
Abstract
We carried out molecular dynamics simulations to investigate the process of formation of ZnO nanoclusters from the gas phase. To describe the interaction between atoms, we used the reactive force field (ReaxFF). We have been studied the influence of cooling rate and concentration of atoms on the physical parameters of obtained nanoparticles. Six computer experiments were conducted with different initial cooling rates and concentrations. Depending on this it is possible different mechanisms of cluster growth. In present work it is shown that the processes of condensation of atoms from the high-temperature gas environment are divided into several stages: nucleation, coalescence and agglomeration, and aggregation and coagulation. It is concluded that the structure, shape, and size of the obtained particles directly depend on the cooling rate of the system. During the analysis it was found that created clusters mainly are formed in three structural phases – amorphous, hexagonal wurtzite, and cubic zincblende.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others

1 Introduction
The development of nanotechnology has attracted great interest to study the properties of nanoparticles and their synthesis. Investigation of the nanoparticle will allow to more fully understand the processes of phase transitions and self-organization in complex disperse systems. Currently, studying of individual nanoparticles is one of the most rapidly developing areas of research in physics, chemistry, and engineering. Huge scientific and practical interest in such research results from the uncommon properties of nanoparticles that are already or will be widely used in the future for the fabrication of miniature electronic devices, and production of new materials.
From all metal oxide nanomaterials, which are now widely used, zinc oxide takes a special place. ZnO is a quite interesting material with a wide range of technical applications. Zinc oxide is a wide-bandgap semiconductor with bandgap E g = 3.37 eV; it is suitable for applications in optoelectronic devices with short wavelengths, such as UV light-emitting diodes and transparent field-effect transistors. It has several favorable properties, including good transparency, high electron mobility, wide bandgap, and strong room temperature luminescence. Zinc oxide crystallizes in two main structures – hexagonal wurtzite and cubic zincblende, with lattice constants (a = 3.24992 Å, c = 5.20658 Å) [1]. Nanopowdered zinc oxide is promising material as a working environment in lasers. Thin films and other nanostructures based on zinc oxide can be used as sensors of gas and biological sensors [2].
The shape of nanoparticles depends on the method and conditions of synthesis. One of the easiest and most popular methods obtaining nanoparticles is gas-phase evaporation of the solid target material in an inert atmosphere, followed by condensation on the surface of the substrate. This method has a rather high efficiency and is widely used in industrial scale. The technology of this method is quite well designed for production of a large number of powdered nonferrous metals such as aluminum, copper, antimony, zinc, palladium, and silver. In this method, the evaporation of material occurs by pulsed laser ablation. In recent years, pulsed laser ablation (PLA) has become a promising nanocluster synthesis technique for photonics, electronics, and medicine. We proposed a method of synthesis of metal oxide nanopowder by pulsed laser ablation of a metal target (Zn, Sn, etc.) in chemically active environment [3]. Laser impulse heats material of metallic target to a high temperature, and atoms evaporate into the background gas. Inert gas contributes reducing of the kinetic energy of evaporated atoms and formation of nanoclusters. Carefully selecting laser radiation parameters and pressure of background gas, we can control structure and particle size. Understanding the physical and chemical processes, which influence on the evolution of nanoparticles in the presence of gas, is extremely important for the further development of this technology.
It is known that the properties of nanoparticles are determined by their structure, and the structure is determined by the process of the growth of nanoparticles. Depending on technological features of the synthesis of particles, processes of the formation can vary significantly. Experimental study of the formation mechanisms of nanoparticles is technically a challenging and time-consuming task because of the small size of these objects. In conditions of experimental synthesis, sometimes it is complicated to study in detail the impact of the basic parameters of synthesis on physical, chemical, and structural properties and outer shape of derived particles. However, computer modeling is an alternative and promising way to study the mechanisms of formation of nanoobjects. Using the methods of computer modeling, we can investigate in detail the processes of synthesis of nanoparticles upon condensation from the gas phase [4, 5]. In this article we considered processes of formation of zinc oxide nanoparticles upon condensation from the gas phase in the oxygen environment by molecular dynamics method. Molecular dynamics simulation has contributed significantly to enhance the fundamental understanding of physical and chemical mechanisms at nanoscale. It can be used to study many important unanswered questions relating to nanoparticles that cannot be directly addressed by continuum approaches.
2 Model and Method
Simulation of the formation processes of nanoparticles, which reproduces the synthesis technology process of thermal saturation and condensation with subsequent formation of nanoclusters, was carried out by molecular dynamics [6] method that is based on numerical solution of Newtonian differential equations of motion for each atom of the system with initial values of speed and coordinates.
where i = 0, …, N, N is the number of atoms in system, m i is mass of i atom, U(r(t)) is potential energy of the system, and r i (t) is coordinate of i atom.
In classical MD simulations, interactions between atoms are described by empirical force fields which are parameterized based upon spectroscopy measurements of small molecules and quantum mechanics calculations. So, central element of the molecular dynamics method is the empirical interatomic potential, through which is carried out calculation of the interaction forces between the particles. Most often, in classical molecular dynamics, particles are presented in the form of point masses. The choice of potential is determined by the character of the problem. Modeling of nanostructures is completely based on the detailed description of particles, which is included in their composition. Therefore, the modeling of the formation of zinc oxide nanoclusters from high-temperature gas phase was conducted using a reactive force field (ReaxFF) that was developed by David Raymand and Adri C.T. van Duin [7].
ReaxFF was designed for a wide range of chemical compounds including ZnO. Common expression for the total energy of the system includes the sum of all terms that describe individual chemical bonds in materials, thus providing accurate modeling of chemical processes:
These partial contributions include bond energies (E bond), under-coordination penalty energies (E under), lone-pair energies (E lp), over-coordination penalty energies (E over), valence angle energies (E val), energy penalty for handling atoms with two double bonds (E pen), torsion angle energies (E tors), conjugated bond energies (E conj), and terms to handle nonbonded interactions, namely van der Waals (E vdWaals) and Coulomb (E Coulomb) interactions. All terms except the last two are bond order dependent, i.e., will contribute more or less depending on the local environment of each atom [7].
Essentially, the reactive force field is an empirical force field derived from a quantum mechanics parameterization [8]. The bond order is directly calculated from an interatomic distance and updated every iteration for all bonded interactions, including covalent bonds, valence, and torsion angles. In addition, the ReaxFF describes nonbonded van der Waals and Coulomb interactions. Such interactions are calculated for all pairs of atoms, and, by incorporating a shielding term, extremely close-range interactions can be modified. Polarization effects also are considered by using a geometry-dependent charge distribution derived from an electronegativity equalization method [9].
For modeling and analysis of the formation processes of ZnO nanoparticles, we have been used Large-scale Atomic/Molecular Massively Parallel Simulator, which was developed by a scientific group from Sandia National Laboratories (USA). Calculations were performed on a computing cluster of the Institute for Applied Problems of Mechanics and Mathematics, which is based on four Intel Xeon multi-core processors under operating system Linux ROCKS.
We carried out the research with two initial configurations. The first configuration composed of four hundred atoms of zinc and the same number of oxygen atoms, and the second one composed of four thousand atoms of zinc and four thousand oxygen atoms, which were uniformly distributed with periodic boundary conditions in a region of space with a volume of V = 8000 nm3. Cell size for these two configurations was the same (200 × 200 × 200 Å3); it was done in order to set two different initial concentrations of atoms (1019, 1020 atoms/cm3). In order to avoid premature unification of atoms at the very early stages of evolution, the average interatomic distance was set greater than the potential cutoff radius.
The direction of the initial velocities was selected randomly. Our system was cooled with certain fixed speed to the desired final temperature T = 300 K. Important aspect of the simulation is coupling the system with a heat bath. Since significant amount of binding energy was being released, when clusters formed, such coupling is necessary in order to avoid unphysical temperature increases. In real experiments this coupling is provided by the inert gas atmosphere. In our computational simulation, the control of temperature was carried out using the Berendsen thermostat method. This method is widely used for molecular dynamics simulations of the large number degrees of freedom. To maintain the temperature system is coupled to an external heat bath with fixed temperature. The velocities are scaled at each step, such that the rate of change of temperature is proportional to the difference in temperature.
Throughout the simulation, system snapshots were saved every 0.5 ps. These images, except visual observation, also had been used to analyze the shape, size, structure, and number of newly formed nanoparticles. It was believed that atoms belong to one cluster, when the distance between them is less than 3 Å. In this work the CNA method was used to analyze the internal structure of clusters [10]. Analysis was carried out layer by layer on samples, which have the typical characteristics for all nanoobjects.
3 Results and Discussion
It is known that the properties of nanoparticles are determined by their structural features, which in its turn are the result of nanoparticle growth. Therefore, we have thoroughly analyzed the structure, shape, and sizes of the investigated nanoclusters.
It was established that grouping of atoms into nanoclusters was carried out rather actively in the first moments of time, and it was accompanied by the formation of a significant number of monomers. At the following moments of time, condensation of already formed nanoobjects was observed that was accompanied by gradual decrease of the number of nanoparticles and increase of their size (Figs. 11.1 and 11.2).

Evolution of a system of 400 O atoms and 400 Zn atoms from (a) t = 0 ns, T = 1800 K to (d) t = 5 ns, T = 300 K

Evolution of a system of 4000 O atoms and 4000 Zn atoms from (a) t = 0 ns, T = 1800 K to (d) t = 5 ns, T = 300 K
The processes of condensation of atoms from the high-temperature gas environment are divided into several stages: nucleation, surface growth, aggregation and coagulation, and coalescence and agglomeration. The first stage is the nucleation process. After termination of this process, the subsequent growth of clusters can proceed according to various mechanisms. The main mechanisms of this process are agglomeration and coalescence. During the nucleation process, there is an association of free atoms into small particles, into so-called dimers and trimers, that leads to a dramatic increase of particles in the system (Fig. 11.3). With further heat removal, small clustered fragments begin to form liquid drops with sizes of a few tens of atoms. This stage is shown in Fig. 11.2b, where a snapshot of trajectory of the system at time 0.3 ns is given. As a result of spontaneous collisions of these liquid drops, up to 0.5 ns, there are formed massive amorphous clustered aggregation.

Changes in the number of monomers in system as a function of time
The next stage after a high nucleation process was the stage of surface growth and coagulation process. At the stage of surface growth, single atoms, dimers, or trimers of the gas phase are joined to the cluster surface, causing its gradual growth. The reverse process is also possible – single atoms are evaporated from the surface of nanocluster. But, mostly, this process was observed in small quantities. It was caused by the intense heat removal, and at the same time decreasing of energy, that is required to overcome the potential barrier for breaking out from the surface of the cluster.
The last stage of the cluster formation was the processes of collision of already existing clusters on the scenario of agglomeration or coalescence. At agglomeration scenario, clusters were combined almost with no change of their original shape. In other words, the result of the agglomeration process was nanoobjects with different shapes that differ from spherical shape (Fig. 11.4). At coalescence scenario, nanoparticles merge together and form a single particle with shape that differ from shape that was before the collision of nanoclusters. Such mechanism is possible on the not very late stages of synthesis, when the temperature is still enough in order for nanoclusters to be in a liquid state. In other words, at low cooling rates, the main growth scenario is agglomeration, and the result was nonspherical shape of obtained nanoclusters, and at higher cooling rates the main scenario is coalescence, and clusters, mostly, had a spherical shape. Also, further, a slight change of the shape of nanoclusters due to changes in the position of individual atoms is possible. Therefore, a shape of newly created objects directly depends on the mechanism of the final stage of formation and, hence, on the cooling rate. So, depending on how quickly the system will be cooled, one or another mechanism will be more important.

Snapshot of a non-spherical cluster
We had been observed change of the main parameters of the system from condensation dynamics, setting different initial cooling rate. Depending on the concentration and cooling rate, it is possible different mechanisms of cluster growth. To study the influence of cooling rate and concentration of atoms on the physical parameters of nanoparticles, it was conducted six computer experiments with different initial cooling rates (0.0364, 0.0036, 0.0003 ps−1) and initial concentrations of atoms (1019, 1020 atoms/cm3). From the simulation results follows that the cooling rate directly affects the number of obtained particles. At reduction of cooling rate in 10 times, the number of obtained nanoparticles decreases approximately in 12 times (Fig. 11.5). Such dependence is quite logical, as, during slow cooling of the system, there is enough kinetic energy in order to chaotically move particles, at the same time, confronted among themselves, forming rather large clusters. These clusters are composed of hundreds of atoms, and they were created by the union between smaller clusters. Speaking about the number of obtained nanoparticles depending on the initial concentration, then obviously, the greater the concentration is, the greater the number of nanogranules will be.

Changes in the cooling rate as a function of number of finite nanoclusters
Observation on resizing of clusters has shown that the formation of stable cluster occurs through the formation of metastable cluster with its subsequent stabilization. In order to analyze the size of the received nanoparticles, we have considered changing the size of the largest cluster from time (Fig. 11.6). From the figure we can see that the cluster size increases with time. This is due to the fact that the potential energy of any cluster is less than the energy in the gas state, and thus, transition of the system to statistically equilibrium state accompanied by a constant reduction of potential energy, and, hence, there is increasing number of bonds in the system, so that there is a gradual clustering. On the basis of curve of potential energy of the system from the time (Fig. 11.7), it can be concluded that there is a separation between nucleation of clusters and their growth. In the first case, the dependence of the potential energy from time describes exponential dependence, and in the second, it has a linear dependence. Analyzing the impact of the rate of condensation on particle size distribution, it can be concluded that by increasing the cooling rate, in system a quite large number of small particles are formed, the average size of which does not exceed a few tens of atoms. At lower rate in system, much less clusters are formed and their average size was several thousand atoms (Fig. 11.8). This is associated to the fact that at such speeds of heat removal, atoms are quickly absorbed by nanoclusters, and in case, even, if they evaporate from the surface of the cluster, they cannot move away from it and again fall in a zone of its attraction.

Size of the largest cluster as a function of time

Changes in the potential energy of the system as a function of time

The average size of nanoclusters as a function of cooling rate
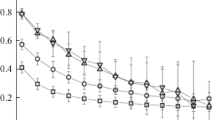
Using the CNA method, the internal morphology of obtained nanoclusters was thoroughly analyzed. During the analysis it was found that created clusters mainly are formed in three structural phases – amorphous, hexagonal wurtzite, and cubic zincblende. Based on results that are represented on Fig. 11.9, it can be said that the most appropriate cooling rate for getting nanoparticles with hexagonal structure is 0.0003 ps−1. From the figure, we can see that at high cooling rates it mainly formed amorphous particles and with decreasing rate, it increases percentage of clusters with structure which differs from amorphous. Namely, with decreasing rate in 10 times, percentage not amorphous clusters is increasing in 14 times. This is because the cooling rate is directly related to a time of simulation, namely, longer time corresponds lower rate of condensation, and, so, for such short time, atoms have not time to form energetically stable and with right internal structure nanoclusters. Therefore, the final structure of the particles directly depends on the cooling rate, and by the controlling of it, it is possible to control character of structure of obtained nanoclusters.

Changes in the structure of obtained nanoparticles as a function of cooling rate
4 Conclusions
In this study, the gas-phase condensation of ZnO nanoclusters was simulated by method of molecular dynamics using reactive force field. In this simulation, the process of synthesis of zinc oxide nanoparticles was conducted with three different cooling rates and two initial concentrations of atoms. It was shown that the structure, shape, and size of the obtained particles depend on the cooling rate and the concentration of atoms. During the analysis it was found that the main scenario, during the transition to lower cooling rates, becomes coalescence of nanoparticles and that created clusters mainly are formed in three structural phases – amorphous, hexagonal wurtzite, and cubic zincblende. So, understanding of the features and growth of nanoparticles can help in the manufacture of nanopowders with fixed size, shape, and structure and accordingly with fixed physicochemical properties.
References
Fierro JLG (2006) Metal oxides chemistry and applications, vol 182. CRC Press, Boca Raton
Zhyrovetsky V, Kovalyuk B, Mocharskyi V, Nikiforov Y, Onisimchuk V, Popovych D, Serednytski A (2013) Modification of structure and luminescence of ZnO nanopowder by the laser shock-wave treatment. Phys Status Solidi 10:1288–1291
Gafiychuk VV, Ostafiychuk BK, Popovych DI, Popovych ID, Serednytski AS (2011) ZnO nanoparticles produced by reactive laser ablation. Appl Surf Sci 257:8396–8401
Bovgyra OV, Bovgyra RV, Kovalenko MV et al (2013) The Density Functional Theory Study of Structural and Electronical Properties of ZnO Clusters. J Nano- Electron Phys 5(1):01027
Bovgyra OV, Bovgyra RV, Popovych DI, Serednytski AS (2015) The Density Functional Theory Study of Electronical Properties of (ZnO)12 Clusters During Gas Adsorption. J Nano- Electron Phys 7(4):04090
Rapaport DC (2004) The art of molecular dynamics simulation. Cambridge University Press, Cambridge
Raymand D, van Duin ACT, Baudin M, Hermansson K (2008) A reactive force field (ReaxFF) for zinc oxide. Surf Sci 602:1020–1031
van Duin ACT, Verners O, Shin Y-K (2013) Reactive force fields: concepts of ReaxFF and applications to hight energy materials. Int J Energ Matter Chem Propul 12:95–118
Mortier WJ, Ghosh SK, Shankar S (1986) Electronegativity-equalization method for the calculation of atomic charges in molecules. J Am Chem Soc 108:4315–4320
Honeycutt JD, Andersen HC (1987) Molecular dynamics study of melting and freezing of small Lennard-Jones clusters. J Phys Chem 91:4950–4963
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper
Cite this paper
Savka, S.S., Popovych, D.I., Serednytski, A.S. (2017). Molecular Dynamics Simulations of the Formation Processes of Zinc Oxide Nanoclusters in Oxygen Environment. In: Fesenko, O., Yatsenko, L. (eds) Nanophysics, Nanomaterials, Interface Studies, and Applications . NANO 2016. Springer Proceedings in Physics, vol 195. Springer, Cham. https://doi.org/10.1007/978-3-319-56422-7_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-56422-7_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56244-5
Online ISBN: 978-3-319-56422-7
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)