCH receptors (Chen et al. 2010). Given the poor outcomes of GSIs in clinical applications, it will be important to identify additional targets that may be used to modulate the Notch pathway. One of this targets is potentially RBPJ which is upregulated in brain CSCs (Xie et al. 2016). Knockdown of RBPJ in CSCs has a stronger effect compared to GSIs in term of proliferation and it significantly increases the life-span of tumor-bearing hosts (Xie et al. 2016). The differences between GSIs treatment and RBPJ knockdown depend on the fact that RBPJ regulates also a Notch-independent transcriptional program and the effect of RBPJ is based on its interaction with CDK9 (cyclin-dependent kinase 9) to support transcriptional elongation (Xie et al. 2016). It must be also noted that a difference in regard to Notch activity in different GBMs cannot be excluded and that this difference is likely dependent on the P53 status; in fact, cells with a mutated P53 background seem to be more sensitive to Notch inhibition compared to cells with a wild type P53 background (Chen et al. 2010). In line with that, P53 wild type GBM cells present with low Notch activity as revealed by GSI treatment and DN-MAML overexpression (Xu et al. 2017).
Gliomas are usually treated by surgical intervention aimed to remove the tumor mass followed by radiotherapy and chemotherapy but, while an initial response to radiotherapy is visible, gliomas are refractory (Grossman and Batara 2004; Furnari et al. 2007), probably associated to radiation resistance of CSCs (Bao et al. 2006). GSIs treatment increases the sensitivity of glioma stem cells to clinical doses of radiation while GOF of active forms of NOTCH1 or NOTCH2 protects them from apoptosis upon radiation (Wang et al. 2010b). Importantly, when CSCs were subjected to NOTCH1 or NOTCH2 knockdown before radiation, they showed a reduced tumorigenic activity in mouse models (Wang et al. 2010b), suggesting that a combined therapy, based on radiotherapy and GSIs may be used as a therapeutic approach. In line with these data, Gilbert and colleagues could show that GSIs treatment significantly reduces the recovery of neurospheres treated with Temozolomide (TMZ), a chemotherapeutic agent used to treat gliomas (Gilbert et al. 2010). Given that the neurospheres number was reduced only when TMZ was added before GSIs, one can ima-gine that Notch signaling, in gliomas, is a mechanism that is activated as part of a resistance upon chemotherapy. Additionally, Gilbert and colleagues could show that combined TMZ and GSIs treatment reduces tumorigenicity in mouse mo-dels (Gilbert et al. 2010).
In summary, although the oncogenic role of Notch is clear, Notch inhibition alone remains ineffective in therapeutic terms. Thus, a combination therapy seems to be highly desirable and targeting the CSCs or preventing the tumor plasticity may lead the way.
4.2.2 Notch in Breast Cancer
Interestingly, Notch signaling has been linked to the triple-negative breast cancer (TNBC), negative for estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) (Foulkes et al. 2010). Similar to the subset of mutations identified in leukemia, Notch activating mutations are found in TNBC at the C-terminal PEST domain of NOTCH1, NOTCH2 and NOTCH3 (Wang et al. 2015). The prolyl-isomerase PIN1 (Peptidylprolyl Cis/Trans Isomerase, NIMA-Interacting 1) is a positive regulator of the Notch signaling pathway (Rustighi et al. 2009) and it supports Notch signaling in TNBC cells by antagonizing the FBXW7-dependent degradation of NICD1 and NICD4 (Rustighi et al. 2014). Overexpression of NUMB, a negative regulator of the Notch signaling pathway, in TNBC cells reduces the epithelial-mesenchymal transition (EMT), a process associated with cancer progression and metastasis and suppresses tumor growth in xenografts mouse models (Zhang et al. 2016a). In line with these observations, NUMB expression is lost in several breast cancer cell lines including lines established from TNBC (Stylianou et al. 2006), as well as in primary samples, leading to increased Notch signaling (Pece et al. 2004).
Mechanistically, Notch signaling regulates cell proliferation in TNBC by directly modulating the expression of CYCLIN D1 (encoded by the CCND1 gene). In fact, NOTCH1 binds to the CCND1 locus and LOF of the Notch ligand JAG1 leads to downregulation of CCND1 associated with cell cycle defects (Cohen et al. 2010).
TNBC frequently presents with alterations in the PI3K/AKT/mTOR (mammalian target of rapamycin) pathway (Lehmann et al. 2011; Banerji et al. 2012; Cancer Genome Atlas 2012) but pharmacological inhibition of this pathway proved to be ineffective. Recently, Bhola and colleagues showed that inhibition of PI3K/mTOR or TORC1/2 (mTOR signaling complex 1/2) in TNBC cells enriches for CSCs and leads to increased expression of NICD1 and JAG1 as well as increased Notch activity (Bhola et al. 2016). Importantly, inhibition of Notch signaling decreases the induction of CSCs upon PI3K/mTOR or TORC1/2 inhibition (Bhola et al. 2016), suggesting a possible combined therapy. In line with this, monoclonal antibodies that prevent Notch signaling activation can reduce tumor growth of TNBC xenografts and increase the efficacy of the chemotherapeutic agent docetaxel in mice (Qiu et al. 2013).
In MCF7 cells (ER+ PR+ HER2-), Notch controls a metabolic switch involved in tumorigenesis (Landor et al. 2011). Mechanistically, this process is controlled by PIM (Proto-Oncogene, Serine/Threonine Kinase) kinases that phosphorylate NOTCH1 increasing both its nuclear localization and activity (Santio et al. 2016). Notch signaling is also upregulated upon anti-estrogen treatment of ER+ patient derived samples and xenografts (Simoes et al. 2015). Additionally, MCF7 cells that undergo EMT upon irradiation, present increased expression of Notch pathway components, namely NOTCH2, DLL4 and JAG1 (Kim et al. 2016). Interestingly, pharmacological inhibition of Notch signaling with GSIs or knockdown of NOTCH2, DLL4 or JAG1 leads to reduced EMT upon radiation of MCF7 cells (Kim et al. 2016), supporting the idea that Notch signaling may contribute to radiation resistance.
In summary, Notch might be a valuable lead target for future therapeutic approaches in TNBC, possibly making use of combined therapies.
4.2.3 Notch in Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDAC) is one of the leading cause of cancer death and it is believed that it develops from pancreatic intraepithelial neoplasia (PanIN). Notch signaling plays a dual role in pancreatic cancer: on one hand it is oncogenic in PDAC whereas it acts as a tumor suppressor in PanIN.
In PDAC, several Notch pathway components are upregulated including NOTCH2, NOTCH3 and JAGGED1 (Miyamoto et al. 2003) whereas, using a conditional pancreatic mouse model based on the expression of the RAS (Rat sarcoma virus oncogene) mutant K-RASG12D (Pdx1-Cre;LSL-Kras G12D), Hanlon and colleagues observed an increase of PanIN upon inactivation of Notch1 (Pdx1-Cre;LSL-Kras G12D;Notch1 flox/flox, (Hanlon et al. 2010)), supporting the tumor suppressive role of Notch signaling in PanIN. This conclusion is further supported by the observation that conditional inactivation of Notch1, in the Ptf1a +/Cre;LSL-Kras +/G12D (Ptf1a +/Cre;LSL-Kras +/G12D;Notch1 flox/flox) background, slightly reduces survival (Mazur et al. 2010a). In contrast, the same study pointed out that Notch2 might play an entirely different role. In fact, its conditional inactivation (Ptf1a +/Cre;LSL-Kras +/G12D;Notch2 flox/flox) leads to decreased PanIN and PDAC development associated with increased survival (Mazur et al. 2010a), suggesting a different and opposing role for the different NOTCH receptors in pancreatic cancer. However, De La O and colleagues observed the opposite in regard to the role of Notch1. Its conditional GOF (Pdx1-CreERT;Rosa26-NICD1) in the Kras G12D background leads to increased PanIN (De La O et al. 2008). These discrepancies are potentially explained by the different genetic approaches used (loss- versus gain-of-function). Thus, it is possible that different NOTCH receptors are involved in different steps of pancreatic tumorigenesis. In line with this hypothesis, conditional LOF of Lunatic Fringe in the Pdx1-Cre;LSL-Kras G12D background (Lfng flox/flox;Pdx1-Cre;LSL-Kras G12D/+), that encodes for an O-fucosylpeptide 3-β-N-acetylglucosaminyltransferase known to modify the epidermal growth factor repeats of NOTCH proteins, caused increased NOTCH3 activation during PDAC initiation and progression but activation of NOTCH1 only at a later time point, suggesting that Lunatic Fringe is a tumor suppressor (Zhang et al. 2016b). It must also be noted that conditional expression of DN-MAML in Kras G12D background (p48-Cre;LSL-Kras G12D;Rosa26 dn-MAML/+), that blocks the canonical activity of all NOTCH receptors, delays PanIN development (Thomas et al. 2014). In agreement with the above, GSIs treatment efficiently blocks Notch signaling and reduces proliferation of both PanIN and PDAC cell lines. GSIs also attenuate PDAC development in mouse models (Plentz et al. 2009). Surprisingly, GSIs treatment of the PDAC mouse model LSL-Kras G12D/+;p53 R172H/+;Pdx-Cre tg/+ only modestly increases survival but, when used in combination with the chemotherapeutic agent gemcitabine, a significant increase in survival is observed (Cook et al. 2012). Similarly, GSIs treatment enhances radiosensitivity in xenografts (Bi et al. 2016). A significant reduction in tumor volume was also observed when anti-DLL4 antibodies, in combination with gemcitabine, were used in pancreatic xenografts models (Yen et al. 2012). Furthermore, genetic inactivation of FBXW7, the E3-ubiquitin ligase that supports the degradation of the NICD, in the p48-Cre;LSL-Kras G12D mouse model increases pancreatic tumorigenesis (Zhang et al. 2016c).
Finally, both JAG2 and NOTCH1 have been linked to cell migration of pancreatic cancer cells but this mechanism does not seem to require Notch downstream signaling as GSIs treatment has no effect on PDAC cell migration (Hu et al. 2015).
In summary, Notch signaling plays a key role in pancreatic cancer and a better dissection of the molecular mechanisms involved in this context may lead to develop more effective therapies.
4.2.4 Notch in Hepatocellular Carcinoma
Notch plays an oncogenic role in hepatocellular carcinoma (HCC). NOTCH1 (Cantarini et al. 2006; Zhu et al. 2017) and NOTCH3 (Hu et al. 2013) are upregulated and inhibition of Notch signaling with antibodies directed against NOTCH2 or JAG1 in a mouse model of liver cancer has a tumor suppressive effect (Huntzicker et al. 2015) while liver specific GOF of NICD2 leads to HCC (Dill et al. 2013). Similar results were observed in mice upon liver specific overexpression of NICD1 and Notch pathway activation is observed in human HCC (Villanueva et al. 2012). Knockdown of NOTCH1 reduces the migration and invasion of HCC cells (Hu et al. 2014) without influencing cell viability (Zhou et al. 2013) and, in line with these data, GSIs treatment reduces invasion of HCC cells but surprisingly also their viability (Zhou et al. 2012), suggesting that cell viability may be regulated by a different member of the NOTCH family.
POFUT1 (protein O-fucosyltransferase 1), a glycosyltransferase that modifies the EGF repeats of NOTCH receptors promoting ligand interaction, is upregulated in HCC and its expression correlates with poor prognosis (Ma et al. 2016). POFUT1 knockdown reduces cell growth, proliferation and migration of HCC cells, associated with reduced activation of the Notch pathway (Ma et al. 2016), suggesting POFUT1 as a possible therapeutic target in HCC. Hyperactivation of the Notch pathway in HCC is also mediated by the upregulation of JAG1, caused by the loss of the transcriptional repressor RUNX3 (Nishina et al. 2011). In addition, RUNX3 also physically interacts with the NICD1/RBPJ complex and decreases its transactivating capacity in HCC cells (Gao et al. 2010). Another study pointed out to a link between IKKα [IκB (inhibitor of kappa B) kinase subunit alpha] and Notch signaling in HCC (Liu et al. 2012). IKKα is upregulated in HCC tumor samples and inactivates the transcription factor FOXA2 (forkhead box A2) by phosphorylation leading to downregulation of NUMB (Liu et al. 2012). Recently, a crosstalk between the Notch and Hippo pathways was described as a mechanism involved in HCC pathogenesis (Kim et al. 2017). Double KO (DKO) of mammalian sterile 20-like kinase 1 and 2 (MST1/2), involved in inhibition of the Hippo pathway by phosphorylation of the transcription factors YAP/TAZ (Yes-associated protein and WW domain containing transcription regulator 1), results in HCC (Song et al. 2010) associated with activation of Notch signaling which forms a positive feedback loop with YAP/TAZ (Kim et al. 2017). GSI treatment leads to reduced HCC in the MST1/2 DKO mouse model and while these data suggest an oncogenic role for Notch signaling in HCC, Wnt pathway plays the opposing role having a tumor suppressive function in HCC (Kim et al. 2017), suggesting the involvement of several different signaling pathways in HCC pathogenesis.
Notch signaling plays a positive role in HCC CSCs as its inhibition reduces their invasion and migration (Luo et al. 2016) and it may also be important in radio-resistance of HCC CSCs. In fact, CD133+ HCC CSCs exhibit upregulation of ADAM17, associated with increased Notch signaling, upon irradiation (Hong et al. 2016).
Of note, some reports also provide evidence for a tumor suppressive role of Notch signaling in HCC. Liver specific deletion of all the three members of the Retinoblastoma protein family [Rb, p107 and p130; triple knockout (TKO) mice] leads to HCC associated with increased expression of Notch pathway components due to upregulation of E2F transcription factors with transactivation capacity (Viatour et al. 2011). Although this suggests that Notch signaling may be an oncogenic driver, GSIs treatment of TKO mice enhances HCC development, revealing a tumor suppressive role of Notch signaling (Viatour et al. 2011). The Sage laboratory could also show that TKO liver progenitors do not show increased expression of Notch1, Hes1, Hey1 or Nrarp Notch target genes, suggesting that dere-gulation of Notch signaling by Rb family members is cell type-specific and occurs during tumor progression (Viatour et al. 2011).
As consequence, Notch signaling may be an important player in HCC and a better comprehension of its function in this disease may lead to significant improvement of the current therapies.
4.2.5 Notch in Lung Cancer
Lung cancer is the leading cause of cancer-associated mortality worldwide. Based on histopathology and molecular characteristics two main subtypes can be distinguished: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC).
SCLC is distinguished from NSCLC by its characteristic small-cell phenotype that reflects its origin from the neuroendocrine lineage. SCLC is highly refractory to chemotherapy. Recent whole-genome sequencing studies of SCLC have identified recurrent mutations in the NOTCH1-4 genes (George et al. 2015), suggesting that Notch needs to be inactivated for SCLC development. As the Notch signaling pathway is a physiological regulator of neuronal and neuroendocrine differentiation, mutations in NOTCH genes are likely responsible for the characteristic neuroendocrine phenotype of SCLC.
In cancer, lineage specification genes often provide survival advantages of which cancer cells become dependent on - similar as they become addicted to - activated oncogenes (Garraway and Sellers 2006). In line with an addiction of SCLC to the neuroendocrine lineage, the Notch target gene ASCL1 (achaete-scute homolog 1), enco-ding for a transcription factor which is physiologically required to establish the lineage of neuroendocrine cells in the lung (Borges et al. 1997), was previously shown to be required for the continued survival of SCLC cells (Osada et al. 2005; Jiang et al. 2009). Thus, in this setting, Notch signaling most likely plays a tumor suppressive role and it would be attractive to reactivate Notch target genes to induce the cancer initiating cells to differentiate into a different lineage to block its malignancy. Only then, conventional chemotherapeutic agents could eliminate this devastating cancer cells.
4.2.6 Notch in Skin Cancer and Melanoma
Counterintuitively, Notch may also play the role of a tumor suppressor in other contexts. Notch signaling has a tumor suppressive function in the skin as conditional inactivation of Notch1 leads to epidermal and corneal hyperplasia followed by the development of skin tumors (Nicolas et al. 2003). Similar results were obtained by skin specific deletion of Notch1, mediated by Pdx1-Cre, using the RAS mutant Kras +/LSL-G12D mouse model (Mazur et al. 2010b). The same study also pointed out a specific tumor suppressive role for Notch1 as genetic depletion of Notch2 does not support carcinogenesis (Mazur et al. 2010b). Demehri and colleagues showed that Notch1 depletion in epidermal keratinocytes induces tumorigenesis in a non-cell autonomous manner (Demehri et al. 2009). Similarly to Notch1 LOF, conditional expression of dn-Maml driven by SM22-Cre in the skin leads to development of cutaneous squamous cell carcinoma [SCC, (Proweller et al. 2006)]. In line with these data, mesenchymal deletion of the Notch signaling effector Rbpj results in skin tumor (Hu et al. 2012). Notch signaling may also play a tumor suppressive role in human skin cancer as several Notch pathway components are downregulated in human basal cell carcinoma [BCC, (Thelu et al. 2002)]. This hypothesis is further supported by the identification of mutations in human NOTCH1 in cutaneous SCC that impair the Notch function (Wang et al. 2011b).
At molecular level, data from keratinocytes and SCC cell lines suggest that NOTCH1 is under the positive control of P53 (Lefort et al. 2007), which is frequently mutated in skin SCC (Backvall et al. 2004). This positive function of P53 is counteracted by EGFR (epidermal growth factor receptor) signaling as its inhibition promotes P53 expression and, consequently, NOTCH1 expression with increased Notch signaling (Kolev et al. 2008). Of note, EGFR inhibition in SCC cells induces differentiation and, when EGFR inhibition is combined with inhibition of the Notch signaling pathway, increased apoptosis is observed (Kolev et al. 2008).
Recently, the involvement of Notch signaling in melanoma has gained attention. NOTCH receptors and ligands as well as Notch effectors are upregulated in melanomas (Balint et al. 2005; Massi et al. 2006) and Notch signaling inhibition, via GSIs or expression of DN-MAML, suppresses melanoma cell growth (Balint et al. 2005). In line with this, GOF of the active form of NOTCH1 increases melanoma cell growth as well as enhances primary melanoma and lung metastasis in adult mice (Balint et al. 2005; Liu et al. 2006). In addition, FBXW7 was found to be mutated in melanoma patients and these mutations compromise the function of FBXW7 protein leading to accumulation of the active form of NOTCH1 (Aydin et al. 2014). At mechanistic level, active NOTCH1 stabilizes the Wnt signaling effector protein β-CATENIN rather than acting through RBPJ . Indeed, LOF of β-CATENIN in melanoma cells mirrors the proliferative defects observed upon LOF of Notch signaling (Balint et al. 2005). Such non-canonical functions of the intracellular active form of Notch affecting other conserved signaling pathways have been recently reviewed (Borggrefe et al. 2016) and are also discussed by Vaccari and colleagues in “Mechanisms of Non-canonical Signaling in Health and Disease: Diversity to Take Therapy up a Notch?” of this book. There is another study by Liu and colleagues showing that NOTCH1 increases melanocyte growth by activating the MAPK and PI3K/AKT signaling pathways (Liu et al. 2006), suggesting that Notch signaling is involved in melanoma by regulating crosstalk with even more signaling pathways.
In conclusion, Notch signaling may be a valuable target also for the treatment of melanoma and skin cancer. However, in the case of skin cancer, this will be particularly challenging because of the tumor-suppressive function of Notch.
5 Mutational Spectrum of Notch Pathway Components
Several mutations involving Notch pathway components have been identified in cancer and genetic disorders as discussed in the previous sections and selectively summarized in Table 1. One striking observation is that the same protein domains are mutated in different diseases (see also “Integration of Drosophila and Human Genetics to Understand Notch Signaling Related Diseases” of this book), suggesting that common molecular mechanisms are probably used to confer pathogenicity.
A number of structure biological studies have fully or partially solved the molecular structure of key Notch signaling components, allowing us to understand the effect of disease-linked mutations in the context of 3D protein structure. For example, mutations occurring in the FBXW7 gene (Fig. 2a and Table 1), encoding for the E3 ubiquitin ligase involved in the degradation of the NICD, are frequently found in melanoma, SCLC and T-ALL. These mutations can compromise the activity of FBXW7, leading to increased protein stability of the NICD and of the other FBXW7 substrates (Aydin et al. 2014). RBPJ has been reported to be mutated in AOS, a genetic disease characterized in most of the patients by terminal limb malformations (Hassed et al. 2012). The AOS-associated missense mutations identified in the RBPJ gene (Fig. 2b) compromise its DNA binding ability (Hassed et al. 2012) and mutations in NOTCH1 and DLL4 have been also identified in AOS patients (Meester et al. 2015; Stittrich et al. 2014). The reader is also referred to “Integration of Drosophila and Human Genetics to Understand Notch Signaling Related Diseases” for an in-depth review of genetic mutations of Notch pathway components. Chromosomal translocations and aberrations involving FBXW7 and RBPJ are also linked to diseases. FBXW7 is translocated in renal cell cancer [RCC; (Kuiper et al. 2009)] while RBPJ in the proximal 4p deletion syndrome (Nakayama et al. 2014).

Examples of pathogenic mutations in key Notch pathway components in the context of protein structure. (a) Structure of the WD40 repeats of FBXW7 (PDB ID, 5V4B). Residues for which mutations have been identified in diseases are indicated. (b) Structure of the transcription factor RBPJ in complex with the DNA (PDB ID, 3IAG). Indicated are residues mutated in AOS. (c) Structure of the NRR of NOTCH1 (PDB ID, 3ETO). Indicated are residues for which mutations have been identified in diseases and that have been functionally analyzed. A: alanine; E: glutamic acid; F: phenylalanine; G: glycine; I: isoleucine; K: lysine; L: leucine; R: arginine; S: serine; V: valine
Mutations occurring in the NOTCH1 gene are clustered in different regions (Table 1). Among them, mutations that occur in the LNR repeats, HD and PEST domains are seen in many types of diseases as well as genetic disorders. Typically, mutations involving the LNR repeats and HD domain lead to disruption of the negative regulatory region (Fig. 2c) and promote ligand-independent cleavage of the receptor, leading to increased Notch signaling. Similarly, mutations that influence the structure of the PEST domain lead to increased half-life of the NICD resulting in aberrant transcriptional activity. Similarly to FBXW7 and RBPJ , also the NOTCH1 gene is subjected to chromosomal translocations that impair its activity (Ellisen et al. 1991).
Thus, there are indeed viable genetic mutations of the Notch receptor or Notch signaling components, that could in future provide even more insights in Notch-related pathologies, not only in the context of development but also in the cancer context.
6 Perspectives
Given the important function of the Notch signaling pathway in cancer as well as in genetic diseases, it will be important to deeper understand its regulation focusing on the molecular basis that characterize this signaling cascade. This approach will allow in the future the development of new and more efficient therapies that can overcome the limitations of the current approaches, primarily the side effects and resistance observed by using GSIs. New cancer therapies might be based on small molecule inhibitors of Notch modulators or Notch pathway components to reactivate the tumor suppressive function or to block the oncogenic activities of the pathway depending on the different pathological contexts. Another fascinating alternative would be the use of antibodies aimed to stimulate or block the activation of NOTCH receptors, an approach that seems to be promising. This can be achieved by using antibodies directed against NOTCH receptors (Aste-Amezaga et al. 2010; Wu et al. 2010; Canalis et al. 2017), ligands (Billiard et al. 2011; Lafkas et al. 2015; Xu et al. 2016; Wang et al. 2017) or the γ-secretase complex (Hayashi et al. 2012). Similar approaches can be employed to modulate the Notch function in macrophages as inflam-mation is one the key processes that drive tumorigenesis. In conclusion, more work is needed to deeply understand the regulation of the Notch signaling pathway and modulate its activity for clinical use.
References
Artavanis-Tsakonas S, Muskavitch MA, Yedvobnick B (1983) Molecular cloning of Notch, a locus affecting neurogenesis in Drosophila melanogaster. Proc Natl Acad Sci U S A 80(7):1977–1981
Aste-Amezaga M, Zhang N, Lineberger JE, Arnold BA, Toner TJ, Gu M, Huang L, Vitelli S, Vo KT, Haytko P, Zhao JZ, Baleydier F, L’Heureux S, Wang H, Gordon WR, Thoryk E, Andrawes MB, Tiyanont K, Stegmaier K, Roti G, Ross KN, Franlin LL, Wang H, Wang F, Chastain M, Bett AJ, Audoly LP, Aster JC, Blacklow SC, Huber HE (2010) Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One 5(2):e9094. https://doi.org/10.1371/journal.pone.0009094
Aster JC, Pear WS, Blacklow SC (2017) The varied roles of Notch in cancer. Annu Rev Pathol 12:245–275. https://doi.org/10.1146/annurev-pathol-052016-100127
Aydin IT, Melamed RD, Adams SJ, Castillo-Martin M, Demir A, Bryk D, Brunner G, Cordon-Cardo C, Osman I, Rabadan R, Celebi JT (2014) FBXW7 mutations in melanoma and a new therapeutic paradigm. J Natl Cancer Inst 106(6):dju107. https://doi.org/10.1093/jnci/dju107
Backvall H, Stromberg S, Gustafsson A, Asplund A, Sivertsson A, Lundeberg J, Ponten F (2004) Mutation spectra of epidermal p53 clones adjacent to basal cell carcinoma and squamous cell carcinoma. Exp Dermatol 13(10):643–650. https://doi.org/10.1111/j.0906-6705.2004.00211.x
Balint K, Xiao M, Pinnix CC, Soma A, Veres I, Juhasz I, Brown EJ, Capobianco AJ, Herlyn M, Liu ZJ (2005) Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J Clin Invest 115(11):3166–3176. https://doi.org/10.1172/JCI25001
Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Pina V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N, Thompson KM, Uribe-Figueroa L, Baselga J, Beroukhim R, Polyak K, Sgroi DC, Richardson AL, Jimenez-Sanchez G, Lander ES, Gabriel SB, Garraway LA, Golub TR, Melendez-Zajgla J, Toker A, Getz G, Hidalgo-Miranda A, Meyerson M (2012) Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486(7403):405–409. https://doi.org/10.1038/nature11154
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444(7120):756–760. https://doi.org/10.1038/nature05236
Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, Gine E, Pinyol M, Royo C, Nadeu F, Conde L, Juan M, Clot G, Vizan P, Di Croce L, Puente DA, Lopez-Guerra M, Moros A, Roue G, Aymerich M, Villamor N, Colomo L, Martinez A, Valera A, Martin-Subero JI, Amador V, Hernandez L, Rozman M, Enjuanes A, Forcada P, Muntanola A, Hartmann EM, Calasanz MJ, Rosenwald A, Ott G, Hernandez-Rivas JM, Klapper W, Siebert R, Wiestner A, Wilson WH, Colomer D, Lopez-Guillermo A, Lopez-Otin C, Puente XS, Campo E (2013) Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 110(45):18250–18255. https://doi.org/10.1073/pnas.1314608110
Bhola NE, Jansen VM, Koch JP, Li H, Formisano L, Williams JA, Grandis JR, Arteaga CL (2016) Treatment of triple-negative breast cancer with TORC1/2 inhibitors sustains a drug-resistant and Notch-dependent cancer stem cell population. Cancer Res 76(2):440–452. https://doi.org/10.1158/0008-5472.CAN-15-1640-T
Bi YL, Min M, Shen W, Liu Y (2016) Numb/Notch signaling pathway modulation enhances human pancreatic cancer cell radiosensitivity. Tumour Biol J Int Soc Oncodev Biol Med 37(11):15145–15155. https://doi.org/10.1007/s13277-016-5311-8
Billiard F, Kirshner JR, Tait M, Danave A, Taheri S, Zhang W, Waite JC, Olson K, Chen G, Coetzee S, Hylton D, Murphy AJ, Yancopoulos GD, Thurston G, Skokos D (2011) Ongoing Dll4-Notch signaling is required for T-cell homeostasis in the adult thymus. Eur J Immunol 41(8):2207–2216. https://doi.org/10.1002/eji.201041343
Bittolo T, Pozzo F, Bomben R, D’Agaro T, Bravin V, Bulian P, Rossi FM, Zucchetto A, Degan M, Macor P, D’Arena G, Chiarenza A, Zaja F, Pozzato G, Di Raimondo F, Rossi D, Gaidano G, Del Poeta G, Gattei V, Dal Bo M (2017) Mutations in the 3’ untranslated region of NOTCH1 are associated with low CD20 expression levels chronic lymphocytic leukemia. Haematologica 102(8):e305–e309. https://doi.org/10.3324/haematol.2016.162594
Blaumueller CM, Qi H, Zagouras P, Artavanis-Tsakonas S (1997) Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell 90(2):281–291
Boonyatecha N, Sangphech N, Wongchana W, Kueanjinda P, Palaga T (2012) Involvement of Notch signaling pathway in regulating IL-12 expression via c-Rel in activated macrophages. Mol Immunol 51(3–4):255–262. https://doi.org/10.1016/j.molimm.2012.03.017
Borges M, Linnoila RI, van de Velde HJ, Chen H, Nelkin BD, Mabry M, Baylin SB, Ball DW (1997) An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature 386(6627):852–855. https://doi.org/10.1038/386852a0
Borggrefe T, Oswald F (2009) The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci 66(10):1631–1646. https://doi.org/10.1007/s00018-009-8668-7
Borggrefe T, Lauth M, Zwijsen A, Huylebroeck D, Oswald F, Giaimo BD (2016) The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFbeta/BMP and hypoxia pathways. Biochim Biophys Acta 1863(2):303–313. https://doi.org/10.1016/j.bbamcr.2015.11.020
Breit S, Stanulla M, Flohr T, Schrappe M, Ludwig WD, Tolle G, Happich M, Muckenthaler MU, Kulozik AE (2006) Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood 108(4):1151–1157. https://doi.org/10.1182/blood-2005-12-4956
Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A (2000) A novel proteolytic cleavage involved in Notch signaling : the role of the disintegrin-metalloprotease TACE. Mol Cell 5(2):207–216
Canalis E, Sanjay A, Yu J, Zanotti S (2017) An antibody to Notch2 reverses the osteopenic phenotype of Hajdu-Cheney mutant male mice. Endocrinology 158(4):730–742. https://doi.org/10.1210/en.2016-1787
Cancer Genome Atlas N (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70. https://doi.org/10.1038/nature11412
Cantarini MC, de la Monte SM, Pang M, Tong M, D’Errico A, Trevisani F, Wands JR (2006) Aspartyl-asparagyl beta hydroxylase over-expression in human hepatoma is linked to activation of insulin-like growth factor and notch signaling mechanisms. Hepatology 44(2):446–457. https://doi.org/10.1002/hep.21272
Chen J, Kesari S, Rooney C, Strack PR, Chen J, Shen H, Wu L, Griffin JD (2010) Inhibition of notch signaling blocks growth of glioblastoma cell lines and tumor neurospheres. Genes Cancer 1(8):822–835. https://doi.org/10.1177/1947601910383564
Cohen B, Shimizu M, Izrailit J, Ng NF, Buchman Y, Pan JG, Dering J, Reedijk M (2010) Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast Cancer Res Treat 123(1):113–124. https://doi.org/10.1007/s10549-009-0621-9
Collins KJ, Yuan Z, Kovall RA (2014) Structure and function of the CSL-KyoT2 corepressor complex: a negative regulator of Notch signaling. Structure 22(1):70–81. https://doi.org/10.1016/j.str.2013.10.010
Contreras AN, Yuan Z, Kovall RA (2015) Thermodynamic binding analysis of Notch transcription complexes from Drosophila melanogaster. Protein Sci 24(5):812–822. https://doi.org/10.1002/pro.2652
Cook N, Frese KK, Bapiro TE, Jacobetz MA, Gopinathan A, Miller JL, Rao SS, Demuth T, Howat WJ, Jodrell DI, Tuveson DA (2012) Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med 209(3):437–444. https://doi.org/10.1084/jem.20111923
D’Agaro T, Bittolo T, Bravin V, Dal Bo M, Pozzo F, Bulian P, Rossi FM, Zucchetto A, Degan M, D’Arena G, Chiarenza A, Zaja F, Pozzato G, Di Raimondo F, Rossi D, Gaidano G, Del Poeta G, Gattei V, Bomben R (2017) NOTCH1 mutational status in chronic lymphocytic leukaemia: clinical relevance of subclonal mutations and mutation types. Br J Haematol. https://doi.org/10.1111/bjh.14843
D’Souza B, Miyamoto A, Weinmaster G (2008) The many facets of Notch ligands. Oncogene 27(38):5148–5167. https://doi.org/10.1038/onc.2008.229
De Keersmaecker K, Lahortiga I, Mentens N, Folens C, Van Neste L, Bekaert S, Vandenberghe P, Odero MD, Marynen P, Cools J (2008) In vitro validation of gamma-secretase inhibitors alone or in combination with other anti-cancer drugs for the treatment of T-cell acute lymphoblastic leukemia. Haematologica 93(4):533–542. https://doi.org/10.3324/haematol.11894
De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, Murtaugh LC (2008) Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A 105(48):18907–18912. https://doi.org/10.1073/pnas.0810111105
del Amo FF, Gendron-Maguire M, Swiatek PJ, Jenkins NA, Copeland NG, Gridley T (1993) Cloning, analysis, and chromosomal localization of Notch-1, a mouse homolog of Drosophila Notch. Genomics 15(2):259–264. https://doi.org/10.1006/geno.1993.1055
Demehri S, Turkoz A, Kopan R (2009) Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 16(1):55–66. https://doi.org/10.1016/j.ccr.2009.05.016
Dill MT, Tornillo L, Fritzius T, Terracciano L, Semela D, Bettler B, Heim MH, Tchorz JS (2013) Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology 57(4):1607–1619. https://doi.org/10.1002/hep.26165
Ducharme V, Guauque-Olarte S, Gaudreault N, Pibarot P, Mathieu P, Bosse Y (2013) NOTCH1 genetic variants in patients with tricuspid calcific aortic valve stenosis. J Heart Valve Dis 22(2):142–149
Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, Sklar J (1991) TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 66(4):649–661
Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, Grunn A, Fangazio M, Capello D, Monti S, Cresta S, Gargiulo E, Forconi F, Guarini A, Arcaini L, Paulli M, Laurenti L, Larocca LM, Marasca R, Gattei V, Oscier D, Bertoni F, Mullighan CG, Foa R, Pasqualucci L, Rabadan R, Dalla-Favera R, Gaidano G (2011) Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med 208(7):1389–1401. https://doi.org/10.1084/jem.20110921
Foffa I, Ait Ali L, Panesi P, Mariani M, Festa P, Botto N, Vecoli C, Andreassi MG (2013) Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med Genet 14:44. https://doi.org/10.1186/1471-2350-14-44
Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, Nikkhah G, Dimeco F, Piccirillo S, Vescovi AL, Eberhart CG (2010) NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28(1):5–16. https://doi.org/10.1002/stem.254
Foldi J, Chung AY, Xu H, Zhu J, Outtz HH, Kitajewski J, Li Y, Hu X, Ivashkiv LB (2010) Autoamplification of Notch signaling in macrophages by TLR-induced and RBP-J-dependent induction of Jagged1. J Immunol 185(9):5023–5031. https://doi.org/10.4049/jimmunol.1001544
Foldi J, Shang Y, Zhao B, Ivashkiv LB, Hu X (2016) RBP-J is required for M2 macrophage polarization in response to chitin and mediates expression of a subset of M2 genes. Protein & cell 7(3):201–209. https://doi.org/10.1007/s13238-016-0248-7
Fortini ME (2002) Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell Biol 3(9):673–684. https://doi.org/10.1038/nrm910
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363(20):1938–1948. https://doi.org/10.1056/NEJMra1001389
Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA (2002) Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev 16(11):1397–1411. https://doi.org/10.1101/gad.991602
Fryer CJ, White JB, Jones KA (2004) Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell 16(4):509–520. https://doi.org/10.1016/j.molcel.2004.10.014
Fung E, Tang SM, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA, Carlesso N, Aster JC, Aikawa M (2007) Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation 115(23):2948–2956. https://doi.org/10.1161/CIRCULATIONAHA.106.675462
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21(21):2683–2710. https://doi.org/10.1101/gad.1596707
Gao J, Chen Y, Wu KC, Liu J, Zhao YQ, Pan YL, Du R, Zheng GR, Xiong YM, Xu HL, Fan DM (2010) RUNX3 directly interacts with intracellular domain of Notch1 and suppresses Notch signaling in hepatocellular carcinoma cells. Exp Cell Res 316(2):149–157. https://doi.org/10.1016/j.yexcr.2009.09.025
Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D (2005) Mutations in NOTCH1 cause aortic valve disease. Nature 437(7056):270–274. https://doi.org/10.1038/nature03940
Garraway LA, Sellers WR (2006) Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer 6(8):593–602. https://doi.org/10.1038/nrc1947
George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, Muller C, Dahmen I, Jahchan NS, Park KS, Yang D, Karnezis AN, Vaka D, Torres A, Wang MS, Korbel JO, Menon R, Chun SM, Kim D, Wilkerson M, Hayes N, Engelmann D, Putzer B, Bos M, Michels S, Vlasic I, Seidel D, Pinther B, Schaub P, Becker C, Altmuller J, Yokota J, Kohno T, Iwakawa R, Tsuta K, Noguchi M, Muley T, Hoffmann H, Schnabel PA, Petersen I, Chen Y, Soltermann A, Tischler V, Choi CM, Kim YH, Massion PP, Zou Y, Jovanovic D, Kontic M, Wright GM, Russell PA, Solomon B, Koch I, Lindner M, Muscarella LA, la Torre A, Field JK, Jakopovic M, Knezevic J, Castanos-Velez E, Roz L, Pastorino U, Brustugun OT, Lund-Iversen M, Thunnissen E, Kohler J, Schuler M, Botling J, Sandelin M, Sanchez-Cespedes M, Salvesen HB, Achter V, Lang U, Bogus M, Schneider PM, Zander T, Ansen S, Hallek M, Wolf J, Vingron M, Yatabe Y, Travis WD, Nurnberg P, Reinhardt C, Perner S, Heukamp L, Buttner R, Haas SA, Brambilla E, Peifer M, Sage J, Thomas RK (2015) Comprehensive genomic profiles of small cell lung cancer. Nature 524(7563):47–53. https://doi.org/10.1038/nature14664
Gilbert CA, Daou MC, Moser RP, Ross AH (2010) Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res 70(17):6870–6879. https://doi.org/10.1158/0008-5472.CAN-10-1378
Grossman SA, Batara JF (2004) Current management of glioblastoma multiforme. Semin Oncol 31(5):635–644
Hanlon L, Avila JL, Demarest RM, Troutman S, Allen M, Ratti F, Rustgi AK, Stanger BZ, Radtke F, Adsay V, Long F, Capobianco AJ, Kissil JL (2010) Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res 70(11):4280–4286. https://doi.org/10.1158/0008-5472.CAN-09-4645
Hansson ML, Popko-Scibor AE, Saint Just Ribeiro M, Dancy BM, Lindberg MJ, Cole PA, Wallberg AE (2009) The transcriptional coactivator MAML1 regulates p300 autoacetylation and HAT activity. Nucleic Acids Res 37(9):2996–3006. https://doi.org/10.1093/nar/gkp163
Hass MR, Liow HH, Chen X, Sharma A, Inoue YU, Inoue T, Reeb A, Martens A, Fulbright M, Raju S, Stevens M, Boyle S, Park JS, Weirauch MT, Brent MR, Kopan R (2015) SpDamID: marking DNA bound by protein complexes identifies Notch-Dimer responsive enhancers. Mol Cell 59(4):685–697. https://doi.org/10.1016/j.molcel.2015.07.008
Hassed SJ, Wiley GB, Wang S, Lee JY, Li S, Xu W, Zhao ZJ, Mulvihill JJ, Robertson J, Warner J, Gaffney PM (2012) RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am J Hum Genet 91(2):391–395. https://doi.org/10.1016/j.ajhg.2012.07.005
Hayashi I, Takatori S, Urano Y, Miyake Y, Takagi J, Sakata-Yanagimoto M, Iwanari H, Osawa S, Morohashi Y, Li T, Wong PC, Chiba S, Kodama T, Hamakubo T, Tomita T, Iwatsubo T (2012) Neutralization of the gamma-secretase activity by monoclonal antibody against extracellular domain of nicastrin. Oncogene 31(6):787–798. https://doi.org/10.1038/onc.2011.265
Hein K, Mittler G, Cizelsky W, Kuhl M, Ferrante F, Liefke R, Berger IM, Just S, Strang JE, Kestler HA, Oswald F, Borggrefe T (2015) Site-specific methylation of Notch1 controls the amplitude and duration of the Notch1 response. Sci Signal 8(369):ra30. https://doi.org/10.1126/scisignal.2005892
Hong SW, Hur W, Choi JE, Kim JH, Hwang D, Yoon SK (2016) Role of ADAM17 in invasion and migration of CD133-expressing liver cancer stem cells after irradiation. Oncotarget 7(17):23482–23497. https://doi.org/10.18632/oncotarget.8112
Hu X, Chung AY, Wu I, Foldi J, Chen J, Ji JD, Tateya T, Kang YJ, Han J, Gessler M, Kageyama R, Ivashkiv LB (2008) Integrated regulation of Toll-like receptor responses by Notch and interferon-gamma pathways. Immunity 29(5):691–703. https://doi.org/10.1016/j.immuni.2008.08.016
Hu B, Castillo E, Harewood L, Ostano P, Reymond A, Dummer R, Raffoul W, Hoetzenecker W, Hofbauer GF, Dotto GP (2012) Multifocal epithelial tumors and field cancerization from loss of mesenchymal CSL signaling. Cell 149(6):1207–1220. https://doi.org/10.1016/j.cell.2012.03.048
Hu L, Xue F, Shao M, Deng A, Wei G (2013) Aberrant expression of Notch3 predicts poor survival for hepatocellular carcinomas. Biosci Trends 7(3):152–156
Hu YJ, Li HY, Qiu KJ, Li DC, Zhou JH, Hu YH, Zhang FM (2014) Downregulation of Notch1 inhibits the invasion of human hepatocellular carcinoma HepG2 and MHCC97H cells through the regulation of PTEN and FAK. Int J Mol Med 34(4):1081–1086. https://doi.org/10.3892/ijmm.2014.1889
Hu Y, Su H, Li X, Guo G, Cheng L, Qin R, Qing G, Liu H (2015) The NOTCH ligand JAGGED2 promotes pancreatic cancer metastasis independent of NOTCH signaling activation. Mol Cancer Ther 14(1):289–297. https://doi.org/10.1158/1535-7163.MCT-14-0501
Huntzicker EG, Hotzel K, Choy L, Che L, Ross J, Pau G, Sharma N, Siebel CW, Chen X, French DM (2015) Differential effects of targeting Notch receptors in a mouse model of liver cancer. Hepatology 61(3):942–952. https://doi.org/10.1002/hep.27566
Iascone M, Ciccone R, Galletti L, Marchetti D, Seddio F, Lincesso AR, Pezzoli L, Vetro A, Barachetti D, Boni L, Federici D, Soto AM, Comas JV, Ferrazzi P, Zuffardi O (2012) Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin Genet 81(6):542–554. https://doi.org/10.1111/j.1399-0004.2011.01674.x
Jiang T, Collins BJ, Jin N, Watkins DN, Brock MV, Matsui W, Nelkin BD, Ball DW (2009) Achaete-scute complex homologue 1 regulates tumor-initiating capacity in human small cell lung cancer. Cancer Res 69(3):845–854. https://doi.org/10.1158/0008-5472.CAN-08-2762
Jung C, Mittler G, Oswald F, Borggrefe T (2013) RNA helicase Ddx5 and the noncoding RNA SRA act as coactivators in the Notch signaling pathway. Biochim Biophys Acta 1833(5):1180–1189. https://doi.org/10.1016/j.bbamcr.2013.01.032
Kao HY, Ordentlich P, Koyano-Nakagawa N, Tang Z, Downes M, Kintner CR, Evans RM, Kadesch T (1998) A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev 12(15):2269–2277
Kapellos TS, Iqbal AJ (2016) Epigenetic control of macrophage polarisation and soluble mediator gene expression during Inflammation. Mediat Inflamm 2016:6591703. https://doi.org/10.1155/2016/6591703
Kent KC, Crenshaw ML, Goh DL, Dietz HC (2013) Genotype-phenotype correlation in patients with bicuspid aortic valve and aneurysm. J Thorac Cardiovasc Surg 146(1):158–165 e151. https://doi.org/10.1016/j.jtcvs.2012.09.060
Kidd S, Kelley MR, Young MW (1986) Sequence of the notch locus of Drosophila melanogaster: relationship of the encoded protein to mammalian clotting and growth factors. Mol Cell Biol 6(9):3094–3108
Kim RK, Kaushik N, Suh Y, Yoo KC, Cui YH, Kim MJ, Lee HJ, Kim IG, Lee SJ (2016) Radiation driven epithelial-mesenchymal transition is mediated by Notch signaling in breast cancer. Oncotarget 7(33):53430–53442. https://doi.org/10.18632/oncotarget.10802
Kim W, Khan SK, Gvozdenovic-Jeremic J, Kim Y, Dahlman J, Kim H, Park O, Ishitani T, Jho EH, Gao B, Yang Y (2017) Hippo signaling interactions with Wnt/beta-catenin and Notch signaling repress liver tumorigenesis. J Clin Invest 127(1):137–152. https://doi.org/10.1172/JCI88486
Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, Gillespie SM, Fernandez D, Ku M, Wang H, Piccioni F, Silver SJ, Jain M, Pearson D, Kluk MJ, Ott CJ, Shultz LD, Brehm MA, Greiner DL, Gutierrez A, Stegmaier K, Kung AL, Root DE, Bradner JE, Aster JC, Kelliher MA, Bernstein BE (2014) An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet 46(4):364–370. https://doi.org/10.1038/ng.2913
Kolev V, Mandinova A, Guinea-Viniegra J, Hu B, Lefort K, Lambertini C, Neel V, Dummer R, Wagner EF, Dotto GP (2008) EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat Cell Biol 10(8):902–911. https://doi.org/10.1038/ncb1750
Kovall RA (2007) Structures of CSL, Notch and Mastermind proteins: piecing together an active transcription complex. Curr Opin Struct Biol 17(1):117–127. https://doi.org/10.1016/j.sbi.2006.11.004
Kovall RA, Hendrickson WA (2004) Crystal structure of the nuclear effector of Notch signaling, CSL, bound to DNA. EMBO J 23(17):3441–3451. https://doi.org/10.1038/sj.emboj.7600349
Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, Gunawardana J, Jenkins C, Cochrane C, Ben-Neriah S, Tan K, Morin RD, Opat S, Sehn LH, Connors JM, Marra MA, Weng AP, Steidl C, Gascoyne RD (2012) Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 119(9):1963–1971. https://doi.org/10.1182/blood-2011-11-391474
Kuiper RP, Vreede L, Venkatachalam R, Ricketts C, Kamping E, Verwiel E, Govaerts L, Debiec-Rychter M, Lerut E, van Erp F, Hoogerbrugge N, van Kempen L, Schoenmakers EF, Bonne A, Maher ER, Geurts van Kessel A (2009) The tumor suppressor gene FBXW7 is disrupted by a constitutional t(3;4)(q21;q31) in a patient with renal cell cancer. Cancer Genet Cytogenet 195(2):105–111. https://doi.org/10.1016/j.cancergencyto.2009.07.001
Kumar V, Palermo R, Talora C, Campese AF, Checquolo S, Bellavia D, Tottone L, Testa G, Miele E, Indraccolo S, Amadori A, Ferretti E, Gulino A, Vacca A, Screpanti I (2014) Notch and NF-kB signaling pathways regulate miR-223/FBXW7 axis in T-cell acute lymphoblastic leukemia. Leukemia 28(12):2324–2335. https://doi.org/10.1038/leu.2014.133
Lafkas D, Shelton A, Chiu C, de Leon Boenig G, Chen Y, Stawicki SS, Siltanen C, Reichelt M, Zhou M, Wu X, Eastham-Anderson J, Moore H, Roose-Girma M, Chinn Y, Hang JQ, Warming S, Egen J, Lee WP, Austin C, Wu Y, Payandeh J, Lowe JB, Siebel CW (2015) Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature 528 (7580):127-131. doi:https://doi.org/10.1038/nature15715
Lake RJ, Grimm LM, Veraksa A, Banos A, Artavanis-Tsakonas S (2009) In vivo analysis of the Notch receptor S1 cleavage. PLoS One 4(8):e6728. https://doi.org/10.1371/journal.pone.0006728
Landor SK, Mutvei AP, Mamaeva V, Jin S, Busk M, Borra R, Gronroos TJ, Kronqvist P, Lendahl U, Sahlgren CM (2011) Hypo- and hyperactivated Notch signaling induce a glycolytic switch through distinct mechanisms. Proc Natl Acad Sci U S A 108(46):18814–18819. https://doi.org/10.1073/pnas.1104943108
Larson Gedman A, Chen Q, Kugel Desmoulin S, Ge Y, LaFiura K, Haska CL, Cherian C, Devidas M, Linda SB, Taub JW, Matherly LH (2009) The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: a report from the children’s oncology group. Leukemia 23(8):1417–1425. https://doi.org/10.1038/leu.2009.64
Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, Devgan V, Lieb J, Raffoul W, Hohl D, Neel V, Garlick J, Chiorino G, Dotto GP (2007) Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev 21(5):562–577. https://doi.org/10.1101/gad.1484707
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121(7):2750–2767. https://doi.org/10.1172/JCI45014
Liefke R, Oswald F, Alvarado C, Ferres-Marco D, Mittler G, Rodriguez P, Dominguez M, Borggrefe T (2010) Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev 24(6):590–601. https://doi.org/10.1101/gad.563210
Lin SE, Oyama T, Nagase T, Harigaya K, Kitagawa M (2002) Identification of new human mastermind proteins defines a family that consists of positive regulators for notch signaling. J Biol Chem 277(52):50612–50620. https://doi.org/10.1074/jbc.M209529200
Liu ZJ, Xiao M, Balint K, Smalley KS, Brafford P, Qiu R, Pinnix CC, Li X, Herlyn M (2006) Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res 66(8):4182–4190. https://doi.org/10.1158/0008-5472.CAN-05-3589
Liu H, Chi AW, Arnett KL, Chiang MY, Xu L, Shestova O, Wang H, Li YM, Bhandoola A, Aster JC, Blacklow SC, Pear WS (2010) Notch dimerization is required for leukemogenesis and T-cell development. Genes Dev 24(21):2395–2407. https://doi.org/10.1101/gad.1975210
Liu M, Lee DF, Chen CT, Yen CJ, Li LY, Lee HJ, Chang CJ, Chang WC, Hsu JM, Kuo HP, Xia W, Wei Y, Chiu PC, Chou CK, Du Y, Dhar D, Karin M, Chen CH, Hung MC (2012) IKKalpha activation of NOTCH links tumorigenesis via FOXA2 suppression. Mol Cell 45(2):171–184. https://doi.org/10.1016/j.molcel.2011.11.018
Logeat F, Bessia C, Brou C, LeBail O, Jarriault S, Seidah NG, Israel A (1998) The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc Natl Acad Sci U S A 95(14):8108–8112
Luo J, Wang P, Wang R, Wang J, Liu M, Xiong S, Li Y, Cheng B (2016) The Notch pathway promotes the cancer stem cell characteristics of CD90+ cells in hepatocellular carcinoma. Oncotarget 7(8):9525–9537. https://doi.org/10.18632/oncotarget.6672
Ma L, Dong P, Liu L, Gao Q, Duan M, Zhang S, Chen S, Xue R, Wang X (2016) Overexpression of protein O-fucosyltransferase 1 accelerates hepatocellular carcinoma progression via the Notch signaling pathway. Biochem Biophys Res Commun 473(2):503–510. https://doi.org/10.1016/j.bbrc.2016.03.062
Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC, Blacklow SC (2006) Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol 26(12):4642–4651. https://doi.org/10.1128/MCB.01655-05
Mancino A, Termanini A, Barozzi I, Ghisletti S, Ostuni R, Prosperini E, Ozato K, Natoli G (2015) A dual cis-regulatory code links IRF8 to constitutive and inducible gene expression in macrophages. Genes Dev 29(4):394–408. https://doi.org/10.1101/gad.257592.114
Mansour MR, Linch DC, Foroni L, Goldstone AH, Gale RE (2006) High incidence of Notch-1 mutations in adult patients with T-cell acute lymphoblastic leukemia. Leukemia 20(3):537–539. https://doi.org/10.1038/sj.leu.2404101
Mansour MR, Duke V, Foroni L, Patel B, Allen CG, Ancliff PJ, Gale RE, Linch DC (2007) Notch-1 mutations are secondary events in some patients with T-cell acute lymphoblastic leukemia. Clin Cancer Res 13(23):6964–6969. https://doi.org/10.1158/1078-0432.CCR-07-1474
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25(12):677–686. https://doi.org/10.1016/j.it.2004.09.015
Masek J, Andersson ER (2017) The developmental biology of genetic Notch disorders. Development 144(10):1743–1763. https://doi.org/10.1242/dev.148007
Massi D, Tarantini F, Franchi A, Paglierani M, Di Serio C, Pellerito S, Leoncini G, Cirino G, Geppetti P, Santucci M (2006) Evidence for differential expression of Notch receptors and their ligands in melanocytic nevi and cutaneous malignant melanoma. Mod Path : an official Journal of the United States and Canadian Academy of Pathology, Inc 19(2):246–254. https://doi.org/10.1038/modpathol.3800526
Mazur PK, Einwachter H, Lee M, Sipos B, Nakhai H, Rad R, Zimber-Strobl U, Strobl LJ, Radtke F, Kloppel G, Schmid RM, Siveke JT (2010a) Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 107(30):13438–13443. https://doi.org/10.1073/pnas.1002423107
Mazur PK, Gruner BM, Nakhai H, Sipos B, Zimber-Strobl U, Strobl LJ, Radtke F, Schmid RM, Siveke JT (2010b) Identification of epidermal Pdx1 expression discloses different roles of Notch1 and Notch2 in murine Kras(G12D)-induced skin carcinogenesis in vivo. PLoS One 5(10):e13578. https://doi.org/10.1371/journal.pone.0013578
McBride KL, Riley MF, Zender GA, Fitzgerald-Butt SM, Towbin JA, Belmont JW, Cole SE (2008) NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum Mol Genet 17(18):2886–2893. https://doi.org/10.1093/hmg/ddn187
McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt TM 3rd (2007) Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg 134(2):290–296. https://doi.org/10.1016/j.jtcvs.2007.02.041
Meester JA, Southgate L, Stittrich AB, Venselaar H, Beekmans SJ, den Hollander N, Bijlsma EK, Helderman-van den Enden A, Verheij JB, Glusman G, Roach JC, Lehman A, Patel MS, de Vries BB, Ruivenkamp C, Itin P, Prescott K, Clarke S, Trembath R, Zenker M, Sukalo M, Van Laer L, Loeys B, Wuyts W (2015) Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome. Am J Hum Genet 97(3):475–482. https://doi.org/10.1016/j.ajhg.2015.07.015
Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, Sriuranpong V, Iso T, Meszoely IM, Wolfe MS, Hruban RH, Ball DW, Schmid RM, Leach SD (2003) Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 3(6):565–576
Mizuhara E, Nakatani T, Minaki Y, Sakamoto Y, Ono Y, Takai Y (2005) MAGI1 recruits Dll1 to cadherin-based adherens junctions and stabilizes it on the cell surface. J Biol Chem 280(28):26499–26507. https://doi.org/10.1074/jbc.M500375200
Mohamed SA, Aherrahrou Z, Liptau H, Erasmi AW, Hagemann C, Wrobel S, Borzym K, Schunkert H, Sievers HH, Erdmann J (2006) Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res Commun 345(4):1460–1465. https://doi.org/10.1016/j.bbrc.2006.05.046
Monsalve E, Ruiz-Garcia A, Baladron V, Ruiz-Hidalgo MJ, Sanchez-Solana B, Rivero S, Garcia-Ramirez JJ, Rubio A, Laborda J, Diaz-Guerra MJ (2009) Notch1 upregulates LPS-induced macrophage activation by increasing NF-kappaB activity. Eur J Immunol 39(9):2556–2570. https://doi.org/10.1002/eji.200838722
Morgan TH (1917) The theory of the gene. Am Nat 51:513–544
Moshkin YM, Kan TW, Goodfellow H, Bezstarosti K, Maeda RK, Pilyugin M, Karch F, Bray SJ, Demmers JA, Verrijzer CP (2009) Histone chaperones ASF1 and NAP1 differentially modulate removal of active histone marks by LID-RPD3 complexes during NOTCH silencing. Mol Cell 35(6):782–793. https://doi.org/10.1016/j.molcel.2009.07.020
Mulligan P, Yang F, Di Stefano L, Ji JY, Ouyang J, Nishikawa JL, Toiber D, Kulkarni M, Wang Q, Najafi-Shoushtari SH, Mostoslavsky R, Gygi SP, Gill G, Dyson NJ, Naar AM (2011) A SIRT1-LSD1 corepressor complex regulates Notch target gene expression and development. Mol Cell 42(5):689–699. https://doi.org/10.1016/j.molcel.2011.04.020
Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R (2000) A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell 5(2):197–206
Nakayama T, Saitsu H, Endo W, Kikuchi A, Uematsu M, Haginoya K, Hino-fukuyo N, Kobayashi T, Iwasaki M, Tominaga T, Kure S, Matsumoto N (2014) RBPJ is disrupted in a case of proximal 4p deletion syndrome with epilepsy. Brain Dev 36(6):532–536. https://doi.org/10.1016/j.braindev.2013.07.009
Nam Y, Weng AP, Aster JC, Blacklow SC (2003) Structural requirements for assembly of the CSL.intracellular Notch1.Mastermind-like 1 transcriptional activation complex. J Biol Chem 278(23):21232–21239. https://doi.org/10.1074/jbc.M301567200
Nam Y, Sliz P, Song L, Aster JC, Blacklow SC (2006) Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 124(5):973–983. https://doi.org/10.1016/j.cell.2005.12.037
Nam Y, Sliz P, Pear WS, Aster JC, Blacklow SC (2007) Cooperative assembly of higher-order Notch complexes functions as a switch to induce transcription. Proc Natl Acad Sci U S A 104(7):2103–2108. https://doi.org/10.1073/pnas.0611092104
Ngondo-Mbongo RP, Myslinski E, Aster JC, Carbon P (2013) Modulation of gene expression via overlapping binding sites exerted by ZNF143, Notch1 and THAP11. Nucleic Acids Res 41(7):4000–4014. https://doi.org/10.1093/nar/gkt088
Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F (2003) Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 33(3):416–421. https://doi.org/10.1038/ng1099
Nishina S, Shiraha H, Nakanishi Y, Tanaka S, Matsubara M, Takaoka N, Uemura M, Horiguchi S, Kataoka J, Iwamuro M, Yagi T, Yamamoto K (2011) Restored expression of the tumor suppressor gene RUNX3 reduces cancer stem cells in hepatocellular carcinoma by suppressing Jagged1-Notch signaling. Oncol Rep 26(3):523–531. https://doi.org/10.3892/or.2011.1336
O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, Look AT (2007) FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med 204(8):1813–1824. https://doi.org/10.1084/jem.20070876
Osada H, Tatematsu Y, Yatabe Y, Horio Y, Takahashi T (2005) ASH1 gene is a specific therapeutic target for lung cancers with neuroendocrine features. Cancer Res 65(23):10680–10685. https://doi.org/10.1158/0008-5472.CAN-05-1404
Oswald F, Tauber B, Dobner T, Bourteele S, Kostezka U, Adler G, Liptay S, Schmid RM (2001) p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol Cell Biol 21(22):7761–7774. https://doi.org/10.1128/MCB.21.22.7761-7774.2001
Oswald F, Kostezka U, Astrahantseff K, Bourteele S, Dillinger K, Zechner U, Ludwig L, Wilda M, Hameister H, Knochel W, Liptay S, Schmid RM (2002) SHARP is a novel component of the Notch/RBP-Jkappa signalling pathway. EMBO J 21(20):5417–5426
Oswald F, Winkler M, Cao Y, Astrahantseff K, Bourteele S, Knochel W, Borggrefe T (2005) RBP-Jkappa/SHARP recruits CtIP/CtBP corepressors to silence Notch target genes. Mol Cell Biol 25(23):10379–10390. https://doi.org/10.1128/MCB.25.23.10379-10390.2005
Oswald F, Rodriguez P, Giaimo BD, Antonello ZA, Mira L, Mittler G, Thiel VN, Collins KJ, Tabaja N, Cizelsky W, Rothe M, Kuhl SJ, Kuhl M, Ferrante F, Hein K, Kovall RA, Dominguez M, Borggrefe T (2016) A phospho-dependent mechanism involving NCoR and KMT2D controls a permissive chromatin state at Notch target genes. Nucleic Acids Res 44(10):4703–4720. https://doi.org/10.1093/nar/gkw105
Outtz HH, Wu JK, Wang X, Kitajewski J (2010) Notch1 deficiency results in decreased inflammation during wound healing and regulates vascular endothelial growth factor receptor-1 and inflammatory cytokine expression in macrophages. J Immunol 185(7):4363–4373. https://doi.org/10.4049/jimmunol.1000720
Palaga T, Buranaruk C, Rengpipat S, Fauq AH, Golde TE, Kaufmann SH, Osborne BA (2008) Notch signaling is activated by TLR stimulation and regulates macrophage functions. Eur J Immunol 38(1):174–183. https://doi.org/10.1002/eji.200636999
Palermo R, Checquolo S, Giovenco A, Grazioli P, Kumar V, Campese AF, Giorgi A, Napolitano M, Canettieri G, Ferrara G, Schinina ME, Maroder M, Frati L, Gulino A, Vacca A, Screpanti I (2012) Acetylation controls Notch3 stability and function in T-cell leukemia. Oncogene 31(33):3807–3817. https://doi.org/10.1038/onc.2011.533
Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo M, Nagase S, Cordon-Cardo C, Parsons R, Zuniga-Pflucker JC, Dominguez M, Ferrando AA (2007) Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 13(10):1203–1210. https://doi.org/10.1038/nm1636
Parks AL, Stout JR, Shepard SB, Klueg KM, Dos Santos AA, Parody TR, Vaskova M, Muskavitch MA (2006) Structure-function analysis of delta trafficking, receptor binding and signaling in Drosophila. Genetics 174(4):1947–1961. https://doi.org/10.1534/genetics.106.061630
Patel U, Rajasingh S, Samanta S, Cao T, Dawn B, Rajasingh J (2017) Macrophage polarization in response to epigenetic modifiers during infection and inflammation. Drug Discov Today 22(1):186–193. https://doi.org/10.1016/j.drudis.2016.08.006
Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J, Baltimore D (1996) Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med 183(5):2283–2291
Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V, Zurrida S, Maisonneuve P, Viale G, Di Fiore PP (2004) Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol 167(2):215–221. https://doi.org/10.1083/jcb.200406140
Plentz R, Park JS, Rhim AD, Abravanel D, Hezel AF, Sharma SV, Gurumurthy S, Deshpande V, Kenific C, Settleman J, Majumder PK, Stanger BZ, Bardeesy N (2009) Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 136(5):1741–1749. e1746. https://doi.org/10.1053/j.gastro.2009.01.008
Porta C, Riboldi E, Ippolito A, Sica A (2015) Molecular and epigenetic basis of macrophage polarized activation. Semin Immunol 27(4):237–248. https://doi.org/10.1016/j.smim.2015.10.003
Pozzo F, Bittolo T, Arruga F, Bulian P, Macor P, Tissino E, Gizdic B, Rossi FM, Bomben R, Zucchetto A, Benedetti D, Degan M, D’Arena G, Chiarenza A, Zaja F, Pozzato G, Rossi D, Gaidano G, Del Poeta G, Deaglio S, Gattei V, Dal Bo M (2016) NOTCH1 mutations associate with low CD20 level in chronic lymphocytic leukemia: evidence for a NOTCH1 mutation-driven epigenetic dysregulation. Leukemia 30(1):182–189. https://doi.org/10.1038/leu.2015.182
Pozzo F, Bittolo T, Vendramini E, Bomben R, Bulian P, Rossi FM, Zucchetto A, Tissino E, Degan M, D’Arena G, Di Raimondo F, Zaja F, Pozzato G, Rossi D, Gaidano G, Del Poeta G, Gattei V, Dal Bo M (2017) NOTCH1-mutated chronic lymphocytic leukemia cells are characterized by a MYC-related overexpression of nucleophosmin 1 and ribosome-associated components. Leukemia 31(11):2407–2415. https://doi.org/10.1038/leu.2017.90
Proweller A, Tu L, Lepore JJ, Cheng L, Lu MM, Seykora J, Millar SE, Pear WS, Parmacek MS (2006) Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res 66(15):7438–7444. https://doi.org/10.1158/0008-5472.CAN-06-0793
Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G, Jares P, Bea S, Gonzalez-Diaz M, Bassaganyas L, Baumann T, Juan M, Lopez-Guerra M, Colomer D, Tubio JM, Lopez C, Navarro A, Tornador C, Aymerich M, Rozman M, Hernandez JM, Puente DA, Freije JM, Velasco G, Gutierrez-Fernandez A, Costa D, Carrio A, Guijarro S, Enjuanes A, Hernandez L, Yague J, Nicolas P, Romeo-Casabona CM, Himmelbauer H, Castillo E, Dohm JC, de Sanjose S, Piris MA, de Alava E, San Miguel J, Royo R, Gelpi JL, Torrents D, Orozco M, Pisano DG, Valencia A, Guigo R, Bayes M, Heath S, Gut M, Klatt P, Marshall J, Raine K, Stebbings LA, Futreal PA, Stratton MR, Campbell PJ, Gut I, Lopez-Guillermo A, Estivill X, Montserrat E, Lopez-Otin C, Campo E (2011) Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 475(7354):101–105. https://doi.org/10.1038/nature10113
Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, Sundaresan T, Pastorino S, Park JK, Mikolaenko I, Maric D, Eberhart CG, Fine HA (2005) Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res 65(6):2353–2363. https://doi.org/10.1158/0008-5472.CAN-04-1890
Qiu M, Peng Q, Jiang I, Carroll C, Han G, Rymer I, Lippincott J, Zachwieja J, Gajiwala K, Kraynov E, Thibault S, Stone D, Gao Y, Sofia S, Gallo J, Li G, Yang J, Li K, Wei P (2013) Specific inhibition of Notch1 signaling enhances the antitumor efficacy of chemotherapy in triple negative breast cancer through reduction of cancer stem cells. Cancer Lett 328(2):261–270. https://doi.org/10.1016/j.canlet.2012.09.023
Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, Aguet M (1999) Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 10(5):547–558
Rampias T, Vgenopoulou P, Avgeris M, Polyzos A, Stravodimos K, Valavanis C, Scorilas A, Klinakis A (2014) A new tumor suppressor role for the Notch pathway in bladder cancer. Nat Med 20(10):1199–1205. https://doi.org/10.1038/nm.3678
Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, Bartoli A, Coaccioli S, Screpanti I, Marconi P (2009) Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood 113(4):856–865. https://doi.org/10.1182/blood-2008-02-139725
Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, Monti S, Vaisitti T, Arruga F, Fama R, Ciardullo C, Greco M, Cresta S, Piranda D, Holmes A, Fabbri G, Messina M, Rinaldi A, Wang J, Agostinelli C, Piccaluga PP, Lucioni M, Tabbo F, Serra R, Franceschetti S, Deambrogi C, Daniele G, Gattei V, Marasca R, Facchetti F, Arcaini L, Inghirami G, Bertoni F, Pileri SA, Deaglio S, Foa R, Dalla-Favera R, Pasqualucci L, Rabadan R, Gaidano G (2012) The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med 209(9):1537–1551. https://doi.org/10.1084/jem.20120904
Rustighi A, Tiberi L, Soldano A, Napoli M, Nuciforo P, Rosato A, Kaplan F, Capobianco A, Pece S, Di Fiore PP, Del Sal G (2009) The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat Cell Biol 11(2):133–142. https://doi.org/10.1038/ncb1822
Rustighi A, Zannini A, Tiberi L, Sommaggio R, Piazza S, Sorrentino G, Nuzzo S, Tuscano A, Eterno V, Benvenuti F, Santarpia L, Aifantis I, Rosato A, Bicciato S, Zambelli A, Del Sal G (2014) Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol Med 6(1):99–119. https://doi.org/10.1002/emmm.201302909
Salat D, Liefke R, Wiedenmann J, Borggrefe T, Oswald F (2008) ETO, but not leukemogenic fusion protein AML1/ETO, augments RBP-Jkappa/SHARP-mediated repression of notch target genes. Mol Cell Biol 28(10):3502–3512. https://doi.org/10.1128/MCB.01966-07
Sanchez-Irizarry C, Carpenter AC, Weng AP, Pear WS, Aster JC, Blacklow SC (2004) Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol Cell Biol 24(21):9265–9273. https://doi.org/10.1128/MCB.24.21.9265-9273.2004
Santio NM, Landor SK, Vahtera L, Yla-Pelto J, Paloniemi E, Imanishi SY, Corthals G, Varjosalo M, Manoharan GB, Uri A, Lendahl U, Sahlgren C, Koskinen PJ (2016) Phos phorylation of Notch1 by Pim kinases promotes oncogenic signaling in breast and prostate cancer cells. Oncotarget 7(28):43220–43238. https://doi.org/10.18632/oncotarget.9215
Simoes BM, O’Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, Alferez DG, Spence K, Santiago-Gomez A, Chemi F, Acar A, Gandhi A, Howell A, Brennan K, Ryden L, Catalano S, Ando S, Gee J, Ucar A, Sims AH, Marangoni E, Farnie G, Landberg G, Howell SJ, Clarke RB (2015) Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep 12(12):1968–1977. https://doi.org/10.1016/j.celrep.2015.08.050
Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, Wang CY, Gao B, Jiang J, Yang Y (2010) Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci U S A 107(4):1431–1436. https://doi.org/10.1073/pnas.0911409107
Southgate L, Sukalo M, Karountzos ASV, Taylor EJ, Collinson CS, Ruddy D, Snape KM, Dallapiccola B, Tolmie JL, Joss S, Brancati F, Digilio MC, Graul-Neumann LM, Salviati L, Coerdt W, Jacquemin E, Wuyts W, Zenker M, Machado RD, Trembath RC (2015) Haploinsufficiency of the NOTCH1 receptor as a cause of adams-oliver syndrome with variable cardiac anomalies. Circ Cardiovasc Genet 8(4):572–581. https://doi.org/10.1161/CIRCGENETICS.115.001086
Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, Lam P, Khromykh A, Iyer RK, Vockley JG, Baveja R, Silva ES, Dixon J, Leon EL, Solomon BD, Glusman G, Niederhuber JE, Roach JC, Patel MS (2014) Mutations in NOTCH1 cause Adams-Oliver syndrome. Am J Hum Genet 95(3):275–284. https://doi.org/10.1016/j.ajhg.2014.07.011
Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, Yuan J, Ware C, MacLean J, Garrett-Engele PW, Kluk M, Laskey J, Haines BB, Moskaluk C, Zawel L, Fawell S, Gilliland G, Zhang T, Kremer BE, Knoechel B, Bernstein BE, Pear WS, Liu XS, Aster JC, Sathyanarayanan S (2014) Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov 4(10):1154–1167. https://doi.org/10.1158/2159-8290.CD-13-0830
Stylianou S, Clarke RB, Brennan K (2006) Aberrant activation of notch signaling in human breast cancer. Cancer Res 66(3):1517–1525. https://doi.org/10.1158/0008-5472.CAN-05-3054
Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T (1995) Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Curr Biol 5(12):1416–1423
Thelu J, Rossio P, Favier B (2002) Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma, psoriasis and wound healing. BMC Dermatol 2:7
Thiel V, Giaimo BD, Schwarz P, Soller K, Vas V, Bartkuhn M, Blatte TJ, Dohner K, Bullinger L, Borggrefe T, Geiger H, Oswald F (2017) Heterodimerization of AML1/ETO with CBFbeta is required for leukemogenesis but not for myeloproliferation. Leukemia 31(11):2491–2502. https://doi.org/10.1038/leu.2017.105
Thomas MM, Zhang Y, Mathew E, Kane KT, Maillard I, Pasca di Magliano M (2014) Epithelial Notch signaling is a limiting step for pancreatic carcinogenesis. BMC Cancer 14:862. https://doi.org/10.1186/1471-2407-14-862
VanderWielen BD, Yuan Z, Friedmann DR, Kovall RA (2011) Transcriptional repression in the Notch pathway: thermodynamic characterization of CSL-MINT (Msx2-interacting nuclear target protein) complexes. J Biol Chem 286(17):14892–14902. https://doi.org/10.1074/jbc.M110.181156
Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C, Zmoos AF, Mazur PK, Schaffer BE, Ostermeier A, Vogel H, Sylvester KG, Thorgeirsson SS, Grompe M, Sage J (2011) Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med 208(10):1963–1976. https://doi.org/10.1084/jem.20110198
Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, Toffanin S, Rodriguez-Carunchio L, Sole M, Thung S, Stanger BZ, Llovet JM (2012) Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 143(6):1660–1669. e1667. https://doi.org/10.1053/j.gastro.2012.09.002
Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, Hu XB, Zheng MH, Liang L, Feng L, Liang YM, Han H (2010a) Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res 70(12):4840–4849. https://doi.org/10.1158/0008-5472.CAN-10-0269
Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, Rich JN, Sullenger BA (2010b) Notch promotes radioresistance of glioma stem cells. Stem Cells 28(1):17–28. https://doi.org/10.1002/stem.261
Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, Pear WS, Schug J, Blacklow SC, Arnett KL, Bernstein BE, Kieff E, Aster JC (2011a) Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci U S A 108(36):14908–14913. https://doi.org/10.1073/pnas.1109023108
Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM, Collisson EA, Gordon PB, Jakkula L, Pennypacker S, Zou Y, Sharma M, North JP, Vemula SS, Mauro TM, Neuhaus IM, Leboit PE, Hur JS, Park K, Huh N, Kwok PY, Arron ST, Massion PP, Bale AE, Haussler D, Cleaver JE, Gray JW, Spellman PT, South AP, Aster JC, Blacklow SC, Cho RJ (2011b) Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A 108(43):17761–17766. https://doi.org/10.1073/pnas.1114669108
Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, Blacklow SC, Liu XS, Aster JC (2014) NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A 111(2):705–710. https://doi.org/10.1073/pnas.1315023111
Wang K, Zhang Q, Li D, Ching K, Zhang C, Zheng X, Ozeck M, Shi S, Li X, Wang H, Rejto P, Christensen J, Olson P (2015) PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a gamma-secretase inhibitor. Clin Cancer Res Off J Am Assoc Can Res 21(6):1487–1496. https://doi.org/10.1158/1078-0432.CCR-14-1348
Wang S, Zhou R, Sun F, Li R, Wang M, Wu M (2017) The two novel DLL4-targeting antibody-drug conjugates MvM03 and MGD03 show potent anti-tumour activity in breast cancer xenograft models. Cancer Lett. https://doi.org/10.1016/j.canlet.2017.09.004
Weng AP, Ferrando AA, Lee W, JPt M, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306(5694):269–271. https://doi.org/10.1126/science.1102160
Wharton KA, Johansen KM, Xu T, Artavanis-Tsakonas S (1985) Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 43(3 Pt 2):567–581
Wilson JJ, Kovall RA (2006) Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell 124(5):985–996. https://doi.org/10.1016/j.cell.2006.01.035
Wongchana W, Palaga T (2012) Direct regulation of interleukin-6 expression by Notch signaling in macrophages. Cell Mol Immunol 9(2):155–162. https://doi.org/10.1038/cmi.2011.36
Wu L, Aster JC, Blacklow SC, Lake R, Artavanis-Tsakonas S, Griffin JD (2000) MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat Genet 26(4):484–489. https://doi.org/10.1038/82644
Wu L, Sun T, Kobayashi K, Gao P, Griffin JD (2002) Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol Cell Biol 22(21):7688–7700
Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, Choy R, Howard P, Kadyk L, Yan M, Zha J, Callahan CA, Hymowitz SG, Siebel CW (2010) Therapeutic antibody targeting of individual Notch receptors. Nature 464(7291):1052–1057. https://doi.org/10.1038/nature08878
Xie Q, Wu Q, Kim L, Miller TE, Liau BB, Mack SC, Yang K, Factor DC, Fang X, Huang Z, Zhou W, Alazem K, Wang X, Bernstein BE, Bao S, Rich JN (2016) RBPJ maintains brain tumor-initiating cells through CDK9-mediated transcriptional elongation. J Clin Invest 126(7):2757–2772. https://doi.org/10.1172/JCI86114
Xu H, Zhu J, Smith S, Foldi J, Zhao B, Chung AY, Outtz H, Kitajewski J, Shi C, Weber S, Saftig P, Li Y, Ozato K, Blobel CP, Ivashkiv LB, Hu X (2012) Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol 13(7):642–650. https://doi.org/10.1038/ni.2304
Xu Z, Wang Z, Jia X, Wang L, Chen Z, Wang S, Wang M, Zhang J, Wu M (2016) MMGZ01, an anti-DLL4 monoclonal antibody, promotes nonfunctional vessels and inhibits breast tumor growth. Cancer Lett 372(1):118–127. https://doi.org/10.1016/j.canlet.2015.12.025
Xu T, Park SS, Giaimo BD, Hall D, Ferrante F, Ho DM, Hori K, Anhezini L, Ertl I, Bartkuhn M, Zhang H, Milon E, Ha K, Conlon KP, Kuick R, Govindarajoo B, Zhang Y, Sun Y, Dou Y, Basrur V, Elenitoba-Johnson KS, Nesvizhskii AI, Ceron J, Lee CY, Borggrefe T, Kovall RA, Rual JF (2017) RBPJ/CBF1 interacts with L3MBTL3/MBT1 to promote repression of Notch signaling via histone demethylase KDM1A/LSD1. EMBO J. https://doi.org/10.15252/embj.201796525
Yatim A, Benne C, Sobhian B, Laurent-Chabalier S, Deas O, Judde JG, Lelievre JD, Levy Y, Benkirane M (2012) NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Mol Cell 48(3):445–458. https://doi.org/10.1016/j.molcel.2012.08.022
Yen WC, Fischer MM, Hynes M, Wu J, Kim E, Beviglia L, Yeung VP, Song X, Kapoun AM, Lewicki J, Gurney A, Simeone DM, Hoey T (2012) Anti-DLL4 has broad spectrum activity in pancreatic cancer dependent on targeting DLL4-Notch signaling in both tumor and vasculature cells. Clin Cancer Res Off J Am Assoc Can Res 18(19):5374–5386. https://doi.org/10.1158/1078-0432.CCR-12-0736
Yuan Z, Praxenthaler H, Tabaja N, Torella R, Preiss A, Maier D, Kovall RA (2016) Structure and function of the Su(H)-Hairless repressor complex, the major Antagonist of Notch signaling in Drosophila melanogaster. PLoS Biol 14(7):e1002509. https://doi.org/10.1371/journal.pbio.1002509
Zhang W, Xu W, Xiong S (2010) Blockade of Notch1 signaling alleviates murine lupus via blunting macrophage activation and M2b polarization. J Immunol 184(11):6465–6478. https://doi.org/10.4049/jimmunol.0904016
Zhang Q, Wang C, Liu Z, Liu X, Han C, Cao X, Li N (2012) Notch signal suppresses Toll-like receptor-triggered inflammatory responses in macrophages by inhibiting extracellular signal-regulated kinase 1/2-mediated nuclear factor kappaB activation. J Biol Chem 287(9):6208–6217. https://doi.org/10.1074/jbc.M111.310375
Zhang J, Shao X, Sun H, Liu K, Ding Z, Chen J, Fang L, Su W, Hong Y, Li H, Li H (2016a) NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget 7(38):61036–61053. https://doi.org/10.18632/oncotarget.11062
Zhang S, Chung WC, Xu K (2016b) Lunatic fringe is a potent tumor suppressor in Kras-initiated pancreatic cancer. Oncogene 35(19):2485–2495. https://doi.org/10.1038/onc.2015.306
Zhang Q, Zhang Y, Parsels JD, Lohse I, Lawrence TS, Pasca di Magliano M, Sun Y, Morgan MA (2016c) Fbxw7 deletion accelerates KrasG12D-Driven pancreatic tumorigenesis via Yap accumulation. Neoplasia 18(11):666–673. https://doi.org/10.1016/j.neo.2016.08.009
Zhou L, Wang DS, Li QJ, Sun W, Zhang Y, Dou KF (2012) Downregulation of the Notch signaling pathway inhibits hepatocellular carcinoma cell invasion by inactivation of matrix metalloproteinase-2 and -9 and vascular endothelial growth factor. Oncol Rep 28(3):874–882. https://doi.org/10.3892/or.2012.1880
Zhou L, Wang DS, Li QJ, Sun W, Zhang Y, Dou KF (2013) The down-regulation of Notch1 inhibits the invasion and migration of hepatocellular carcinoma cells by inactivating the cyclooxygenase-2/Snail/E-cadherin pathway in vitro. Dig Dis Sci 58(4):1016–1025. https://doi.org/10.1007/s10620-012-2434-7
Zhu B, Sun L, Luo W, Li M, Coy DH, Yu L, Yu W (2017) Activated Notch signaling augments cell growth in hepatocellular carcinoma via up-regulating the nuclear receptor NR4A2. Oncotarget 8(14):23289–23302. https://doi.org/10.18632/oncotarget.15576
Zhu YM, Zhao WL, Fu JF, Shi JY, Pan Q, Hu J, Gao XD, Chen B, Li JM, Xiong SM, Gu LJ, Tang JY, Liang H, Jiang H, Xue YQ, Shen ZX, Chen Z, Chen SJ (2006) NOTCH1 mutations in T-cell acute lymphoblastic leukemia: prognostic significance and implication in multifactorial leukemogenesis. Clin Cancer Res 12(10):3043–3049. https://doi.org/10.1158/1078-0432.CCR-05-2832
Acknowledgements
We are grateful to Dr. Rhett A. Kovall (University of Cincinnati, USA) for providing us with Fig. 2 and to Drs. Shinya Yamamoto and Jose L. Salazar (Baylor College of Medicine, Houston, USA) and Francesca Ferrante (University of Giessen, Germany) for critical reading. This work was supported by the collaborative research grant TRR81, the Excellence Cluster for Cardio Pulmonary System (ECCPS) and the University of Giessen (Germany).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Giaimo, B.D., Borggrefe, T. (2018). Introduction to Molecular Mechanisms in Notch Signal Transduction and Disease Pathogenesis. In: Borggrefe, T., Giaimo, B. (eds) Molecular Mechanisms of Notch Signaling. Advances in Experimental Medicine and Biology, vol 1066. Springer, Cham. https://doi.org/10.1007/978-3-319-89512-3_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-89512-3_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-89511-6
Online ISBN: 978-3-319-89512-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)