
Abstract
Given the complex etiology of age-related macular degeneration (AMD), treatments are developed to target intermediate/late stages of the disease. Unfortunately, the design of therapies for early stages of the disease is limited by our understanding of the mechanisms involved in the formation of basal deposits and drusen, the first clinical signs of AMD. During the last decade, the identification of common and rare alleles in complement genes as risk AMD variants in addition to the presence of active complement components in basal deposits and drusen has provided compelling evidence that the complement system plays a key role in the pathobiology of AMD. However, the mechanisms for complement activation in AMD are unknown. Here we propose that the activation of the complement system is a consequence of alterations in the aged extracellular matrix (ECM) of the retinal pigment epithelium (RPE)/Bruch’s membrane (BrM), which favors the anchoring of complement C3b generated by convertase-independent cleavage of C3 via tick-over and produces a chronic activation of the alternative complement pathway.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others

Keywords
- AMD
- Complement
- Tick-over
- C3
- RPE
- Bruch’s membrane
- Drusen
- Basal deposits
- C3(H2O)
- C3(H2O)Bb-convertase
- Anticomplement drugs
1 Introduction
AMD is the most common cause of vision loss in elderly people in developed countries. Given the diverse etiology of AMD, treatments are developed to target intermediate and late stages of the disease, and there is no effective therapy for early AMD (Miller 2013a). The development of anti-VEGF therapies for choroidal neovascularization has been a major advance, and the AREDS trial showed some benefit of antioxidant supplementation for slowing AMD progression in patients with intermediate disease, but therapies for the more common atrophic form of AMD are necessary to prevent vision loss (Evans and Lawrenson 2012; Miller 2013b).
The first clinical sign of AMD is the formation of deposits and drusen between the basal lamina of the RPE and the BrM (Sarks et al. 1999). Although the mechanisms of deposit formation are not fully understood, some authors support the hypothesis that drusen is an inflammatory process (Mullins et al. 2000; Hageman et al. 2001; Anderson et al. 2002). Indeed, genetic variants in complement genes, such as CFH, C3, CFB, CFI, C9, and C2, have been associated with AMD (Hageman et al. 2005; Seddon et al. 2013). Further, active complement components have been identified in basal deposits and drusen, but their functional role in drusen formation remains to be defined (Mullins et al. 2000; Crabb et al. 2002; Wang et al. 2010; Garland et al. 2014).
Studies in mouse models also demonstrate a key role for the complement in the formation of basal laminar deposits (Fu et al. 2007; Ding et al. 2014, Fernandez-Godino 2015 #389; Garland et al. 2014). Thus, understanding the role of the complement system in early stages of AMD may help to design complement-modulating therapies that prevent the progression of drusen to geographic atrophy or neovascular AMD.
2 Activation of the Alternative Complement Pathway via Tick-Over Process
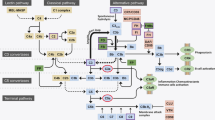
The complement system is a part of the innate immune system usually activated by external insults via three pathways: classical, lectin, and alternative (Ricklin et al. 2010). However, the complement system has the potential to be damaging to host tissues; hence, it is tightly regulated by complement-regulating proteins , such as CFH, CFI, DAF, etc. (Fig. 4.1) (Ricklin et al. 2010).

Schematic representation of the alternative complement pathway and tick-over process. C3 is cleaved to C3a and C3b, which forms C3-convertase unless it is inactivated by CFH and CFI. C3 is spontaneously hydrolyzed to C3(H2O) via tick-over process. C3(H2O) is functionally similar to C3b and binds CFB to form the C3(H2O)Bb-convertase, which is not recognized by CFH for inactivation
Under normal conditions, the alternative complement pathway can be activated by the tick-over process; C3 can be cleaved by convertase-independent proteolysis or physiological hydrolysis of its internal thioester, generating free C3b or C3(H2O) respectively, and C3a (Lachmann and Halbwachs 1975; Pangburn et al. 1981; Nilsson and Nilsson Ekdahl 2012) (Fig. 4.1). C3(H2O) is functionally similar to C3b and presents high affinity to adsorb surfaces, such as lipids or biomaterials (Pangburn et al. 1981; Andersson et al. 2005; Bexborn et al. 2008), and it has been demonstrated to bind ECM proteins, such as collagen IV, fibronectin, and especially laminin (Leivo and Engvall 1986). C3(H2O) deposited on surfaces binds CFB, which is cleaved to Bb and Ba by CFD, creating a C3(H2O)Bb-convertase capable of binding complement receptors and activating the alternative pathway (Fig. 4.1) (Nilsson and Nilsson Ekdahl 2012), but that cannot be regulated by CFH and CFI (Andersson et al. 2005; Bexborn et al. 2008). Indeed, it has been postulated that CFB preferentially binds C3b/C3(H2O) bound to foreign surfaces, while CFH has major affinity for C3b bound to host surfaces to prevent C3b deposition in autologous tissue (Atkinson and Farries 1987). It is possible that changes in the BrM due to aging favor the anchoring of C3b/C3(H2O) to its ECM, which would enhance the local activation of the alternative complement pathway.
3 The Role of the Tick-Over Pathway in Basal Deposit Formation in AMD
With age, the BrM undergoes changes in structure and composition, and there is an accumulation of membranous debris, proteins, and lipids (Sarks et al. 1999; Curcio and Johnson. 2013). In AMD, material secreted by the RPE, especially collagen, is accumulated in the region of the macula and causes the thickening of the BrM and the formation of basal laminar deposits under the RPE (Hogan and Alvarado 1967; Reale et al. 2009). Given the affinity of C3(H2O) to adsorb biosurfaces, the tick-over process may play an important role in the formation of basal deposits in dry AMD. We think that the abnormal structure/composition of the aged BrM, as well as the accumulation of proteins and lipids, may favor the anchoring of C3(H2O) generated by the physiological hydrolysis of C3. C3(H2O) would be recognized by CFB to form a C3(H2O)Bb-convertase that is deposited to the BrM in a stable manner and triggers the chronic activation of complement through the alternative pathway. Changes in extracellular matrix cause RPE cells to make basal deposits and activate the alternative complement pathway (Fernandez-Godino et al. 2018). This process generates more C3b and creates a positive feedback loop that results in increased release of the anaphylatoxin C3a, which initiates a local inflammatory process (Hindmarsh and Marks 1998; Harboe and Mollnes 2008). To support this hypothesis, C3a has been detected in basal deposits and drusen and has been reported to cause deposit formation by RPE cells in vitro (Nozaki et al. 2006; Fernandez-Godino et al. 2015). Moreover, we have shown that C3a produced locally by RPE cells stimulates deposit formation, suggesting that C3a is a potential target for therapeutic intervention (Fernandez-Godino et al. 2015).
4 Complement-Modulating Drugs to Treat AMD
The role of the complement system in deposit formation has led to the development of several anticomplement drugs, mostly directed to block complement activation through the alternative pathway, which are being tested in clinical trials but have not had significant success to date (Ricklin and Lambris 2013; Garcia Filho et al. 2014; Yehoshua et al. 2014). For example, drugs that block the terminal complement component C5 administered systemically have not shown any benefit in dry AMD patients (Garcia Filho et al. 2014; Yehoshua et al. 2014). Phase II trials using lampalizumab showed that inhibition of CFD reduced geographic atrophy only in some patients, although Phase III trials have been initiated (NCT02247531, NCT02247479).
There is controversy about which complement pathway is first initiated in AMD. Because C3 plays a central role in the activation of complement system through any of the three pathways, it is an attractive target for complement-modulating therapy administered locally. Compstatin is a peptide that inhibits complement activation by binding C3 and interfering with the formation of C3-convertase and C3 cleavage (Mastellos et al. 2015). The clinical efficacy of compstatin for AMD has been demonstrated in Phase I studies in patients (Mastellos et al. 2015). Further, POT-4, a compstatin analog with improved activity, was developed, but did not show efficacy in Phase II clinical trials (Clin Trial NCT01603043) (Qu et al. 2013).
In conclusion, complement-modulating therapies have shown some promising results; however, the design of novel drugs is limited by our understanding of the process of complement activation in AMD . Also, it remains unclear if regulation of the complement system locally or systemically is needed for the most effective treatment of AMD or at what stage of disease these treatments can be most usefully applied.
5 C3(H2O)Bb-Convertase and Response to Complement-Modulating Therapies
As previously mentioned, C3(H2O) generated by the hydrolysis of C3 is functionally similar to C3b because it can bind complement receptors and form C3(H2O)Bb- convertase (Nilsson and Nilsson Ekdahl 2012). However, the epitope recognized by CFH in C3b is not exposed in C3(H2O); thus, the formation of C3(H2O)Bb-convertase cannot be inhibited via CFH (Andersson et al. 2005; Bexborn et al. 2008). Alternatively, C3(H2O) can be inactivated by CFI and another cofactor, but the inactivation rate is much slower than the normal inactivation of C3b (Pangburn et al. 1981).
Differential activation of C3 via the alternative pathway or tick-over in AMD also has important consequences in response to complement-modulating drugs. Compstatin does not block or prevent the convertase-independent cleavage of C3 (Mastellos et al. 2015), which would explain the low efficiency of this drug in AMD patients. Thus, regarding therapies to treat AMD, an alternative approach may be needed to block local complement activation in basal deposits . Our hypothesis for the activation of complement system via tick-over caused by degeneration of ECM/BrM opens new avenues for the development of pharmaceutical compounds never considered for AMD to date, for example, regulators of the ECM synthesis and turnover, which have shown to be key for other human diseases like cancer, fibrosis, or cardiovascular disease (Tziakas et al. 2005; Overall and Kleifeld 2006; Sivakumar and Das 2008). Further, the local administration of complement-modulating drugs in combination with ECM-modulating drugs could avoid or at least delay the progression of the disease to legal blindness in dry AMD patients without compromising the whole immune system or vital biological processes.
References
Anderson DH, Mullins RF, Hageman GS et al (2002) A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol 134:411–431
Andersson J, Ekdahl KN, Lambris JD et al (2005) Binding of C3 fragments on top of adsorbed plasma proteins during complement activation on a model biomaterial surface. Biomaterials 26:1477–1485
Atkinson JP, Farries T (1987) Separation of self from non-self in the complement system. Immunol Today 8:212–215
Bexborn F, Andersson PO, Chen H et al (2008) The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb). Mol Immunol 45:2370–2379
Crabb JW, Miyagi M, Gu X et al (2002) Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A 99:14682–14687
Curcio CA, Johnson M (2013) Structure, function, and pathology of Bruch’s membrane. Retina 1:465–481
Ding JD, Kelly U, Groelle M et al (2014) The role of complement dysregulation in AMD mouse models. Adv Exp Med Biol 801:213–219
Evans JR, Lawrenson JG (2012) Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst Rev 11:CD000254
Fernandez-Godino R, Garland DL, Pierce EA (2015) A local complement response by RPE causes early-stage macular degeneration. Hum Mol Genet 24:5555–5569
Fernandez-Godino R, Bujakowska KM, Pierce EA (2018) Hum Mol Genet 27(1):147–159
Fu L, Garland D, Yang Z et al (2007) The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum Mol Genet 16:2411–2422
Garcia Filho CA, Yehoshua Z, Gregori G et al (2014) Change in drusen volume as a novel clinical trial endpoint for the study of complement inhibition in age-related macular degeneration. Ophthalmic Surg Lasers Imaging Retina 45:18–31
Garland DL, Fernandez-Godino R, Kaur I et al (2014) Mouse genetics and proteomic analyses demonstrate a critical role for complement in a model of DHRD/ML, an inherited macular degeneration. Hum Mol Genet 23:52–68
Hageman GS, Luthert PJ, Victor Chong NH et al (2001) An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res 20:705–732
Hageman GS, Anderson DH, Johnson LV et al (2005) A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A 102:7227–7232
Harboe M, Mollnes TE (2008) The alternative complement pathway revisited. J Cell Mol Med 12:1074–1084
Hindmarsh EJ, Marks RM (1998) Complement activation occurs on subendothelial extracellular matrix in vitro and is initiated by retraction or removal of overlying endothelial cells. J Immunol 160:6128–6136
Hogan MJ, Alvarado J (1967) Studies on the human macula. IV. Aging changes in Bruch’s membrane. Arch Ophthalmol 77:410–420
Lachmann PJ, Halbwachs L (1975) The influence of C3b inactivator (KAF) concentration on the ability of serum to support complement activation. Clin Exp Immunol 21:109–114
Leivo I, Engvall E (1986) C3d fragment of complement interacts with laminin and binds to basement membranes of glomerulus and trophoblast. J Cell Biol 103:1091–1100
Mastellos DC, Yancopoulou D, Kokkinos P et al (2015) Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Investig 45:423–440
Miller JW (2013a) Age-related macular degeneration revisited–piecing the puzzle: the LXIX Edward Jackson memorial lecture. Am J Ophthalmol 155(1–35):e13
Miller JW (2013b) Age-related macular degeneration revisited–piecing the puzzle: the LXIX Edward Jackson memorial lecture. Am J Ophthalmol 155(1–35):e13
Mullins RF, Russell SR, Anderson DH et al (2000) Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J (Official Publication of the Federation of American Societies for Experimental Biology) 14:835–846
Nilsson B, Nilsson Ekdahl K (2012) The tick-over theory revisited: is C3 a contact-activated protein? Immunobiology 217:1106–1110
Nozaki M, Raisler BJ, Sakurai E et al (2006) Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A 103:2328–2333
Overall CM, Kleifeld O (2006) Tumour microenvironment – opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer 6:227–239
Pangburn MK, Schreiber RD, Muller-Eberhard HJ (1981) Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med 154:856–867
Qu H, Ricklin D, Bai H et al (2013) New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology 218:496–505
Reale E, Groos S, Eckardt U et al (2009) New components of ‘basal laminar deposits’ in age-related macular degeneration. Cells Tissues Organs 190:170–181
Ricklin D, Lambris JD (2013) Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol 190:3831–3838
Ricklin D, Hajishengallis G, Yang K et al (2010) Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11:785–797
Sarks SH, Arnold JJ, Killingsworth MC et al (1999) Early drusen formation in the normal and aging eye and their relation to age related maculopathy: a clinicopathological study. Br J Ophthalmol 83:358–368
Seddon JM, Yu Y, Miller EC et al (2013) Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet 45:1366–1370
Sivakumar P, Das AM (2008) Fibrosis, chronic inflammation and new pathways for drug discovery. Inflamm Res 57:410–418
Tziakas DN, Chalikias GK, Hatzinikolaou HI et al (2005) Levosimendan use reduces MMP-2 in patients with decompensated heart failure. Cardiovasc Drugs Ther 19:399–402
Wang L, Clark ME, Crossman DK et al (2010) Abundant lipid and protein components of drusen. PLoS One 5:e10329
Yehoshua Z, de Amorim Garcia Filho CA, Nunes RP et al (2014) Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology 121:693–701
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this paper

Cite this paper
Fernandez-Godino, R. (2018). Alterations in Extracellular Matrix/Bruch’s Membrane Can Cause the Activation of the Alternative Complement Pathway via Tick-Over. In: Ash, J., Anderson, R., LaVail, M., Bowes Rickman, C., Hollyfield, J., Grimm, C. (eds) Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology, vol 1074. Springer, Cham. https://doi.org/10.1007/978-3-319-75402-4_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-75402-4_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75401-7
Online ISBN: 978-3-319-75402-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)