
Abstract
Type 2 diabetes (T2D) has attained a pandemic status with more than half a billion cases expected by 2030; and many having cardiovascular complications, with main hallmark of these events, the presence of systemic (chronic) inflammation. Systemic inflammation is in turn characterized by a changed haematological system, including a pathologic coagulation system, endothelial dysfunction and ultimately vascular complications. This chapter discusses the pathogenesis of T2D, and how it is interlinked with cardiovascular disease and inflammation. Literature is reviewed that shows the inflammatory nature of the T2D, how this inflammatory profile and pathological inflammatory markers, affects the coagulation system, and how it plays a role in the impaired vascular function, which is a fundamental characteristic of T2D. As part of the pathogenesis we discuss the considerable literature showing that both hypercoagulability and hypofibrinolysis are present in a large number of inflammatory and vascular diseases, including T2D. We discuss novel methods to monitor and study manifestations of both hypercoagulation and hypofibrinolysis in T2D. We conclude by suggesting that the multifaceted nature of the condition suggests a patient-orientated approach is followed where both traditional and novel methods should be equally explored in the monitoring of T2D patients.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Type 2 diabetes
- Hypercoagulation
- Hypofibrinolysis
- Inflammatory markers
- Cardiovascular complications
- Clot structure
1 Introduction
Type 2 diabetes (T2D) have reached pandemic status with more than half a billion cases expected by 2030 [1]. Comorbidities of both obesity and T2D include cardiovascular disease , cancer and neuropsychiatric disorders [2]. Cardiovascular disease in particular, is one of the most common diabetes-associated complications , as well as a leading cause for death in these patients [3]. Important cardiovascular events include myocardial infarction and stroke [4] and the main hallmark of these events are the presence of systemic (chronic) inflammation . Systemic inflammation is in turn characterized by a changed haematological system, including a pathologic coagulation system [5,6,7,8,9], endothelial dysfunction [10] and ultimately vascular complications .
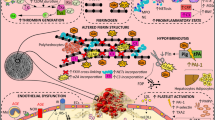
This chapter reviews and discusses literature that shows the inflammatory nature of the condition, how this inflammatory profile affects the coagulation system, including hyercoagulation and mechanisms of impaired clot lyses; and finally shows how these changes lead to the pathological and impaired vascular function , which is a fundamental characteristic of T2D (see Fig. 17.1).

The inflammatory nature of type 2 diabetes
2 Markers of Systemic Inflammation and Cardiovascular Disease (CVD)
As systemic (chronic) inflammation plays a fundamental role in many chronic conditions including T2D and CVD [11,12,13,14,15] and because CVD and vascular complications are a fundamental part of the ethiology of T2D, a quick review of the various dysregulated inflammatory markers in CVD follows. Dysregulated inflammatory markers like C-reactive protein (CRP), along with IL-2, IL-6, IL-8, TNF-α, NOS, PGE2, the COX-family and thromboxane A2 and NFκB, all belong to the cluster of general inflammation markers that are changed in systemic inflammation and CVD. In this chapter the focus will therefore be on the above-mentioned inflammatory markers, although there are others that also play important roles in inflammation .
CRP is a leading inflammatory biomarker for CVD [16,17,18,19,20,21,22,23] andis produced by the liver hepatocytesunder regulatory control from circulating cytokines , in particular IL-6 andtumour necrosis factor-α [19, 24]. Because it is increased in the presence of inflammation , it is used to screen for inflammation , particularly high-sensitivity C-reactive protein (hsCRP), adds prognostic information in CVD [19, 20, 25].
The interleukins are a cytokine group that is well-known to be upregulated in inflammation [26]. Interleukin 1 Receptor 1 (IL1R1) and its ligand, IL1β, are unregulated in CVD and infection [27]. IL-1β is also known to be present in autoimmune conditions and contributes to several chronic diseases, including atherosclerosis [28,29,30]. IL-6 regulates the immune response, haemopoiesis, the acute phase response, inflammation [31] and the central nervous system [31, 32]. Its expression is high and transiently unregulated in nearly all pathophysiological inflammatory conditions and also in autoimmune diseases [33, 34]. IL-8 is also a well-known circulating inflammatory cytokine [35, 36]. Macrophages and other cell types such as epithelial cells, airway smooth muscle cells and endothelial cells produce IL-8.
Tumour necrosis factor- α (TNF-α) is a cell signalling cytokine involved in inflammation and is one of the cytokines that make up the acute phase reaction, and its primary function is to regulate immune cells [37,38,39,40].TNF-α dysregulation plays an important role in the development of metabolic syndrome features, including dyslipidaemia and altered glucose tolerance, and is therefore is an important cytokine in the development and maintenance of systemic inflammation [39]. Vascular endothelial cells also respond to TNF-α by undergoing pro-inflammatory changes, which ultimately promote thrombosis [41, 42].
Another important marker of inflammation is the nitric oxide synthases (NOS) family. They are synthesized by many cell types involved in immunity and is also well known for its role in systemic inflammation and cardiovascular disease [43,44,45,46]. It is also crucial in maintaining cardiovascular homeostasis [45] and a modulator of vascular disease [47]. In CVD, endothelium damage induced by atherosclerosis leads to the reduction in bioactivity of endothelial NO synthase (eNOS ) with subsequent impaired release of NO and ultimately leads to a cascade of oxidation-sensitive mechanisms in the arterial wall [47, 48]. In a comprehensive review, Costa and co-workers discussed the 3 NOS isoforms, neuronal NOS (nNOS or NOS 1), endothelial NOS (eNOS or NOS 3), and an inducible NOS (iNOS or NOS 2). eNOS is considered the main isoform involved in the control of the vascular function , however, the role of nNOS in vascular homeostasis andcardiovascular disorders such as hypertension and atherosclerosis has recently come to light [43].
Prostaglandins (PGs) have two derivatives, namely prostacyclins and thromboxanes and are critical mediators of inflammation [49,50,51,52,53,54] Cyclooxygenases (COXs) are the biosynthetic enzymes of PGs.PGE2 (which inhibits platelet activation and is also an effective vasodilator ), and thromboxane (Tx)A2 (TXA2); and is synthesized via three sequential enzymatic reactions: The first step being arachidonic acid (AA) release from membrane phospholipids by phospholipase A2 (cPLA2); then, AA is converted into the unstable endoperoxide intermediates PGG2 and PGH2 by cyclooxygenase-1 (COX-1) or COX-2 [55]. Markers like COX-1 and -2 and prostaglandin E2 are all closely connected and also play a prominent role in inflammation and CVD [56]. As mentioned before, TXA2 is also a product from COX [51, 57] and is a vasoconstrictor, and a potent hypertensive agent that also facilitates platelet aggregation. Both PGE2 and TXA2 are therefore key role-players in inflammation and CVD.
NF-κB is a protein complex that is activated by pro-inflammatory cytokines such as interleukin 1 (IL-1) and TNFα [58] and the chronic activation or dysregulation of NF-κB signalling is the central to the pathogenesis of many diseases, including CVD [59, 60]. The activity of NF-κB in the canonical pathway results in up-regulation of pro-inflammatory (TNFα, IL-6 and IL-8) and pro-thrombotic [MMPs and TF (tissue factor)] mediators, which are known to be pro-atherogenic [60].
Central to the dysregulation of the mentioned (and other) markers of inflammation is the resulting oxidative stress and ROS generation, which plays crucial roles in both inflammation and CVD [61,62,63]. In CVD there is an imbalance between the antioxidant defence mechanism and ROS production and this leads to oxidative stress [63,64,65]. Ultimately, oxidative stress, is a strong pro-thrombotic factor [66], and the hallmark of inflammation is a prothrombotic prevalence and this translates to hypercoagulation . Inflammation causes hypercoagulation (which is a pro-thrombotic state) because of an elevated expression of the above-mentioned markers, and also elevated expression of the prothrombotic molecules like plasminogen activator inhibitor-1, tissue factor (TF) and increased platelet activation [67,68,69,70]. TF is the main trigger of the coagulation cascade; by binding Factor VIIa it activates Factor IX and Factor X, thereby resulting in fibrin formation [71, 72]. Increased fibrinogen and pathological fibrin formation are key in the development of a hypercoagulable state during inflammation .
If we take a closer look at the pathology in T2D, we see that the primary cause of death in T2D patients, is CVD and it is 2–4× times higher in people with T2D compared with those who are non-diabetic [73]. It is thus noteworthy that patients with T2D have an increased risk of atherothrombotic events [74]. Also, T2D can be classified as an inflammatory condition, due to upregulation of different inflammatory markers [18].
3 Type 2 Diabetes and Its Relation with Cardiovascular Disease
The pathogenesis of T2D, and how it is interlinked with CVD and inflammation , is summarized below:
-
There is an intimate relationship between inflammation and metabolism, including glucose, fat and cholesterol metabolism [75].
-
T2D is known to be one common risk factors for CVD [63], and both obesity and T2D are associated with a state of chronic low-level inflammation [18, 76, 77] and cardiovascular complications [78, 79].
-
Patients with CVD and T2D have increased circulating inflammatory markers [80] and a number of systematic reviews have shown the association between inflammatory markers, such as CRP, IL-1β, IL-6, TNF-α, IL-4, or IL-10, and cardio-metabolic diseases (e.g. T2D) [15, 81,82,83,84,85,86]. TNF-α e.g. has emerged as a key cytokine that influences intermediary metabolism [39].
-
Oxidative stress plays an important role in T2D and it has a critical impact on the development and progression of vascular pathologies, including atherosclerosis and diabetic vasculopathy [64].
-
Endothelial dysfunction is implicated in the pathogenesis of vascular disease seen in T2D [10]; and central to this dysfunction is microvascular complications which are related to oxidative stress , and inflammation , all factors traditionally associated with the pathogenesis of vascular damage seen in CVD [87].
-
In T2D there is a decreased fibrinolysis, increased thrombin generation, and platelet hyperactivity.
-
In T2D there is elevated levels of circulating TF and this is a biomarker for the severity of microvascular disease in these individuals [67, 72, 88].
4 Hypercoagulability and Hypofibrinolysis in Type 2 Diabetes
Recently, we have reviewed in great detail the considerable literature showing that both hypercoagulability and hypofibrinolysis are present in a large number of inflammatory and vascular diseases [89] (e.g. [90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123]). We have also shown that in T2D, fibrin structure is fundamentally changed, and that both erythrocytes and platelets are affected by oxidative stress and circulating up regulated inflammatory markers [6,7,8,9, 124,125,126,127]. Also see Table 17.1 for selected references for the co-occurrence of hypercoagulation and hypofibrinolysis in diabetes; adjusted from [89].
Because T2D is associated with both a hypofibrinolytic and hypercoagulable state , both these pathologies are of crucial importance in the overarching mechanism for increased cardiovascular risk in this population . This forms the basis of the pathology related to, and involved in atherothrombotic complications , which are the main cause of mortality in T2D. This inflammatory state in T2D presents itself as premature atherosclerosis , increased platelet reactivity and activation of coagulation factors, with associated hypofibrinolysis . Ultimately all of these pathologies together contribute to increased cardiovascular risk in this population [128].
Except for the pathological levels of inflammatory markers in T2D leading to ROS generation and oxidative stress that we discussed in the previous paragraphs, a number of factors have been implicated in impaired fibrin clot lyses are:
-
Altered structure of the fibrin (ogen), including glycation and oxidation, resulting in a more compact clot with thinner fibres and increased branching that are more difficult to lyse [74, 129, 130].
-
Increased incorporation of antifibrinolytic proteins (e.g. plasminogen inhibitor and complement C3 into the clot [131, 132] with both proteins having antifibrinolytic activities [74].
-
Higher levels of plasminogen activator inhibitor-1 (PAI-1), which causes a pathological fibrinolytic process, because of a decreased plasmin generation [128]. PAI-1 has been found in blood from patients with T2D and in other conditions associated with insulin resistance [133,134,135]. IncreasedPAI-1 in blood is also associated with a tendency toward venous thrombosis and pulmonary embolism [135], and is associated with a decreased fibrinolytic activity or hypofibrinolysis [136]. This hypofibrinolysis are also related to insulin resistance [137]. Schneider and co-workers in 2004 already suggested that an increase in PAI-1 in vessel walls might predispose to acceleration of atherosclerosis and development of plaques with specific characteristics rendering them vulnerable to rupture [138]. Glycation of plasminogen in T2D also directly affects fibrinolysis by decreasing plasmin generation and reducing protein-specific activity [74].
-
Elevated glucose levels result in increased plasminogen glycation, which affects protein clearance [139]. Tissue plasminogen activator (tPA) mediates plasminogen conversion to plasmin. Binding of tPA to fibrin typically increases the catalytic conversion of plasminogen to plasmin while simultaneously localizing plasmin generation to the site of thrombus formation, thus preventing systemic plasmin generation [74]. Therefore, hypofibrinolysis in T2D is also the result of glycation of plasminogen leads to both decreased plasmin generation and lower catalytic efficiency of plasmin activity [74].
All of the above, result in an inhibition of the fibrinolytic process and together with the known hypercoagulability contribute to the development of (specially ischemic) cardiovascular disease in T2D [74].
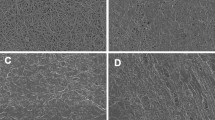
Two of the more novel methods to study clot structure in inflammatory conditions, including T2D, is thromboelastography (TEG ) that shows both clot formation and clot lyses, as well as scanning electron microscopy (SEM) that gives visual information regarding the structure of the actual clot. These two techniques are grouped under the general term, visco-elastic techniques, and together with inflammatory marker analysis, can give valuable information in an individualized patient-orientated approach, when treating individuals with T2D. For a background on the technique, see various publications of Vance Nielsen’s group [140,141,142,143,144,145,146]. Table 17.2 shows the typical parameters that show clot formation and lyses with TEG , and Fig. 17.2 shows examples of healthy and aberrant T2D fibrin clot structures. In a typical healthy individual, we see a spaghetti-like fibrin network with elongated fibrin fibres (for additional examples of healthy fibrin fibres (see https://1drv.ms/f/s!AgoCOmY3bkKHgkFy7q1sVsxRv_2s) [147]. In T2D, plasma with added thrombin forms a clot with finer fibre structure and areas of thick matted areas [6, 9, 124, 126, 148, 149]. Such a pathologic finer fibrin structure might be the cause of the known hypofibrinolytic clot in T2D, where the denser clot areas, together with the netted areas may also lead to the characteristic a hypercoagulable state in T2D. We have also previously found that in T2D, the TEG results vary considerably, depending of the individual clot parameters. This condition is extremely complex, and therefore we have suggested a individualized approach, using not only traditional pathology tests, but also novel methods like SEM and TEG to monitor patient wellness [125].

(a) Fibrin clot from plasma of a healthy individual; (b) Fibrin clot from plasma of a patient with type 2 diabetes . Clots were created by adding thrombin to plasma
5 Conclusion
T2D is probably one of the most complex inflammatory conditions that clinicians need to treat, particularly due to the complex cardiovascular involvement. The mechanisms of both hypercoagulation and aberrant clot lyses in T2D are of great importance in the treatment of the condition. Furthermore, the multifaceted nature of the condition suggests that we follow a patient-orientated approach and educate clinicians to use e.g. TEG as an additional method for disease monitoring. Only by closely following each individual patient’s progress with a variety of research and traditional laboratory pathology methods will we ensure the healthiness of this vulnerable population . The most important strategy is to manage systemic inflammation , and the resulting cardiovascular pathology ; only then will we be able to reduce the T2D pandemic.
References
Klein MS, Shearer J (2016) Metabolomics and type 2 diabetes: translating Basic research into clinical application. J Diabetes Res 2016:3898502. doi:10.1155/2016/3898502
Narayanaswami V, Dwoskin LP (2016) Obesity: current and potential Pharmacotherapeutics and targets. Pharmacol Ther 170:116–147. doi:10.1016/j.pharmthera.2016.10.015
Schnell O, Rydén L, Standl E, Ceriello A (2016) Current perspectives on cardiovascular outcome trials in diabetes. Cardiovasc Diabetol 15(1):139. doi:10.1186/s12933-016-0456-8
Rahmani S, Nakanishi R, Budoff MJ (2016) Imaging atherosclerosis in diabetes: current state. Curr Diab Rep 16(11):105. doi:10.1007/s11892-016-0799-2
Pretorius E, Bester J (2016) Viscoelasticity as a measurement of clot structure in poorly controlled type 2 diabetes patients: towards a precision and personalized medicine approach. Oncotarget 7:50895–50907. doi:10.18632/oncotarget.10618
Buys AV, Van Rooy MJ, Soma P, Van Papendorp D, Lipinski B, Pretorius E (2013) Changes in red blood cell membrane structure in type 2 diabetes: a scanning electron and atomic force microscopy study. Cardiovasc Diabetol 12(1):25. doi:10.1186/1475-2840-12-25
Pretorius E (2013) The adaptability of red blood cells. Cardiovasc Diabetol 12:63. doi:10.1186/1475-2840-12-63
Pretorius E, Bester J, Vermeulen N, Lipinski B, Gericke GS, Kell DB (2014) Profound morphological changes in the erythrocytes and fibrin networks of patients with hemochromatosis or with hyperferritinemia, and their normalization by iron chelators and other agents. PlosOne. doi:10.1371/journal.pone.0085271. eCollection 2014, 10.1371/journal.pone.0085271
Pretorius E, Lipinski B, Bester J, Vermeulen N, Soma P (2013) Albumin stabilizes fibrin fiber ultrastructure in low serum albumin type 2 diabetes. Ultrastruct Pathol 37(4):254–257. doi:10.3109/01913123.2013.778929
Dhananjayan R, Koundinya KS, Malati T, Kutala VK (2016) Endothelial dysfunction in type 2 diabetes mellitus. Indian J Clin Biochem 31(4):372–379. doi:10.1007/s12291-015-0516-y
Frostegard J (2013) Immunity, atherosclerosis and cardiovascular disease. BMC Med 11:117. doi:10.1186/1741-7015-11-117
Gregor MF, Hotamisligil GS (2011) Inflammatory mechanisms in obesity. Annu Rev Immunol 29:415–445. doi:10.1146/annurev-immunol-031210-101322
Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352(16):1685–1695. doi:10.1056/NEJMra043430
Ross R (1999) Atherosclerosis--an inflammatory disease. N Engl J Med 340(2):115–126. doi:10.1056/nejm199901143400207
Ruiz-Canela M, Bes-Rastrollo M, Martínez-González MA (2016) The role of dietary inflammatory index in cardiovascular disease, metabolic syndrome and mortality. Int J Mol Sci 17(8). doi:10.3390/ijms17081265
Berk BC, Weintraub WS, Alexander RW (1990) Elevation of C-reactive protein in “active” coronary artery disease. Am J Cardiol 65(3):168–172
de Beer FC, Hind CR, Fox KM, Allan RM, Maseri A, Pepys MB (1982) Measurement of serum C-reactive protein concentration in myocardial ischaemia and infarction. Br Heart J 47(3):239–243
Lin N, Shi JJ, Li YM, Zhang XY, Chen Y, Calder PC, Tang LJ (2016) What is the impact of n-3 PUFAs on inflammation markers in type 2 diabetic mellitus populations?: a systematic review and meta-analysis of randomized controlled trials. Lipids Health Dis 15:133. doi:10.1186/s12944-016-0303-7
Ridker PM (2016) From C-reactive protein to interleukin-6 to interleukin-1: moving upstream to identify novel targets for Atheroprotection. Circ Res 118(1):145–156. doi:10.1161/circresaha.115.306656
Ridker PM (2016) A test in context: high-sensitivity C-reactive protein. J Am Coll Cardiol 67(6):712–723. doi:10.1016/j.jacc.2015.11.037
Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH (1998) Plasma concentration of C-reactive protein and risk of developing peripheral vascular disease. Circulation 97(5):425–428
Ridker PM, Hennekens CH, Buring JE, Rifai N (2000) C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 342(12):836–843. doi:10.1056/nejm200003233421202
Soeki T, Sata M (2016) Inflammatory biomarkers and atherosclerosis. Int Heart J 57(2):134–139. doi:10.1536/ihj.15-346
Ridker PM (2009) C-reactive protein: eighty years from discovery to emergence as a major risk marker for cardiovascular disease. Clin Chem 55(2):209–215. doi:10.1373/clinchem.2008.119214
Qureshi WT, Rana JS, Yeboah J, Bin Nasir U, Al-Mallah MH (2015) Risk stratification for primary prevention of coronary artery disease: roles of C-reactive protein and coronary artery calcium. Curr Cardiol Rep 17(12):110. doi:10.1007/s11886-015-0666-9
Bester J, Pretorius E (2016) Effects of IL-1beta, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci Rep 6:32188. doi:10.1038/srep32188
Beaulieu LM, Lin E, Mick E, Koupenova M, Weinberg EO, Kramer CD, Genco CA, Tanriverdi K, Larson MG, Benjamin EJ, Freedman JE (2014) Interleukin 1 receptor 1 and interleukin 1beta regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol 34(3):552–564. doi:10.1161/atvbaha.113.302700
Burger D, Dayer JM, Palmer G, Gabay C (2006) Is IL-1 a good therapeutic target in the treatment of arthritis? Best Pract Res Clin Rheumatol 20(5):879–896. doi:10.1016/j.berh.2006.06.004
Dinarello CA (2005) Blocking IL-1 in systemic inflammation. J Exp Med 201(9):1355–1359. doi:10.1084/jem.20050640
Dinarello CA (2011) A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol 41(5):1203–1217. doi:10.1002/eji.201141550
Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M (2012) IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (London, England: 1979) 122(4):143–159. doi:10.1042/cs20110340
Rose-John S (2015) The soluble interleukin-6 receptor and related proteins. Best Pract Res Clin Endocrinol Metab 29(5):787–797. doi:10.1016/j.beem.2015.07.001
Hashizume M, Mihara M (2011) The roles of interleukin-6 in the pathogenesis of rheumatoid arthritis. Arthritis 2011:765624. doi:10.1155/2011/765624
Wolf J, Rose-John S, Garbers C (2014) Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine 70(1):11–20. doi:10.1016/j.cyto.2014.05.024
Rabinovich A, Cohen JM, Kahn SR (2015) Predictive value of markers of inflammation in the postthrombotic syndrome: a systematic review: inflammatory biomarkers and PTS. Thromb Res 136(2):289–297. doi:10.1016/j.thromres.2015.06.024
Ueland T, Gullestad L, Nymo SH, Yndestad A, Aukrust P, Askevold ET (2015) Inflammatory cytokines as biomarkers in heart failure. Clin Chim Acta 443:71–77. doi:10.1016/j.cca.2014.09.001
Esposito E, Cuzzocrea S (2009) TNF-alpha as a therapeutic target in inflammatory diseases, ischemia-reperfusion injury and trauma. Curr Med Chem 16(24):3152–3167
LaDuca JR, Gaspari AA (2001) Targeting tumor necrosis factor alpha. New drugs used to modulate inflammatory diseases. Dermatol Clin 19(4):617–635
Popa C, Netea MG, van Riel PL, van der Meer JW, Stalenhoef AF (2007) The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res 48(4):751–762. doi:10.1194/jlr.R600021-JLR200
Sfikakis PP (2010) The first decade of biologic TNF antagonists in clinical practice: lessons learned, unresolved issues and future directions. Curr Dir Autoimmun 11:180–210. doi:10.1159/000289205
Bradley JR (2008) TNF-mediated inflammatory disease. J Pathol 214(2):149–160. doi:10.1002/path.2287
Swadzba J, Iwaniec T, Musial J (2011) Increased level of tumor necrosis factor-alpha in patients with antiphospholipid syndrome: marker not only of inflammation but also of the prothrombotic state. Rheumatol Int 31(3):307–313. doi:10.1007/s00296-009-1314-8
Costa ED, Rezende BA, Cortes SF, Lemos VS (2016) Neuronal nitric oxide synthase in vascular physiology and diseases. Front Physiol 7:206. doi:10.3389/fphys.2016.00206
Gielis JF, Lin JY, Wingler K, Van Schil PE, Schmidt HH, Moens AL (2011) Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic Biol Med 50(7):765–776. doi:10.1016/j.freeradbiomed.2010.12.018
Hermann M, Flammer A, Luscher TF (2006) Nitric oxide in hypertension. J Clin Hypertens (Greenwich) 8(12 Suppl 4):17–29
Laroux FS, Lefer DJ, Kawachi S, Scalia R, Cockrell AS, Gray L, Van der Heyde H, Hoffman JM, Grisham MB (2000) Role of nitric oxide in the regulation of acute and chronic inflammation. Antioxid Redox Signal 2(3):391–396. doi:10.1089/15230860050192161
Napoli C, de Nigris F, Williams-Ignarro S, Pignalosa O, Sica V, Ignarro LJ (2006) Nitric oxide and atherosclerosis: an update. Nitric Oxide 15(4):265–279. doi:10.1016/j.niox.2006.03.011
Ignarro LJ, Napoli C (2004) Novel features of nitric oxide, endothelial nitric oxide synthase, and atherosclerosis. Curr Atheroscler Rep 6(4):281–287
Andreasson K (2010) Emerging roles of PGE2 receptors in models of neurological disease. Prostaglandins Other Lipid Mediat 91(3–4):104–112. doi:10.1016/j.prostaglandins.2009.04.003
Cimino PJ, Keene CD, Breyer RM, Montine KS, Montine TJ (2008) Therapeutic targets in prostaglandin E2 signaling for neurologic disease. Curr Med Chem 15(19):1863–1869
Egan KM, Wang M, Fries S, Lucitt MB, Zukas AM, Pure E, Lawson JA, FitzGerald GA (2005) Cyclooxygenases, thromboxane, and atherosclerosis: plaque destabilization by cyclooxygenase-2 inhibition combined with thromboxane receptor antagonism. Circulation 111(3):334–342. doi:10.1161/01.cir.0000153386.95356.78
Ganesh T (2013) Prostanoid receptor EP2 as a therapeutic target. J Med Chem. doi:10.1021/jm401431x
Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K (2005) Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci Off J Soc Neurosci 25(44):10180–10187. doi:10.1523/jneurosci.3591-05.2005
Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, Andreasson K (2010) The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. J Immunol (Baltimore, Md: 1950) 184(12):7207–7218. doi:10.4049/jimmunol.0903487
Yang G, Chen L (2016) An update of microsomal prostaglandin E synthase-1 and PGE2 receptors in cardiovascular health and diseases. Oxid Med Cell Longev:5249086. doi:10.1155/2016/5249086
Mesaros C, Blair IA (2016) Mass spectrometry-based approaches to targeted quantitative proteomics in cardiovascular disease. Clin Proteomics 13:20. doi:10.1186/s12014-016-9121-1
Belton O, Byrne D, Kearney D, Leahy A, FitzGerald DJ (2000) Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation 102(8):840–845
Lawrence T (2009) The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1(6):a001651. doi:10.1101/cshperspect.a001651
Panday A, Inda ME, Bagam P, Sahoo MK, Osorio D, Batra S (2016) Transcription factor NF-kappaB: an update on intervention strategies. Arch Immunol Ther Exp 64(6):463–483. doi:10.1007/s00005-016-0405-y
Van der Heiden K, Cuhlmann S, Luong le A, Zakkar M, Evans PC (2010) Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci (London, England: 1979) 118(10):593–605. doi:10.1042/cs20090557
Lakshmi SV, Padmaja G, Kuppusamy P, Kutala VK (2009) Oxidative stress in cardiovascular disease. Indian J Biochem Biophys 46(6):421–440
Mooradian AD (2016) Therapeutic Targeting of cellular stress to prevent cardiovascular disease: a review of the evidence. Am J Cardiovasc Drugs 17(2):83–95. doi:10.1007/s40256-016-0199-7
Panth N, Paudel KR (2016) Reactive oxygen species: a key hallmark of cardiovascular disease. Adv Med 2016:9152732
Byon CH, Heath JM, Chen Y (2016) Redox signaling in cardiovascular pathophysiology: a focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol 9:244–253. doi:10.1016/j.redox.2016.08.015
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39(1):44–84. doi:10.1016/j.biocel.2006.07.001
Leone A (2016) Markers of atherosclerotic disease: what do they mean? Current opinion and future trends. Curr Pharm Des 22(1):7–17
Cerletti C, Tamburrelli C, Izzi B, Gianfagna F, de Gaetano G (2012) Platelet-leukocyte interactions in thrombosis. Thromb Res 129(3):263–266. doi:10.1016/j.thromres.2011.10.010
Esmon CT (1999) Possible involvement of cytokines in diffuse intravascular coagulation and thrombosis. Baillieres Best Pract Res Clin Haematol 12(3):343–359
Lester PA, Diaz JA, Shuster KA, Henke PK, Wakefield TW, Myers DD (2012) Inflammation and thrombosis: new insights. Front Biosci (Schol Ed) 4:620–638
Samad F, Ruf W (2013) Inflammation, obesity, and thrombosis. Blood 122(20):3415–3422. doi:10.1182/blood-2013-05-427708
Breitenstein A, Camici GG, Tanner FC (2009) Tissue factor: beyond coagulation in the cardiovascular system. Clin Sci (Lond, England 1979) 118(3):159–172. doi:10.1042/cs20080622
Meerarani P, Moreno PR, Cimmino G, Badimon JJ (2007) Atherothrombosis: role of tissue factor; link between diabetes, obesity and inflammation. Indian J Exp Biol 45(1):103–110
Yoo JY, Kim SS (2016) Probiotics and prebiotics: present status and future perspectives on metabolic disorders. Forum Nutr 8(3):173. doi:10.3390/nu8030173
Ajjan RA, Gamlen T, Standeven KF, Mughal S, Hess K, Smith KA, Dunn EJ, Anwar MM, Rabbani N, Thornalley PJ, Philippou H, Grant PJ (2013) Diabetes is associated with posttranslational modifications in plasminogen resulting in reduced plasmin generation and enzyme-specific activity. Blood 122(1):134–142. doi:10.1182/blood-2013-04-494641
Ruiz-Núñez B, Dijck-Brouwer DA, Muskiet FA (2016) The relation of saturated fatty acids with low-grade inflammation and cardiovascular disease. J Nutr Biochem 36:1–20. doi:10.1016/j.jnutbio.2015.12.007
Esser N, Paquot N, Scheen AJ (2015) Inflammatory markers and cardiometabolic diseases. Acta Clin Belg 70(3):193–199. doi:10.1179/2295333715y.0000000004
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115(5):1111–1119. doi:10.1172/jci25102
Can U, Buyukinan M, Guzelant A, Ugur A, Karaibrahimoglu A, Yabanciun S (2016) Investigation of the inflammatory biomarkers of metabolic syndrome in adolescents. J Pediatr Endocrinol Metab 29(11):1277–1283. doi:10.1515/jpem-2016-0136
Rani V, Deep G, Singh RK, Palle K, Yadav UC (2016) Oxidative stress and metabolic disorders: pathogenesis and therapeutic strategies. Life Sci 148:183–193. doi:10.1016/j.lfs.2016.02.002
Maiorino MI, Bellastella G, Giugliano D, Esposito K (2016) Cooling down inflammation in type 2 diabetes: how strong is the evidence for cardiometabolic benefit? Endocrine. doi:10.1007/s12020-016-0993-7
Chacón MR, Vendrell J, Miranda M, Ceperuelo-Mallafré V, Megía A, Gutiérrrez C, Fernández-Real JM, Richart C, Garcia-España A (2007) Different TNFalpha expression elicited by glucose in monocytes from type 2 diabetes mellitus patients. Atherosclerosis 194(2):e18–e25. doi:10.1016/j.atherosclerosis.2006.12.011
Kampoli AM, Tousoulis D, Briasoulis A, Latsios G, Papageorgiou N, Stefanadis C (2011) Potential pathogenic inflammatory mechanisms of endothelial dysfunction induced by type 2 diabetes mellitus. Curr Pharm Des 17(37):4147–4158
Mandosi E, Fallarino M, Gatti A, Carnovale A, Rossetti M, Lococo E, Buchetti B, Filetti S, Lenti L, Morano S (2010) Atorvastatin downregulates monocyte CD36 expression, nuclear NFkappaB and TNFalpha levels in type 2 diabetes. J Atheroscler Thromb 17(6):539–545
Prasad P, Tiwari AK, Kumar KM, Ammini AC, Gupta A, Gupta R, Thelma BK (2007) Association of TGFbeta1, TNFalpha, CCR2 and CCR5 gene polymorphisms in type-2 diabetes and renal insufficiency among Asian Indians. BMC Med Genet 8:20. doi:10.1186/1471-2350-8-20
Song J, Chen S, Liu X, Duan H, Kong J, Li Z (2015) Relationship between C-reactive protein level and diabetic retinopathy: a systematic review and meta-analysis. PLoS One 10(12):e0144406. doi:10.1371/journal.pone.0144406
Tehrani DM, Wong ND (2016) Integrating biomarkers and imaging for cardiovascular disease risk assessment in diabetes. Curr Cardiol Rep 18(11):105. doi:10.1007/s11886-016-0789-7
Škrha J, Šoupal J, Škrha J, Prázný M (2016) Glucose variability, HbA1c and microvascular complications. Rev Endocr Metab Disord 17(1):103–110. doi:10.1007/s11154-016-9347-2
Buchs AE, Kornberg A, Zahavi M, Aharoni D, Zarfati C, Rapoport MJ (2004) Increased expression of tissue factor and receptor for advanced glycation end products in peripheral blood mononuclear cells of patients with type 2 diabetes mellitus with vascular complications. Exp Diabesity Res 5(2):163–169. doi:10.1080/15438600490424325
Kell DB, Pretorius E (2015) The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr Biol 7:24–52. doi:10.1039/C4IB00173G
Aksu G, Ozturk C, Kavakli K, Genel F, Kutukculer N (2007) Hypercoagulability: interaction between inflammation and coagulation in familial Mediterranean fever. Clin Rheumatol 26(3):366–370. doi:10.1007/s10067-006-0334-y
Anžej S, Božič M, Antovič A, Peternel P, Gašperšič N, Rot U, Tratar G, Stegnar M (2007) Evidence of hypercoagulability and inflammation in young patients long after acute cerebral ischaemia. Thromb Res 120(1):39–46. doi:10.1016/j.thromres.2006.08.005
Borissoff JI, Spronk HMH, ten Cate H (2011) The hemostatic system as a modulator of atherosclerosis. N Engl J Med 364(18):1746–1760
Chertok-Shacham E, Ishay A, Lavi I, Luboshitzky R (2008) Biomarkers of hypercoagulability and inflammation in primary hyperparathyroidism. Med Sci Monit 14(12):CR628–CR632
Choi G, Schultz MJ, Levi M, van der Poll T (2006) The relationship between inflammation and the coagulation system. Swiss Med Wkly 136(9–10):139–144
Chu AJ (2005) Tissue factor mediates inflammation. Arch Biochem Biophys 440(2):123–132. doi:10.1016/j.abb.2005.06.005
Cicala C, Cirino G (1998) Linkage between inflammation and coagulation: an update on the molecular basis of the crosstalk. Life Sci 62(20):1817–1824
Conway DSG, Buggins P, Hughes E, Lip GYH (2004) Predictive value of indexes of inflammation and hypercoagulability on success of cardioversion of persistent atrial fibrillation. Am J Cardiol 94(4):508–510. doi:10.1016/j.amjcard.2004.04.070
Coughlin SR (2000) Thrombin signalling and protease-activated receptors. Nature 407(6801):258–264. doi:10.1038/35025229
Dusitanond P, Eikelboom JW, Hankey GJ, Thom J, Gilmore G, Loh K, Yi Q, CJM K, Langton P, van Bockxmeer FM, Baker R, Jamrozik K (2005) Homocysteine-lowering treatment with folic acid, cobalamin, and pyridoxine does not reduce blood markers of inflammation, endothelial dysfunction, or hypercoagulability in patients with previous transient ischemic attack or stroke: a randomized substudy of the VITATOPS trial. Stroke 36(1):144–146. doi:10.1161/01.STR.0000150494.91762.70
Esmon CT (2005) The interactions between inflammation and coagulation. Br J Haematol 131(4):417–430. doi:10.1111/j.1365-2141.2005.05753.x
Esmon CT (2013) Molecular circuits in thrombosis and inflammation. Thromb Haemost 109(3):416–420. doi:10.1160/Th12-08-0634
Esmon CT (2014) Targeting factor Xa and thrombin: impact on coagulation and beyond. Thromb Haemost 111(4):625–633. doi:10.1160/Th13-09-0730
Gupta A, Watkins A, Thomas P, Majer R, Habubi N, Morris G, Pansari K (2005) Coagulation and inflammatory markers in Alzheimer’s and vascular dementia. Int J Clin Pract 59(1):52–57. doi:10.1111/j.1742-1241.2004.00143.x
Gurbel PA, Bliden KP, Kreutz RP, Dichiara J, Antonino MJ, Tantry US (2009) The link between heightened thrombogenicity and inflammation: pre-procedure characterization of the patient at high risk for recurrent events after stenting. Platelets 20(2):97–104. doi:10.1080/09537100802687666
Hatoum OA, Binion DG (2005) The vasculature and inflammatory bowel disease: contribution to pathogenesis and clinical pathology. Inflamm Bowel Dis 11(3):304–313
Karabudak O, Ulusoy RE, Erikci AA, Solmazgul E, Dogan B, Harmanyeri Y (2008) Inflammation and hypercoagulable state in adult psoriatic men. Acta Derm Venereol 88(4):337–340. doi:10.2340/00015555-0456
Kitchens CS, Erkan D, Brandão LR, Hahn S, James AH, Kulkarni R, Pericak-Vance M, Vance J, Ortel TL (2011) Thrombotic storm revisited: preliminary Diagnostic criteria suggested by the thrombotic storm study group. Am J Med 124(4):290–296. doi:10.1016/j.amjmed.2010.10.018
Kon ZN, Brown EN, Grant MC, Ozeki T, Burris NS, Collins MJ, Kwon MH, Poston RS (2008) Warm ischemia provokes inflammation and regional hypercoagulability within the heart during off-pump coronary artery bypass: a possible target for serine protease inhibition. Eur J Cardiothorac Surg 33(2):215–221. doi:10.1016/j.ejcts.2007.11.008
Levi M, ten Cate H (1999) Disseminated intravascular coagulation. N Engl J Med 341(8):586–592. doi:10.1056/Nejm199908193410807
Levi M, van der Poll T (2010) Inflammation and coagulation. Crit Care Med 38(2 Suppl):S26–S34. doi:10.1097/CCM.0b013e3181c98d21
Levi M, van der Poll T (2013) Disseminated intravascular coagulation: a review for the internist. Intern Emerg Med 8(1):23–32. doi:10.1007/s11739-012-0859-9
Levi M, van der Poll T, Buller HR (2004) Bidirectional relation between inflammation and coagulation. Circulation 109(22):2698–2704. doi:10.1161/01.CIR.0000131660.51520.9A
Libby P, Simon DI (2001) Inflammation and thrombosis: the clot thickens. Circulation 103(13):1718–1720
Marcucci R, Gori AM, Giannotti F, Baldi M, Verdiani V, Del Pace S, Nozzoli C, Abbate R (2006) Markers of hypercoagulability and inflammation predict mortality in patients with heart failure. J Thromb Haemost: JTH 4(5):1017–1022. doi:10.1111/j.1538-7836.2006.01916.x
Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454(7203):428–435
Petäjä J (2011) Inflammation and coagulation. An overview. Thromb Res 127(Suppl 2):S34–S37. doi:10.1016/S0049-3848(10)70153-5
Tantry US, Bliden KP, Suarez TA, Kreutz RP, Dichiara J, Gurbel PA (2010) Hypercoagulability, platelet function, inflammation and coronary artery disease acuity: results of the Thrombotic RIsk Progression (TRIP) study. Platelets 21(5):360–367. doi:10.3109/09537100903548903
Thor M, Yu A, Swedenborg J (2002) Markers of inflammation and hypercoagulability in diabetic and nondiabetic patients with lower extremity ischemia. Thromb Res 105(5):379–383
Tomobe K (2007) The relation between adiponectin and four hypercoagulable, inflammatory biomarkers during normal pregnancy. Dokkyo J med Sci 34(2):69–77
Turpie AGG, Esmon C (2011) Venous and arterial thrombosis--pathogenesis and the rationale for anticoagulation. Thromb Haemost 105(4):586–596. doi:10.1160/TH10-10-0683
Van de Wouwer M, Collen D, Conway EM (2004) Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol 24(8):1374–1383. doi:10.1161/01.ATV.0000134298.25489.92
van der Poll T, de Boer JD, Levi M (2011) The effect of inflammation on coagulation and vice versa. Curr Opin Infect Dis 24(3):273–278. doi:10.1097/QCO.0b013e328344c078
Yoshida H, Granger DN (2009) Inflammatory bowel disease: a paradigm for the link between coagulation and inflammation. Inflamm Bowel Dis 15(8):1245–1255. doi:10.1002/Ibd.20896
Lipinski B, Pretorius E (2012) Novel pathway of iron-induced blood coagulation: implications for diabetes mellitus and its complications. Pol Arch Med Wewn 122(3):115–122
Pretorius E, Bester J (2016) Viscoelasticity as a measurement of clot structure in poorly controlled type 2 diabetes patients: towards a precision and personalized medicine approach. Oncotarget 7(32):50895–50907. doi:10.18632/oncotarget.10618
Pretorius E, Kell DB (2014) Diagnostic morphology: biophysical indicators for iron-driven inflammatory diseases. Integr Biol 6:486–510. doi:10.1039/c4ib00025k
Pretorius E, Oberholzer HM, van der Spuy WJ, Swanepoel AC, Soma P (2011) Qualitative scanning electron microscopy analysis of fibrin networks and platelet abnormalities in diabetes. Blood Coagul Fibrinolysis 22(6):463–467. doi:10.1097/MBC.0b013e3283468a0d
Alzahrani SH, Ajjan RA (2010) Coagulation and fibrinolysis in diabetes. Diab Vasc Dis Res 7(4):260–273. doi:10.1177/1479164110383723
Henschen-Edman AH (2001) Fibrinogen non-inherited heterogeneity and its relationship to function in health and disease. Ann N Y Acad Sci 936:580–593
Pieters M, van Zyl DG, Rheeder P, Jerling JC, Loots du T, van der Westhuizen FH, Gottsche LT, Weisel JW (2007) Glycation of fibrinogen in uncontrolled diabetic patients and the effects of glycaemic control on fibrinogen glycation. Thromb Res 120(3):439–446. doi:10.1016/j.thromres.2006.10.016
Dunn EJ, Philippou H, Ariëns RA, Grant PJ (2006) Molecular mechanisms involved in the resistance of fibrin to clot lysis by plasmin in subjects with type 2 diabetes mellitus. Diabetologia 49(5):1071–1080. doi:10.1007/s00125-006-0197-4
Hess K, Alzahrani SH, Mathai M, Schroeder V, Carter AM, Howell G, Koko T, Strachan MW, Price JF, Smith KA, Grant PJ, Ajjan RA (2012) A novel mechanism for hypofibrinolysis in diabetes: the role of complement C3. Diabetologia 55(4):1103–1113. doi:10.1007/s00125-011-2301-7
Auwerx J, Bouillon R, Collen D, Geboers J (1988) Tissue-type plasminogen activator antigen and plasminogen activator inhibitor in diabetes mellitus. Arteriosclerosis (Dallas, Tex) 8(1):68–72
Jotic A, Milicic T, Covickovic Sternic N, Kostic VS, Lalic K, Jeremic V (2015) Decreased insulin sensitivity and impaired fibrinolytic activity in type 2 diabetes patients and nondiabetics with ischemic stroke. Int J Endocrinol:934791. doi:10.1155/2015/934791
Schneider DJ, Sobel BE (2012) PAI-1 and diabetes: a journey from the bench to the bedside. Diabetes Care 35(10):1961–1967. doi:10.2337/dc12-0638
Vague P, Juhan-Vague I, Aillaud MF, Badier C, Viard R, Alessi MC, Collen D (1986) Correlation between blood fibrinolytic activity, plasminogen activator inhibitor level, plasma insulin level, and relative body weight in normal and obese subjects. Metab Clin Exp 35(3):250–253
Chen YQ, Su M, Walia RR, Hao Q, Covington JW, Vaughan DE (1998) Sp1 sites mediate activation of the plasminogen activator inhibitor-1 promoter by glucose in vascular smooth muscle cells. J Biol Chem 273(14):8225–8231
Schneider DJ, Hayes M, Wadsworth M, Taatjes H, Rincon M, Taatjes DJ, Sobel BE (2004) Attenuation of neointimal vascular smooth muscle cellularity in atheroma by plasminogen activator inhibitor type 1 (PAI-1). J Histochem Cytochem 52(8):1091–1099. doi:10.1369/jhc.4A6260.2004
Hatton MW, Southward SM, Ross-Ouellet B, Richardson M, Winocour PD (1995) Comparative metabolism of plasminogen glycoforms I and II in the alloxan-diabetic rabbit. Am J Phys 269(6 Pt 1):E1017–E1023
Nielsen VG (2008) Beyond cell based models of coagulation: analyses of coagulation with clot “lifespan” resistance-time relationships. Thromb Res 122(2):145–152. doi:10.1016/j.thromres.2007.09.003
Nielsen VG, Audu P, Cankovic L, Lyerly RT 3rd, Steenwyk BL, Armstead V, Powell G (2007) Qualitative thrombelastographic detection of tissue factor in human plasma. Anesth Analg 104(1):59–64. doi:10.1213/01.ane.0000248223.05152.a1
Nielsen VG, Boyer LV (2015) Iron and carbon monoxide attenuate degradation of plasmatic coagulation by Crotalus atrox venom. Blood Coagul Fibrinolysis 27(5):506–510. doi:10.1097/mbc.0000000000000440
Nielsen VG, Cerruti MA, Valencia OM, Amos Q (2016) Decreased snake venom metalloproteinase effects via inhibition of enzyme and modification of fibrinogen. Biometals 29(5):913–919. doi:10.1007/s10534-016-9963-z
Nielsen VG, Kirklin JK, Holman WL, Steenwyk BL (2009) Clot lifespan model analysis of the effects of warfarin on thrombus growth and fibrinolysis: role of contact protein and tissue factor initiation. ASAIO J 55(1):33–40. doi:10.1097/MAT.0b013e318190c1a9
Nielsen VG, Losada PA (2016) Direct inhibitory effects of carbon monoxide on six venoms containing Fibrinogenolytic metalloproteinases. Basic Clin Pharmacol Toxicol 120(2):207–212. doi:10.1111/bcpt.12654
Nielsen VG, Matika RW (2016) Effects of iron and carbon monoxide on lachesis muta muta venom-mediated degradation of plasmatic coagulation. Hum Exp Toxicol. doi:10.1177/0960327116661401
Strydom MA, Bester J, Mbotwe S, Pretorius E (2016) The effect of physiological levels of South African puff adder (Bitis arietans) snake venom on blood cells: an in vitro model. Sci Rep 6:35988. doi:10.1038/srep35988
Pretorius E, Bester J, Vermeulen N, Alummoottil S, Soma P, Buys AV, Kell DB (2015) Poorly controlled type 2 diabetes is accompanied by significant morphological and ultrastructural changes in both erythrocytes and in thrombin-generated fibrin: implications for diagnostics. Cardiovasc Diabetol 14:30. doi:10.1186/s12933-015-0192-5
Soma P, Pretorius E (2015) Interplay between ultrastructural findings and atherothrombotic complications in type 2 diabetes mellitus. Cardiovasc Diabetol 14:96. doi:10.1186/s12933-015-0261-9
Aras R, Sowers JR, Arora R (2005) The proinflammatory and hypercoagulable state of diabetes mellitus. Rev Cardiovasc Med 6(2):84–97
Banga JD (2002) Coagulation and fibrinolysis in diabetes. Semin Vasc Med 2(1):75–86. doi:10.1055/s-2002-23098
Beijers HJBH, Ferreira I, Spronk HMH, Bravenboer B, Dekker JM, Nijpels G, ten Cate H, Stehouwer CDA (2012) Impaired glucose metabolism and type 2 diabetes are associated with hypercoagulability: potential role of central adiposity and low-grade inflammation--the Hoorn Study. Thromb Res 129(5):557–562. doi:10.1016/j.thromres.2011.07.033
Carr ME (2001) Diabetes mellitus: a hypercoagulable state. J Diabet Complicat 15(1):44–54
Grant PJ (2007) Diabetes mellitus as a prothrombotic condition. J Intern Med 262(2):157–172. doi:10.1111/j.1365-2796.2007.01824.x
Tripodi A, Branchi A, Chantarangkul V, Clerici M, Merati G, Artoni A, Mannucci PM (2011) Hypercoagulability in patients with type 2 diabetes mellitus detected by a thrombin generation assay. J Thromb Thrombolysis 31(2):165–172. doi:10.1007/s11239-010-0506-0
Ye Y, Perez-Polo JR, Aguilar D, Birnbaum Y (2011) The potential effects of anti-diabetic medications on myocardial ischemia-reperfusion injury. Basic Res Cardiol 106(6):925–952. doi:10.1007/s00395-011-0216-6
Alzahrani SH, Hess K, Price JF, Strachan M, Baxter PD, Cubbon R, Phoenix F, Gamlen T, Ariëns RAS, Grant PJ, Ajjan RA (2012) Gender-specific alterations in fibrin structure function in type 2 diabetes: associations with cardiometabolic and vascular markers. J Clin Endocrinol Metab 97(12):E2282–E2287. doi:10.1210/jc.2012-2128
Bochenek M, Zalewski J, Sadowski J, Undas A (2013) Type 2 diabetes as a modifier of fibrin clot properties in patients with coronary artery disease. J Thromb Thrombolysis 35(2):264–270. doi:10.1007/s11239-012-0821-8
Cucuianu M, Fekete T, Marcusiu C, Mosler R, Dutu A (1984) Fibrinolysis in diabetes mellitus. Role of overweight and hypertriglyceridemia. Med Interne 22(3):171–177
Dunn EJ, Ariëns RAS, Grant PJ (2005) The influence of type 2 diabetes on fibrin structure and function. Diabetologia 48(6):1198–1206. doi:10.1007/s00125-005-1742-2
Konieczynska M, Fil K, Bazanek M, Undas A (2014) Prolonged duration of type 2 diabetes is associated with increased thrombin generation, prothrombotic fibrin clot phenotype and impaired fibrinolysis. Thromb Haemost 111(4):685–693. doi:10.1160/th13-07-0566
Walus-Miarka M, Wolkow P, Cyganek K, Mirkiewicz-Sieradzka B, Malecki MT, Undas A (2012) Altered fibrin-clot properties are associated with retinopathy in type 2 diabetes mellitus. Diabete Metab 38(5):462–465. doi:10.1016/j.diabet.2012.03.007
Yano Y, Kitagawa N, Gabazza EC, Morioka K, Urakawa H, Tanaka T, Katsuki A, Araki-Sasaki R, Hori Y, Nakatani K, Taguchi O, Sumida Y, Adachi Y (2003) Increased plasma thrombin-activatable fibrinolysis inhibitor levels in normotensive type 2 diabetic patients with microalbuminuria. J Clin Endocrinol Metab 88(2):736–741. doi:10.1210/jc.2002-020691
de Villiers S, Swanepoel A, Bester J, Pretorius E (2015) Novel Diagnostic and monitoring tools in stroke: an individualized patient-centered precision medicine approach. J Atheroscler Thromb 23:493–504. doi:10.5551/jat.32748
Ethical Approval Disclosure
Ethical approval was granted at the University of Pretoria (UP) (Human Ethics Committee: Faculty of Health Sciences): E Pretorius. (EP was previously employed at UP).
Conflict of Interest
The author has nothing to disclose.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Pretorius, E. (2017). Mechanisms of Hypercoagulation and Aberrant Clot Lyses in Type 2 Diabetes. In: Kartha, C., Ramachandran, S., Pillai, R. (eds) Mechanisms of Vascular Defects in Diabetes Mellitus. Advances in Biochemistry in Health and Disease, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-319-60324-7_17
Download citation
DOI: https://doi.org/10.1007/978-3-319-60324-7_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60323-0
Online ISBN: 978-3-319-60324-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)