
Abstract
A quantitative understanding of the principle factors that govern their geochemical behavior is required to employ boron and its isotopes as geochemical tracers of any vapor-, liquid- or melt-mediated process in the Earth’s interior. Feedback between experiments and computational predictions are required to gain insight into the processes driving isotope partitioning. This chapter comprises methods and results of selected experimental studies and first principles atomistic modeling techniques aimed at determining and predicting temperature-, pressure-, and pH-dependent B-isotope fractionation among B-bearing geomaterials.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

3.1 Introduction
One of the fundamental tasks in Geosciences is unraveling the transport of Earth’s materials and their interactions with the dynamic, evolving Earth system. For any sophisticated description of geochemical cycles, knowledge of the distribution of elements and isotopes among geomaterials is a prerequisite to understand the underlying mechanisms and laws that govern their interactions. The light element boron is highly fluid-mobile and tends to strongly partition into the fluid phase during fluid-rock interaction processes (e.g., Mottl and Holland 1978). The two stable isotopes, 10B and 11B, differ in their atomic mass by 10%. Consequently, boron isotopes strongly fractionate during geological processes, thereby leading to natural δ11B-variations of about 100‰ (Barth 1993; Palmer and Swihart 2002). Therefore, B isotopes have great potential for modeling geochemical cycles and are widely used as geochemical tracers to unravel processes operating at the surface, as well as in the deeper crust and mantle (e.g., Leeman and Sisson 2002; Palmer and Swihart 2002).
Arc magmas, for example, are typically enriched in B relative to fresh mid-ocean ridge basalts and mantle (e.g., Ryan and Langmuir 1993). In addition, the boron concentration in arc magmas often systematically decreases with increasing distance to the underlying old and cold subducting plates (e.g., Rosner et al. 2003). This indicates continuous dehydration within these slabs during the breakdown of (OH, B)-bearing minerals (e.g., micas, serpentine) with increasing depth. Furthermore, 11B preferentially fractionates into hydrous fluids relative to the co-existing minerals, and arc magmas are successively depleted in the heavy 11B with progressing subduction (e.g., Ishikawa and Nakamura 1994). Both observations indicate that B is transported from subducted sediments and/or altered oceanic crust into the magma’s source region by a fluid or melt. As those reservoirs show different B concentrations and δ11B-values (Palmer and Swihart 2002), B and its isotopes are ideal geochemical tracers for tracking the source and to quantify the mass flux from the slab into the mantle wedge and finally to the surface. Details of a large body of work on this topic are presented in this book (De Hoog and Savov 2017).
Another important natural process, which strongly fractionates B isotopes, is the interaction of seawater with various solid marine components (e.g., biogenic carbonates, clays, oxides) at near surface conditions. The isotopic composition of seawater is governed by the B isotope exchange between the two main aqueous B-species, B(OH)3 and [B(OH)4]−, which can be expressed as:
This reaction in turn controls the isotopic composition of geomaterials in contact with seawater through fractionation during surface adsorption and/or a structural exchange mechanism. As the B-isotopic fractionation factor α with
strongly depends on pH, paleo-pH values of seawater can be reconstructed from the B-isotopic composition of biogenic carbonates in marine sediments (e.g., Hemming and Hanson 1992; Spivack et al. 1993; Gaillardet and Allègre 1995; Sanyal et al. 1995; Palmer et al. 1998). Three chapters within this book (Branson 2017; Rae 2017; McCulloch 2017).
The use of such carbonate archives is a key to the understanding of past and present climate changes (e.g., Lea 2003), whereas the first example is used to expand geochemical models in subduction-zone regimes (e.g., Marschall et al. 2007; Konrad-Schmolke and Halama 2014). Both, either require the assumption of isotope equilibrium or quantitative knowledge of pH-, temperature-, and pressure-dependent equilibrium B-isotope fractionation among the various phases of interest in order to identify disequilibrium.
The establishment of B-isotope fractionation data has progressed via two different routes, namely by laboratory experiments and by computational modeling. In the first part of this chapter, we present methods and results from selected experimental studies, designed to determine B-isotopic fractionation data at low to high temperatures in the systems vapor–liquids–solids–melts. In the second part, we present the modeling methodology behind the first-principles simulation approach to the ab initio prediction of equilibrium isotope fractionation between phases at high temperatures and pressures recently developed by Kowalski and Jahn (2011) and Kowalski et al. (2013). For various systems and conditions, B-isotopic fractionation data are calculated and compared with results from the experimental studies.
3.2 Notations
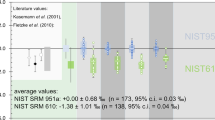
The B-isotope composition of a phase is generally given as per mil variations relative to the B isotopic standard NIST 951 boric acid according to
For the B-isotopic exchange reaction
the B-isotopic fractionation between two phases is denoted by the isotopic fractionation factor α, an equilibrium coefficient defined as:
or as:
The α factor, rather than Δ11B, is used in thermodynamic modeling of isotopic fractionation because α is equal to the equilibrium constant (K) in the Gibbs free energy equation
of the exchange reaction, with ΔG (Gibbs free energy), ΔH (reaction enthalpy), ΔS (entropy), R (universal gas constant), and T (temperature). For ideal gas reactions, ln K follows ~1/T (in kelvin) for phases containing hydroxyl groups, at low temperatures (below room temperature) and is roughly proportional to 1/T 2 at high temperatures relevant for geological environments.
The isotopic fractionation factors Δ11B and α are related by:
Only for strong isotopic fractionation and large δ11B-values the approximation (3.8) does not match within the range of experimental uncertainties of δ11B (see later). Combining Eqs. 3.6 and Eq. (3.8), the isotope fractionation factor α, expressed in terms of δ11B, is defined as:
3.3 Controls of B-Isotopic Fractionation
Due to the rather low experimentally derived diffusivities of B in melts of haplogranitic compositions (Chakroborty et al. 1993), it can be assumed that kinetic B-isotope exchange is insignificant at melt-crystallization temperatures. This is in strong contrast to the behavior of lithium and its isotopes, for which significant kinetic isotopic fractionation is observed at temperatures above 800 °C (Dohmen et al. 2010). Owing to the limited amount of experimental data on high-T diffusion-induced B-isotope fractionation—which strongly awaits further future high-temperature studies—B-isotope fractionation is regarded here as solely resulting from low-T surface adsorption processes (e.g., smectites) and from equilibrium isotope exchange. Generally, equilibrium isotopic fractionation is driven by differences in the chemical potentials between coexisting phases, which may be represented by the energetic differences in the respective bonding environments. Equilibrium isotope fractionation usually decreases as temperature increases, following the approximation ln α ~ 1/T 2. For many solid–solid or solid–fluid systems, it has been shown experimentally and by modeling that isotopic fractionation between two phases follows a simple rule: the lighter isotope is preferentially incorporated in the phase where it is at higher coordination. This is accompanied with longer cation-anion bond lengths and weaker bond strength (e.g., Zheng 1993; Schauble 2004; Wunder et al. 2006). Owed to its small ionic radius ([3]r = 0.01Å, [4]r = 0.11Å after Shannon 1976) B occurs either in trigonal or tetrahedral coordination with oxygen, whereby 11B prefers trigonal coordination and 10B the tetrahedral coordination (Palmer and Swihart 2002). In addition to the concept of coordination-driven isotope fractionation there is also significant isotopic fractionation between phases of the same coordination, e.g., tourmaline—boric acid (Palmer et al. 1992; Meyer et al. 2008) both with trigonal B-coordination (some tourmaline also have [4]B, see later). This indicates that beside the often as “first-order criteria” defined coordination rule, also second-order effects, like differences in bond strengths, geometries, energetics, and electronic environments of the local atomic structures of different phases have to be considered. Therefore, structural characterisation of the B-environment in all phases of interest is a prerequisite for the understanding of the principle mechanism and for reliable modeling of B-isotope fractionation in the different systems.
3.4 B-Speciation in Liquids, Vapor, and Melts
3.4.1 B-Speciation in Aqueous Fluids
B in seawater mainly forms two aqueous species which are in a pH-dependent equilibrium (Eq. 3.1): the neutral trigonal boric acid B(OH)3 and the larger tetrahedral borate anion [B(OH)4]− (Hershey et al. 1986). At atmospheric conditions, the predominant B species is B(OH)3 at low pH and [B(OH)4]− at high pH (Vengosh et al. 1991; Sanchez-Valle et al. 2005). At high aqueous B-concentrations (>0.5 mol/L) other minor B species may be present, e.g., polyborates and borates NaB(OH)4, [MgB(OH)4]+, [CaB(OH)4]+ (Christ and Harder 1978), and [B3O3(OH)4]−, [B3O3(OH)5]2− (Kakihana et al. 1977). Hershey et al. (1986) determined the fraction of B(OH)3 and [B(OH)4]− species present at ambient conditions as a function of pH. For determination of aqueous B-species in crustal and mantle wedge fluids, Schmidt et al. (2005) studied B-bearing fluids of various compositions in situ at temperatures between 22 and 600 °C and pressures from 0.1 MPa to about 2 GPa using Raman spectroscopy and an externally heated hydrothermal diamond anvil cell (Fig. 3.1a). In acidic fluids, they found B(OH)3 as the main species with traces of HBO2, whose amount weakly increases with temperature and decreases with the addition of NaCl (Fig. 3.1b, e). In alkaline solutions, ring-like polyboric ions [B3O3(OH)4]−, [B4O5(OH)4]2−, [B5O6(OH)4]−, which occur together with B(OH)3 and [B(OH)4]− at low pressures and temperatures, completely depolymerise with inceasing temperature, and the amount of B(OH)3 increases relative to [B(OH)4]− (Fig. 3.1c,d). In basic fluids, the [B(OH)4]− to B(OH)3 ratio increases with increasing pressure.

a Schematic diagram of the central portion (not to scale) of an externally heated hydrothermal diamond anvil cell (courtesy of Christian Schmidt, GFZ Potsdam). In situ Raman spectra of fluids for three different systems: b H2O + H3BO3; c, d H2O + Na2B4O7; e H2O + H3BO3 + NaCl (modified after Schmidt et al. 2005). Additionally, the structures of the various B-species dissolved in fluids are shown (green boron; red oxygen; blue hydrogen) according to Liu and Tossel (2005)
In summary, Schmidt et al. (2005) showed that B(OH)3 is the predominant aqueous B-species over a wide range of pressure-temperature-pH conditions in slab-related fluids and that the aqueous [B(OH)4]− species may become more important in near-neutral to basic fluids at greater depth. Full quantification of the relative proportions of the different B-species from the Raman spectra (Schmidt et al. 2005) would require scattering coefficients of individual species, which are not readily available. Additionally, the aqueous B-species are completely unknown for temperatures above 600 °C and pressures above 2.0 GPa. These are shortcomings, when modeling high pressure and temperature B-isotope fractionation (see later). It should also be kept in mind that due to the high fluid solubilities of alkalis, Si, Al and particularly light elements like Li and Be at high pressures and temperatures (Manning 2004), natural fluids might exhibit other B-species associated with the fluid-compatible elements, more complex than boric acid and the anionic tetrahedral borate.
3.4.2 Boron Speciation in Vapor
To the best of our knowledge, no direct measurements of B-species in vapor are available. Speculations about B speciation in vapor are based on experimental data on B-isotopic vapor - liquid fractionation determined in the T-range 400–450 °C for seawater-like H2O–NaCl model systems at near-neutral pH conditions (see below, Spivack et al. 1990; Liebscher et al. 2005). The Δ11Bvapor-liquid-values are small; hence, due to the coordination rule, this suggests similar B(OH)3-speciation in the conjugated aqueous fluid and vapor pairs at the experimental conditions. It is furthermore assumed that B in vapor exhibits the same pH-dependent characteristics as in aqueous fluids: B(OH)3 is predominant at acidic to neutral conditions, while [B(OH)4]− is only significantly abundant at strongly basic pH. In the system H2O–NaCl, the hydrolysis reaction H2O + NaCl = HCl + NaOH exerts control on the pH. At ambient conditions hydrolysis is small and H2O–NaCl solutions have near neutral pH. At higher temperatures the concentrations of HCl and NaCl increase. In liquid–vapor systems HCl fractionates into the vapor and the conjugated liquid becomes slightly enriched in NaOH (Shumolvich et al. 2002). Consequently, according to Vakulenko et al. (1989) at 390–470 °C, and 21.7–35.5 MPa, the vapor becomes more acidic (quench pH = 5–6) and the liquid more basic (quench pH = 8–9). This process may influence the boron speciation of the liquid to contain slightly more [B(OH)4]− than the coexisting vapor (see later Chap. 5.1).
3.4.3 Boron Speciation in Melt
Boron is only a minor component in natural magmas, but can be significantly enriched by several weight percent of B2O3 in S-type granites evolved from evaporitic metasediments (Pichavant et al. 1988), or during late magmatic or pegmatitic stages of granitic intrusions (e.g., London 1997; Thomas et al. 2003). These stages are known to be associated with highly complex internal processes, which may involve more than one phase, i.e., by separation of immiscible silica- and (hydrous) borate-rich liquids (Thomas et al. 2000; Veksler et al. 2002; see also Trumbull and Slack 2017, this book). Boron is the smallest of the network-forming cations in glasses and melts with a high ionic charge. It strongly affects the physical and chemical properties in melts (e.g., glass transition temperature Tg, density, thermal expansion coefficient) and therefore has also broad applications in the glass industry (e.g., Scholze 1988). Information on B-speciation in melts and glasses come from neutron diffraction, infrared (IR), Raman, and nuclear magnetic resonance (NMR) spectroscopy. In borosilicate melts and glasses two distinct structural units of boron exist: quasi-planar [BO3]3− triangles and [BO4]5− tetrahedra. Both species tend to polymerise and form numerous combinations with network-forming components, i.e., [SiO4]4−, [AlO4]5−. The proportions of 3- and 4-fold coordinations in melts and glasses depend on several factors including composition (e.g., Geisinger et al. 1988; Zhong and Bray 1989; Schmidt et al. 2004; Wu and Stebbins 2013), temperature (e.g., Smirnov et al. 2005; Manara et al. 2009; Wu and Stebbins 2010, 2013), and pressure (e.g., Schmidt et al. 2004; Edwards et al. 2014). Furthermore, the fraction of tetrahedral B significantly increases below the glass transition temperature (Michel et al. 2013). The structural modification during quenching of a B-rich silicate melt changes B from a network-modifier to a network-forming cation, and might explain the low viscosity of B-bearing melts. As the glass transition temperature depends on the cooling rate, the latter also affects the B-coordination. In binary alkali borate melts the addition of up to 35 mol% alkali oxide to a B2O3-glass results in a decrease of the [3]B/[4]B-ratio (Bray and O’Keefe 1963; Zhong and Bray 1989). At higher alkali contents tetrahedral diborate-complexes depolymerise again and form trigonal B, which is coordinated by one nonbridging oxygen (NBO) with an associated alkali cation. For glasses along the albite (Ab, NaAlSi3O8)—reedmergnerite (Rd, NaBSi3O8) join, the [BO3]/[BO4]-ratio increases from about 0.3 for an Al-free Rd-glass to 0.6 for a glass of Ab80Rd20-composition (Geisinger et al. 1988). This indicates that the presence of Al destabilizes the tetrahedral B units. This trend of an increasing [3]B/[4]B-ratio was also determined for anhydrous melts of increasing A/CNK-value (A = Al2O3; CNK = sum of oxides of charge-balancing cations like CaO, Na2O, K2O; all in mole) varying from peralkaline (A/CNK < 1) to peraluminous (A/CNK > 1) compositions. Additionally, increasing temperature shifts the reaction [BO4] ↔ [BO3] + NBO to the right (Wu and Stebbins 2010, 2013). Anhydrous boroaluminosilicate glasses synthesized at ambient pressure contain about 2% of tetrahedral species, the amount of which increases to approximately 6% upon hydration with 4.4 wt% water (Schmidt et al. 2004). In the same study, anhydrous glasses synthesized at 1.0 and 2.0 GPa have increased fractions of tetrahedral [BO4] to 3 and 7, respectively. Spectroscopic measurements of B speciation in three natural rhyolite glasses by Tonarini et al. (2003) revealed 74–92% of B in trigonal sites. Using a highly sophisticated in situ method of high-pressure 11B NMR spectroscopy, Edwards et al. (2014) studied the trigonal [BO3] to tetrahedral [BO4] conversion in B-bearing glasses with increasing pressure, and found that the coordination change proceeds via a trigonal bipyramid as precursor phase.
In summary, in natural melts, the amount of tetrahedral boron units is generally small and only slightly increases with higher water content, increasing amounts of alkalis, or at high pressure and low temperature, which might be relevant for the melts of deeply subducted young slabs.
3.5 Experimental Determination of B-Isotope Fractionation
Here we present methods and some examples on the experimental determination of B-isotope fractionation between different phases. Firstly, we résumé experimental studies on vapor–liquid–melt fractionation at medium to high temperatures. A clear distinction needs to be made between diffusive fractionation at the crystal surface, which often represents disequilibrium, and equilibrium exchange due to bulk processes. We, thus, divide the second section of mineral–fluid fractionation into two different parts: (1) solid–aqueous fluid fractionation related to surface B adsorption at low temperatures, and (2) equilibrium solid–fluid fractionation at medium to high temperatures.
3.5.1 Vapor–Liquid–Melt Boron Isotope Fractionation
3.5.1.1 Vapor–Liquid Boron Isotope Fractionation
The B concentration and B-isotopic composition of natural hydrothermal fluids may be altered by various processes, including vapor–liquid separation. Numerous studies on fluid inclusions (e.g., Roedder 1979; Reynolds and Beane 1985; Hedenquist et al. 1998) have confirmed that liquid brines and low-density vapor can coexist across large sections through the altered oceanic crust and also around shallow crustal plutons. The distribution of B between coexisting liquid and vapor has been described for several natural geothermal systems (Smith et al. 1987; Chiodini et al. 1988; Glover 1988; Leeman et al. 1992). At low temperatures B fractionates preferentially into the liquid. D liquid/vaporB and C liquidB /C vaporB decreases with temperature and varies from ~100 to ~10 between 100 and 300 °C (Leeman et al. 1992), and approaches unity at around 450 °C, 34–38 MPa (Spivack et al. 1990). At T > 500 °C B fractionates into the vapor phase with D liquid/vaporB = 0.84 at 528 °C, 57 MPa (Audétat et al. 1998) and 0.2 at 800 °C, 100 MPa (Schatz et al. 2004).
Investigating the dependence of D liquid/vaporB on pressure and salinity and for determination of B-isotope fractionation between liquid and vapor at subcritical conditions, Liebscher et al. (2005) conducted experiments in a large-volume Ti autoclave (Fig. 3.2a), which enabled quasi-isobaric recovery of coexisting liquid and vapor. Liquid and vapor pairs were analyzed separately by ICP-AES for Na and B and by positive thermal ionization mass spectroscopy (p-TIMS) for their B isotopic composition. Experiments were performed in the system H2O–NaCl–B2O3 along the 400 and 450 °C isotherms, ranging from 23 to 28 MPa and from 38 to 42 MPa, respectively. The starting fluids had a neutral pH (at room temperature), contained 200 μg/g B of natural seawater isotopic composition (~39.5‰), and NaCl concentrations that correspond to the composition at the critical point of the respective isotherms. Boron liquid–vapor fractionation is generally small and increases from 1.0 at the critical points with increasing opening of the H2O–NaCl solvus (Fig. 3.2b). The maximum values at extrapolated salt saturated conditions are D liquid/vaporB = 2.7 (at 400 °C) and 1.8 (at 450 °C).

Experimental data on vapor–liquid fractionation of B and its isotopes in the system H2O–NaCl–B2O3 at high-salinity conditions according to Liebscher et al. (2005). a Schematic drawing of a large-volume Ti-autoclave to extract immiscible fluids; b B concentration in coexisting vapor–liquid pairs along the 450 and 400 °C isotherms; c B isotope fraction between coexisting vapor and liquid along the 450 and 400 °C isotherms, with additional low-salinity data (green symbols) of Spivack et al. (1990); d extrapolation of derived Δ11Bvapor-liquid at 450 and 400 °C to salt-saturated conditions as a function of the distance to the critical pressure. For comparison, vapor–liquid Li-isotopic fractionation data are also shown (Liebscher et al. 2007). All figures are modified after Liebscher et al. (2005)
B-isotope compositions of coexisting vapor and liquid from natural geothermal systems induced by phase separation at 170–260 °C were studied by Leeman et al. (1992). They determined Δ11Bvapor-liquid in the range of about 1–3‰, with more pronounced 11B-enrichment in the vapor phase at low temperatures. In experiments within systems of high salinity described above (Liebscher et al. 2005), B-isotope fractionation between vapor and liquid always yielded fractionation of 11B into the vapor phase, analogous to data from the natural systems (Leeman et al. 1992). However, the experimentally derived Δ11Bvapor-liquid values are smaller and range from +0.2(± 0.7) to +0.9(± 0.5)‰ at 450 °C and from +0.1(± 0.6) to +0.7(± 0.6)‰ at 400 °C (Fig. 3.2c), without any obvious T-effect on Δ11Bvapor-liquid. At extrapolated salt-saturated conditions, calculations resulted in Δ11Bvapor-liquid values of +1.5(± 0.7)‰ at 450 °C and +1.3(± 0.6)‰ at 400 °C (Fig. 3.2d). The experimental data perhaps indicate an increase of vapor - liquid B-isotope fractionation with decreasing pressure during increasing opening of the solvus (Fig. 3.2c). The same trend of an increasingly positive isotope fractionation with opening of the solvus was also observed for vapor–fluid (Fig. 3.2d) from experiments in the system LiCl–H2O at 400 °C/20–28 MPa (Liebscher et al. 2007). The weak B-isotopic fractionation suggests a similar trigonal speciation of B in vapor and the liquid. Increasing fractionation might be explained by a slight increase of the [B(OH)4]− species in the liquid with decreasing pressure (Liebscher et al. 2005) as the pH of the liquid relative to that of the conjugated vapor increases (Vakulenko et al. 1989, see above Chap. 4.1). From these experimental results it was concluded that fluid phase separation is able to significantly alter the B isotope signature of hydrous fluids in open hydrothermal systems. However, due to the low value of Δ11Bvapor-liquid, processes other than vapor–liquid separation might dominate the B isotope geochemistry in oceanic hydrothermal fluids (Liebscher et al. 2005). The B-isotope vapor–liquid fractionation data for high-salinity brines (Liebscher et al. 2005) slightly differ from Δ11Bvapor-liquid values determined for low-salinity vapor–fluid systems near the critical point (Fig. 3.2c) of seawater in an earlier experimental study (Spivack et al. 1990). They found that Δ11Bvapor-liquid = + 0.4(± 0.7)‰ (at 34.6 MPa, 425 °C) and −0.4(± 0.7)‰ at 37.9 MPa, 450 °C, which suggests that within the analytical and experimental uncertainties, the fractionation is negligible (Spivack et al. 1990). Thus Spivack et al. (1990) concluded that phase separation in high-temperature geothermal systems is unlikely to have a significant effect on the B-isotopic composition of liquid and vapor.
3.5.1.2 Aqueous Fluid–Melt B-Isotope Fractionation
Hervig et al. (2002) have experimentally investigated B-isotope fractionation between hydrous fluids and silicate melt of basaltic and rhyolitic composition over a pressure–temperature range of 110–500 MPa, 750–1100 °C, which are conditions above the critical point; thus the fluid is defined here as including gas and liquid. The experiments were performed in a piston-cylinder press and an internally heated rapid-quench hydrothermal apparatus, using B-bearing glass chips (containing approximately 2000 μg/g B) and water in a 1:1 ratio as starting materials, sealed in Au or AuPd capsules. The B concentration and B-isotopic composition of the starting and run-product glasses were analyzed by secondary ion mass spectrometry (SIMS); B contents and δ11Bfluid of coexisting fluids were calculated by mass balance considerations.
The partitioning of B between coexisting fluid and melt (D fluid/meltB ) is 0.33–0.54 at 950–1100 °C, 110–170 MPa for the system fluid–basaltic melt and 1.19–1.20 at 750–850 °C, 500 MPa for fluid–rhyolitic melt. All experiments on B-isotopic fractionation between melt and aqueous fluid (Δ11Bmelt-fluid) showed significant preferential partitioning of 11B into the hydrous fluid over the silicate melts. For the basaltic system, Δ11Bmelt-fluid values were determined to be: −5.2(± 1.6)‰ (at 950 °C, 140 MPa), −3.6(± 2.0)‰ (at 1080 °C, 110 MPa), −1.3(± 3.4)‰ (at 1100 °C, 170 MPa); and for the rhyolitic system: −7.1(± 2.0)‰ (at 750 °C, 500 MPa) and −4.4(± 2.0)‰ (at 850 °C, 500 MPa). Extrapolating these Δ11Bmelt-fluid values to low temperatures, the extent of melt–fluid B-isotope fractionation corresponds to that known for the B-isotope fractionation between the 4- and 3-fold coordinated phases illite–fluid (Williams et al. 2001) and mica–fluid, respectively (Wunder et al. 2005, see also later Fig. 3.10). Therefore, Hervig et al. (2002) concluded that the large Δ11Bmelt-fluid results from significant differences in B coordination between the two coexisting phases, i.e., that B mostly exists as tetrahedral [BO4] in the melts and as trigonal B(OH)3 species in the fluids. However, this is in contradiction to the accepted understanding described above (Sect. 3.4.3), in which [BO3] is the predominant B species in melts, particularly in peraluminous rhyolitic compositions, which have at most 26% of the B at tetrahedral sites, as determined for rhyolitic glasses (Tonarini et al. 2003); corresponding rhyolitic melts are expected to have an even lower proportion of [BO4]. Therefore, the rational of the strong B-isotopic melt–fluid fractionation at magmatic temperatures observed in the experimental study of Hervig et al. (2002) is not fully understood. Future experimental studies should be completed, for example, to investigate possible kinetically driven B-isotopic fractionation processes, which are known to be significant for lithium and its isotopes (Tomascak et al. 2016).
3.5.2 Solid–Fluid B-Isotope Fractionation
3.5.2.1 Low-Temperature Solid–Aqueous Fluid B-Isotope Fractionation
Diffusive reequilibration of solids with an isotopically distinct B-bearing aqueous fluid is the usual approach to study low-temperature solid–fluid B-isotope fractionation. In most of these exchange experiments, B is added to the solid in a combination of (1) weak adsorption to the mineral’s surface and to interlayer positions (in clay minerals) and/or (2) boron incorporation at distinct structural positions by coupled substitutions. The adsorption of weakly bonded B species strongly depends on fluid-pH and charge of the surface. It is therefore essential to characterize the reaction mechanism during adsorption of dissolved boron, for understanding B-isotope fractionation in low-T regimes.
3.5.2.1.1 Carbonate–Fluid B-Isotope Fractionation
In many studies, the low-temperature B-isotope fractionation in the system carbonate–fluid has been attributed to the exclusive adsorption of the charged borate ion [B(OH)4]− (e.g., Hemming et al. 1995; Sanyal et al. 2000). Some more recent results suggest more complex schemes of adsorption (Tossel 2006; Klochko et al. 2009): after surface adsorption of B(OH)3 and/or [B(OH)4]− (stage 1), reaction of the two B species with HCO3 − proceeds in an intermediate stage forming an amorphous carbonate surface layer (stage 2); structural incorporation of trigonal and/or tetrahedral species is completed in the final stage 3.
Boron is reported as a typical trace element in natural carbonates, with concentrations in the order of ~1 to ~100 μg/g (e.g., Furst et al. 1976; Vengosh et al. 1989; Hemming and Hanson 1992; Branson 2017; Marschall 2017, this book). Low-temperature experiments indicate that at ambient conditions and near neutral pH, B uptake by aragonite is greater by a factor of 3–5 compared to calcite; additionally, the presence of Mg in the parent solution results in an increased B incorporation into calcite compared to Mg-free solutions (Hemming et al. 1995). Experiments by Sanyal et al. (2000) yielded an increased incorporation of B into calcite with increasing pH, which was explained by greater [B(OH)4]− uptake, due to the increased [B(OH)4]−/B(OH)3−ratio in the more basic fluids. 11B MAS NMR investigations of Sen et al. (1994) show that in synthetic calcite boron occurs principally in trigonal coordination, whereas it is tetrahedrally coordinated in synthetic aragonite. For calcite it was assumed that [BO3]3− replaces [CO3]2− in a coupled substitution; on the other hand, the structural role of [BO4]5− in aragonite was ambiguous (Sen et al. 1994). These results are contrasted by NMR-investigations of Klochko et al. (2009), who observed mixtures of trigonally and tetrahedrally coordinated B in almost equal abundances in both natural biogenic calcite and aragonite. In a calcite-aragonite co-precipitation study under controlled pH (7.4–9.5), temperature (5–25 °C) and precipitation rate, Mavromatis et al. (2015) showed with 11B MAS NMR that B in aragonite is mostly (≥85%) tetrahedrally coordinated. In contrast, proportions of trigonally and tetrahedrally coordinated B are highly variable in calcite, with the amount of [4]B increasing with precipitation rate. These data indicate that adsorbed surface [B(OH)4]− mostly preserves its coordination upon structural incorporation into aragonite, but can undergo a structural coordination change during incorporation into calcite. Recent first-principles calculations (Belan et al. 2016) indicate substitution of [CO3]2− anions by [BO2(OH)]2− and [B(OH)4]− groups in calcite, and mainly by [B(OH)4]− species in aragonite. Synchrotron X-ray spectroscopic investigations clearly show that B is hosted solely as trigonal BO3 in foraminiferal calcite (Amphistegina lessonii) and that B-concentration exhibits banding on the micrometer length scale (Branson et al. 2015). Using Atomic Force Microscopy (AFM), Ruiz-Agudo et al. (2012) showed that calcite growth rates are site-specific and strongly depend on pH and B supersaturation within the fluid. For further details see the book chapters by Branson (2017), Rae (2017), and McCulloch (2017).
Hemming et al. (1995) were the first to experimentally investigate B-isotopic fractionation between carbonates and fluids during growth of synthetic carbonates (calcite, aragonite, (Ca,Mg)-carbonate) from solutions at ambient pressure–temperature conditions and a constant pH of approximately 8. The yielded B-isotopic carbonate–fluid fractionation (Δ11Bcarbonate-fluid = −16.5(± 0.7)‰) was indistinguishable among the three different carbonates, indicating no dependence of B-isotope fractionation on carbonate structure and chemistry. The determined Δ11Bcarbonate-fluid is in good agreement with B-isotope fractionation observed between modern marine carbonates and seawater with an average pH of 8.2 (Hemming and Hanson 1992). The B-isotopic composition of carbonate corresponded to that of the calculated B-isotopic composition of charged [B(OH)4]− species in the experimental fluid. Hence, the experimental data suggest adsorption of [B(OH)4]− to the crystal surface combined with preferential 10B fractionation as the dominant influence on B uptake in carbonates. Sanyal et al. (2000) experimentally investigated the dependence of the B-isotopic composition of calcite on the pH of seawater at ambient pressure and temperature conditions. For the pH-range of 7.9–8.6, which approximately covers modern and ancient seawater, B incorporation into calcite strongly increased with increasing pH (pH = 7.9, ~34 µg/g; pH = 8.3, ~48 µg/g; pH = 8.6, ~84 µg/g). The experimental results show a clear relationship between δ11B of precipitated calcite and the pH of seawater (pH = 7.9, δ11B = −20.3(± 0.3)‰; pH = 8.3, δ11B = −17.9(± 0.4)‰; pH = 8.6, δ11B = −14.8(± 0.6)‰). These data are consistent with former conclusions (Hemming et al. 1995), determining [B(OH)4]−as the dominant species adsorbed on the carbonate surface in contact with seawater. Compared to results from pH-dependent B-isotope exchange experiments by cultivation of foraminifera (Orbulina universa) in contact with seawater (Sanyal et al. 1996), the B-isotopic fractionation between inorganic calcite and seawater (Sanyal et al. 2000) is lower by approximately 1‰, which indicates the presence of a small biogenic effect on B-isotope carbonate–seawater fractionation. The extent of such a biogenic effect might vary for different biogenic carbonate species (Sanyal et al. 1996).
In summary, small changes in the acidity of the oceans as a result of changing atmospheric CO2 levels will significantly change the pH with respect to the proportions of B(OH)3 and [B(OH)4]− species dissolved in seawater. From the experimental studies (Hemming et al. 1995; Sanyal et al. 1996, 2000), it is obvious that the B-isotopic composition of carbonates in marine sediments is a powerful tool to reconstruct paleo-ocean pH and ultimately past concentrations of atmospheric CO2. However, data on B coordination in aragonite and calcite are partly conflicting, which in turn affects the interpretation of measured δ11B for natural carbonates. This calls for further detailed studies on both structural B incorporation and surface-related mechanisms of B adsorption to different carbonates (calcite, aragonite, carbonates of various solid solutions).
3.5.2.1.2 Clay Mineral–Fluid B-Isotope Fractionation
Palmer et al. (1987) proposed that incorporation of B in clay minerals (e.g., illite) is initialized by adsorption of trigonal B(OH)3 to its surface. In a second step, which includes B-isotope fractionation, the coordination of the B species changes from trigonal to pseudo-tetrahedral at the clay’s surface. During ongoing exchange, the adsorbed tetrahedral species (enriched in 10B) is then incorporated into the Al-silicate structure, without further isotope fractionation (Palmer et al. 1987). This concept of adsorption mechanism has been modified because more recent studies (e.g., Singh and Mattigod 1992; Goldberg et al. 1996; Williams et al. 2001; Środoň and McCarty 2008) show that adsorption is a function of the charge density in non-homogenous structures, such as phyllosilicates, and the temperature- and pH-dependent mineral’s zero point of charge (charge density on the surface is zero). Expandable clay minerals have different kind of charged surfaces: edge(+), exterior(−), basal(−), to which the weak B-adsorption of different aqueous B-species is strongly dependent on fluid pH. For example, at low pH (below 7), the hydroxyl-group of boric acid may absorb to the H+ on the negatively charged exterior and basal siloxane surfaces; above pH 7 the anionic species [B(OH)4]− should become prominent to adsorb to the positively charged clay’s edges.
In clastic sediments, B is hosted predominantly in the clay mineral illite, which can contain orders of magnitude more B (up to several hundred μg/g) than other common diagenetic minerals (e.g., smectites, carbonates). Boron in clays is located at three different structural positions: (1) at low temperatures <120 °C clay minerals adsorb B to their surface by weak bonding (see above); (2) in expanded smectite minerals boron might also be adsorbed at interlayer positions as both trigonal and tetrahedral species, perhaps reflecting the composition of the fluid (Williams et al. 2007); (3) in smectite and illite B substitutes for Si by a coupled substitution at structural tetrahedral positions within the layers of the sheet silicates (Spivack and Edmond 1987).
Schwarcz et al. (1969) were the first to study B-isotope clay–fluid fractionation by B adsorption on clays during interaction in contact with synthetic seawater at 23 °C. Fractionation factors α were calculated to be 0.957–0.963 for reversible and 0.970–0.974 for irreversible adsorption, respectively. Based on the interpretation of Schwarcz et al. (1969) that the real adsorption process is partly reversible, and the observation that adsorbed B on clays is isotopically light, the assumption of a pH control on the isotope composition of adsorbed B arose. The pH (pH-range: 6.65–8.45) and temperature (T-range: 5–40 °C) controls over B-isotopic fractionation during B adsorption from seawater on marine clay-bearing sediments were studied experimentally by Palmer et al. (1987). In the investigated T-range (at constant pH) the B-isotope fractionation was almost invariant; with increasing pH (at constant temperature) the B-isotope fractionation decreased as [B(OH)4]− becomes the dominant aqueous species (see below, Fig. 3.10). For the investigated conditions, the clay’s surface-adsorbed B is 23.4–34.3‰ lighter than the boron dissolved in the coexisting seawater (δ11B = + 39.61 ± 0.04‰; Foster et al. 2010). These values are much larger than the adsorption on carbonates at the same pH and temperature (Hemming et al. 1995).
According to Williams and Hervig (2005), the illitization of smectite represents a dissolution/recrystallization process. The clay mineral δ11B values, therefore, vary during crystal growth as the fluid B composition or temperature change (Clauer et al. 2014). Williams et al. (2001) investigated the B-isotopic change during illitization of smectite under conditions simulating burial digenesis. Experiments were performed at 300 and 350 °C at 100 MPa with smectite as a solid starting material and B-bearing fluid containing 1000 μg/g B, sealed in Au-tubes in a hydrothermal apparatus for different run-durations of up to 150 days. After the experiments, the solids were washed in water and mannitol solution (0.1 N) to facilitate removal of exterior surface-adsorbed B (Tonarini et al. 1997). Adsorbed interlayer B was removed by cation exchange (Moore and Reynolds 1997). Analyses of the B-isotopic composition of the solids were done by SIMS and negative Thermal Ionization Mass Spectrometry (n-TIMS); the B-isotopic composition of the fluids was not measured, but determined from mass-balance calculations. Equilibrium was determined by monitoring the oxygen isotopes of the mineral and fluid, as the equilibrium oxygen isotope fractionation of illite as a function of temperature is well known (Longstaffe 1987). Since B substitution requires breaking of Si–O–bonds, it follows that B is at equilibrium when O is at equilibrium with the fluid. Williams et al. (2001) interpreted their results on B isotope clay–fluid fractionation as follows: during early stages of smectite recrystallization, aqueous B may be adsorbed onto the clay’s exterior surfaces and some may be incorporated into the smecitite’s interlayer site where the dominant aqueous B species varies according to the layer charge distribution. As smectite recrystallizes to illite, interlayer collapse occurs during ordering (Altaner and Ylagen 1997). Eventually, B is substituted in tetrahedral sites of illite replacing Si (in competition with Al3+) in a coupled substitution. The B-isotopic illite–fluid fractionation of well-ordered illite resulted in a Δ11Billite-fluid = −13‰ (at 350 °C) and −16‰ (at 300 °C). The long-duration experiments represent fractionation between the trigonal aqueous B (quench pH of the fluid was 6.0, thus the fluid mainly contained B(OH)3 species) and structural tetrahedral B, and is a linear function of temperature. B-isotopic characterization of natural and synthetic clays with various degrees of smectite–illite ordering revealed that trigonal interlayer boron is isotopically heavier than tetrahedral boron by up to 40‰. Williams et al. (2007) observed that the intracrystalline B-isotope fractionation for clays (Δ11Btetrahedral-interlayer = δ11Btetrahedral−δ11Binterlayer) is strongly dependent on the degree of clay ordering and thus on the temperature (or reaction time) of recrystallization. Accordingly, Williams et al. (2007) proposed a single-mineral geothermometer approximated by the equation T (°C) = (Δ11Btetrahedral-interlayer + 30)/0.05, which however, only applies to samples in which the interlayer B species is in equilibrium with the silicate (closed system).
3.5.2.1.3 Boron-Isotope Fractionation in Other Low-Temperature Solid–Fluid Systems
Isotopic fractionation between the B(OH)3 and [B(OH)4]− species has been studied experimentally by Kakihana et al. (1977) at low temperatures by adsorption of B from boric acid solution to an ion-exchange resin. The results were used for the theoretical determination of reduced partition function ratios to calculate B-isotope fractionation between trigonal and tetrahedral sites. The calculated fractionation factor α to for Eq. (3.1) decreases from 1.0206 to 1.0177 for the temperature range 0 to 60 °C, with α (Kakihana et al. 1977). A larger fractionation factor α of 1.028 (± 0.001) at 25 °C was determined experimentally by Byrne et al. (2006) using spectrophotometric absorbance measurements of aqueous solutions, which is in good agreement with theoretical assessments (e.g., Zeebe 2005; Liu and Tossel 2005; Rustad et al. 2010a, see below).
Oi et al. (1997) found isotopic fractionation between trigonal and tetrahedral species to vary between 18 and 22‰ for different kinds of hydrocarbon-based ion-exchange resins. They concluded that smaller fractionation results from the adsorption of some trigonal B-species as well as the dominant tetrahedral species, with different resins adsorbing more or less trigonal boron. All the resins studied were hydrocarbon-based, and adsorption was assumed to occur via B(OH)2(OC)2 clusters (Oi et al. 1997).
Synthesis experiments on B-bearing minerals at the low temperature of 25 °C from aqueous boric acid–sodium hydroxide solutions at various pH conditions (Oi et al. 1991) resulted in the formation of sassolite (B(OH3) at low pH (5.4–5.9), and borax (Na2(B4O5(OH)4·3H2O) at neutral to high pH (7.4–11.5). B-isotope analyses of sassolite and water, both containing B in trigonal coordination, showed B isotopic fractionation (Δ11Bsassolite-fluid = + 4.3‰ at pH = 5.9 and +0.6‰ at pH = 5.5), with preferential fractionation of 11B into the solid phase. Analyses of borax, which contains B in both trigonal and tetrahedral coordination in equal amounts, showed significant pH-dependent fractionation (Δ11Bborax-fluid = −12.6 to +13.8‰) in the pH-range 7.0–11.5, with 11B fractionating into the fluid phase at pH ≤ 8.7 and 11B fractionating into borax at pH ≥ 10.3 (Oi et al. 1991).
Vengosh et al. (1992) studied B-isotope variations in brines and coprecipitated salt minerals (gypsum, halite) and potassium-magnesium sulfate minerals during evaporation of seawater at 25 °C. They found similar B-isotope fractionation data as for smectite–fluid (Palmer et al. 1987), which was interpreted as coprecipitation of [B(OH)4]− species with the salts during evaporation of seawater (see below, Table 3.1 and Fig. 3.10).
Strong pH-dependent B-isotope solid–fluid fractionation at 25 °C during surface adsorption was determined by Lemarchand et al. (2007) for goethite (α-FeOOH): Δ11Bgoethite-fluid = −40 to 0‰ in the pH-range ~8–10 and birnessite (K0.1MnO2.2·0.9H2O): Δ11Bbirnesite-fluid = −15 to +23‰ in the pH-range ~8–10.8. Maximum boron adsorption on both of the hydroxide/oxide surfaces occurred at 8 < pH < 9. Adsorption and the subsequent B-isotope fractionation were rationalized by the formation of trigonal and tetrahedral boron inner-sphere complexes on the goethite surface, and tetrahedral inner-sphere and trigonal outer and inner-sphere complexes on the birnessite surface (Lemarchand et al. 2007).
The strong influence of charge properties at a mineral’s surface on B-isotope solid–fluid fractionation was experimentally shown for various (hydr)oxide–fluid systems by Elmaci et al. (2015). For the manganese oxides birnesite (K0.3MnO2·1.8H2O) and cryptomelane (K0.1MnO2·0.5H2O) both possessing negatively charged surfaces, [10B(OH)4]− is preferentially fractionated into the solution (pH = 8.5). For [Mg0.58Al0.42(OH)2](NO3)0.42·0.6H2O, a Mg–Al–layered double hydroxide, selective [10B(OH)4]− adsorption was favored by the positively charged layers of the solid at a fluid-pH of 9.5–10.
3.5.2.2 High-Temperature Solid–Aqueous Fluid B-Isotope Fractionation
Some solid phases, e.g., tourmaline, are known to exhibit very low diffusivities. Due to this refractory nature, low-temperature diffusive reequilibrium experiments are generally precluded. In the high-temperature B-isotope fractionation experiments for mica–fluid and tourmaline–fluid (both discussed below), the solids were synthesized in situ from their components in the presence of a coexisting B-bearing fluid. During this procedure, B is incorporated structurally as a main component at distinct crystallographic positions. In the following, boron adsorbed at the crystal surface is neglected, as its volume concentration is dominating over the amount of boron at the low surface area for solids synthesized at high temperatures (≥350 °C).
3.5.2.2.1 Mica–Fluid B-Isotope Fractionation
White mica is ubiquitous in metamorphic rocks of both igneous and sedimentary origin, and is stable to depths of 300 km in cold subducting slabs where, with progressive subduction, its modal amount continuously decreases and its composition shifts towards phengitic mica (Schmidt 1996). White mica is an important carrier of fluid-mobile trace elements including B, Li, and large ion lithophile elements. Because of its high modal abundance, white mica dominates the B-budget in most tourmaline-free rocks and its B-isotopic composition can be considered as representative of the bulk rock (Peacock and Hervig 1999). Measuring the B-concentration in subduction zone minerals (excluding tourmaline and other boron minerals), white mica contains the highest amount, with up to 269 μg/g, followed by lawsonite, biotite, chlorite, and amphibole all having B concentrations below 100 μg/g (Domanik et al. 1993; Marschall et al. 2006). The maximum B-incorporation capacity in white mica is given for boromuscovite, with end member composition KAl2[BSi3O10](OH)2, containing approximately 9 wt% B2O3. Boromuscovite was chosen as representative of B-bearing white mica in the experiments on B isotope mica–fluid fractionation performed by Wunder et al. (2005), which was determined using three different approaches: (a) synthesis of boromuscovite from gels with boric acid solutions (weakly acidic conditions with B(OH)3 as the major B-bearing species in the fluid); (b) synthesis of boromuscovite from an oxide mixture and a 7.6 M KOH-solution (strongly basic conditions with [B(OH)4]−species in the fluid); (c) time-dependent exchange experiments lasting up to 28 days with B(OH)3-bearing aqueous solutions, whose initial isotopic composition was far off the expected equilibrium value using the previously synthesized boromuscovite with known isotopic composition (Fig. 3.3a, b). Experiments using approaches (a) and (c) were performed in a piston cylinder press at 3.0 GPa, 500 °C and at 3.0 GPa, 700 °C; experiments using approach (b) were done in a hydrothermal apparatus at 0.4 GPa, 400 °C and at 0.4 GPa, 500 °C. The solids were characterized by powder X-ray diffraction followed by Rietveld refinement and by transmission electron microscopy (TEM) using electron energy-loss spectroscopy (EELS) for B/K-ratio determination. Both the solid run products and coexisting fluids were characterized by p-TIMS for their B-isotopic composition. All experiments produced only boromuscovite with traces of quartz in some of the experiments. The boromuscovite consisted mainly of the 2M1 polytype (approximately 90%) and approximately 10% of the 1 M polytype (see Chap. 6.3.2 below, for the small effect that different mica-polytypes have on B-isotope fractionation). The synthesis approach (a) and the time-dependent experiments (c) resulted in approximately the same fractionations (Fig. 3.3a, b): Δ11Bmica-fluid = −10.9 ± 1.3‰ (at 3.0 GPa, 500 °C) and −6.5 ± 0.4‰ (at 3.0 GPa, 700 °C). Experiments at 0.4 GPa that used the strongly basic fluid produced significantly less fractionation Δ11Bmica-fluid = −7.4 ± 1.0‰ (at 400 °C) and −4.8 ± 1.0‰ (at 500 °C), reflecting the reduced fractionation effect, when large amounts of boron in basic fluids are tetrahedrally coordinated. From these results, Wunder et al. (2005) proposed that the B-isotopic signature in volcanic arcs might result from continuous dehydration of micas, acting as important B carrier during subduction.

a, b Experimental data on mica—fluid B-isotope fractionation slightly modified after Wunder et al. (2005) for experiments performed at 500 and 700 °C at weakly acidic conditions. Black circles show isotopic composition of exchange experiments at their starting conditions; orange circles denote the measured Δ11Bmica-fluid for time-dependent experiments. Results from synthesis experiments (in dark blue) fit within error; c the thermal structure of the Kamchatka subduction zone used in the thermodynamic model; d the distribution of the B-isotopic fractionation values in the subducting plate (modified after Konrad-Schmolke and Halama 2014) using fractionation data after Wunder et al. (2005)
Marschall et al. (2007) modeled the release of B in a progressively dehydrating altered oceanic crust during subduction. The derived information on the B-concentration of rocks and fluids at different stages of the P-T-path of the subducting slab was combined with T-dependent B-isotope fractionation data (Wunder et al. 2005). Assuming low-pH fluids (i.e., trigonally coordinated B in the fluid), this resulted in a model of the B-isotopic evolution of subducting rocks and the released fluids from greenschist to eclogite conditions (Marschall et al. 2007). Konrad-Schmolke and Halama (2014) extended this model by combining thermodynamic modeling of phase relations at subduction conditions for the Kamchatkan subduction zone (Fig. 3.3c), with T-dependent mica–fluid B-isotope fractionation (Wunder et al. 2005), in order to quantify the B concentration and B-isotopic composition of coexisting fluid and solid phases during devolatilization, fluid migration and fluid-rock interaction in a subducting slab (Fig. 3.3d). Their derived data provides insights into element cycles, and their model can be used to constrain metamorphic processes and determine thermo-mechanical parameters for subduction zone models (Konrad-Schmolke and Halama 2014). For more details on this topic see De Hoog and Savov (2017).
3.5.2.2.2 Tourmaline—Fluid B-Isotope Fractionation
Tourmaline forms in various geochemical environments that have undergone diagenetic, metamorphic, metasomatic, or magmatic processes over a wide range of bulk compositions and has a large P–T-stability field ranging from near-surface to mantle conditions (e.g., Dutrow and Henry 2011; van Hinsberg et al. 2011). Negligible diffusion of elements through tourmaline’s structure results in the preservation of growth and sector zoning (van Hinsberg and Schumacher 2007). Its refractory behavior has allowed tourmaline to be successfully used for geothermometry (e.g., van Hinsberg and Schumacher 2007), geobarometry (Berryman et al. 2015), for provenance studies (e.g., Dutrow and Henry 2011), as a monitor for fluid-rock interactions in metamorphic rocks (e.g., von Goerne et al. 2001), as well as in tourmaline-bearing fluid–melt–rock systems (e.g., Trumbull and Chaussidon 1999). Tourmaline’s B-isotope composition, which can also be affected by growth zoning (van Hinsberg and Marschall 2007), extends its applicability to include its use as a geochemical tracer for geological mass transfer, fluid origin and evolution, and even for evaluating the source and genesis of ore deposits (Marschall and Jiang 2011; Slack and Trumbull 2011).
Tourmaline’s compositional variability is due to its complex crystal chemical structure [9]X[6]Y [6]3 Z6([4]T6O18)([3]BO3)3(O,OH)3(O,OH,F). B in tourmaline is situated at two different structural positions: trigonal-planar [3]B and tetrahedral [4]T-site. At the latter, B is incorporated via exchange with Si by various coupled substitutions (Kutzschbach et al. 2016a). In most tourmalines, B is exclusively three-fold coordinated; however, some natural [4]B-bearing tourmalines with up to 1.23 [4]B per formula unit (pfu) have also been described (e.g., Kalt et al. 2001; Hughes et al. 2004; Marschall et al. 2004; Ertl et al. 2007), mostly from high-pressure environments and for Al-rich stoichiometries. According to Ertl et al. (2008), it is possible to have a maximum of 3 [4]B pfu at the T-site of tourmaline; they also concluded that the amount of [4]B increases with decreasing temperature and increasing pressure. Kutzschbach et al. (2016a) achieved the incorporation of up to 2.53 ± 0.25 pfu [4]B into olenitic tourmaline synthesized at 4.0 GPa, 700 °C, which is the highest B concentration ever detected in either synthetic or natural tourmaline.
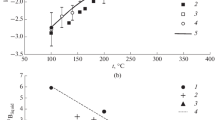
Two experimental studies on B-isotope fractionation between tourmaline and aqueous fluid exist (Palmer et al. 1992; Meyer et al. 2008). Both report an enrichment of 11B in the fluid phase under all conditions. Palmer et al. (1992) determined the fractionation of B isotopes between dravitic tourmaline and fluid in hydrothermal experiments at 350–750 °C and 50–200 MPa. The resulting Δ11Btour-fluid, [−5.88 to −8.35‰ at 50 MPa, 450–350 °C; −2.34 to −7.76‰ at 100 MPa, 750–350 °C; −2.24 to −6.50‰ at 200 MPa, 750–350 °C] display a decrease in fractionation with increasing temperature and pressure (Fig. 3.4a). Meyer et al. (2008) determined the fractionation of B isotopes between dravitic tourmaline and fluid in hydrothermal experiments between 400 and 700 °C at 200 MPa and at 500 °C, 500 MPa. Tourmaline was crystallized from a mix of oxides and boric acid in the presence of water that contained B in excess. In one series of experiments, the fluid to tourmaline B-concentration ratio C fluidB /C tourB was 9 after the run; in another series it was 0.1 to demonstrate the effect that fractional crystallization has on the B isotope tourmaline–fluid distribution (see below). All experiments produced tourmaline as the only B-bearing solid, along with traces of quartz and talc whose B contents are negligible. Solid samples were characterized by electron microprobe (EMP) and by X-ray diffraction with Rietveld refinement; the isotopic composition of both solids and fluids, were determined by p-TIMS. Rietveld refinements on tourmaline structure revealed no significant amounts of tetrahedrally coordinated B (Meyer et al. 2008). For experiments where C fluidB /C tourB was 9, a consistent T-dependent B-isotope fractionation, valid from 400 to 700 °C, is approximated by Δ11Btour-fluid = −4.20 · (1000/T[K]) + 3.52; R2 = 0.77 (Fig. 3.4a). In contradiction to Palmer et al. (1992), no pressure dependence was observed and the Δ11Btour-fluid values of Meyer et al. (2008) [−2.7 ± 0.5‰ at 400 °C; −0.8 ± 0.5‰ at 700 °C] are lower by approximately 2 to 4‰ [−4.1‰ at 450 °C, 200 MPa; −2.6 ± 0.3‰ at 750 °C, 200 MPa, see Fig. 3.4a]. Experiments where C fluidB /C tourB was 0.1 showed a significantly larger apparent fractionation of up to −4.7‰ at 600 °C (Fig. 3.4a). In one of these runs, the isotopic composition of handpicked tourmaline crystals of different sizes varied by 1.3‰. This was interpreted as a result of fractional crystallization of B isotopes during tourmaline growth due to the small B reservoir of the fluid relative to tourmaline. The effect is eliminated or minimized in experiments with high amounts of excess B in the fluid. It is, therefore, suggested that values given by the above relationship using a high-B-excess reservoir in the fluid represent true equilibrium fractionation. Marschall et al. (2009) confirmed this interpretation by B-isotope SIMS-measurements of the same synthesized tourmalines. Tourmaline from an experiment with low excess B had significant B-isotope zonation with 11B/10B increasing in the growth direction of the crystals (Fig. 3.4b, c). Trend, magnitude, and absolute values strongly support results from the high-B-excess isotope fractionation experiments. Furthermore, the closed system B-isotopic evolution of the experimental fluid, modeled by Rayleigh fractionation, is in excellent agreement with the measured B-isotope composition of the run-product fluid (Fig. 3.4d). In the experiments of Palmer et al. (1992) only small amounts of B excess (~10%) were present in the fluid; thus, one may hypothesize that fractional crystallization also occurred in their experiments, creating stronger fractionation just as in the low-excess B experiments of Meyer et al. (2008). On the basis of the adsorption mechanism presented for clays (Palmer et al. 1987, see above), Palmer et al. (1992) suggested a modified version aimed at explaining the slight B-isotope fractionation and its P dependence for the system tourmaline–aqueous fluid, where both phases have exclusively trigonal [3]B. In contrast to clay minerals, in this scheme no coordination-change from trigonal to tetrahedral species occurs after surface adsorption of the trigonal-B species. Instead, the coordination changes to a pseudo-trigonal coordinated adsorption species at tourmaline’s surface; this mechanism should be favored at higher pressure. Palmer et al. (1992) also envisaged kinetic effects as a possible reason for the P dependence of fractionation, leaving the observed effect not fully constrained. Moreover, Palmer et al. (1992) detected a small amount of tetrahedrally-coordinated B in their tourmalines by MAS-NMR. However, only significant amounts of tetrahedral B in tourmaline would cause a clear increase in Δ11Btour-fluid (Kowalski et al. 2013) and the small observed [4]B concentrations cannot account for the variation of approximately 1–2‰ between low and high P experiments (Palmer et al. 1992). For synthetic olenitic tourmaline formed at 700 °C, 4.0 GPa with significant amounts of [4]B, preliminary data by Kutzschbach et al. (2016b) reveal an intracrystalline B-isotope fractionation with Δ11B [3]( [4]B− B) = δ11B[3] − δ11B[4] of +8.4‰, which is in agreement with computationally determined values of B-isotope fractionation for [4]B-bearing tourmaline–fluid (Kowalski et al. 2013; see below).

a Experimental data on tourmaline–fluid B-isotope fractionation after Palmer et al. (1992) at various pressures (red) and after Meyer et al. (2008) for two different fluid/tourmaline B-concentration ratios (C fluidB /C tourB = 9, in blue; C fluidB /C tourB = 0.1, in violet). Note that the apparent fractionation from the small B-excess experiments are in the range of those proposed by Palmer et al. (1992) or even larger. b Reflected light microscope image of tourmaline from a C fluidB /C tourB = 0.1 experiment (#CM2, 600 °C) of Meyer et al. (2008). SIMS spots are visible; numbers refer to measured δ11B values. c B-isotope profile along the growth direction (root to tip) determined from SIMS analyses of various tourmaline crystals of #CM2. Arrows show fractionation (according to Meyer et al. 2008) between the starting material and early-stage tourmaline in the root (No. 1), and final fluid and late-stage grown tourmaline at the tip (No. 2), respectively. d Evolution of B-isotopic composition of fluid and tourmaline modeled by Rayleigh fractionation for conditions of experiment #CM2. Note that the theoretical δ11B value of −10.14‰ calculated from the T-dependent fractionation value of Meyer et al. (2008) is in very good agreement with the value of −10.0 ± 0.3‰ measured after the experimental run. b–d are slightly modified after Marschall et al. (2009)
Furthermore, according to Kowalski et al. (2013), tourmaline–fluid B-isotope fractionation might vary for different tourmaline members due to differences in the B–O distances of the trigonal planar groups of different tourmaline (see later Fig. 3.7a).
3.6 Ab Intio Prediction of B-Isotope Fractionation
With the tremendous increase in the availability of computational power that rises by at least a factor of 100 per decade (www.top500.org), the ab initio methods of quantum chemistry, namely density functional theory (DFT), become very popular investigation tools in various research fields, including isotope geochemistry (e.g., Schauble 2004; Kowalski et al. 2013; Jahn and Kowalski 2014). Therefore, reliable ab initio computational methods have been established recently and were successfully applied to predict the isotope fractionation factors among various crystalline solids (Driesner 1997; Yamaji et al. 2001; Schauble 2004; Domagal-Goldman and Kubicki 2008; Hill and Schauble 2008; Méheut et al. 2007, 2009; Schauble et al. 2009; Zeebe 2009, 2010; Hill et al. 2010; Rustad et al. 2010a, b; Kowalski and Jahn 2011; Kowalski et al. 2013; Feng et al. 2014; Méheut and Schauble 2014) and recently also between solids and aqueous solutions (e.g., Zeebe 2005; Otake et al. 2008; Domagal-Goldman and Kubicki 2008; Hill and Schauble 2008; Rustad et al. 2008; Zeebe 2009, 2010; Hill et al. 2010; Rustad et al. 2010a, b; Kowalski and Jahn 2011; Markland and Berne 2012; Kowalski et al. 2013; Fujii et al. 2015; Pinilla et al. 2015; Dupuis et al. 2015). Many of these studies have shown that such calculations are essential for our understanding of the geochemical mechanisms responsible for emerged isotope fractionation and signatures from experiments and nature.
The majority of the ab initio studies focus on the computation of stable isotope fractionation between simple crystalline phases and aqueous solution represented by ion-aqua complexes, whereas calculations of continuous fluids are less abundant. The aqueous solution species are usually represented by an isolated cluster containing the fractionating species and a hydration shell (e.g., Zeebe 2005; Domagal-Goldman and Kubicki 2008; Hill and Schauble 2008; Zeebe 2009, 2010; Hill et al. 2010; Rustad et al. 2010a; b). However, it is known that the distribution of cation coordination and cation–oxygen bond lengths can significantly influence the fractionation of isotopes (Bigeleisen and Mayer 1947). These structural properties of materials are determined by the dynamics of the considered system or its change under compression (Jahn and Wunder 2009; Wunder et al. 2011; Kowalski and Jahn 2011) and can have strong impact on isotope exchange. For instance, considering fractionation of Li isotopes, Kowalski and Jahn (2011) have demonstrated that a cation-hydration shell cluster is not an appropriate representation of aqueous solutions at high pressure (P > 2 GPa). Only a few published ab initio studies account for the dynamical effects on the isotope fractionation in fluids (Rustad and Bylaska 2007; Kowalski and Jahn 2011; Kowalski et al. 2013). Rustad and Bylaska (2007) considered boron equilibrium isotope fractionation between B(OH)3 and [B(OH)4]− species in aqueous solution. They performed ab initio molecular dynamic simulations of these fluids and calculated the 11B/10B isotope fractionation coefficient from the vibrational density of states obtained through the Fourier transform of the velocity auto-correlation function. Although the obtained fractionation factor of 0.86 was substantially different from the experimental value of 1.028, the experiment and theory were reconciled by computation of the harmonic frequencies on a set of the quenched ionic configurations extracted from the molecular dynamics trajectory. Using a similar approach together with the “single atom approximation” of Bigeleisen and Mayer (1947), in which the isotope fractionation factor was derived from the force constants acting on Li cation, Kowalski and Jahn (2011) computed the high temperature Li isotope fractionation factors between complex minerals (spodumene, staurolite and mica) and aqueous fluid. It is worth noticing that because these simulations were performed using periodic boundary conditions, the computed solids and fluids were treated as continuous media, which allowed for correct accounting for the pressure effects.
With the steadily increasing access to the computing power it becomes possible to apply other methods of quantum chemistry to derive the isotope fractionation factors. For instance, Marland and Berne (2012), Pinila et al. (2014) and Dupuis et al. (2015) used path integral molecular dynamics technique to investigate the fractionation of H and O isotopes in water. We thus expect that further similar studies will be performed in the next years.
3.6.1 Computational Approach
3.6.1.1 The “Single Atom Approximation” Following Bigeleisen and Mayer (1947)
Following the pioneering study of Urey (1947) it is well accepted that the mass-dependent equilibrium fractionation of isotopes is driven by the change in molecular and crystalline vibration frequencies resulting from the different masses of isotopes. The principal parameter which describes the equilibrium isotope fractionation is the so-called isotopic reduced partition function ratio, known also as β factor, which describes the fractionation of isotopes between given species and an ideal monoatomic gas. In the harmonic approximation it is given by the following formula (Bigeleisen and Mayer 1947; Urey 1947; Chacko et al. 2001):
where u i = hν i /k B T, h is the Planck constant, k B is the Boltzmann constant, ν i is the vibrational frequency of the ith degree of freedom, N dof is the number of degrees of freedom and the asterisk marks the heavier isotope. For N being the number of atoms in the considered system N dof is equal to 3 N-5 for diatomic molecule, 3 N-6 for multiatomic molecules and 3 N for crystals. The fractionation factor between two substances A and B, α A-B is computed as the ratio of the relevant β factors, which is well approximated by the differences in the β factors:
The calculation of the β factor requires thus only knowledge of the vibrational frequencies that can be measured or computed ab initio (Jahn and Kowalski 2014). The only problem on the computational site is that the calculation of the whole vibrational spectra of complex minerals or fluids requires substantial computational resources and, therefore, is currently limited to systems containing no more than a few dozens of atoms. Many of the current studies thus focus on computation of simple crystalline solids and ion-aqua complexes, accounting for the closest hydration shells only.
Kowalski and Jahn (2011) noticed that by computing only the force constants acting upon the fractionating element, by using for instance the “single atom approximation” of Bigeleisen and Mayer (1947), instead of the full phonon spectrum required by Eq. (3.10), one can dramatically reduce the computational demand by a factor of ~N, where N is the number of the atoms constituting the computed system (supercell). Kowalski and Jahn (2011) proposed such a method for an efficient computing of the high temperature isotope fractionation factors between complex materials such as fluids and crystalline solids. The β factor (Eq. 3.12) can be then approximated as:
where A i are the three force constants acting on the isotopic atom and Δm = m * −m, where m and m* are the masses of the lighter and heavier isotopes. This approach refers to the “single atom approximation” (Bigeleisen and Mayer 1947), because the computation of the β factors using Eq. (3.12) only requires the knowledge of properties of the fractionating element. However, the validity criteria restrict the usage of Eq. (3.12) to frequencies ν [cm−1] smaller than 1.39 T [K] (Kowalski et al. 2013). Because of this difficulty, Kowalski et al. (2013) introduced a modification of the “single atom approximation” showing that the error of the computed fractionation factors can be substantially reduced if one uses the three frequencies ν i derived from the force constants acting on the fractionating element ν i = A i /4π2m—called pseudofrequencies—and combined this with Eq. (3.10) for the calculations. Kowalski et al. (2013) provided a theoretical justification for this method, and tested and applied it successfully to B isotope systems, including cases of tourmaline (dravite, olenite), mica, grandidierite, serendibite, and aqueous solutions, which will be discussed in the next sections.
3.6.1.2 Computational Technique
The calculations reported here were performed by Kowalski et al. (2013), MacGregor et al. (2013) and Belley et al. (2014). These studies are complemented by the discussion of the ab initio data of Zeebe (2005) and Rustadt and Bylaska (2007). The calculations of pseudofrequencies and β factors for solids and aqueous solutions were performed following the procedure outlined in Kowalski et al. (2013) by applying density functional theory (DFT), which is currently the most efficient method allowing for quantum-mechanical treatment of extended many-particle systems. The calculations were performed with the planewave DFT code CPMD (Marx and Hutter 2000), which is especially suited for ab initio molecular dynamics simulations of fluids, and with the BLYP exchange-correlation functional (Lee et al. 1988). The norm-conserving Goedecker pseudopotentials were applied to mimic the presence of the core electrons (Goedecker et al. 1996). One advantage of using the BLYP functional is that it usually gives harmonic frequencies that most closely resemble the observed frequencies (e.g., Finley and Stephens 1995; Alecu et al. 2010). The energy cut-off for the plane wave basis set was set to 70 Ryd for geometry relaxations and molecular dynamics simulations and to 140 Ryd for static computation of the force constants and the vibrational pseudofrequencies. The periodic boundary conditions were applied for crystalline solids as well as aqueous solutions in order to preserve the continuity of the media. The force constants and resulting pseudofrequencies were computed using the finite displacement scheme. In case of the crystal structures these calculations were performed with all atomic positions relaxed to the equilibrium positions. The calculations are restricted to a single phonon wave vector using large supercells, thus avoiding an expensive calculation of full phonon dispersion. Due to the restrictions of CPMD code and the applied “single atom approximation”, the calculations were performed at Γ-point. Blanchard et al. (2009) have shown that selection of a representative Γ-point, such as the mean value Γ-point (Baldereschi 1973), could improve the accuracy of the calculations. This procedure has been validated at high temperatures (> 400 °C) for Mg-bearing materials (Schauble 2011) and Fe-bearing minerals (Blanchard et al. 2009).
The computation of the force constants and frequencies in case of the aqueous species were performed after relaxing the positions of all the atoms constituting the B-carrying molecule (B(OH)3 or [B(OH)4]−) to the equilibrium positions, but keeping all other atomic positions fixed. The 10 ps long molecular dynamics trajectories of systems consisting of 64 H2O molecules and one B(OH)3 or [B(OH)4]− molecule were obtained for different temperatures of 1000, 800 and 600 K and a pressure of 0.5 GPa. These parameters closely resemble the experimental conditions of Wunder et al. (2005) and Meyer et al. (2008). The ab initio molecular dynamics simulations were performed for fixed temperature and volume using the Car–Parrinello scheme (Car and Parrinello 1985) and the pressure of aqueous solution was calculated according to the equation of state of Wagner and Pruss (2002). The temperature during each run was controlled by a Nosé–Hoover chain thermostat (Nosé and Klein 1983; Hoover 1985). The β factors were computed on the snapshots extracted uniformly along the molecular dynamics trajectories using an interval of 0.1 ps.
3.6.1.3 Error Estimation Technique
An important aspect of reliable predictions is an estimation of error of the computed fractionation factors, which unfortunately is often omitted in many modeling studies. As shown by Kowalski and Jahn (2011) and Kowalski et al. (2013) the errors in the computed value of the β and Δ fractionation factors can be estimated from an average error of vibrational frequencies computed using the chosen DFT method. For instance, Finley and Stephens (1995), Menconi and Tozer (2002) and Alecu et al. (2010) estimated the errors originating from using different DFT functionals in the calculations of vibrational frequencies of small molecules. According to these studies, the BLYP functional systematically overestimates the harmonic frequencies by 3.5% with a deviation from the mean offset of 1%. Therefore, we expect that using BLYP functional the β and Δ values are systematically overestimated by 7% and that in addition there is a 2% error in the derived β factors. All the values presented here are thus provided together with errors estimated in this way. Similar errors result from using other functionals or even more sophisticated and computationally demanding post-Hartree–Fock methods such as MP2 (Finley and Stephens 1995; Alecu et al. 2010).
3.6.2 Computation of Fractionation Factors
3.6.2.1 Vapor
Kowalski et al. (2013) compared their results for the gas phase B(OH)3 and [B(OH)4]− molecules with the published data of Zeebe (2005), where the same DFT BLYP functional was used. The computed vibrational frequencies that are affected by the different B isotope substitutions are in good agreement with earlier theoretical predictions and show similar agreement with the experimental measurements. Figure 3.5a reports the resulting β factors, together with the results of Zeebe (2005). The β factors are smaller by approximately 1‰ at 600–1000 K than the values of Zeebe (2005), although the difference between β factors of B(OH)3 and [B(OH)4]− remains nearly identical in both sets of calculations, which points to a systematic error. The discrepancy between the two sets of calculations is substantially reduced when the results obtained with more complete basis set 6-311 + G(d,p) are used (Zeebe 2005). This shows that the here utilized plane-wave based DFT approach provides adequate vibrational frequencies and the resulting isotope fractionation factors are reliable.

a Gas phase β factors for B(OH)3 (solid blue line) and [B(OH)4]− (dashed red lines); b gas phase B(OH)3–[B(OH)4]− fractionation factors. The thick lines represent calculations of Kowalski et al. (2013), and the thin lines represent the results derived from data computed by Zeebe (2005); c computed aqueous solution β factors for B(OH)3 (solid blue lines) and [B(OH)4]− (dashed red lines); d aqueous solution B(OH)3–[B(OH)4]− fractionation factors. The thick lines represent the results computed by Kowalski et al. (2013). The thin lines show the results of Sanchez-Valle et al. (2005) obtained using harmonic frequencies. The dotted line represents the corrected Sanchez-Valle et al. (2005) results. The correction is made according to Rustad et al. (2010a). Figure modified after Kowalski et al. (2013)
3.6.2.2 Aqueous Fluid
Computed β factors for B(OH)3 and [B(OH)4]− in aqueous solutions according to Kowalski et al. (2013) are shown in Fig. 3.5c. The presented functions, β = 1 + 2.416 104/T 2−5.823 108/T 4 for B(OH)3 and β = 1 + 1.772 104/T 2−4.234 108/T 4 for [B(OH)4]− were fitted to the computed ab initio values. According to the assessment of uncertainties of the computational method (such as DFT exchange-correlation functional) and the “single atom approximation” (Kowalski et al. 2013), the errors of the computed values are T dependent with larger uncertainties at lower temperatures: 27% (at T = 300 K), 16% (at T = 600 K), 11% (at T = 1000 K). The high uncertainties of computed β factors at ambient conditions are also shown by Rustad et al. (2010a, their Fig. 3.2) from the spread of fractionation factors calculated using various computational methods.
The β factor of B(OH)3-bearing fluid is substantially larger than the one for the [B(OH)4]− and not significantly different from the results obtained for the isolated molecule (Fig. 3.5a). This reflects the substantial difference in the B–O bond lengths exhibited by the two considered B species. For the isolated molecules Kowalski et al. (2013) determined the B–O bond length of 1.40 Å and 1.51 Å for B(OH)3 and [B(OH)4]−, respectively. Compared to experimental data by Sanchez-Valle et al. (2005), for B(OH)3 both studies give nearly identical β factor; however, for [B(OH)4]− Kowalski et al. (2013) predicted a lower value. This discrepancy is caused by the incorrect assignment of a major vibrational mode of [B(OH)4]- in the force field by Sanchez-Valle et al. (2005) as discussed by Rustad and Bylaska (2007) and shown by Kowalski et al. (2013). The accordingly corrected results of Sanchez-Valle et al. (2005), which are in good agreement with the prediction of Kowalski et al. (2013), are plotted in Fig. 3.5d.
Liebscher et al. (2005) measured the fractionation of boron isotopes between liquid and vapor systems and found a positive Δ11Bliquid-vapour of +0.1 to +1.3‰ at 400 °C and of +0.2 to +1.5‰ at 450 °C (see Fig. 3.2). The calculations reported in Fig. 3.5 indicate Δ11Bliquid-vapour = + 1.0 ± 1.9‰ and +1.5 ± 2.2‰ for temperature of 400 and 450 °C, respectively. These values are thus well consistent with the experimental data.
3.6.2.3 Pressure Dependence of the Fractionation Factor in Aqueous Fluids
Kowalski and Jahn (2011) have shown that for P > 2 GPa the β factor of Li in aqueous solution increases with increasing pressure. Kowalski et al. (2013) observed the same for B(OH)3 and [B(OH)4]− in aqueous fluid, which is consistent with the experimentally observed variation in shift of the vibrational frequencies of different B species with increasing pressure (Sanchez-Valle et al. 2005; Schmidt et al. 2005). The computed β-1 values for B(OH)3 fluid show a linear dependence in pressure, β-1 = 23.6 + 0.28P (P in GPa). Such a relationship is expected as β-1∝ν 2 ~ ν 2 0 + 2ν 0 Δν (Schauble 2004) and Δν is a linear function of P (Sanchez-Valle et al. 2005; Schmidt et al. 2005). As illustrated in Fig. 3.6c, in case of [B(OH)4]−, the pressure-dependence is linear up to P ~ 2–3 GPa but it becomes less steep at higher pressures. Kowalski et al. (2013) have shown that these predictions are consistent with the measured P-dependent vibrational frequency shifts of Sanchez-Valle et al. (2005). Such a good agreement with the experimental data shows that ab initio calculations can be successfully used in derivation of the pressure dependence of the fractionation factors and the pressure-induced vibrational frequencies shifts. Moreover, first principles calculations can also be useful in extrapolation of the experimental values for β and Δν to more extreme conditions, which are difficult to be reached by experimental techniques.

a Computed pressure dependent β factor of neutral fluid (B(OH)3) at T = 1000 K. Dashed line represents the linear regression fit to the calculated values (squares); b computed change of β factor as a function of pressure (blue squares) compared to the results derived from the pressure-induced frequency shifts of the 666 and 1454 cm−1 vibrational modes measured by Sanchez-Valle et al. (2005) (dashed black line); c computed pressure dependent β factor of strongly basic fluid ([B(OH)4]−) at T = 1000 K. Dashed line is the regression fit to the calculated values (squares); d computed change of the β factor with pressure compared to the increase in the β factor derived from the frequency shift of the 975 cm−1 line measured by Sanchez-Valle et al. (2005) (black dashed line). Figure modified after Kowalski et al. (2013)
3.6.3 Computationally Determined Mineral–Fluid B-Isotope Fractionation
Kowalski et al. (2013), MacGregor et al. (2013) and Belley et al. (2014) used the computational approach outlined above to investigate the fractionation between boron-bearing fluids and minerals, such as dravite, olenite, boromuscovite, prismatine, grandidierite and serendibite. The aim of these studies was to investigate the mechanisms driving the fractionation process, including the role of coordination and the B–O bond length to better understand measured B-isotopic signatures. Below we discuss each case separately.
3.6.3.1 Tourmaline–Neutral Fluid B-Isotope Fractionation
Meyer et al. (2008) measured the boron isotope fractionation between tourmaline, represented by dravite, and pH-neutral fluid at T = 400–700 °C and P = 0.2 GPa (see Fig. 3.4a). The calculated fractionation curve and the experimental data of Meyer et al. (2008) are given in Fig. 3.7a. Calculations reproduce the measurements within the computational accuracy. According to Kowalski et al. (2013) the dravite–fluid fractionation is small because both, dravite and neutral fluid, contain boron in BO3 units. Kowalski et al. (2013) also predicted a small but positive fractionation between olenite and aqueous fluid. It is assumed that the calculated olenite–fluid fractionation differs from that of dravite–fluid due to shorter B–O bond lengths for olenite (1.378 Å) versus 1.397 Å for dravite (Kowalski et al. 2013).

a B-isotopic fractionation between tourmaline and aqueous fluid. The solid line represents the fractionation between dravite and B(OH)3 neutral fluid computed by Kowalski et al. (2013). The dashed line represents the prediction for the fractionation between olenite (without tetrahedral boron) and neutral fluid. The squares represent the experimental data measured for the dravite–fluid system by Meyer et al. (2008); b B-isotope fractionation factors between boromuscovite and basic aqueous fluid (fluid containing [B(OH)4]−). The lines represent the values calculated by Kowalski et al. (2013) assuming the presence of boron species in the form of [B(OH)4]− only (solid line) and admixture of 10% of B(OH)3 in the fluid (dashed line). The squares represent the experimental data (Wunder et al. 2005); c B-isotope fractionation factors between boromuscovite and aqueous fluid. The data points are the values measured by Wunder et al. (2005). The thick lines represent the fractionation factor computed assuming ambient pressure (solid line) and P = 3 GPa accounting for compression and thermal expansion (dashed line). The dotted lines represent the fractionation factors obtained assuming different admixture of [4]B species (represented by [B(OH)4]− with abundance indicated in the figure) to the fluid. The blue regions represent the computational uncertainty. Figure modified after Kowalski et al. (2013)
Beside trigonally-coordinated boron, tourmaline can incorporate boron also at the tetrahedral site replacing Si in a coupled substitution (e.g., Ertl et al. 1997). Calculations of intra-site B-isotope fractionation \( \left( {\Delta^{11} {\text{B}}_{{\left( {^{[3]} {\text{B}} -^{[4]} {\text{B}}} \right)}} } \right) \) between trigonal and tetrahedral boron sites in dravite resulted in +8.9 ± 1.7‰ (Kowalski et al. 2013) at 600 °C, which is in good agreement to experimental data (Kutzschbach et al. 2016b, see above).
3.6.3.2 Boromuscovite–Strongly Basic Fluid B-Isotope Fractionation
Wunder et al. (2005) measured the isotopic signatures of synthesized boromuscovite that consisted of polytypes, 1 M (~10%) and 2M1 (~90%). As a comparison with these data, Kowalski et al. (2013) derived β factors for both polytypes and used their weighted average for the analysis. Both polytypes of mica have similar mean B–O bond lengths [1.515 Å (1 M) and 1.525 Å (2M1)], and the calculations show that they are characterized by similar β factors with difference in (β−1) no larger than 3% (see Fig. 3.9). In order to investigate the impact of the B–O bond length on the fractionation, Kowalski et al. (2013) compared the fractionation between boromusovite, that generally has B in tetrahedral coordination, and a strongly basic fluid containing boron as [B(OH)4]−. The results of the calculations and the experimental data from the system boromuscovite–strongly basic fluid (Wunder et al. 2005) are shown in Fig. 3.7b. The calculations predict a negative fractionation between mica and the [B(OH)4]− fluid, in line with the experimental findings. This result seems reasonable, because the mean B–O bond length of mica is larger than that of aqueous [B(OH)4]− (1.513 Å).
Although the data from calculations and experiments agree within errors, the experimental data might indicate a slightly stronger fractionation. Kowalski et al. (2013) explained this discrepancy by admixture of aqueous [3]B species in the experiment. The presence of as little as 10% of B(OH)3 in the measured basic fluid brings predictions and experiments to good agreement (Fig. 3.7b). The experiments were performed with a “strongly basic” fluid (Wunder et al. 2005), yet, its exact composition, especially the amount of [3]B species at the conditions of the experiments, was unknown. Nevertheless, good agreement between the prediction and the experimental data exists, which indicates that the strongly basic fluid was dominated by [B(OH)4]− species, in line with previous studies (e.g., Zeebe 2005).
3.6.3.3 Boromuscovite–Neutral Fluid B-Isotope Fractionation
The strong B-isotopic fractionation between boromuscovite and neutral fluid results from different coordination of boron in boromuscovite ([4]B) and neutral fluid ([3]B). Comparison of data from experiments performed at 3.0 GPa (Wunder et al. 2005) with the predicted fractionation shows a difference of ~2.5‰ (Fig. 3.7c). The effect of difference in thermal expansion and compressibility between boromuscovite and fluid was shown to be not responsible for the discrepancy (Kowalski et al. 2013). However, the comparison of calculated results with the experimental data (Kowalski et al. 2013) suggests that the fractionation between boromuscovite and fluid is the same as between B(OH)3 and [B(OH)4]− fluids, which is at odds with the non-negligible and negative fractionation between boromuscovite and a strongly basic fluid (Fig. 3.7b). As the β factor derived for boromuscovite seems to be reliable, this points towards another, unaccounted effect that led to the decrease of the B-isotope fractionation between mica and neutral fluid in the high-pressure experiments of Wunder et al. (2005). One possible solution for the discrepancy is a non-negligible amount of [4]B in the neutral solution at 3.0 GPa. This is in line with the studies of Schmidt et al. (2005), who detected admixture of significant amounts of [4]B species in near-neutral, B(OH)3-dominated fluid at high pressures (see Fig. 3.1). Assuming a presence of 15–30% [4]B species in the fluid at 3.0 GPa and a β factor of these species similar to that of [B(OH)4]−, the fractionation factor between boromuscovite and B(OH)3 in the aqueous fluid decreases, bringing theory and experiment to much better agreement (Fig. 3.7c). If this interpretation is correct, it suggests that boron isotope fractionation could be used to gather information on the B-speciation in aqueous fluids.
3.6.4 B-Isotope Fractionation Among Minerals
The fractionation of B isotopes among solids has received considerable attention (e.g., Hervig et al. 2002; Marschall 2005; Klemme et al. 2011; MacGregor et al. 2013; Belley et al. 2014). From a theoretical point of view, Kowalski et al. (2013) investigated the mica–tourmaline system in which the two coexisting minerals have boron in different coordination, [3]B in tourmaline and [4]B in mica. This results in large B isotope fractionation between these two minerals, which has been investigated by analyzing natural coexisting pairs of tourmaline and mica (Klemme et al. 2011, and references herein). Comparison of experimental data with measurements from natural samples and the computed T-dependent fractionation curve are given in Fig. 3.8. The fractionation factors predicted by Kowalski et al. (2013) are consistent with the natural data (Klemme et al. 2011), which are both larger (taking the absolute values) than the experimental data (Wunder et al. 2005; Meyer et al. 2008). As discussed in Sect. 3.6.3, this discrepancy can be resolved by assuming that in the experiments on mica–fluid B-isotope fractionation (Wunder et al. 2005) the fluid contained a significant admixture of [4]B species. The experimental mica–tourmaline fractionation factor corrected for the presence of [4]B species in the high-pressure fluids results in a better consistency with the natural data (Fig. 3.8).

Boron isotope fractionation factors between mica and tourmaline. The solid line represents the computed fractionation factor between B-muscovite and dravite. The uncertainties are indicated by the blue area. The dashed red line is the experimental fractionation factor determined by combining the data of Wunder et al. (2005) and Meyer et al. (2008). The experimental error is ± 2‰. The dotted red line is the experimental fractionation factor corrected for the presence of [4]B species in the neutral fluid in the experiments of Wunder et al. (2005). The squares represent the data on natural samples (Klemme et al. 2011). Figure modified after Kowalski et al. (2013)
The mineral serendibite (ideally Ca4(Mg6Al6)O4[Si6B3Al3O36]) contains boron in tetrahedral coordination. The B-isotope difference of peak metamorphic serendibite and coexisting uvitic tourmaline in the Central Metasedimentary Belt in the Grenville Province of Quebec was determined to be −6.3‰ (Belley et al. 2014), which is consistent with the fractionation factor from ab initio calculations using the method of Kowalski et al. (2013).
Kowalski et al. (2013) have demonstrated that the β factor not only depends on boron coordination but is also strongly correlated with the B–O bond length. Phases with trigonal-coordinated B and short B–O bond lengths have higher β factors than [4]B-containing phases and long B–O bond lengths (Fig. 3.9). This observation was used to explain the measured 11B-enrichment of grandidierite (containing structural [3]B) and 11B-depletion of prismatine (containing structural [4]B) relative to tourmaline (MacGregor et al. 2013). At a temperature of 623 °C, the calculated fractionation factor between grandidierite (B–O bond length of 1.368 Å) and dravitic tourmaline (B–O bond length of 1.385 Å) is +1.8 ± 1.1‰, whereas for the system tourmaline–prismatine the calculations resulted in a B-isotopic fractionation of −6.4 ± 1.3‰. For both systems these data are in good agreement to measured B-isotope mineral–mineral fractionation of +3.3 ± 0.8‰ for coexisting natural grandidierite–tourmaline and −5.0 ± 1.4‰ for tourmaline–prismatine pairs (MacGregor et al. 2013).

β factor at T = 1000 K for various boron-bearing species as a function of the B–O bond length. Blue circles and red triangles represent the values obtained for [3]B and [4]B species, respectively. Figure modified after Kowalski et al. (2013)
3.7 Conclusions
Table 3.1 and Fig. 3.10 summarize the results of experimentally determined B-isotope fractionation data for selected systems presented in this chapter. Due to the discrepancies between assumed B-species in (rhyolitic) melts and the measured large B-isotopic melt–fluid fractionation (Hervig et al. 2002), these data are assumed here to represent disequilibrium fractionation. Therefore, further experimental investigation is greatly needed, which should be complemented by the evaluation of possible kinetic B-isotope fractionation at high temperatures, which might produce considerable effects. Furthermore, inconsistencies in the results on B-incorporation mechanisms for calcite and aragonite and the not-fully understood variations in Δ11B values for different solid–fluid systems (e.g., smectite and carbonates), alert us to the potential role of often poorly known surface-dependent processes in controlling subsequent B-isotope fractionation for different minerals in low-temperature diffusive experiments. Thus, further experimental investigation is needed here too, e.g., by innovative methods like AFM (Ruiz-Agudo et al. 2012), to develop a robust mechanistic knowledge of B incorporation into minerals during growth. Additionally, it is obvious from Fig. 3.10 that there is a lack of experimental data in the T-range 40–350 °C. Further experiments at these temperatures are needed to fill the gap between existing high- and low-T data and to better understand the structural controls on B-isotope fractionation during diagenesis, low-T metamorphism, low-T hydrothermal activity and volcanic exhalation. Generally, it would seem that using experiments with a well-understood B-incorporation mechanism is the favorable approach for acquiring more reliable fractionation data and for avoiding surface-related effects and diffusion problems potentially-occurring in the low-temperature reequilibration experiments.

Compilaton of selected experimental data on vapor, melt, solid–fluid B-isotope fractionation. Fractionation data for systems with contrasting B-coordination (i.e., [4]B vs. [3]B) are marked by a blue background. Possibly, this does not hold for the melt–fluid (for more details see text). Pink color mark fractionation data for systems with uniform B-coordination. Borax (containing mixed [3]B and [4]B)–fluid data (in green) show a strong variation in fractionation due to variable experimental fluid pH (7, with mainly [3]B, to 11.5, with mainly [4]B). Smectite–fluid B-isotope fractionation (in orange) as result of B-surface adsorption strongly depend on pH. The numbers correspond to the references given in Table 3.1
Recently performed ab initio calculations have the potential to obtain T- and P-dependent fractionation factors for given materials, which are successfully used not only to complement the experimental data but also to assist their correct interpretation and to derive constraints on unmeasured systems. The computed B-isotope fractionation factors can also help in explaining discrepancies between contrasting measurements and provide valuable insight into the boron speciation in fluids. In consideration of the continuous increase in the efficiency of computing resources and performance of quantum chemistry software, it is concluded that ab initio computational geochemistry will flourish in the coming decades, and that computer-aided first-principles simulations will become standard research tools in isotope geochemistry. They assist in the interpretation of experimental data and the understanding of B behavior in natural settings.
References
Alecu IM, Zheng J, Zhao Y, Truhlar DG (2010) Computational thermochemistry: scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J Chem Theory Comput 6:2872–2887
Altaner SP, Ylagen RF (1997) Comparison of structural models of mixed-layer illite/smectite and reaction mechanisms of smectite illitization. Clays Clay Miner 45:517–533
Audétat A, Günther D, Heinrich CA (1998) Formation of a magmatic-hydrothermal ore deposit: Insights with LA-ICP-MS analysis of fluid inclusions. Science 279:2091–2094
Balan E, Pietrucci F, Gervais C, Blanchard M, Schott J, Gaillardet J (2016) First-principles study of boron speciation in calcite and aragonite. Geochim Cosmochim Acta. doi:10.1016/j.gca.2016.07.026. (in press)
Baldereschi A (1973) Mean-value point in the Brillouin zone. Phys Rev B 7:5212–5215
Barth S (1993) Boron isotope variations in nature: a synthesis. Geol Rundschau 82:640–651
Belley PM, Grice JD, Fayek M, Kowalski PM, Grew ES (2014) A new occurrence of the borosilicate serendibite in tourmaline-bearing calc-silicate rocks, Portage-du-Fort Marble, Grenville Province, Québec: evolution of boron isotopes and tourmaline compositions in a metamorphic context. Can Mineral 52:595–615
Berryman EJ, Wunder B, Wirth R, Rhede D, Schettler G, Franz G, Heinrich W (2015) An experimental study on K and Na incorporation in dravitic tourmaline and insight into the origin of diamandiferous tourmaline from the Kokchetav Massif Kazakhstan. Contrib Mineral Petrol 129:28
Bigeleisen J, Mayer MG (1947) Calculation of equilibrium constants for isotopic exchange reactions. J Chem Phys 15:262–267
Blanchard M, Poitrasson F, Méheut M, Lazzeri M, Mauri F, Balan E (2009) Iron isotope fractionation between pyrite (FeS2), hematite (Fe2O3) and siderite (FeCO3): a first-principles density functional theory study. Geochim Cosmochim Acta 73:6565–6578
Branson O, Kaczmarek K, Redfern SAT, Misra S, Langer G, Tyliszcczak T, Bijma J, Elderfield H (2015) The coordination and distribution of B in foraminiferal calcite. Earth Planet Sci Lett 416:67–72
Branson O (2017) Boron incorporation into marine CaCO3. In: Marschall HR, Foster GL (eds) Advances in isotope geochemistry. Boron isotopes—the fifth element. Springer, Heidelberg
Bray PJ, O’Keefe JG (1963) Nuclear magnetic resonance investigations of the structure of alkali borate glasses. Phys Chem Glasses 4:37–46
Byrne RH, Yao W, Klochko K, Tossel JA, Kaufman AJ (2006) Experimental evaluation of the isotopic exchange equilibrium 10B(OH)3 + 11B(OH) −4 = 11B(OH)3 + 10B(OH) −4 in aqueous solution. Deep Sea Res I 53:684–688
Car R, Parrinello M (1985) Unified approach for molecular dynamics and density-functional theory. Phys Rev Lett 55:2471–2474
Chacko T, Cole DR, Horita J (2001) Equilibrium oxygen, hydrogen and carbon isotope fractionation factors applicable to geologic systems. Rev Mineral Geochem 43:1–81
Chakraborty S, Dingwell DB, Chaussidon M (1993) Chemical diffusivity of boron in melts of haplogranitic composition. Geochim Cosmochim Acta 57:1741–1751
Chiodini G, Comodi P, Giaquinto S (1988) Ammonia and boric acid in steam and water. Experimental data from geothermal wells in the Phlegrean Fields, Naples, Italy. Geothermics 17:711–718
Christ CL, Harder H (1978) Boron. In: Wedephol KH (ed) Handbook of Geochemistry, vol II/1. Springer, Berlin, pp 5E1–5O3
Clauer N, Williams LB, Fallick AE (2014) Genesis of nanometric illite crystals elucidated by light-element (hydrogen, lithium, boron and oxygen) isotope tracing and K-Ar and Rb-Sr dating. Chem Geol 383:26–50
De Hoog JCM, Savov IP (2017) Boron as tracer of subduction zone processes. In: Marschall HR, Foster GL (eds) Advances in isotope geochemistry. Boron isotopes—the fifth element. Springer, Heidelberg
Dohmen R, Kasemann SA, Coogan L, Chakraborty S (2010) Diffusion of Li in olivine. Part I: experimental observations and multi species diffusion model. Geochim Cosmochim Acta 74:274–292
Domagal-Goldman SD, Kubicki JD (2008) Density functional theory predictions of equilibrium isotope fractionation of iron due to redox changes and organic complexation. Geochim Cosmochim Acta 72:5201–5216
Domanik KJ, Hervig RL, Peacock SM (1993) Beryllium and boron in subduction zone minerals: an ion microprobe study. Geochim Cosmochim Acta 57:4997–5010
Driesner T (1997) The effect of pressure on deuterium–hydrogen fractionation in high-temperature water. Science 277:791–794
Dupuis R, Benoit M, Nardin E, Méheut M (2015) Fractionation of silicon isotopes in liquids: the importance of configural disorder. Chem Geol 396:239–254
Dutrow BL, Henry DJ (2011) Tourmaline: a geological DVD. Elements 7:301–306
Edwards T, Endo T, Walton JH, Sen S (2014) Observation of the transition state for pressure-induced BO3 → BO4 conversion in glass. Science 345:1027–1029
Elmaci G, Icten O, Nedim Ay A, Zümreoglu-Karan B (2015) Boron isotopic fractionation in aqueous boric acid solution over synthetic minerals: effect of layer and surface charge on fractionation factor. Appl Clay Sci 107:117–121
Ertl A, Pertlik F, Bernhardt H-J (1997) Investigations on olenite with excess boron from the Koralpe, Styria, Austria. Anz Abt I 124:3–10
Ertl A, Hughes JM, Prowatke S, Ludwig T, Brandstätter F, Körner W, Dyar MD (2007) Tetrahedrally coordinated boron in Li-bearing olenite from “mushroom” tourmaline from Momeik, Myanmar. Can Mineral 45:891–899
Ertl A, Tillmanns E, Ntaflos T, Francis C, Giester G, Körner W, Hughes JM, Lengauer C, Prem M (2008) Tetrahedrally coordinated boron in Al-rich tourmaline and its relationship to the pressure-temperature conditions of formation. Eur J Mineral 20:881–888
Feng C, Qin T, Huang S, Wu Z, Huang F (2014) First-principles investigations of equilibrium calcium isotope fractionation between clinopyroxene and Ca-doped orthopyroxene. Geochim Cosmochim Acta 143:132–142
Fijii T, Pringle EA, Chaussidon M, Moynier F (2015) Isotope fractionation of Si in protonation/deprotonation reaction of silicic acid: a new pH proxy. Geochim Cosmochim Acta 168:193–205
Finley JW, Stephens PJ (1995) Density functional theory calculations of molecular structures and harmonic vibrational frequencies using hybrid density functionals. J Mol Struct 357:225–235
Foster GL, Pogge von Strandmann PAE, Rae JWB (2010) Boron and magnesium isotopic composition of seawater. Geochem Geophys Geosys 11:Q08015. doi:10.1029/2010GC003201
Furst M, Lowestam HA, Burnett DS (1976) Radiographic study of the distribution of boron in recent mollusc shells. Geochim Cosmochim Acta 40:1381–1386
Gaillardet J, Allègre CJ (1995) Boron isotopic composition of coral: seawater or diagenesis record? Earth Planet Sci Lett 136:665–676
Geisinger KL, Oestrike R, Navrotsky A, Turner GL, Kirkpatrick RJ (1988) Thermochemistry and structure of glasses along the join NaAlSi3O8–NaBSi3O8. Geochim Cosmochim Acta 52:2405–2414
Glover RB (1988) Boron distribution between liquid and vapour in geothermal fluids. In: Proceedings 10th New Zealand Geothermal Workshop, New Zealand
Goedecker S, Teter M, Hutter J (1996) Separable dual-space Gaussian pseudopotentials. Phys Rev B 54:1703–1710
Goldberg S, Forster HS, Lesch SM, Heick EL (1996) Influence of anion competition on boron adsorption by clays and soils. Soil Sci 161:99–103
Hedenquist JW, Arribas A, Reynolds TJ (1998) Evolution of an intrusion-centered hydrothermal system, far southeast-Lepanto porphyry and epithermal Cu–Au deposits, Phillipines. Econ Geol 93:373–404
Hemming NG, Hanson GN (1992) Boron isotopic composition and concentration in modern marine carbonates. Geochim Cosmochim Acta 56:537–543
Hemming NG, Reeder RJ, Hanson GN (1995) Mineral-fluid partitioning and isotopic fractionation of boron in synthetic calcium carbonate. Geochim Cosmochim Acta 59:371–379
Hershey JP, Fernandez M, Milne PJ, Millero FJ (1986) The ionization of boric acid in NaCl, Na–Ca–Cl, Na–Mg–Cl solutions at 25 °C. Geochim Cosmochim Acta 50:143–148
Hervig RL, Moore GM, Williams LB, Peacock SM, Holloway JR, Roggensack K (2002) Isotopic and elemental partitioning of boron between hydrous fluid and silicate melt. Am Mineral 87:769–774
Hill PS, Schauble EA (2008) Modeling the effects of bond environment on equilibrium iron isotope fractionation in ferricaquo–chloro complexes. Geochim Cosmochim Acta 72:1939–1958
Hill PS, Schauble EA, Young ED (2010) Effects of changing solution chemistry on Fe3+/Fe2+ isotope fractionation in aqueous FeCl solutions. Geochim Cosmochim Acta 74:6669–6689
Hoover WG (1985) Canonical dynamics equilibrium phase-space distributions. Phys Rev A 31:1695–1697
Hughes JM, Ertl A, Dyar MD, Grew E, Wiedenbeck M, Brandstätter F (2004) Structural and chemical response to varying [4]B content in zoned Fe-bearing olenite from Koralpe, Austria. Can Mineral 38:447–454
Ishikawa T, Nakamura E (1994) Origin of the slab component in arc lavas from across-arc variation of B and Pb isotopes. Nature 370:205–208
Jahn S, Kowalski PM (2014) Theoretical approaches to structure and spectroscopy of Earth materials. In: Henderson GS, Neuville DR, Downs RT (eds) Spectroscopic methods, vol 78. Rev Mineral Geochem, chap 17. Mineralogical Society of America, Washington DC, pp 691–743
Jahn S, Wunder B (2009) Lithium speciation in aqueous fluids at high P and T studied by ab initio molecular dynamics and consequences for Li isotope fractionation between minerals and fluids. Geochim Cosmochim Acta 73:5428–5434
Kakihana H, Kotaka M, Satoh S, Nomura M, Okamoto M (1977) Fundamental studies on the ion-exchange separation of boron isotopes. Bull Chem Soc Japan 50:158–163
Kalt A, Schreyer W, Ludwig T, Prowatke S, Bernhardt H-J, Ertl A (2001) Complete solid solution between magnesium schorl and lithium excess-boron olenite in pegmatite from the Koralpe (eastern Alps, Austria). Eur J Mineral 13:1191–1205
Klemme S, Marschall HR, Jacob DE, Prowatke S, Ludwig T (2011) Trace-element partitioning and boron isotope fractionation between white mica and tourmaline. Can Mineral 49:165–176
Klochko K, Cody GD, Tossell JA, Dera P, Kaufman AJ (2009) Re-evaluation boron speciation in biogenic calcite and aragonite using 11B MAS NMR. Geochim Cosmochim Acta 73:1890–1900
Konrad-Schmolke M, Halama R (2014) Combined thermodynamic-geochemical modeling in metamorphic geology: boron as tracer of fluid-rock interaction. Lithos 208:393–414
Kowalski PM, Jahn S (2011) Prediction of equilibrium Li isotope fractionation between minerals and aqueous solutions at high P and T: an efficient ab initio approach. Geochim Cosmochim Acta 75:6112–6123
Kowalski PM, Wunder B, Jahn S (2013) Ab initio prediction of equilibrium boron isotope fractionation between minerals and aqueous fluids at high P and T. Geochim Cosmochim Acta 101:285–301
Kutzschbach M, Wunder B, Rhede D, Koch-Müller M, Ertl A, Giester G, Heinrich W, Franz G (2016a) Tetrahedral boron in natural and synthetic HP/UHP tourmaline: evidence from Raman spectroscopy, EMPA, and single-crystal XRD. Am Mineral 101:93–104
Kutzschbach M, Wunder B, Trumbull RB, Meixner A, Heinrich W, Franz G (2016b) The effect of tetrahedral B on the B isotope fractionation between olenitic tourmaline and fluid. In: Abstract, EMC2016-conference, Rimini, Italy
Lea DW (2003) Elemental and isotopic proxies of past ocean temperatures. In: Elderfield H, Holland HD, Turekian KK (eds) The oceans and marine geochemistry. Elsevier, Oxford, p 365
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Leeman WP, Sisson VB (2002) Geochemistry of boron and its petrological applications. In: Grew ES, Anovitz LM (eds) Boron: mineralogy, petrology and geochemistry, vol 33, 2nd edn. Mineralogical Society of America, Washington DC, pp 645–708
Leeman WP, Vocke RD, McKibben MA (1992) Boron isotopic fractionation between coexisting vapor and liquid in natural geothermal systems. In: Kharaka YF, Maest AS (eds) Proc 7th Int Sympos on water-rock interaction-WRI-7. Balkema, Rotterdam, pp 1007–1010
Lemarchand E, Schott J, Gaillardet J (2007) How surface complexes impact boron isotope fractionation: evidence from Fe and Mn oxides sorption experiments. Earth Planet Sci Lett 260:277–296
Liebscher A, Meixner A, Romer RL, Heinrich W (2005) Liquid-vapor fractionation of boron and boron-isotopes: Experimental calibration at 400 °C/23 MPa to 450 °C/42 MPa. Geochim Cosmochim Acta 69:5693–5704
Liebscher A, Meixner A, Romer RL, Heinrich W (2007) Experimental calibration of the vapor-liquid phase relations and lithium isotope fractionation in the system H2O–LiCl at 400 °C/20–28 MPa. Geofluids 7:369–375
Liu Y, Tossell JA (2005) Ab initio molecular orbital calculations for boron isotope fractionations on boric acids and borates. Geochim Cosmochim Acta 69:3995–4006
London D (1997) Estimating abundances of volatile and other mobile components in evolved silicate melts through mineral-melt equilibria. J Petrol 38:1691–1706
Longstaffe FJ (1987) Stable isotope studies of diagenetic processes. In: Kyser K (ed) Short course in stable isotope geochemistry of low-temperature fluids. Mineralogical Association of Canada, Saskatoon, pp 187–257
MacGregor JR, Grew ES, De Hoog JCM, Harley SL, Kowalski PM, Yates MG, Carson CJ (2013) Boron isotopic composition of tourmaline, prismatine, and grandidierite from granulite facies paragneisses in the Larsemann Hills, Prydz Bay, East Antarctica: evidence for a non-marine evaporite source. Geochim Cosmochim Acta 123:261–283
Manara D, Grandjean A, Neuville DR (2009) Advances in understanding the structure of borosilicate glasses: a Raman spectroscopy study. Am Mineral 94:777–784
Manning CE (2004) The chemistry of subduction-zone fluids. Earth Planet Sci Lett 223:1–16
Markland TE, Berne BJ (2012) Unraveling quantum mechanical effects in water using isotopic fractionation. Proc Natl Acad Sci USA 109:7988–7991
Marschall HR (2005) Lithium, beryllium, and boron in high-pressure metamorphic rocks from Syros (Greece). PhD thesis, University of Heidelberg, Heidelberg, Germany, 411 pp. www.ub.uni-heidelberg.de/archiv/5634
Marschall HR, Jiang S-Y (2011) Tourmaline isotopes: no element left behind. Elements 7:313–319
Marschall HR, Ertl A, Hughes JM, McCommon C (2004) Metamorphic Na- and OH-rich disordered dravite with tetrahedral boron associated with omphacite, from Syros, Greece: chemistry and structure. Eur J Mineral 16:817–823
Marschall HR, Altherr R, Ludwig T, Kalt A, Gméling K, Kasztovszky Z (2006) Partitioning and budget of Li, Be and B in high-pressure metamorphic rocks. Geochim Cosmochim Acta 70:4750–4769
Marschall HR, Altherr R, Rüpke L (2007) Squeezing out the slab—modeling the release of Li, Be and B during progressive high-pressure metamorphism. Chem Geol 239:323–335
Marschall HR, Meyer C, Wunder B, Ludwig T, Heinrich W (2009) Experimental boron isotope fractionation between tourmaline and fluid: confirmation from in situ analyses by secondary ion mass spectrometry and from Rayleigh fractionation modelling. Contrib Miner Petrol 158:675–681
Marx D, Hutter J (2000) Ab initio molecular dynamics: theory and implementation. In: Grotendorst J (ed) Modern methods and algorithms of quantum chemistry. NIC, FZ Jülich, pp 301–449, CPMD code: J. Hutter et al. see: www.cpmd.org
Mavromatis V, Montouillout V, Noireaux J, Gaillardet J, Schott J (2015) Characterization of boron incorporation and speciation in calcite and aragonite from co-precipitation experiments under controlled pH, temperature and precipitation rate. Geochim Cosmochim Acta 150:299–313
McCulloch MT (2017) Boron isotopic and element constraints on coral calcification. In: Marschall HR, Foster GL (eds) Advances in isotope geochemistry. Boron isotopes—the fifth element. Springer, Heidelberg
Méheut M, Schauble EA (2014) Silicon isotope fractionation in silicate minerals: insights from first-principles models of phyllosilicates, albite and pyrope. Geochim Cosmochim Acta 134:137–154
Méheut M, Lazzeri M, Balan E, Mauri F (2007) Equilibrium isotopic fractionation in the kaolinite, quartz, water system: prediction from first-principles density-functional theory. Geochim Cosmochim Acta 71:3170–3181
Méheut M, Lazzeri M, Balan E, Mauri F (2009) Structural control over equilibrium silicon and oxygen isotopic fractionation; a first principles density functional theory study. Chem Geol 258:28–37
Menconi G, Tozer DJ (2002) Diatomic bond lengths and vibrational frequencies: assessment of recently developed exchange-correlation functionals. Chem Phys Lett 360:38–46
Meyer C, Wunder B, Meixner A, Romer RL, Heinrich W (2008) Boron-isotope fractionation between tourmaline and fluid: an experimental re-investigation. Contrib Miner Petrol 156:259–267
Michel F, Cormier L, Lombard P, Beuneu B, Galoisy L, Calas G (2013) Mechanisms of boron coordination change between borosilicate glasses and melts. J Non-Cryst Solids 379:169–176
Moore DM, Reynolds RC (1997) X-ray diffraction and the identification and analysis of clay minerals, 2nd edn. Oxford University Press, New York, p 378
Mottl MJ, Holland HD (1978) Chemical exchange during hydrothermal alteration of basalt by seawater - I Experimental results for major and minor components of seawater. Geochim Cosmochim Acta 42:1103–1115
Nosé S, Klein ML (1983) Constant pressure molecular dynamics for molecular systems. Mol Phys 50:1055–1076
Oi T, Kato J, Ossaka T, Karihana H (1991) Boron isotope fractionation accompanying boron mineral formation from aqueous boric acid-sodium hydroxide solutions at 25 °C. Geochem J 25:377–385
Oi T, Shimazaki H, Ishii R, Hosoe M (1997) Boron isotope fractionation in liquid chromatography with boron-specific resins as column packing material. Sep Sci Technol 32:1821–1834
Otake T, Lasaga AC, Ohmoto H (2008) Ab initio calculations for equilibrium fractionations in multiple sulfur isotope systems. Chem Geol 249:357–376
Palmer MR, Swihart GH (2002) Boron isotope geochemistry: an overview. In: Grew ES, Anovitz LM (eds) Boron: mineralogy, petrology and geochemistry Vol 33, 2nd edn. Reviews in Mineralogy, Mineralogical Society of America, Washington, DC, pp 709–744
Palmer MR, Spivack AJ, Edmond JM (1987) Temperature and pH controls over isotopic fractionation during adsorption of boron on marine clays. Geochim Cosmochim Acta 51:2319–2323
Palmer MR, London D, Morgan VIGB, Babb HA (1992) Experimental determination of fractionation of 11B/10B between tourmaline and aqueous vapor: a temperature- and pressure-dependent isotopic system. Chem Geol 101:123–129
Palmer MR, Pearson PN, Cobb SJ (1998) Reconstructing past ocean pH-depth profiles. Science 282:1468–1471
Peacock SM, Hervig RL (1999) Boron isotopic composition of subduction-zone metamorphic rocks. Chem Geol 160(4):281–290
Pichavant M, Kontak DJ, Briquen L, Herrera JV, Clark AH (1988) The Miocene-Pliocene Macusani volcanics, SE Peru II. Geochemistry and origin of a felsic peraluminous magma. Contrib Miner Petrol 100:325–338
Pinilla C, Blanchard M, Balan E, Ferlat G, Vuilleumier R, Mauri F (2014) Equilibrium fractionation of H and O isotopes in water from path integral molecular dynamics. Geochim Cosmochim Acta 135:203–216
Pinilla C, Blanchard M, Balan E, Natarajan SK, Vuilleumier R, Mauri F (2015) Equilibrium magnesium isotope fractionation between aqueous Mg2+ and carbonate minerals: Insights from path integral molecular dynamics. Geochim Cosmochim Acta 163:126–139
Rae JWB (2017) Boron isotopes in foraminifera. In: Marschall HR, Foster GL (eds) Advances in isotope geochemistry. Boron isotopes—the fifth element. Springer, Heidelberg
Reynolds TJ, Beane RE (1985) Evolution of hydrothermal fluid characteristics at the Santa Rita, New Mexico, porphyr copper deposit. Econ Geol 80:1328–1347
Roedder E (1979) Fluid inclusions as samples of ore fluids. In: Barnes HL (ed) Geochemistry of hydrothermal ore deposits, 2nd edn. Wiley, New York, pp 684–737
Rosner M, Erzinger J, Franz G, Trumbull RB (2003) Slab-derived boron isotope signatures in arc volcanic rocks from the Central Andes and evidence for boron isotope fractionation during progressive slab dehydration. Geochem Geophys Geosys 4:1–25
Ruiz-Agudo E, Putnis CV, Kowacz M, Ortega-Huertas M, Putnis A (2012) Boron incorporation into calcite during growth: implications for the use of boron in carbonates as a pH proxy. Earth Planet Sci Lett 345:9–17
Rustad JR, Bylaska EJ (2007) Ab initio calculation of isotopic fractionation in B(OH)3 (aq) and B(OH) −4 (aq). J Am Chem Soc 129:2222–2223
Rustad JR, Nelmes SL, Jackson VE, Dixon DA (2008) Quantum-chemical calculations of carbon-isotope fractionation in CO2(g), aqueous carbonate species, and carbonate minerals. J Phys Chem A 112(3):542–555
Rustad JR, Bylaska EJ, Jackson VE, Dixon DA (2010a) Calculation of boron-isotope fractionation between B(OH)3(aq) and B(OH) −4 (aq). Geochim Cosmochim Acta 74:2843–2850
Rustad JR, Casey WH, Yin QZ, Bylaska EJ, Felmy AR, Bogatko SA, Jackson VE, Dixon DA (2010b) Isotopic fractionation of Mg2+(aq), Ca2+(aq), and Fe2+(aq) with carbonate minerals. Geochim Cosmochim Acta 74:6301–6323
Ryan JG, Langmuir CH (1993) The systematics of boron abundances in young volcanic rocks. Geochim Cosmochim Acta 57:1489–1498
Sanchez-Valle C, Reynard B, Daniel I, Lecuyer C, Martinez I, Chervin J-C (2005) Boron isotopic fractionation between minerals and fluids: new insights from in situ high pressure-high temperature vibrational spectroscopic data. Geochim Cosmochim Acta 69:4301–4313
Sanyal A, Hemming NG, Hanson GN, Broecker WS (1995) Evidence for a higher pH in the glacial ocean from boron isotopes in foraminifera. Nature 373:234–236
Sanyal A, Hemming NG, Broecker WS, Lea DW, Spero HJ, Hanson GN (1996) Oceanic pH control on the boron isotopic composition: evidence from culture experiments. Paleooceanography 11:513–517
Sanyal A, Nugent M, Reeder RJ, Bijma J (2000) Seawater pH control on the boron isotopic composition of calcite: evidence from inorganic calcite precipitation experiments. Geochim Cosmochim Acta 64:1551–1555
Schatz OJ, Dolejš D, Stix J, Williams-Jones AE, Layne GD (2004) Partitioning of boron among melt, brine and vapor in the system haplogranite-H2O–NaCl at 800 °C and 100 MPa. Chem Geol 210:135–147
Schauble EA (2004) Applying stable isotope fractionation to new systems. In: Johnson CM, Beard BL, Albarede F (eds) Geochemistry of non-traditional stable isotopes, vol 55. Rev Mineral Geochem, chap 3. Mineralogical Society of America, Washington DC, pp 65–111
Schauble EA (2011) First-principles estimates of equilibrium magnesium isotope fractionation in silicate, oxide, carbonate and hexaaquamagnesium(2+) crystals. Geochim Cosmochim Acta 75:844–869
Schauble EA, Méheut M, Hill PS (2009) Combining metal stable isotope fractionation theory with experiments. Elements 5:369–374
Schmidt MW (1996) Experimental constraints on recycling of potassium from subducted oceanic crust. Science 272:1927–1930
Schmidt BC, Zotov N, Dupree R (2004) Structural implications of water and boron dissolution in albite glass. J Non-Cryst Solids 37:207–219
Schmidt C, Thomas R, Heinrich W (2005) Boron speciation in aqueous fluids at 22 to 600 °C and 0.1 MPa to 2 GPa. Geochim Cosmochim Acta 69:275–281
Scholze H (1988) Glas. Springer, Berlin, 339 pp
Schwarcz HP, Agyei EK, McMullen CC (1969) Boron isotopic fractionation during clay adsorption from sea-water. Earth Planet Sci Lett 6:1–5
Sen S, Stebbins JF, Hemming NG, Ghosh B (1994) Coordination environments of B impurities in calcite and aragonite polymorphs: A 11B MAS NMR study. Am Mineral 79:819–825
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst A 32:751–767
Shmulovich K, Heinrich W, Möller P, Dulski P (2002) Experimental determination of REE fractionation between liquid and vapour in the system NaCl–H2O and CaCl2–H2O up to 450 °C. Contrib Miner Petrol 144:257–273
Singh SPN, Mattigod SV (1992) Modeling boron adsorption on kaolinite. Clays Clay Miner 40:192–205
Slack JF, Trumbull RB (2011) Tourmaline as a recorder of ore-forming processes. Elements 7:321–326
Smirnov SZ, Thomas VG, Demin SP, Drebushchak VA (2005) Experimental study of boron solubility and speciation in the Na2O–B2O3–SiO2–H2O system. Chem Geol 223:16–34
Smith CL, Ficklin WH, Thompson JM (1987) Concentrations of arsenic, antimony, and boron in steam and steam condensate at the Geyers, California. J Volcanol Geotherm Res 32:329–341
Spivack AJ, Edmond JM (1987) Boron isotope exchange between seawater and the oceanic crust. Geochim Cosmochim Acta 51:1033–1041
Spivack AJ, Berndt ME, Seyfried WE Jr (1990) Boron isotope fractionation during supercritical phase separation. Geochim Cosmochim Acta 54:2337–2339
Spivack AJ, You CF, Smith HJ (1993) Foraminifera boron isotope ratios as a proxy for surface ocean pH over the past 21 Myr. Nature 363:149–151
Środoň J, McCarty DK (2008) Surface area and layer charge of smectite from CEC and EGME/H2O retention measurements. Clays Clay Miner 56:142–161
Thomas R, Webster JD, Heinrich W (2000) Melt inclusions in pegmatitic glass: complete miscibility between silicate melts and hydrous fluids at low pressure. Contrib Miner Petrol 139:394–401
Thomas R, Förster H-J, Heinrich W (2003) The behaviour of boron in a peraluminious granite-pegmatite system and associated hydrothermal solutions: a melt and fluid-inclusion study. Contrib Miner Petrol 144:457–472
Tomascak PB, Magna T, Dohmen R (2016) Advances in lithium isotope geochemistry, Springer, Berlin, 195 pp
Tonarini S, Pennisi M, Leeman WP (1997) Precise boron isotopic analysis of complex silicate (rock) samples using alkali carbonate fusion and ion-exchange separation. Chem Geol 142:129–137
Tonarini S, Forte C, Petrini R, Ferrara G (2003) Melt/biotite 11B/10B isotopic fractionation and the boron local environment in the structure of volcanic glasses. Geochim Cosmochim Acta 67:1863–1873
Tossel JA (2006) Boric acid adsorption on humic acids: ab initio calculation of structures, stabilities, 11B NMR and 11B, 10B isotopic fractionations of surface complexes. Geochim Cosmochim Acta 70:5089–5103
Trumbull RB, Chaussidon M (1999) Chemical and boron isotopic composition of magmatic and hydrothermal tourmalines from Sinceni granite-pegmatite system in Swaziland. Chem Geol 153:125–137
Trumbull RB, Slack JF (2017) Boron isotopes in the continental crust: granites, pegmatites and related hydrothermal systems. In: Marschall HR, Foster GL (eds) Advances in isotope geochemistry. Boron isotopes—the fifth element. Springer, Heidelberg
Urey HC (1947) The thermodynamic properties of isotopic substances. J Chem Soc 562–581
Vakulenko AG, Alekhin YV, Razina MV (1989) Solubility and thermodynamic properties of alkali chlorides in steam. In: Proceedings II International Symposium “Properties of water and steam”, Prague, pp 395–401
van Hinsberg VJ, Marschall HR (2007) Boron isotope and light element sector zoning in tourmalines: Implications for the formation of B-isotopic signatures. Chem Geol 238:141–148
van Hinsberg VJ, Schumacher JC (2007) Using estimated thermodynamic properties to model accessory phases in the cases of tourmaline. J Metamorph Geol 25:769–779
van Hinsberg VJ, Henry DJ, Dutrow BL (2011) Tourmaline as a petrological forensic mineral. A unique recorder of its geologic past. Elements 7:327–332
Veksler IV, Dorfman AM, Dingwell DB, Zotov N (2002) Element partitioning between immiscible borosilicate liquids: A high-temperature centrifuge study. Geochim Cosmochim Acta 66:2603–2614
Vengosh A, Chivas AR, McCulloch MT (1989) Direct determination of boron and chlorine isotopic compositions in geological materials by negative thermal-ionization mass spectrometry. Chem Geol 79:333–343
Vengosh A, Kolodny Y, Starinsky A, Chivas AR, McCulloch M (1991) Coprecipitation and isotopic fractionation in boron in modern biogenic carbonates. Geochim Cosmochim Acta 55:2901–2910
Vengosh A, Starinsky A, Kolodny Y, Chivas AR, Raab M (1992) Boron isotope variations during fractional evaporation of sea water: new constraints on the marine vesurs nonmarine debate. Geology 20:799–802
von Goerne G, Franz G, Heinrich W (2001) Synthesis of tourmaline solid solutions in the system Na2O–MgO–Al2O3–SiO2–B2O3–H2O–HCl and the distribution of Na between tourmaline and fluid at 300–700 °C and 200 MPa. Contrib Miner Petrol 141:160–173
Wagner W, Pruss A (2002) The IAPWS formulation 1995 for the thermodynamic properties of ordinary water substance for general and scientific use. J Phys Chem Ref Data 31:387–535
Williams LB, Hervig RL (2005) Lithium and boron isotopes in illite-smectite: the importance of crystal size. Geochim Cosmochim Acta 69:5705–5716
Williams LB, Hervig RL, Holloway JR, Hutcheon I (2001) Boron isotope geochemistry during diagenesis. Part I Experimental determination of fractionation during illitization of smectite. Geochim Cosmochim Acta 65:1769–1782
Williams LB, Turner A, Hervig RL (2007) Intracrystalline boron isotope partitioning in illite-smectite: testing the geothermometer. Am Mineral 92:1958–1965
Wu J, Stebbins JF (2010) Quench rate and temperature effects on boron coordination in aluminoborosilicates. J Non-Cryst Solids 356:2097–2108
Wu J, Stebbins JF (2013) Temperature and modifier cation field strength effects on aluminoborosilicate glass network structure. J Non-Cryst Solids 362:73–81
Wunder B, Meixner A, Romer RL, Wirth R, Heinrich W (2005) The geochemical cycle of boron: constraints from boron isotope partitioning experiments between mica and fluid. Lithos 84:206–216
Wunder B, Meixner A, Romer RL, Heinrich W (2006) Temperature-dependent isotopic fractionation of lithium between clinopyroxene and high-pressure hydrous fluids. Contrib Miner Petrol 151:112–120
Wunder B, Romer RL, Meixner A, Jahn S (2011) Li-isotope silicate fluid fractionation: pressure dependence and influence of the bonding environment. Eur J Miner 23:333–342
Yamaji K, Makita Y, Watanabe H, Sonoda A, Kanoh H, Hirotsu T, Ooi K (2001) Theoretical estimation of lithium isotopic reduced partition function ratio for lithium ions in aqueous solution. J Phys Chem A 105:602–613
Zeebe RE (2005) Stable boron isotope fractionation between dissolved B(OH)3 and B(OH) −4 . Geochim Cosmochim Acta 69:2753–2766
Zeebe RE (2009) Hydration in solution is critical for stable oxygen isotope fractionation between carbonate ion and water. Geochim Cosmochim Acta 73:5283–5291
Zeebe RE (2010) A new value for the stable oxygen isotope fractionation between dissolved sulfate ion and water. Geochim Cosmochim Acta 74:818–828
Zheng Y-F (1993) Oxygen isotope fractionation in SiO2 and Al2SiO5 polymorphs: effect of crystal structure. Eur J Mineral 5:651–658
Zhong J, Bray PJ (1989) Change of coordination in alkali borate glasses, and mixed alkali effects as elucidatd by NMR. J Non-Cryst Solids 111:67–76
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Kowalski, P.M., Wunder, B. (2018). Boron Isotope Fractionation Among Vapor–Liquids–Solids–Melts: Experiments and Atomistic Modeling. In: Marschall, H., Foster, G. (eds) Boron Isotopes. Advances in Isotope Geochemistry. Springer, Cham. https://doi.org/10.1007/978-3-319-64666-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-64666-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64664-0
Online ISBN: 978-3-319-64666-4
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)