
Abstract
The cells of the osteoblast lineage (osteoblasts, osteocytes, and bone lining cells) have multiple stage-specific functions in the skeleton, including formation of the bone matrix. They arise from pluripotent mesenchymal progenitors, which can also give rise to adipocytes and chondrocytes. The functions carried out by the osteoblast lineage are regulated by both paracrine and endocrine factors. During bone formation, osteoblasts produce the collagenous matrix (osteoid), which becomes mineralized through the deposition and accumulation of bioapatite mineral crystals; the latter is dependent on the availability of inorganic phosphate, which is regulated by the osteoblast lineage through production of alkaline phosphatase, and other non-collagenous proteins. During osteoid deposition, osteoblasts can become embedded within their own matrix, and differentiate further, to become interconnected osteocytes. Alternatively, they may remain on the surface as bone lining cells, which have the potential to become reactivated into matrix-producing osteoblasts. The osteoblast lineage also supports osteoclast formation and thereby regulates bone resorption through the production of receptor activator of NF-ΚB ligand (RANKL), colony-stimulating factor 1 (CSF-1), and osteoprotegerin (OPG), a decoy receptor for RANKL. This versatile cell lineage therefore not only forms bone but can also controls its removal, which makes understanding the stage-specific roles of the osteoblast lineage crucial for targeting many skeletal conditions, such as osteoporosis, osteogenesis imperfecta, and other pathological conditions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Key Points-
The osteoblast lineage includes pluripotent precursors, preosteoblasts, osteoblasts, osteocytes, and bone lining cells.
-
Osteoblasts are the cells responsible for formation of the collagen-rich bone matrix (osteoid) which becomes mineralized by the deposition and accumulation of mineral crystals.
-
Mineralization of the bone matrix is regulated by proteins produced by the osteoblast lineage including alkaline phosphatase and non-collagenous proteins in the bone matrix.
-
Osteoblast lineage cells also control the differentiation of osteoclasts through their production of receptor activator of NF-ΚB ligand (RANKL), macrophage colony-stimulating factor (M-CSF), and osteoprotegerin (OPG).
-
Bone lining cells have the potential to be a source of osteoblast precursors.
Introduction to the Osteoblast Lineage: Multiple Stage-Specific Functions
Osteoblasts are specialized mesenchymal-derived cells that produce and deposit the collagenous bone matrix and regulate the mineralization of that matrix by their production of additional non-collagenous proteins. The osteoblast lineage includes not only these bone-forming osteoblasts but also their pluripotent and lineage-committed precursors, bone lining cells, and matrix-embedded osteocytes (Fig. 1.1). Each of these stages of the osteoblast lineage has distinct functions, morphologies, particular locations relative to the bone surface, and increasingly well-defined markers of differentiation (noted on Fig. 1.1 and discussed below).

Stages of the osteoblast lineage. The osteoblast lineage arises from pluripotent mesenchymal progenitors, capable of differentiating into adipocytes or into chondrocytes or osteoblasts. Commitment to the osteoblast lineage is determined by expression of transcription factors including Runx2 and osterix. Once osteoblasts become mature, they deposit collagen type I-rich matrix (osteoid) as a template for bioapatite mineral deposition and express alkaline phosphatase (ALP), osteopontin, and osteocalcin, proteins that regulate bone mineralization. Osteoblasts then undergo one of three fates: (1) apoptosis, (2) remain on the bone surface as bone lining cells, or (3) become embedded within their collagenous bone matrix as “osteoid-osteocytes,” which then become terminally differentiated osteocytes. Osteocytes also regulate the mineralization of the bone matrix through their production of DMP-1, MEPE, and sclerostin. Bone lining cells appear to be capable of reactivation to become active osteoblasts or osteoblast precursors
The different stages of osteoblast differentiation allow these cells to perform three major functions that determine skeletal structure (noted on Fig. 1.1 and discussed below): (1) production of bone matrix (osteoid), (2) regulation of osteoid mineralization by production of non-collagenous proteins, and (3) support of osteoclast formation. In addition, osteoblast lineage cells produce paracrine factors, such as IL-6 family cytokines, parathyroid hormone-related protein (PTHrP), and contact-dependent molecules such as EphrinB2, that regulate their own differentiation and activity [1,2,3]. Osteoblasts have also been suggested to act as “reversal” cells, allowing communication between osteoclasts and osteoblasts, during the bone remodeling process [4]. The functions of the osteoblast lineage are not limited to the control of bone structure. They also regulate the hematopoietic stem cell niche [5, 6], contribute to hematopoietic malignancies [7], and to B cell homeostasis [8]. The osteoblast lineage also has endocrine functions in phosphate homeostasis [9] and glucose metabolism [10]. This chapter will focus on describing the stages of osteoblast differentiation and the functions of the lineage that regulate bone structure and bone matrix composition.
Osteoblast Differentiation and the Stages of the Osteoblast Lineage
Osteoblast Precursors
The osteoblast lineage arises from pluripotent mesenchymal progenitors. In vitro, these cells can be induced to differentiate into other mesenchymal origin cells such as chondrocytes, adipocytes, myoblasts, or fibroblasts [11] (Fig. 1.1). In vivo, bone marrow-derived mesenchymal progenitors have a more restricted future, being capable of differentiating into chondrocytes, osteoblasts, and adipocytes [12]. The location of these cells in the marrow has been refined by cell lineage-tracing studies (using genetically altered mice with fluorescent tags that are retained throughout differentiation) to be in close association with vascular structures [13]. This provided support for much earlier studies proposing that the pericyte, a cell found wrapped around endothelial cells, can behave as an osteoblast progenitor [14, 15]. Pericytes in different tissues appear to behave in an organ-specific manner, dictated by their anatomy and position; only bone marrow-residing pericytes appear capable of becoming osteoblasts [12]. This illustrates the importance of the microenvironment in determining differentiation. For more details, the reader is directed to a recent focused review on the identity of osteoblast progenitor populations [16].
The source of osteoblast progenitors is not restricted to the bone marrow pericytes. During embryonic bone development, perichondrial cells were identified as precursors giving rise to osteoblasts on trabecular bone [17]. This has been confirmed by lineage-tracing studies, which also identified these precursors as entering the marrow space with invading blood vessels and thereby contributing to both bone development and fracture healing [18]. Similar observations have been made that differentiated hypertrophic chondrocytes at the growth plate can “transdifferentiate” into osteoblasts during development and fracture healing [19], again confirming much earlier in vitro work [20]. Lineage tracing studies have also suggested that bone lining cells [21] and recently embedded osteocytes [22] can act as osteoblast precursors, although the latter remain highly controversial. It is likely that lining cells are already committed to the lineage, rather than having the potential to differentiate in chondrocytes or adipocytes. This suggests that there are multiple sources of osteoblast progenitors in vivo, with differentiation that is both context- and location-specific.
Commitment of precursors to the osteoblast lineage is controlled by the expression of a range of transcription factors. Absolutely essential for the commitment to the preosteoblast stage are runt-related transcription factor 2 (Runx2) and osterix [23, 24]. Other transcription factors including activating transcription factor 4 (ATF4) [25], activator protein 1 (AP-1) [26], and CCAAT/enhancer-binding proteins β and δ (C/EBPβ and C/EBPδ) [27] promote the transition to matrix-producing osteoblasts.
Since osteoblasts and adipocytes are derived from common precursors, many of these transcription factors also inhibit mesenchymal progenitor commitment to adipogenesis [26, 28]. Alternatively, transcription factors such as peroxisome proliferation-activated receptor γ (PPARγ) [29] and CCAAT/enhancer-binding protein α (C/EBPα) [30] promote differentiation into adipocytes. This inverse relationship between osteoblast and adipocyte differentiation was first observed in cell culture [31]. This has also been described in vivo in genetically altered mouse models, either where high osteoblast numbers are associated with low marrow adipocyte volume [26] or where low osteoblast numbers are associated with high marrow adipocyte volume [32,33,34]. Similar reciprocal regulation has been made in animal models of ovariectomy-induced bone loss [35]. There are exceptions to this, such as the C3H/HeJ mouse strain which has high bone mass [36] and high marrow adiposity [37]. Reciprocal regulation of osteoblasts and adipocytes has also been observed clinically: increased marrow adiposity is associated with age-related osteoporosis [38]. Understanding the relationships between osteoblast and adipocyte commitment remains an area of active research, since it may allow the development of additional treatments to increase bone mass.
The osteoblast precursor can also give rise to chondrocytes; this is important in the context of developmental and pediatric bone growth, and fracture healing, and may be of relevance for methods to repair joint cartilage. The osteoblast commitment transcription factors Runx2 and osterix not only promote osteoblast commitment but also stimulate the final stage of chondrocyte differentiation prior to vascular invasion in endochondral ossification [39,40,41]. Reciprocal regulation of chondrogenesis versus osteoblastogenesis from the same common precursor has also been suggested [42], as described above for adipocytes, but mechanisms controlling this have not yet been identified.
Matrix-Forming Osteoblasts
Mature matrix-forming osteoblasts are characterized by a cuboidal morphology and are located in groups with extensive cell-cell contact [43,44,45]. Osteoblasts are also located in close apposition to the bone surface; this indicates that as they differentiate to this stage, these cells must migrate, probably in groups to the bone surface, likely in response to coupling factors produced by osteoclasts or other cells within the basic multicellular unit [46,47,48]. There are two exceptions to this. During skeletal development, osteoblasts can form bone de novo (without a surface to work on), and during endochondral ossification, calcified cartilage serves as a template on which osteoblasts deposit bone.
At the electron microscope level, matrix-forming osteoblasts exhibit abundant endoplasmic reticulum, in line with their major function as factories for production of type I collagen, the main component of the osteoid matrix (see below). Matrix-producing osteoblasts also express a range of non-collagenous proteins. These include proteins involved in regulating the incorporation of mineral into the osteoid matrix (alkaline phosphatase [49], osteocalcin [50], and osteopontin [50]) and receptors that regulate their response to factors influencing their further differentiation and function, such as receptors for IL-6 family cytokines [33, 51] or the receptor used by parathyroid hormone (PTH) and PTH-related protein (PTHrP), PTH1R [52]. The mechanisms of matrix production and mineralization will be discussed below.
When their production of osteoid matrix is complete, mature osteoblasts undergo one of three fates: [1] remain on the surface of bone as less metabolically active bone lining cells, [2] die by apoptosis, or [3] become entrapped within the osteoid matrix and, as the osteoid is mineralized, further differentiate to become osteocytes (Fig. 1.1).
Osteocytes
Osteocytes are embedded within the bone matrix during the process of bone formation, and through their extensive dendritic processes and their fluid-filled network of communicating channels, they sense and respond to mechanical strain and microdamage to bone. They are the most abundant cells in bone by far, forming a highly complex cellular communication network through the bone matrix with a total of ~3.7 trillion connections throughout the adult skeleton [53].
How osteoblasts become embedded into the bone matrix remains unknown. The manner in which osteoblasts become osteocytes has been described as “encased,” “buried,” and “merged” into the matrix suggesting that the manner of transformation may depend on the type of bone formed [54]. It is possible that the type of bone being made (woven vs lamellar) or mode of ossification as well as location (periosteal/endocortical/trabecular) can determine how an osteoblast becomes embedded into the matrix. There are no specific signals made by the osteoblast that have been found to directly control this process. When an osteoblast transitions into the recently secreted matrix (osteoid) to become an osteocyte (Fig. 1.1) they are termed “osteoid-osteocytes” [55]. The most striking difference between osteoblasts and osteoid-osteocytes is the morphological change that occurs during this transition. The cuboidal morphology of the osteoblast changes into a less cuboidal cell which eventually transforms into a smaller cell body with many dendritic cellular projections characteristic of osteocytes. Upon mineralization of the osteoid, the ultrastructure of the osteocyte changes in line with its reduced protein-production capacity, including reduced endoplasmic reticulum and Golgi apparatus [56].
Differentiated osteocytes reside within lacunae in the bone matrix and form an extensive intercellular network throughout the bone matrix and regulate both bone formation and resorption. Cell contact is a notable feature of this network [53], as is the ability of these cells to sense and respond to mechanical load and microdamage [57]. In addition to controlling osteoblast activity on the bone surface by the release of local factors such as sclerostin [58], and oncostatin M [33], osteocytes regulate mineralization of the bone matrix by expressing factors such as dentin matrix protein 1 (DMP-1) [59] and matrix extracellular phosphoglycoprotein (MEPE) [60] and act in an endocrine manner to control phosphate homeostasis by their release of fibroblast growth factor 23 (FGF23) [61] (refer also to Chap. 3 (Basic Aspects of Osteocyte Function)).
Bone Lining Cells
Osteoblasts that do not become terminally differentiated osteocytes or undergo apoptosis remain on the bone surface to become flattened bone lining cells. Lining cells are characterized by flat nuclei and the ability to synthesize only small amounts of protein and, like other cells of the osteoblast lineage, connect with each other via gap junctions [62].
Although long regarded as a protective cell population covering the bone surface that is “resting,” or “quiescent”, bone lining cells, like osteoblasts, express receptors for endocrine and paracrine agents. Their contraction from the bone surface in response to PTH [63] was suggested to allow osteoclasts access to the bone surface [64]. It has been suggested that this lifting of the bone lining cell layer occurs not only in response to PTH treatment but also at the commencement of bone remodeling to generate a temporary canopy [65]. Such a canopy was previously suggested as a mechanism that encloses the bone remodeling activity, separating it from the rest of the bone marrow microenvironment [66], thereby providing a controlled locale in which osteoblast lineage cells, osteoclasts, and other contributing marrow cells, may exchange factors. This canopy is also closely associated with blood vessels, which can thereby readily provide both osteoblast and osteoclast precursors for the bone remodeling process [67, 68].
In addition to forming a canopy, bone lining cells are capable of reactivation to form active matrix-producing osteoblasts. This was first hypothesized when intermittent PTH administration increased osteoblast number on the bone surface without increasing osteoblast proliferation [69]. This mechanism has now been verified by lineage-tracing studies where intermittent administration of PTH reactivated quiescent lining cells to mature osteoblast in vivo [70]. Such reactivation of lining cells has also been demonstrated after mechanical loading [71] and after treatment with anti-sclerostin, a therapeutic stimulus of bone formation [72]. This reactivation is in addition to the proposal that these cells form a proliferating progenitor population during adulthood [21] and may provide a more rapidly inducible partially differentiated source of osteoblast precursors.
Bone Formation: Osteoid Production and its Mineralization
Bone is a heterogenous compound material. The mineral phase, in the form of modified hydroxyapatite (bioapatite) crystals, contributes about two-thirds of its weight. The remaining organic matrix consists largely of type I collagen (~90%) [73, 74], with small amounts of lipid (~2%), ~5% non-collagenous proteins, proteoglycans, and water [75]. Non-collagenous proteins within the bone matrix include substances that act as signaling molecules (such as transforming growth factor β (TGFβ) and insulin-like growth factor 1 (IGF1)) and substances that regulate mineralization (such as osteocalcin and DMP-1).
While a range of cell types are capable of depositing mineral, particularly in cell culture conditions or in pathological circumstances (such as vascular calcifications), it is only the osteoblast that is capable of bone formation. Osteoblasts are responsible for the deposition of bone matrix on a range of surfaces and in a number of different contexts. During endochondral bone formation, osteoblasts deposit bone on a cartilage template. This process occurs both in skeletal development and in fracture healing. In these instances, osteoblasts attach to the cartilage template and deposit osteoid, which becomes mineralized, according to processes described below. During intramembranous bone development, bone is formed directly by mesenchymal precursors with no underlying template. This process occurs largely during skeletal development, particularly of the calvarial bones, and occurs during the formation of the periosteal collar at the diaphysis (midshaft) of bones that form by endochondral ossification. During bone remodeling, bone mass is maintained by osteoblasts that form sufficient bone to replace bone that was recently removed by osteoclasts. In contrast, during bone growth, periosteal expansion occurs by modeling, where osteoblasts form bone on a bone surface that has not been previously resorbed. There are also pathological conditions, where bone is formed in locations where it is not normally found, e.g., in heterotopic ossifications in the muscle in the context of injury [76] or in rare genetic conditions [77]. In all of these processes, bone formation occurs as follows.
Osteoblasts do not produce “bone” per se, but synthesize a collagen-rich osteoid matrix. The osteoid matrix serves as a template for the subsequent deposition of mineral in the form of bioapatite which contributes to the hardness of bone. The balance between osteoid and mineral content determines bone strength: essentially, the collagen provides flexibility, while the mineral provides hardness. The process of mineralization is controlled by non-collagenous proteins produced by late-stage osteoblasts and osteocytes. We will describe each of these processes in turn (Fig. 1.2).

The process of bone matrix production and mineralization. Mature osteoblasts on the bone surface deposit newly formed matrix, known as osteoid (1), largely comprised of type I collagen (a triple helical structure). After collagen deposition, the matrix becomes progressively mineralized by the accumulation of hydroxyapatitic bioapatite crystals (2). This mineralization process has two phases. Within ~5–10 days, osteoid undergoes rapid primary mineralization, and over subsequent weeks, months, and years, secondary mineralization occurs. The bioapatite crystals grow and accumulate carbonate in the more mature regions of bone, and collagen fibers become more condensed (compact) presumably due to steric hindrance caused by the presence of the growing crystals
Osteoid Deposition
When osteoid is deposited by osteoblasts, it has two potential forms depending on its collagen orientation and speed of production: woven and lamellar bone. During bone development and fracture healing, woven bone is deposited rapidly: this substance contains disordered, seemingly randomly oriented collagen fibers. In contrast, lamellar bone is highly organized. Fibers are more slowly deposited, predominantly oriented longitudinally, and create a defined, ordered structure [78]. Collagen fibers in lamellar bone are oriented in perpendicular planes in adjacent lamellae [79], adding strength of the substance. The loose structure and random orientation of woven bone suggest that it is mechanically weaker than lamellar bone. This has been tested in human fetal bone, where younger, more woven bone was associated with lower elasticity and lower resistance to penetration (microhardness) [80].
How osteoblasts are instructed to form either woven or lamellar bone is not known, but ultra-high voltage electron microscopy studies suggest that even during lamellar bone formation, collagen fibers are deposited sparsely and randomly, but as the osteoblast becomes more distant due to further deposition, the fibres begin to reoirent parallel to the direction of growth and become thicker [81]. This suggests that as-yet unidentified events after initial collagen secretion may be responsible for the woven or lamellar nature of bone. Adding to these observations, live cell imaging of osteoblasts engineered to deposit fluorescent-labeled collagen has recently revealed that osteoblasts constantly move during the collagen assembly process, and actively exert forces on the fibrils, physically shaping the collagen matrix and potentially guiding the formation of osteocyte lacunae [82].
Type I collagen comprises a triple-helix structure of two α1 and one α2 polypeptide chain [83]. In osteoblasts, single pro-α chains are synthesized in the endoplasmic reticulum, which assemble into procollagen triple helices, and are released by exocytosis into the extracellular space, where the N- and C-termini are cleaved, allowing the formation of fibrils [84]. Multiple intracellular posttranslational modifications, including hydroxylation of proline and lysine residues, and glycosylation, stabilize the collagen triple helical structure [85]. After secretion, collagen fibers are stabilized and bone is strengthened further by the formation of inter- and intra-molecular cross-links, through the action of lysyl oxidase [86]. Other modifications such as advanced glycation adversely affect the mechanical properties of the bone matrix, particularly during ageing [87]. Defects not only in the proteins coding collagen itself but also in the many different aspects of collagen fibril assembly, including collagen folding, secretion, cross-linking, and posttranslational modifications, have been described in the diverse family of skeletal fragilities observed in osteogenesis imperfecta [88].
Matrix Mineralization
After collagen is deposited, it becomes progressively mineralized by the accumulation of bioapatite crystals. This mineralization process has two phases. Within ~5–10 days, osteoid undergoes rapid primary mineralization, and over subsequent weeks, months, and years, secondary mineralization occurs [89]. During primary mineralization, the tissue usually reaches ~50–70% of its final mineral content [90, 91]. During secondary mineralization, mineral continues to accumulate at a slower rate [92], the crystals beome larger [89], and carbonate is substituted for phosphate groups within the matrix [93, 94]. In addition, as mineral is deposited, the surrounding collagen fibers of bone also change, becoming more compact, possibly in response to the growing crystals [93, 94] (Fig. 1.2).
The final stage of mineralization achieved in the bone substance varies locally within the bone matrix and depends on the species, sex, age, and anatomical location of the bone [95]. Mineralization involves the release of matrix vesicles, which are cell-derived extracellular membrane-enclosed particles of poorly crystalline bioapatite mineral [96, 97]. The mineral crystals become ordered (a process termed nucleation) by a process driven by contact with collagen, local availability of calcium and phosphate, and by apatite nucleators such as DMP-1 and osteopontin [98, 99]. The importance of phosphate-regulating proteins is clearly illustrated by the association of human and murine genetic insufficiencies in phosphate regulators with impaired bone mineralization [100,101,102].
Mineralization initiation, accrual, and crystal maturation are controlled, not only by apatite nucleators but also by a range of multifunctional non-collagenous proteins secreted by mature osteoblasts and osteocytes. Osteoblasts and osteocytes express proteins that support mineralization such as alkaline phosphatase, PHOSPHO1, phosphate-regulating neutral endopeptidase, X-linked (PHEX), and bone sialoprotein/integrin-binding sialoprotein. Osteoblasts and osteocytes also express proteins that inhibit mineralization, such as osteocalcin [103], MEPE, and PC-1 (Enpp1) [104]. An illustration of the fine control exerted by osteoblasts on mineralization is their ability to control local levels of inorganic phosphate through alkaline phosphatase (ALP) and plasma cell membrane glycoprotein-1 (PC-1). Hydroxyapatite nucleation depends on a high ratio of inorganic phosphate (Pi), which promotes mineralization, to inorganic pyrophosphate (PPi), which inhibits it. Alkaline phosphatase (ALP) positively regulates this balance by hydrolyzing PPi to form the Pi required for hydroxyapatite crystal nucleation; insufficiency of ALP leads to poor mineralization, as observed in individuals with hypophosphatasia [100]. In contrast, PC-1 inhibits mineralization by producing inorganic pyrophosphate; insufficiency of PC-1 therefore leads to excessive mineralization [104].
The Osteoblast Lineage Supports Osteoclast Formation, Attachment, and Bone Resorption
The function of the osteoblast lineage is not restricted to bone formation. Osteoblast lineage cells also control the differentiation of osteoclasts, the cells responsible for bone resorption. There are three major ways in which cells of the osteoblast lineage carry out this role: (1) by producing RANKL and OPG in response to paracrine and endocrine agents, (2) by releasing chemoattractants that draw osteoclast precursors to the bone surface, and (3) by preparing the bone surface for osteoclast attachment. We will discuss each of these actions in turn.
Production of RANKL and OPG
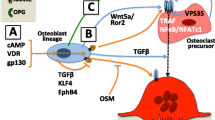
A range of locally acting cytokines, including interleukin-11 (IL-11), prostaglandin E2, PTHrP, and oncostatin M, stimulate osteoclast formation, but do not achieve this by direct action on osteoclast precursor themselves. Instead, these agents, and endocrine factors like PTH and 1,25-dihydroxyvitamin D, stimulate osteoclast formation indirectly, by acting on osteoblast lineage cells to stimulate expression of RANKL and CSF-1 (M-CSF), two regulatory molecules that are both required for osteoclastogenesis [105,106,107,108,109,110]. It is the interaction of RANKL with its receptor (RANK), expressed on the cell surface of mononuclear hemopoietic osteoclast precursors, that triggers osteoclast formation (Fig. 1.3).

The osteoblast lineage supports osteoclastogenesis. Osteoblast lineage cells control the differentiation of osteoclasts in response to paracrine and endocrine agents and locally acting cytokines such as vitamin D, interleukin-6 (IL-6), oncostatin M (OSM), and parathyroid hormone (PTH) / parathyroid hormone-related protein (PTHrP). These agents and factors act on the osteoblast lineage to stimulate expression of RANKL and M-CSF which each promote osteoclast formation. M-CSF is soluble. Receptors for both RANKL and M-CSF are expressed on the cell surface of mononuclear hemopoietic osteoclast precursors. Direct contact between membrane-bound RANKL and its membrane-bound receptor (RANK) triggers osteoclast formation. Osteoblast lineage cells also express a decoy receptor for RANKL, known as osteoprotegerin (OPG), which blocks the interaction of RANKL and RANK. Through their modulation of RANKL and OPG expression, osteoblasts can precisely regulate the formation of osteoclasts. Osteocytes also express RANKL, but the mechanism by which this reaches the osteoclast precursors remains undefined
The necessity for RANKL and RANK for osteoclastogenesis was demonstrated by the generation of genetically altered mice that lack either RANKL or RANK and exhibited a lack of osteoclasts and severe osteopetrosis [111, 112]. Osteoblast lineage cells also express a soluble protein that is a non-signaling decoy receptor for RANKL, known as osteoprotegerin (OPG). OPG acts as a “brake” on osteoclast differentiation by blocking the interaction of RANKL and RANK [113, 114], and through modulation of RANKL and OPG expression, osteoblasts can precisely regulate the formation of osteoclasts.
RANKL is expressed at all stages of osteoblast differentiation, including in precursors, matrix-producing osteoblasts, bone lining cells, and osteocytes [115]. RANKL production is not exclusive to osteoblast lineage cells. T-cells and natural killer (NK)-cells also express RANKL and are capable of promoting osteoclast formation [116, 117]. It appears that expression of RANKL by T-cells is dispensable for normal bone development and maintenance [118]. In contrast, in mice that lack RANKL in the osteoblast lineage, severe osteopetrosis is observed [119]. However, the most important stage in osteoblast differentiation for production of RANKL is not known, and whether the key source of RANKL is the osteocyte, the bone lining cell, or the preosteoblast remains controversial [21, 119,120,121,122]. One important concept to consider is that direct contact between the RANKL-expressing osteoblast lineage cells and the RANK-expressing haemopoietic osteoclast precursors is absolutely required for osteoclast formation in vitro [123, 124], and the same situation is likely to be true in vivo (Fig. 1.3). While recombinant soluble RANKL certainly promotes osteoclast formation from precursors in vitro [125], and in vivo [126], there remains no convincing evidence that soluble RANKL, produced by osteoblast lineage cells, can substitute for the membrane form, nor is there any convincing evidence of a physiological role for circulating RANKL. This means it is important to consider the location of the osteoblast lineage cells most likely to support osteoclast formation. Cells in the marrow, or in direct contact with it, such as osteoblast precursors and bone lining cells, rather than embedded osteocytes, are more likely to come into contact with osteoclast precursors, and therefore more likely to support osteoclast formation in normal remodeling. It has been difficult to understand how osteocytes, from within the matrix, could control RANKL availability to osteoclast precursors in the bloodstream through a contact-dependent mechanism although it has been suggested that osteocyte processes extend into the marrow space [127]. However, even when osteocytes were cultured in direct contact with osteoclast precursors and stimulated with appropriate stimuli, only binucleated “osteoclasts” formed [120].
RANKL production by osteoblast-lineage cells is also stimulated by microdamage within the bone matrix. Microdamage or microcracks are small defects in the bone matrix that occur in both pathological conditions and with normal skeletal loading [128]. Experimental loading, which causes a higher level of microdamage, initiates bone resorption [129], and indeed, resorption and replacement of the bone compromised by this damage is one of the important mechanical functions of bone remodeling [128]. It has been suggested that the microdamage site “steers” those osteoclasts already functioning on the bone surface toward the site of damage [130]. Microdamage within the bone is sensed by osteocytes, which are terminally differentiated osteoblasts that reside within the bone matrix, and sense changes in pressure within the matrix. Anatomical studies of rat bone in which microcracks were induced by ex vivo loading demonstrated that osteocytes located near to microcracks are more likely to be apoptotic compared to sites more distant to the microcrack [131]. Mechanical loading of human bone ex vivo and of rat bone in vivo increases osteocyte apoptosis [132, 133], and osteocytes surrounding the dying cell increase their production of RANKL to initiate resorption [134]. In support of this, short-term deletion of osteocytes in vivo resulted in a rapid increase in expression of RANKL mRNA in the bone, presumably by osteoblast lineage cells, and an increase in osteoclast formation [135].
Another factor produced by the osteoblast lineage and required for osteoclast formation is CSF-1/M-CSF [136, 137]. Together, RANKL and CSF-1 are all that is required to support osteoclast formation from bone marrow precursors in vitro. Just as observed in RANKL null mice, mutant mice lacking CSF-1 also exhibit severe osteopetrosis due to lack of osteoclast formation [138]. While RANKL is membrane bound and acts to promote osteoclast precursor fusion, CSF-1 is secreted by osteoblasts and promotes osteoclast precursor proliferation [139].
Release of Chemoattractants
Another mechanism by which osteoblasts control osteoclast differentiation is by controlling the movement of osteoclast precursors toward each other (allowing fusion) and to the bone surface (allowing attachment) through their release of chemoattractants. These factors may be deposited in the bone matrix itself during bone formation; they may be released by active osteoblasts or may be released from apoptotic osteocytes. Some bone matrix-derived factors, suggested to act as chemoattractants for monocytic osteoclast precursors, include osteocalcin, fetuin-A, and collagen-I fragments [140]. Thus, attraction of osteoclast precursors to the bone surface may be determined by the specific content of the bone to be resorbed; this is supported by studies of ageing bone. As bone ages, collagen-I is isomerized, and aged bone, which has a higher ratio of α/β collagen isomers, supports the formation of many more osteoclasts in vitro than younger bone [141], supporting a role for matrix constituents, deposited by osteoblasts, in the control of osteoclast formation.
Production of a range of chemokines (including stromal-derived factor-1 (SDF-1/CXCL12); chemokine-ligands 3, 5, and 7 (CCL3, CCL5, CCL7) [142]; chemokine (C-X-C motif) ligand 1 (CXCL1) [143]; and monocyte chemoattractant protein-1 (MCP-1) [144]) by osteoblast-lineage cells is stimulated by osteoclastogenic factors including the cytokines interleukin-1β (IL-1β), tumor necrosis factor α (TNF-α), and PTHrP. Such factors have been shown in vitro to act on osteoclast precursors (monocyte macrophages) to stimulate their chemotaxis and fusion [143, 145, 146], and it is likely that they have similar roles in vivo.
Preparing the Bone Surface for Osteoclast Attachment and Resorption
To commence resorption, the multinucleated osteoclast attaches to the bone matrix via the interaction of integrins with arginine-glycine-aspartic acid (RGD) sequences in non-collagenous matrix proteins including osteopontin and bone sialoprotein [147]. These proteins were laid down by osteoblasts during the previous cycle of bone formation. So, at some distance, it could be said that osteoblasts regulate osteoclast attachment by their control of the bone matrix itself. Intriguingly, mice lacking bone sialoprotein or osteopontin demonstrate, respectively, reduced osteoclast surface and reduced response to osteoclastogenic stimuli [148, 149]. However, this appears to be an indirect result of the reduced osteoblast numbers (and therefore reduced osteoblast-derived RANKL and M-CSF), or a requirement for intracellular osteoclastic osteopontin [150], rather than it relating to attachment to the bone matrix. Further work is required to determine how the bone matrix itself regulates osteoclast attachment; however, it should be noted that this is unlikely to be a method that precisely controls bone resorption, given the time delay between bone formation and subsequent resorption; more likely it is a mechanism that may exist in different types of bone that are responsible for biological variation in the level of bone resorption.
Concluding Remarks
The osteoblast lineage includes a range of cell types: multipotent precursors, matrix-producing osteoblasts, osteocytes, and bone lining cells; each of these stages of the lineage has distinct functions which we are only beginning to fully understand. The most well-known role of the osteoblast lineage is the production of bone matrix and the control of its mineralization by non-collagenous proteins. The osteoblast lineage controls both the progression of differentiation of its own lineage and the formation of osteoclasts, the cells that resorb bone. As such the lineage is central to the control of bone mass, both by forming it and by controlling its destruction.
References
Sims NA. Cell-specific paracrine actions of IL-6 family cytokines from bone, marrow and muscle that control bone formation and resorption. Int J Biochem Cell Biol. 2016;79:14–23.
Tonna S, Sims NA. Talking among ourselves: paracrine control of bone formation within the osteoblast lineage. Calcif Tissue Int. 2014;94(1):35–45.
Martin TJ, Sims NA. Integrating endocrine and paracrine influences on bone: lessons from parathyroid hormone and parathyroid homrone-related protein. In: Thakker RW, Whyte MP, Eisman JA, Igarashi T, editors. Genetics of bone biology and skeletal disease. New York: Academic Press; 2013.
Abdelgawad ME, Delaisse J-M, Hinge M, Jensen PR, Alnaimi RW, Rolighed L, et al. Early reversal cells in adult human bone remodeling: osteoblastic nature, catabolic functions and interactions with osteoclasts. Histochem Cell Biol. 2016;145(6):603–15.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–6.
Askmyr M, Sims NA, Martin TJ, Purton LE. What is the true nature of the osteoblastic hematopoietic stem cell niche? Trends Endocrinol Metab. 2009;20(6):303–9.
Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–7.
Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci U S A. 2008;105(44):16976–81.
Fukumoto S, Martin TJ. Bone as an endocrine organ. Trends Endocrinol Metab. 2009;20(5):230–6.
Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456–69.
Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008;2(4):313–9.
Sacchetti B, Funari A, Remoli C, Giannicola G, Kogler G, Liedtke S, et al. No identical "mesenchymal stem cells" at different times and sites: human committed progenitors of distinct origin and differentiation potential are incorporated as adventitial cells in microvessels. Stem Cell Rep. 2016;6(6):897–913.
Kalajzic Z, Li H, Wang LP, Jiang X, Lamothe K, Adams DJ, et al. Use of an alpha-smooth muscle actin GFP reporter to identify an osteoprogenitor population. Bone. 2008;43(3):501–10.
Brighton CT, Lorich DG, Kupcha R, Reilly TM, Jones AR, Woodbury RA 2nd. The pericyte as a possible osteoblast progenitor cell. Clin Orthop Relat Res. 1992;275:287–99.
Doherty MJ, Ashton BA, Walsh S, Beresford JN, Grant ME, Canfield AE. Vascular pericytes express osteogenic potential in vitro and in vivo. J Bone Min Res: Off J Am Soc Bone Min Res. 1998;13(5):828–38.
Bianco P, Robey PG. Skeletal stem cells. Development. 2015;142(6):1023–7.
Colnot C, Lu C, Hu D, Helms JA. Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev Biol. 2004;269(1):55–69.
Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell. 2010;19(2):329–44.
Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014;10(12):e1004820.
Roach HI. Trans-differentiation of hypertrophic chondrocytes into cells capable of producing a mineralized bone matrix. Bone Miner. 1992;19(1):1–20.
Matic I, Matthews BG, Wang X, Dyment NA, Worthley DL, Rowe DW, et al. Quiescent bone lining cells are a major source of osteoblasts during adulthood. Stem Cells. 2016;34(12):2930–42.
Torreggiani E, Matthews BG, Pejda S, Matic I, Horowitz MC, Grcevic D, et al. Preosteocytes/osteocytes have the potential to dedifferentiate becoming a source of osteoblasts. PLoS One. 2013;8(9):e75204.
Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, et al. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999;13(8):1025–36.
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29.
Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology: implication for coffin-Lowry syndrome. Cell. 2004;117(3):387–98.
Sabatakos G, Sims NA, Chen J, Aoki K, Kelz MB, Amling M, et al. Overexpression of DeltaFosB transcription factor(s) increases bone formation and inhibits adipogenesis. Nat Med. 2000;6(9):985–90.
Gutierrez S, Javed A, Tennant DK, van Rees M, Montecino M, Stein GS, et al. CCAAT/enhancer-binding proteins (C/EBP) beta and delta activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J Biol Chem. 2002;277(2):1316–23.
Komori T. Regulation of osteoblast differentiation by transcription factors. J Cell Biochem. 2006;99(5):1233–9.
Christy RJ, Yang VW, Ntambi JM, Geiman DE, Landschulz WH, Friedman AD, et al. Differentiation-induced gene expression in 3T3-L1 preadipocytes: CCAAT/enhancer binding protein interacts with and activates the promoters of two adipocyte-specific genes. Genes Dev. 1989;3(9):1323–35.
Tanaka T, Yoshida N, Kishimoto T, Akira S. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 1997;16(24):7432–43.
Beresford JN, Bennett JH, Devlin C, Leboy PS, Owen ME. Evidence for an inverse relationship between the differentiation of adipocytic and osteogenic cells in rat marrow stromal cell cultures. J Cell Sci. 1992;102(Pt 2):341–51.
Sims NA, Clement-Lacroix P, Da Ponte F, Bouali Y, Binart N, Moriggl R, et al. Bone homeostasis in growth hormone receptor-null mice is restored by IGF-I but independent of Stat5. J Clin Invest. 2000;106(9):1095–103.
Walker EC, McGregor NE, Poulton IJ, Solano M, Pompolo S, Fernandes TJ, et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest. 2010;120(2):582–92.
Poulton IJ, McGregor NE, Pompolo S, Walker EC, Sims NA. Contrasting roles of leukemia inhibitory factor in murine bone development and remodeling involve region-specific changes in vascularization. J Bone Miner Res. 2012;27(3):586–95.
Martin RB, Chow BD, Lucas PA. Bone marrow fat content in relation to bone remodeling and serum chemistry in intact and ovariectomized dogs. Calcif Tissue Int. 1990;46(3):189–94.
Chen C, Kalu DN. Strain differences in bone density and calcium metabolism between c3h/hej and c57bl/6j mice. Bone. 1999;25(4):413–20.
Horowitz MC, Berry R, Holtrup B, Sebo Z, Nelson T, Fretz JA, et al. Bone marrow adipocytes. Adipocytes. 2017;6(3):193–204.
Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology. 2001;2(3):165–71.
Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, et al. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn. 1999;214(4):279–90.
Kim IS, Otto F, Zabel B, Mundlos S. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. 1999;80(2):159–70.
Nishimura R, Wakabayashi M, Hata K, Matsubara T, Honma S, Wakisaka S, et al. Osterix regulates calcification and degradation of chondrogenic matrices through matrix metalloproteinase 13 (MMP13) expression in association with transcription factor Runx2 during endochondral ossification. J Biol Chem. 2012;287(40):33179–90.
Komori T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem Cell Biol. 2018;149(4):313–23.
Ecarot-Charrier B, Glorieux FH, van der Rest M, Pereira G. Osteoblasts isolated from mouse calvaria initiate matrix mineralization in culture. J Cell Biol. 1983;96(3):639–43.
Abe Y, Akamine A, Aida Y, Maeda K. Differentiation and mineralization in osteogenic precursor cells derived from fetal rat mandibular bone. Calcif Tissue Int. 1993;52(5):365–71.
Gerber I, ap Gwynn I. Influence of cell isolation, cell culture density, and cell nutrition on differentiation of rat calvarial osteoblast-like cells in vitro. Eur Cell Mater. 2001;2:10–20.
Martin TJ, Sims NA. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med. 2005;11(2):76–81.
Sims NA, Martin TJ. Coupling signals between the osteoclast and osteoblast: how are messages transmitted between these temporary visitors to the bone surface? Front Endocrinol (Lausanne). 2015;6:41.
Sims NA, Martin TJ. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep. 2014;3:481.
Aubin JE. Advances in the osteoblast lineage. Biochem Cell Biol. 1998;76(6):899–910.
Stein GS, Lian JB, Owen TA. Relationship of cell growth to the regulation of tissue-specific gene expression during osteoblast differentiation. FASEB J. 1990;4(13):3111–23.
Bellido T, Stahl N, Farruggella TJ, Borba V, Yancopoulos GD, Manolagas SC. Detection of receptors for interleukin-6, interleukin-11, leukemia inhibitory factor, oncostatin M, and ciliary neurotrophic factor in bone marrow stromal/osteoblastic cells. J Clin Invest. 1996;97(2):431–7.
Aubin JE, Liu F, Malaval L, Gupta AK. Osteoblast and chondroblast differentiation. Bone. 1995;17(2 Suppl):77S–83S.
Buenzli PR, Sims NA. Quantifying the osteocyte network in the human skeleton. Bone. 2015;75:144–50.
Franz-Odendaal TA, Hall BK, Witten PE. Buried alive: how osteoblasts become osteocytes. Dev Dyn. 2006;235(1):176–90.
Palumbo C, Palazzini S, Marotti G. Morphological study of intercellular junctions during osteocyte differentiation. Bone. 1990;11(6):401–6.
Dudley HR, Spiro D. The fine structure of bone cells. J Biophys Biochem Cytol. 1961;11(3):627–49.
Schaffler MB, Cheung WY, Majeska R, Kennedy O. Osteocytes: master orchestrators of bone. Calcif Tissue Int. 2014;94(1):5–24.
van Bezooijen RL, Roelen BA, Visser A, Van der Wee-Pals L, de Wilt E, Karperien M, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199(6):805–14.
Toyosawa S, Shintani S, Fujiwara T, Ooshima T, Sato A, Ijuhin N, et al. Dentin matrix protein 1 is predominantly expressed in chicken and rat osteocytes but not in osteoblasts. J Bone Min Res: Off J Am Soc Bone Min Res. 2001;16(11):2017–26.
Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, et al. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem. 2003;278(3):1998–2007.
Fukumoto S. FGF23-FGF receptor/klotho pathway as a new drug target for disorders of bone and mineral metabolism. Calcif Tissue Int. 2016;98(4):334–40.
Miller SC, de Saint-Georges L, Bowman BM, Jee WS. Bone lining cells: structure and function. Scanning Microsc 1989;3(3):953–960; discussion 60-1.
Jones SJ, Boyde A. Experimental study of changes in osteoblastic shape induced by calcitonin and parathyroid extract in an organ culture system. Cell Tissue Res. 1976;169(4):449–65.
Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption--a hypothesis. Calcif Tissue Int. 1981;33(4):349–51.
Hauge EM, Qvesel D, Eriksen EF, Mosekilde L, Melsen F. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J Bone Min Res: Off J Am Soc Bone Min Res. 2001;16(9):1575–82.
Rasmussen HH, Bordier P. The physiological basis of metabolic bone disease Williams and Wilkins. Baltimore: Waverley Press; 1974.
Eriksen EF, Eghbali-Fatourechi GZ, Khosla S. Remodeling and vascular spaces in bone. J Bone Min Res: Off J Am Soc Bone Min Res. 2007;22(1):1–6.
Kristensen HB, Andersen TL, Marcussen N, Rolighed L, Delaisse JM. Increased presence of capillaries next to remodeling sites in adult human cancellous bone. J Bone Miner Res. 2013;28(3):574–85.
Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology. 1995;136(8):3632–8.
Kim SW, Pajevic PD, Selig M, Barry KJ, Yang JY, Shin CS, et al. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J Bone Min Res: Off J Am Soc Bone Min Res. 2012;27(10):2075–84.
Chow JW, Wilson AJ, Chambers TJ, Fox SW. Mechanical loading stimulates bone formation by reactivation of bone lining cells in 13-week-old rats. J Bone Min Res: Off J Am Soc Bone Min Res. 1998;13(11):1760–7.
Kim SW, Lu Y, Williams EA, Lai F, Lee JY, Enishi T, et al. Sclerostin antibody administration converts bone lining cells into active osteoblasts. J Bone Min Res: Off J Am Soc Bone Min Res. 2017;32(5):892–901.
Fonseca H, Moreira-Gonçalves D, Coriolano H-JA, Duarte JA. Bone quality: the determinants of bone strength and fragility. Sports Med. 2014;44(1):37–53.
Ural A, Vashishth D. Hierarchical perspective of bone toughness–from molecules to fracture. Int Mater Rev. 2014;59(5):245–63.
Fratzl P, Gupta H, Paschalis E, Roschger P. Structure and mechanical quality of the collagen–mineral nano-composite in bone. J Mater Chem. 2004;14(14):2115–23.
Genet F, Kulina I, Vaquette C, Torossian F, Millard S, Pettit AR, et al. Neurological heterotopic ossification following spinal cord injury is triggered by macrophage-mediated inflammation in muscle. J Pathol. 2015;236(2):229–40.
Convente MR, Chakkalakal SA, Yang E, Caron RJ, Zhang D, Kambayashi T, et al. Depletion of mast cells and macrophages impairs heterotopic ossification in an Acvr1(R206H) mouse model of Fibrodysplasia Ossificans Progressiva. J Bone Miner Res. 2018;33(2):269–82.
Weiner S, Wagner HD. The material bone: structure-mechanical function relations. Annu Rev Mater Sci. 1998;28(1):271–98.
Giraud-Guille MM. Twisted plywood architecture of collagen fibrils in human compact bone osteons. Calcif Tissue Int. 1988;42(3):167–80.
Su X, Feng Q, Cui F, Zhu X. Microstructure and micromechanical properties of the mid-diaphyses of human fetal femurs. Connect Tissue Res. 1997;36(3):271–86.
Hosaki-Takamiya R, Hashimoto M, Imai Y, Nishida T, Yamada N, Mori H, et al. Collagen production of osteoblasts revealed by ultra-high voltage electron microscopy. J Bone Miner Metab. 2016;34(5):491–9.
Lu Y, Kamel-El Sayed SA, Wang K, Tiede-Lewis LM, Grillo MA, Veno PA, et al. Live imaging of type I collagen assembly dynamics in osteoblasts stably expressing GFP and mCherry-Tagged collagen constructs. J Bone Min Res. 2018;33(6):1166–82.
Brodsky B, Ramshaw JA. The collagen triple-helix structure. Matrix Biol. 1997;15(8–9):545–54.
Leblond CP. Synthesis and secretion of collagen by cells of connective tissue, bone, and dentin. Anat Rec. 1989;224(2):123–38.
Viguet-Carrin S, Garnero P, Delmas P. The role of collagen in bone strength. Osteoporos Int. 2006;17(3):319–36.
Oxlund H, Barckman M, Ørtoft G, Andreassen TT. Reduced concentrations of collagen cross-links are associated with reduced strength of bone. Bone. 1995;17(4, Supplement):S365–S71.
Vashishth D, Gibson GJ, Khoury JI, Schaffler MB, Kimura J, Fyhrie DP. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone. 2001;28(2):195–201.
Forlino A, Marini JC. Osteogenesis imperfecta. Lancet. 2016;387(10028):1657–71.
Glimcher M. The natuer of the mineral phase in bone: biological and clinical implications. In: Avioli L, Krane S, editors. Metabolic bone disease, vol. 23. San Diego: Academic Press; 1998. p. 50.
Ruffoni D, Fratzl P, Roschger P, Klaushofer K, Weinkamer R. The bone mineralization density distribution as a fingerprint of the mineralization process. Bone. 2007;40(5):1308–19.
Boivin G, Meunier PJ. The degree of mineralization of bone tissue measured by computerized quantitative contact microradiography. Calcif Tissue Int. 2002;70(6):503–11.
Fuchs RK, Allen MR, Ruppel ME, Diab T, Phipps RJ, Miller LM, et al. In situ examination of the time-course for secondary mineralization of Haversian bone using synchrotron Fourier transform infrared microspectroscopy. Matrix Biol. 2008;27(1):34–41.
Vrahnas C, Pearson TA, Brunt AR, Forwood MR, Bambery KR, Tobin MJ, et al. Anabolic action of parathyroid hormone (PTH) does not compromise bone matrix mineral composition or maturation. Bone. 2016;93:146–54.
Vrahnas C, Buenzli PR, Pearson TA, Pennypacker BL, Tobin MJ, Bambery KR, et al. Differing effects of parathyroid hormone, alendronate and Odanacatib on bone formation and on the mineralisation process in intracortical and endocortical bone of ovariectomized rabbits. Calcif Tissue Int. 2018;103(6):625–37.
Boskey AL. Bone composition: relationship to bone fragility and antiosteoporotic drug effects. Bonekey Rep. 2013;2:447.
Anderson HC. Molecular biology of matrix vesicles. Clin Orthop Relat Res. 1995;314:266–80.
Hoshi K, Ozawa H. Matrix vesicle calcification in bones of adult rats. Calcif Tissue Int. 2000;66(6):430–4.
Rohde M, Mayer H. Exocytotic process as a novel model for mineralization by osteoblasts in vitro and in vivo determined by electron microscopic analysis. Calcif Tissue Int. 2007;80(5):323–36.
Stanford CM, Jacobson PA, Eanes ED, Lembke LA, Midura RJ. Rapidly forming apatitic mineral in an osteoblastic cell line (UMR 106-01 BSP). J Biol Chem. 1995;270(16):9420–8.
Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev. 1994;15(4):439–61.
Quarles LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab. 2003;285(1):E1–9.
Holm E, Aubin JE, Hunter GK, Beier F, Goldberg HA. Loss of bone sialoprotein leads to impaired endochondral bone development and mineralization. Bone. 2015;71:145–54.
Boskey AL, Gadaleta S, Gundberg C, Doty SB, Ducy P, Karsenty G. Fourier transform infrared microspectroscopic analysis of bones of osteocalcin-deficient mice provides insight into the function of osteocalcin. Bone. 1998;23(3):187–96.
Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002;99(14):9445–9.
Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139(3):1329–37.
Horwood NJ, Elliott J, Martin TJ, Gillespie MT. Osteotropic agents regulate the expression of osteoclast differentiation factor and osteoprotegerin in osteoblastic stromal cells. Endocrinology. 1998;139(11):4743–6.
Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J Immunol. 2002;169(6):3353–62.
Liu BY, Guo J, Lanske B, Divieti P, Kronenberg HM, Bringhurst FR. Conditionally immortalized murine bone marrow stromal cells mediate parathyroid hormone-dependent osteoclastogenesis in vitro. Endocrinology. 1998;139(4):1952–64.
Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–49.
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20(3):345–57.
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402(6759):304–9.
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13(18):2412–24.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–19.
Tsuda E, Goto M, Mochizuki S, Yano K, Kobayashi F, Morinaga T, et al. Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun. 1997;234(1):137–42.
Kartsogiannis V, Zhou H, Horwood NJ, Thomas RJ, Hards DK, Quinn JM, et al. Localization of RANKL (receptor activator of NF kappa B ligand) mRNA and protein in skeletal and extraskeletal tissues. Bone. 1999;25(5):525–34.
Horwood NJ, Kartsogiannis V, Quinn JM, Romas E, Martin TJ, Gillespie MT. Activated T lymphocytes support osteoclast formation in vitro. Biochem Biophys Res Commun. 1999;265(1):144–50.
Soderstrom K, Stein E, Colmenero P, Purath U, Muller-Ladner U, de Matos CT, et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci U S A. 2010;107(29):13028–33.
Danks L, Komatsu N, Guerrini MM, Sawa S, Armaka M, Kollias G, et al. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann Rheum Dis. 2016;75(6):1187–95.
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–41.
Chia LY, Walsh NC, Martin TJ, Sims NA. Isolation and gene expression of haematopoietic-cell-free preparations of highly purified murine osteocytes. Bone. 2015;72:34–42.
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–4.
Fumoto T, Takeshita S, Ito M, Ikeda K. Physiological functions of osteoblast lineage and T cell-derived RANKL in bone homeostasis. J Bone Min Res: Off J Am Soc Bone Min Res. 2014;29(4):830–42.
Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley JM, et al. Osteoblastic cells are involved in osteoclast formation. Endocrinology. 1988;123(5):2600–2.
Suda T, Takahashi N, Martin TJ. Modulation of osteoclast differentiation. Endocr Rev. 1992;13(1):66–80.
Quinn JM, Itoh K, Udagawa N, Hausler K, Yasuda H, Shima N, et al. Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J Bone Miner Res. 2001;16(10):1787–94.
Tomimori Y, Mori K, Koide M, Nakamichi Y, Ninomiya T, Udagawa N, et al. Evaluation of pharmaceuticals with a novel 50-hour animal model of bone loss. J Bone Min Res: Off J Am Soc Bone Min Res. 2009;24(7):1194–205.
Kamioka H, Honjo T, Takano-Yamamoto T. A three-dimensional distribution of osteocyte processes revealed by the combination of confocal laser scanning microscopy and differential interference contrast microscopy. Bone. 2001;28(2):145–9.
Parfitt AM. Targeted and nontargeted bone remodeling: relationship to basic multicellular unit origination and progression. Bone. 2002;30(1):5–7.
Mori S, Burr DB. Increased intracortical remodeling following fatigue damage. Bone. 1993;14(2):103–9.
Martin RB. Targeted bone remodeling involves BMU steering as well as activation. Bone. 2007;40(6):1574–80.
Verborgt O, Tatton NA, Majeska RJ, Schaffler MB. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? J Bone Min Res: Off J Am Soc Bone Min Res. 2002;17(5):907–14.
Noble BS, Peet N, Stevens HY, Brabbs A, Mosley JR, Reilly GC, et al. Mechanical loading: biphasic osteocyte survival and targeting of osteoclasts for bone destruction in rat cortical bone. Am J Physiol Cell Physiol. 2003;284(4):C934–43.
Mann V, Huber C, Kogianni G, Jones D, Noble B. The influence of mechanical stimulation on osteocyte apoptosis and bone viability in human trabecular bone. J Musculoskelet Neuronal Interact. 2006;6(4):408–17.
Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone. 2012;50(5):1115–22.
Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5(6):464–75.
Van Wesenbeeck L, Odgren PR, MacKay CA, D'Angelo M, Safadi FF, Popoff SN, et al. The osteopetrotic mutation toothless (tl) is a loss-of-function frameshift mutation in the rat Csf1 gene: evidence of a crucial role for CSF-1 in osteoclastogenesis and endochondral ossification. Proc Natl Acad Sci U S A. 2002;99(22):14303–8.
Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW Jr, Ahmed-Ansari A, Sell KW, Pollard JW, et al. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci U S A. 1990;87(12):4828–32.
Marks SC Jr. Morphological evidence of reduced bone resorption in osteopetrotic (op) mice. Am J Anat. 1982;163(2):157–67.
Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med. 1999;190(12):1741–54.
Malone JD, Teitelbaum SL, Griffin GL, Senior RM, Kahn AJ. Recruitment of osteoclast precursors by purified bone matrix constituents. J Cell Biol. 1982;92(1):227–30.
Henriksen K, Leeming DJ, Byrjalsen I, Nielsen RH, Sorensen MG, Dziegiel MH, et al. Osteoclasts prefer aged bone. Osteoporos Int. 2007;18(6):751–9.
Yu X, Huang Y, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase-9 (MMP-9) activity, and collagen transmigration. J Bone Min Res: Off J Am Soc Bone Min Res. 2003;18(8):1404–18.
Onan D, Allan EH, Quinn JM, Gooi JH, Pompolo S, Sims NA, et al. The chemokine Cxcl1 is a novel target gene of parathyroid hormone (PTH)/PTH-related protein in committed osteoblasts. Endocrinology. 2009;150(5):2244–53.
Zhu JF, Valente AJ, Lorenzo JA, Carnes D, Graves DT. Expression of monocyte chemoattractant protein 1 in human osteoblastic cells stimulated by proinflammatory mediators. J Bone Miner Res. 1994;9(7):1123–30.
Yu X, Huang Y, Collin-Osdoby P, Osdoby P. CCR1 chemokines promote the chemotactic recruitment, RANKL development, and motility of osteoclasts and are induced by inflammatory cytokines in osteoblasts. J Bone Min Res: Off J Am Soc Bone Min Res. 2004;19(12):2065–77.
Li X, Qin L, Bergenstock M, Bevelock LM, Novack DV, Partridge NC. Parathyroid hormone stimulates osteoblastic expression of MCP-1 to recruit and increase the fusion of pre/osteoclasts. J Biol Chem. 2007;282(45):33098–106.
Ross FP, Teitelbaum SL. alphavbeta3 and macrophage colony-stimulating factor: partners in osteoclast biology. Immunol Rev. 2005;208:88–105.
Malaval L, Wade-Gueye NM, Boudiffa M, Fei J, Zirngibl R, Chen F, et al. Bone sialoprotein plays a functional role in bone formation and osteoclastogenesis. J Exp Med. 2008;205(5):1145–53.
Ihara H, Denhardt DT, Furuya K, Yamashita T, Muguruma Y, Tsuji K, et al. Parathyroid hormone-induced bone resorption does not occur in the absence of osteopontin. J Biol Chem. 2001;276(16):13065–71.
Chellaiah MA, Kizer N, Biswas R, Alvarez U, Strauss-Schoenberger J, Rifas L, et al. Osteopontin deficiency produces osteoclast dysfunction due to reduced CD44 surface expression. Mol Biol Cell. 2003;14(1):173–89.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Vrahnas, C., Sims, N.A. (2020). Basic Aspects of Osteoblast Function. In: Leder, B., Wein, M. (eds) Osteoporosis. Contemporary Endocrinology. Humana, Cham. https://doi.org/10.1007/978-3-319-69287-6_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-69287-6_1
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-319-69286-9
Online ISBN: 978-3-319-69287-6
eBook Packages: MedicineMedicine (R0)