
Abstract
Hereditary Tyrosinemia type I (HT1) is clinically mainly characterised by severe liver disease. Most patients present in their first months of life with liver failure, but others can present later with issues of compensated cirrhosis, renal tubulopathy or acute intermittent porphyria. If patients survive the acute phase with liver failure or if they present later with compensated cirrhosis, they often develop hepatocellular carcinoma early but also later in life. The course of the disease changed after the introduction of 2-(2 nitro-4-3 trifluoro-methylbenzoyl)-1, 3-cyclohexanedione (NTBC), which blocks the tyrosine degradation pathway at an earlier step. Therefore, the toxic products did not accumulate anymore and all clinical problems resolved. However, the risk (although clearly decreased) for developing liver cancer remained, especially if NTBC treatment is initiated late, a slow decrease of the tumor marker α-fetoprotein is seen or if the α-fetoprotein concentrations remain just above the normal range. A rise of α-fetoprotein in these HT1 patients is more or less pathognomonic for liver cancer. Although hepatoblastoma development occurs in HT1 patients, most HT1 patients develop hepatocellular carcinoma (HCC) or a mixed type of carcinoma consisting of HCC and hepatoblastoma. Due to the small risk of liver cancer development, screening for liver cancer (especially HCC) is still recommended in HT1 patients using regular measures of α-fetoprotein and imaging. Ultrasound is mostly the modality of choice for surveillance, because it is widely available, it does not use radiation and is noninvasive. When a suspicious lesion is present, the higher sensitivity of MRI could be used for characterization and staging of lesions. At this moment, no HCC development in pre-symptomatically treated patients is reported. These different situations could possibly indicate that NTBC can prevent the start of the development of HCC when initiated early, but can’t stop the development of HCC if it is prescribed at a later stage, stressing the importance of early diagnosis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Hereditary Tyrosinmia type 1 (HT1) is caused by an enzymatic defect in the tyrosine degradation pathway. Due to a genetic defect in the enzyme fumarylacetoacetate hydrolase (FAH), its substrate fumarylacetoacetate (FAA) could not be metabolized into fumarate and acetoacetate. In this way, FAA and associated metabolites such as its precursor maleylacetoacetate and alternative degradation products succinylacetoacetate and succinylacetone are accumulating. The accumulation of these toxic products especially causes severe liver dysfunction. This liver dysfuntion secondarily causes a decrease in various enzyme systems including 4-hydroxy phenylpyruvate dioxygenase, phenylalanine hydroxylase and methionine adenosyltransferase leading to high blood concentrations of phenylalanine, tyrosine and methionine in clinically presenting and dietary treated HT1 patients. Treatment with 2-(2 nitro-4-3 trifluoro-methylbenzoyl)-1, 3-cyclohexanedione (NTBC) resolved most of the clinical problems by placing a new metabolic block upstream of the primary enzymatic defect. However, treatment with NTBC leads to increased blood tyrosine and phenylalanine concentrations as these products are just above the new metabolic block, making dietary restriction of both large neutral amino acids necessary.
2 Epidemiology of Hepatocellular Carcinoma
In various countries, HT1 is diagnosed pre-symptomatically with newborn screening by using succinylacetone in blood spot. In countries without a screening program or with a program using tyrosine as the biomarker in newborn screening, HT1 patients can present clinically with heterogeneuous symptoms mainly affecting the liver, kidney and peripheral nerves or otherwise pre-symptomatically due to affected siblings (Hadzic and Vara 2015; van Spronsen et al. 1994). Three different groups of HT1 patients are identified, based on the timing of diagnosis: very early (onset of symptoms <2 months), early (2–6 months) and late presenting form (> 6 months) (van Spronsen et al. 1994). The first group that presents at the hospital within 2 months of life is mostly suffering from acute liver failure. The prognosis of these patients is rather poor, with a 2 year survival probability of 29%. In the patients presenting between 2 and 6 months and after 6 months of life, renal tubulopathy and porphyria like syndrome are also seen next to the liver problems. The later the patients present, the better their survival probability is as they mostly survive the acute initial phase. However, the liver in all these surviving patients may show cirrhosis already or proceed towards cirrhosis and therefore these patients have a high risk for developing hepatocellular carcinoma (HCC) later on (Russo et al. 2001; van Spronsen et al. 1989, 1994, 2005).
Worldwide, HCC is the most common primary hepatic malignancy, mostly occurring after the onset chronic liver disease. The world leading causes of HCC in adults are chronic Hepatitis B and C infection and alcohol abuses. These risk factors lead to the formation and progression of cirrhosis, which is present in 80–90% of patients with HCC (El-Serag 2011; Forner et al. 2012). Overall, the 5 year cumulative risk for the development of HCC in patients with cirrhosis ranges between 5% and 30% depending on the cause, region or ethnic group and stage of cirrhosis (El-Serag 2012).
When only treated with a tyrosine and phenylalanine restricted diet (before introduction of NTBC) (almost) all HT1 patients who survived the first critical period of life developed cirrhosis and in this group of patients, the incidence of HCC was really high, probably up to 37% (van Spronsen et al. 1994; Weinberg et al. 1976). This incidence of HCC in HT1 patients is higher than in other cirrhotic liver diseases and considerably above the incidence of HCC in cirrhotic liver of adults (El-Serag 2012; Weinberg et al. 1976). Due to the high incidence of HCC and its severity, HCC development is the main cause of death of HT1 patients who survived the critical first period (Weinberg et al. 1976). The peak incidence for tumor development in HT1 patients is between 4 and 5 years old, however, the variation is large with malignant transformation after long periods of time (Weinberg et al. 1976).
Most of the malignancies in HT1 patiënts are HCC, however, development of hepatoblastoma is reported at least once in a HT1 patient (Nobili et al. 2010), and next to this one of our patients had a mixed type of HCC and hepatoblastoma. Hepatoblastoma is common among young children and has a favourable outcome compared to HCC. Therefore, a neoplastic mass in HT1 patients is likely, but not definitely HCC. It is important to distinguish between both, because of the difference in treatment and outcome.
The drug or NTBC was released in 1992 (Lindstedt et al. 1992). By placing a metabolic block upstream from the primary enzymatic defect, accumulation of the toxic metabolites was prevented and clinical symptoms, such as liver failure and renal involvement, resolved. Next to this, the incidence of HCC decreased tremendously (Holme and Lindstedt 1998; Larochelle et al. 2012). However, patients receiving NTBC are still at risk for developing HCC, especially when initiation of treatment with NTBC was late due to delayed diagnosis or unavailability of NTBC, a slow decrease of the tumor marker α-fetoprotein (AFP) or an AFP level that remains just above the normal range (<10 ug/L) (Koelink et al. 2006; van Ginkel et al. 2015; van Spronsen et al. 2005). So far, no HCC development in presymptomatically treated patients is reported. These different situations could possibly indicate that NTBC can prevent the start of the development of HCC if initiated early, but can’t stop the development of HCC when it is prescribed at later stage, stressing the importance of early diagnosis.
3 Etiology of Hepatocellular Carcinoma
Due to the enzyme deficiency in HT1 (FAH deficiency), the substrate for FAA, accumulates. This FAA is partly converted into succinylacetoacetate and afterwards in succinylacetone. The accumulating FAA in the cells where it is generated (hepatocytes and cells in the proximal tubule of the kidneys) causes oxidative stress by reacting with glutathione and sulfhydryl groups of proteins. Especially this FAA, but maybe also its precursor, maleylacetoacetate, has shown to be mutagenic and to cause chromosomal instability as well as lead to cell cycle arrest and apoptosis, leading first to cirrhosis in the liver and to HCC formation later on (Bergeron et al. 2006; Jorquera and Tanguay 2001).
Microscopic features in the liver of HT1 patients will reveal micronodulair cirrhosis that will develop to macronodulair cirrhosis (Dehner et al. 1989). Therefore, the chronic hepatic phase usually shows mixed micro- and macro-nodular cirrhosis with minimal ductular proliferation and mild lymphoplasmacytic infiltrates within the fibrous septa. Varying degrees of steatosis are also seen and may show variation within a nodule and/or between nodules. The most significant histological feature is again the foci of dysplasia (large and small cell types) and/or HCC (Schady et al. 2015; Weinberg et al. 1976).
4 Diagnosis of Hepatocellular Carcinoma
4.1 Clinical Presentation
In theory, HCC can give complaints ranging from fatigue due to anemia to abdominal distress due to the increasing mass or a bleeding in the liver due to tumour rupture. However, in general, patients are detected before clinical symptoms arise due to several screening mechanisms such as laboratory investigation and imaging, which are described below.
4.2 Laboratory Parameters
The main laboratory parameter suggestive of HCC development is AFP. AFP is a glycoprotein that typically is very high just after birth. However, AFP concentrations in HT1 patients are at birth already much higher than the AFP concentrations of healthy infants (Hostetter et al. 1983). These AFP concentrations stay higher than normal during infancy in untreated HT1 patients due to the liver regeneration that already starts during fetal life (Hostetter et al. 1983). So far, high AFP levels at diagnosis have not been related to any prognostic value for initial recovery of acute physical complaints or development of HCC later on.
After initiation of adequate treatment, consisting of NTBC and tyrosine and restricted diet, hepatotoxic metabolites should not accumulate anymore and AFP levels should decrease more or less exponentially to normal levels (<10 μg/L) within 1 year like it is shown in the figure published by Koelink et al. in 1996 (de Laet et al. 2013; Koelink et al. 2006) (Fig. 9.1).

Tyrosinemia type 1 patients with proven liver cancer, all showing a clear increase in AFP concentrations. Year 0 represents birth, ● start NTBC treatment; patient 4 started with NTBC treatment before the first AFP concentration was measured. ▲ diagnosis of liver cancer.
The tumormarker AFP has been used as a marker for HCC for many years. AFP concentrations could rise in various circumstances, for example in chronic hepatitis with reactivation, but without HCC. On the other hand it could be normal when there is only a small HCC (Paul et al. 2007). Furthermoref, in other diseases with a high risk of developing HCC (e.g., hepatitis B and C), up to 44% of patients diagnosed with HCC show no clear increase in AFP levels (Beale et al. 2008; Giannelli et al. 2007). Due to this, the reported sensitivity of AFP is variable across different studies.
However, in HT1 patients, AFP was considered to be a reliable marker for liver cancer development. Until recently, all reported HT1 patients with proven HCC and hepatoblastoma had exhibited a well-defined rise in AFP levels and a clear lesion at imaging (de Laet et al. 2013; Koelink et al. 2006; Nobili et al. 2010). But recently, a case study showed that HCC in HT1 patients is not always accomplished with the rise in AFP, like it is shown in Fig. 9.2 published by van Ginkel et al. in 2015 (van Ginkel et al. 2015) (Fig. 9.2). Therefore the search for other reliable markers is necessary. At this moment, no other markers have been investigated extensively, although promising results were seen with Lectin-reactive alfa-fetoprotein (Baumann et al. 2006). Unfortunately, this did not get follow-up so far.

Course of AFP from diagnosis until 1 year after liver transplantation (histologically proven HCC). After an initial decline in AFP, AFP levels stabilised around 25 μg/L. After liver transplantation AFP further decreased to normal values. This is in contrast with the reference patients who almost immediately reached normal values (0–10 μg/L)
4.3 Imaging Modalities
Many patients with tyrosinaemia show signs of cirrhosis on imaging. In severe cirrhosis the size of the liver can change with (1) hypertrophy of the caudate lobe and lateral segments of the left lobe (segment 2 and 3) and (2) atrophy of the posterior segments of the right lobe (segment 6 and 7). Other findings are nodularity of the surface and heterogeneity or nodular aspect of the parenchyma. Signs of portal hypertension like splenomegaly, portosystemic collaterals and ascites can be present (Dubois et al. 1996).
The three major goals of imaging in patients with Tyrosinaemia is the screening for lesions; the characterization of the lesion, and a change in the characterization or the volume. Ultrasound is mostly the modality of choice for surveillance, because it is widely available, it does not use radiation and is noninvasive. Ultrasound has a good sensitivity and specificity for the detection of HCC in cirrhotic patients with a sensitivity of 60–80% and a specificity of 45–94 % particularly depending on the experience of the performer. For the purpose of surveillance, Computed Tomography (CT) is less useful due to the radiation exposure. Magnetic Resonance Imaging (MRI) has a high sensitivity in the detection of HCC (89–100%), especially for smaller lesions between 1 and 2 cm. However, the higher sensitivity of MRI is mainly used for characterization and staging of lesions that are suspicious for HCC, caused by the higher costs and lower availability of MRI (Ayuso et al. 2012; Bolondi 2003; Burrel et al. 2003).
The sonographic appearance of HCC is nonspecific. Small lesions are often hypoechoic to the liver parenchyma, but isoechoic or hyperechoic lesions can occur. The lesions that are larger are often heterogeneous with hypoechoic and hyperechoic areas. The hyperechoic areas can represent fat or more acute hemorrhage, whereas the anechoic areas can be seen representing necrosis or hemorrhage of older date. The lesions can have a thin hypoechoic halo corresponding with a capsule. Doppler examination can show high-velocity arterial flow. In practice, in can be difficult to distinguish small tumors from the nodular pattern of the parenchyma of the cirrhotic liver. Benign lesions like regenerative and dysplastic nodules are frequently present in cirrhotic livers and can be indistinguishable from small HCC (Ayuso et al. 2012; Chung et al. 2011; Helmberger et al. 1999).
On MRI the typical appearance of HCC is a mass with slightly hyperintense signal on the T2-weighted images. On the T1-weighted images the signal intensity is more variable. Heterogeneous signal intensity is often seen in larger lesions due to fat, hemorrhage, necrosis or calcifications. After intravenous gadolinium contrast admission HCC typically shows arterial enhancement with wash-out in the portal venous phase. Capsules show enhancement in the delayed phase. The diffusion weighted images often demonstrate a high signal intensity at the higher B-value and a hypointens appearance on the apparent diffusion coefficient corresponding to diffusion restriction. Regenerative nodules are isointense or slightly hyperintense on the T1 weighted images and isointense or hypointense on the T2 weighted images. A distinction between HCC and regenerative nodules can be made, as regenerative nodules do not show arterial enhancement and portal venous wash-out. Because dysplastic nodules can be low or high grade they a show a variable appearance. The low grade resembles regenerative nodules while the high grade resembles HCC (Chung et al. 2011; Helmberger et al. 1999; Saar and Kellner-Weldon 2008).
If using CT, on unenhanced CT HCC is a hypoattenuating mass, which can show areas with different attenuation as a result of fat, hemorrhage, necrosis and calcification. After contrast administration HCC typically shows the same enhancing characteristics on CT as on MRI with arterial enhancement and wash-out in the portal venous phase. If present, the capsule shows enhancement in the delayed phase (Chung et al. 2011; Helmberger et al. 1999; Saar and Kellner-Weldon 2008).
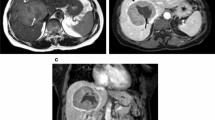
To conclude, current recommendations for the screening for HCC in HT1 therefore consist of regular AFP and ultrasound examinations. A rise in AFP is suggestive of HCC development. However, a new nodule on ultrasound with stable AFP should also be considered to be suggestive of HCC (van Ginkel et al. 2015). If nodules are present in the liver, further imaging preferably with MRI should be done. An example of this surveillance and staging protocol with the radiological appearance of HCC on ultrasound and MRI is shown in Fig. 9.3.

Imaging features from a case presenting with HCC, images provided by courtesy of dr. S. McGuirk from the Birmingham Children’s Hospital, UK. (a) US demonstrates a hypoechoic lesion in the right liver lobe (histologically proven HCC). (b) On the coronal T2 weighted the lesions demonstrates a hyperintens signal. On the T1weighted images the lesion shows arterial enhancement (c) with wash-out in the portal venous phase (d)
5 Treatment
Before introduction of NTBC, the incidence of HCC in HT1 patients was really high. Therefore, in that time, the important question was not whether, but when should liver transplantation be performed. During that time, liver transplantation was the only possible option for metabolic correction and long-term survival (Salt et al. 1992; van Spronsen et al. 1989, 1995; Weinberg et al. 1976). This changed after the introduction of NTBC. Due to NTBC, the incidence of HCC diminished and, therefore, the need for transplantation in HT1 patients decreased as well. Due to the required major abdominal surgery, post-operative complications and life long immunosuppression with risks for secondary malignant diseases, liver transplantation is currently in most centers only an option in patients not responding on NTBC or who develop HCC despite treatment with NTBC. However, as NTBC is costly, some countries seem to prefer liver transplantation that in the long run may be less expensive (personal communication). Therefore, studies that compare the outcome of patients treated with NTBC versus liver transplantation are needed.
Pediatric liver transplantation is done under various circumstances, with biliary atresia still being the main indication for liver transplantation in children. Since the first liver transplantation in 1953 this treatment option has changed to one with an excellent long term outcome, having high short and long term survival rates (Hackl et al. 2015). Liver graft survival has improved as well to 80% at 5 years and an estimated half-life of 13 years (Yazigi 2013).
In HT1 patients, recent articles about the outcome after liver transplantation are limited, as most of the manuscripts are published before 2000, mostly describing patients before introduction of NTBC. At that time, the survival after transplantation was relatively good with survival rates around 90% (Paradis 1996; Weinberg et al. 1976), although post-operative complications such as non-functioning of the graft due to artery thrombosis and graft rejection were seen (Weinberg et al. 1976). More recently, Arnon et al. showed in a large cohort of patients that HT1 patients have an excellent outcome after transplantation with a 5 year survival rate over 90% (Arnon et al. 2011). However, a clear and continuous decrease in the need for liver transplantation has been seen after introduction of NTBC (Arnon et al. 2011).
Abbreviations
- AFP:
-
Alpha feto protein
- CT:
-
Computed tomography
- FAA:
-
Fumarylacetoacetate
- FAH:
-
Fumarylacetoacetate hydrolase
- HCC:
-
Hepatocellular carcinoma
- HT1:
-
Hereditary tyrosinemia type 1
- MRI:
-
Magnetic resonance imaging
- NTBC:
-
2-(2 nitro-4-3 trifluoro-methylbenzoyl)-1, 3-cyclohexanedione
References
Arnon R, Annunziato R, Miloh T, Wasserstein M, Sogawa H, Wilson M,…( Kerkar N (2011) Liver transplantation for hereditary tyrosinemia type I: analysis of the UNOS database. Pediatr Transplant 15(4): 400–405. doi:10.1111/j.1399–3046.2011.01497.x [doi]
Ayuso C, Rimola J, Garcia-Criado A (2012) Imaging of HCC. Abdom Imaging 37(2):215–230. doi:10.1007/s00261-011-9794-x [doi]
Baumann U, Duhme V, Auth MK, McKiernan PJ, Holme E (2006) Lectin-reactive alpha-fetoprotein in patients with tyrosinemia type I and hepatocellular carcinoma. J Pediatr Gastroenterol Nutr 43(1):77–82. doi:10.1097/01.mpg.0000228112.29359.f8
Beale G, Chattopadhyay D, Gray J, Stewart S, Hudson M, Day C,… Reeves H (2008) AFP, PIVKAII, GP3, SCCA-1 and follisatin as surveillance biomarkers for hepatocellular cancer in non-alcoholic and alcoholic fatty liver disease. BMC Cancer 8: 200-2407-8-200. doi:10.1186/1471-2407-8-200; 10.1186/1471-2407-8-200
Bergeron A, Jorquera R, Orejuela D, Tanguay RM (2006) Involvement of endoplasmic reticulum stress in hereditary tyrosinemia type I. J Biol Chem 281(9):5329–5334
Bolondi L (2003) Screening for hepatocellular carcinoma in cirrhosis. J Hepatol 39(6):1076–1084
Burrel M, Llovet JM, Ayuso C, Iglesias C, Sala M, Miquel R,… Barcelona Clinic Liver Cancer Group (2003) MRI angiography is superior to helical CT for detection of HCC prior to liver transplantation: an explant correlation. Hepatology (Baltimore, Md), 38(4), 1034–1042. 10.1053/jhep.2003.50409
Chung EM, Lattin GE Jr, Cube R, Lewis RB, Marichal-Hernandez C, Shawhan R, Conran RM (2011) From the archives of the AFIP: pediatric liver masses: radiologic-pathologic correlation. part 2. malignant tumors. Radiographics: A Rev Publ Radiol Soc N Am Inc 31(2):483–507. doi:10.1148/rg.312105201
de Laet C, Dionisi-Vici C, Leonard JV, McKiernan P, Mitchell G, Monti L,…, Spiekerkotter U (2013) Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 8: 8–1172–8-8. doi: 10.1186/1750-1172-8-8
Dehner LP, Snover DC, Sharp HL, Ascher N, Nakhleh R, Day DL (1989) Hereditary tyrosinemia type I (chronic form): pathologic findings in the liver. Hum Pathol 20(2):149–158
Dubois J, Garel L, Patriquin H, Paradis K, Forget S, Filiatrault D et al (1996) Imaging features of type 1 hereditary tyrosinemia: a review of 30 patients. Pediatr Radiol 26(12):845–851
El-Serag HB (2011) Hepatocellular carcinoma. N Engl J Med 365(12):1118–1127. doi:10.1056/NEJMra1001683 [doi]
El-Serag HB (2012) Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 142(6):1264–1273.e1. doi:10.1053/j.gastro.2011.12.061
Forner A, Llovet JM, Bruix J (2012) Hepatocellular carcinoma. Lancet (London, England) 379(9822):1245–1255. doi:10.1016/S0140-6736(11)61347-0
Giannelli G, Fransvea E, Trerotoli P, Beaugrand M, Marinosci F, Lupo L,… Antonaci S (2007) Clinical validation of combined serological biomarkers for improved hepatocellular carcinoma diagnosis in 961 patients. Clin Chim Acta; Int J Clin Chem, 383(1–2): 147–152. 10.1016/j.cca.2007.05.014
Hackl C, Schlitt HJ, Melter M, Knoppke B, Loss M (2015) Current developments in pediatric liver transplantation. World J Hepatol 7(11):1509–1520. doi:10.4254/wjh.v7.i11.1509
Hadzic N, Vara R (2015) Neonatal screening for hereditary tyrosinaemia: are we there yet? Arch Dis Child 100(8):720–721. doi:10.1136/archdischild-2015-308265
Helmberger TK, Ros PR, Mergo PJ, Tomczak R, Reiser MF (1999) Pediatric liver neoplasms: a radiologic-pathologic correlation. Eur Radiol 9(7):1339–1347. 90091339.330 [pii]
Holme E, Lindstedt S (1998) Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione). J Inherit Metab Dis 21(5):507–517
Hostetter MK, Levy HL, Winter HS, Knight GJ, Haddow JE (1983) Evidence for liver disease preceding amino acid abnormalities in hereditary tyrosinemia. N Engl J Med 308(21):1265–1267. doi:10.1056/NEJM198305263082105
Jorquera R, Tanguay RM (2001) Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalities and genomic instability. Hum Mol Genet 10(17):1741–1752
Koelink CJ, van Hasselt P, van der Ploeg A, van den Heuvel-Eibrink MM, Wijburg FA, Bijleveld CM, van Spronsen FJ (2006) Tyrosinemia type I treated by NTBC: how does AFP predict liver cancer? Mol Genet Metab 89(4):310–315. doi:10.1016/j.ymgme.2006.07.009
Larochelle J, Alvarez F, Bussieres JF, Chevalier I, Dallaire L, Dubois J,… Mitchell GA (2012) Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in quebec. Mol Genet Metab 107(1–2):49–54. doi:10.1016/j.ymgme.2012.05.022; 10.1016/j.ymgme.2012.05.022
Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B (1992) Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 340(8823):813–817. doi:0140-6736(92)92685-9 [pii]
Nobili V, Jenkner A, Francalanci P, Castellano A, Holme E, Callea F, Dionisi-Vici C (2010) Tyrosinemia type 1: metastatic hepatoblastoma with a favorable outcome. Pediatrics 126(1):e235–e238. doi:10.1542/peds.2009-1639; 10.1542/peds.2009-1639
Paradis K (1996) Tyrosinemia: the quebec experience. Clinical and investigative medicine. Med Clin Exp 19(5):311–316
Paul SB, Gulati MS, Sreenivas V, Madan K, Gupta AK, Mukhopadhyay S, Acharya SK (2007) Evaluating patients with cirrhosis for hepatocellular carcinoma: Value of clinical symptomatology, imaging and alpha-fetoprotein. Oncology 72(Suppl 1):117–123
Russo PA, Mitchell GA, Tanguay RM (2001) Tyrosinemia: a review. Pediatr Dev Pathol: Off J Soc Pediatr Pathol Paediat Pathol Soc 4(3):212–221
Saar B, Kellner-Weldon F (2008) Radiological diagnosis of hepatocellular carcinoma. Liver Int: Off J Int Assoc Study Liver 28(2):189–199. doi:10.1111/j.1478-3231.2007.01655.x
Salt A, Barnes ND, Rolles K, Calne RY, Clayton PT, Leonard JV (1992) Liver transplantation in tyrosinaemia type 1: the dilemma of timing the operation. Acta Paediatr (Oslo, Norway : 1992) 81(5):449–452
Schady DA, Roy A, Finegold MJ (2015) Liver tumors in children with metabolic disorders. Transl Pediatr 4(4):290–303. doi:10.3978/j.issn.2224-4336.2015.10.08
van Ginkel WG, Gouw AS, van der Jagt EJ, de Jong KP, Verkade HJ, van Spronsen FJ (2015) Hepatocellular carcinoma in tyrosinemia type 1 without clear increase of AFP. Pediatrics 135(3):e749–e752. doi:10.1542/peds.2014-1913
van Spronsen FJ, Berger R, Smit GP, de Klerk JB, Duran M, Bijleveld CM et al (1989) Tyrosinaemia type I: orthotopic liver transplantation as the only definitive answer to a metabolic as well as an oncological problem. J Inherit Metab Dis 12(Suppl 2):339–342
van Spronsen FJ, Bijleveld CM, van Maldegem BT, Wijburg FA (2005) Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro- methylbenzoyl)-1, 3-cyclohexanedione treatment. J Pediatr Gastroenterol Nutr 40(1):90–93
van Spronsen FJ, Smit GP, Wijburg FA, Thomasse Y, Visser G, Heymans HS (1995) Tyrosinaemia type I: considerations of treatment strategy and experiences with risk assessment, diet and transplantation. J Inherit Metab Dis 18(2):111–114
van Spronsen FJ, Thomasse Y, Smit GP, Leonard JV, Clayton PT, Fidler V, … Heymans HS (1994) Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology (Baltimore, Md.) 20(5):1187–1191. doi:S027091399400340X [pii]
Weinberg AG, Mize CE, Worthen HG (1976) The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J Pediatr 88(3):434–438
Yazigi NA (2013) Long term outcomes after pediatric liver transplantation. Pediatr Gastroenterol Hepatol Nutr 16(4):207–218. doi:10.5223/pghn.2013.16.4.207
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
van Ginkel, W.G., Pennings, J.P., van Spronsen, F.J. (2017). Liver Cancer in Tyrosinemia Type 1. In: Tanguay, R. (eds) Hereditary Tyrosinemia. Advances in Experimental Medicine and Biology, vol 959. Springer, Cham. https://doi.org/10.1007/978-3-319-55780-9_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-55780-9_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55779-3
Online ISBN: 978-3-319-55780-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)