
Abstract
The Von Hippel–Lindau (VHL) disease is an autosomal dominant disorder characterized by the predisposition for multiple tumors caused by germline mutations in the tumor suppressor gene VHL. This disease is associated with a high morbidity and mortality and presents a variable expression, with different phenotypes from family to family, affecting different organs during the lifetime. The main manifestations of VHL are hemangioblastomas of the central nervous system and retina, renal carcinomas and cysts, bilateral pheochromocytomas, cystic and solid tumors of the pancreas, cystadenomas of the epididymis, and endolymphatic sac tumors. The discovery of any of the syndrome components should raise suspicion of this disease and other stigmas must then be investigated. Due to the complexities associated with management of the various VHL manifestation, the diagnosis and the follow-up of this syndrome is a challenge in the clinical practice and a multidisciplinary approach is needed. The particular relevance to endocrinologists is the detection of pheochromocytomas in 35% and islet cell tumors in 17% of VHL patients, which can be associated with hypertension, hypoglycemia, cardiac arrhythmias, and carcinoid syndrome. The purpose of this review is to define the Von Hippel–Lindau syndrome addressing its clinical aspects and classification, the importance of genetic counseling and to propose a protocol for clinical follow-up.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Von Hippel–Lindau (VHL) disease is an autosomal dominant syndrome, first reported in the twentieth century, characterized by the predisposition for multiple tumors caused by mutations in the VHL gene [1].
In 1904, Eugene von Hippel, a German ophthalmologist, described some cases of retinal angiomatosis. After 20 more years, in 1926, Lindau, a Swedish pathologist, established the relationship between the cerebellar and retinal lesions. In the coming reports, the full spectrum of the disease was established including retinal, visceral, and central nervous system (CNS) manifestations, under the eponym of Von Hippel–Lindau disease.
This is a rare disease, nowadays, perfectly established as having a genetic origin, with an incidence of 1/36,000 in the general population, but with a high penetrance in the affected families, reaching 90% at 65 years of age [2, 3], which justifies the high risk of developing its related diseases in the individuals that carry the mutation.
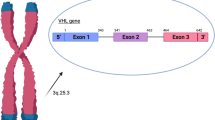
The Von Hippel–Lindau disease is an autosomal dominant disorder that implies a genetic alteration consisting in the loss of the tumor suppressor function of the VHL gene [4, 5] located in chromosome 3 (3p25.3). The protein encoded by this gene participates in the oxygen-sensing system. It takes hypoxia-inducible factors (HIF) for degradation when oxygen attains normal levels [6]. In normal situations, the VHL protein is activated only when there is hypoxia. However, when mutated it becomes inactive. When hypoxic conditions take place (or when the VHL protein is not active due to mutations or deletion) HIF-1 accumulates and translocates to the nucleus where they activate the transcription of angiogenic and erythropoietic factors. It is a simple adaptation process to hypoxia [7]. When this process becomes active in response to other conditions that are not hypoxia, it may start a tumorigenic process.
Besides this HIF-dependent mechanism of action, the VHL protein participates in tumorigenesis by other processes: cell senescence control, microtubule stabilization, and maintenance of ciliar structure and function [8,9,10]. One recognized function of the VHL protein is to maintain Mad2 which is a key regulator of mitotic progression [11]. Mad2 prevents the formation of chromosomal instability that is responsible for aneuploidy (abnormal chromosomal numbers). When the VHL gene is mutated, cells have low levels of Mad2 [12].
Mutations in the VHL gene increase the risk of tumor formation in several organs. Usually, the mutation process affects only one allele. It is necessary that the other allele is inactivated (loss of heterozygosity or deletion) for the VHL disease to occur.
The disease presents a variable expression, with different phenotypes from family to family, affecting many different organs and systems during the lifetime, including malignant and benign tumors. Approximately 20% of the cases are caused by de novo mutations and hence do not have family history [13]. It is a syndrome associated with high morbidity and mortality, which justifies the importance of genetic screening [14].
The purpose of this review is to define the Von Hippel–Lindau syndrome, addressing its clinical aspects and classification, the importance of genetic counseling, and to propose a protocol for clinical follow-up.
Clinical presentation and management
The clinical presentation of this syndrome is variable with different phenotypes in different families or even in the same family. The main manifestations are: hemangioblastomas of the CNS and retina, renal carcinomas, renal cysts, bilateral pheochromocytomas, cystic and solid tumors of the pancreas, cystadenomas of the epididymis, and endolymphatic sac tumors. It is rare to find all of the manifestations of the disease in one patient; in familial cases, up to 50% of patients have only one manifestation of the syndrome. Clinical manifestations depend on the type of mutation [15]; patients with truncating mutations or deletions usually have type 1 VHL disease (without pheochromocytoma), whereas in type 2 VHL disease (the type with pheochromocytomas) missense mutations are the rule [16]. Patients with nonsense or frameshift mutations have a higher risk of clear-cell renal cell carcinomas (ccRCC) and hemangioblastomas. The disease phenotype is age dependent, with an almost complete penetrance at 65 years [2, 17].
VHL disease is a syndrome associated with high morbidity and mortality, having a median life expectancy of 50 years. The most frequent causes of death are complications of cerebellar hemangioblastomas (53%) and renal cell carcinoma metastases (32%) [2]. The diagnostic criteria for isolated cases are the existence of two hemangioblastomas (CNS or retina) or a hemangioblastoma coupled with a visceral manifestation. In familial cases, the presence of only one manifestation is enough for the diagnosis [17].
Hemangioblastomas in the central nervous system
The hemangioblastomas of the CNS are the most frequent lesions in VHL disease. It presents between 60 and 84% of the cases [13]. The average age of diagnosis is 29 years, mostly presented as single tumor (58%) [2]. Multiple tumors are always associated with the VHL syndrome [18]. The lesions are almost exclusively limited to the brainstem, cerebellum, and spinal cord. In a study of 225 patients with VHL syndrome presenting hemangioblastomas, 1921 tumors were identified, of which 36% were present in the spinal cord, 12% in the cauda equina, 44% in the cerebellum, 7% in the brainstem, and 1% were supratentorial. No reliable threshold for tumor size could predict symptom development and need for treatment [19]. The diagnosis of central nervous system lesions is usually obtained by computerized tomography (CT) and preferably by magnetic resonance imaging (MRI). The signs and symptoms are variable, depending on the location of the tumor. If cerebellar, the patient may experience headache, dizziness, gait and speech disturbances, nystagmus, diplopia, ptosis, dysmetry, ninth cranial nerve palsy, and paroxysmal hypertension. Erythrocytosis is a rare condition that may be present in a subset of patients secondary to constitutive activation of erythropoietin caused by impaired degradation of the hypoxia-inducible factor (HIF) [20]. If a spinal cord involvement exists, symptoms may include neurogenic pain, multiple sensory deficits, proprioceptive changes, paraparesis, paresthesia, muscle atrophy, and medullar hypertonicity [17].
The tumors of the CNS are benign tumors that can remain latent for indefinite time. Whenever accelerated growth occurs with the consequent mass effect, the appropriate treatment consists of either stereotactic radiosurgery or craniospinal radiation surgery [13, 18, 19]. Radiation therapy is indicated for the treatment of lesions not amenable to complete excision and also as an attempt to avoid multiple surgeries [21,22,23]. Surgery can provide the complete remission of the tumor or at least improvement of the symptoms in 88% of the cases. The presence of a peritumoral cyst, adjacent to the CNS hemangioblastoma, underlies significant neurologic morbidity and even mortality [24]. So, the development of such peritumoral cysts in VHL patients frequently requires surgical intervention. The hemangioblastomas’ burden in VHL and particularly the contribution of peritumoral cysts for that burden is generally associated with germline partial VHL gene deletions [24].
Hemangioblastomas in the retina
Retinal capillary hemangiomas are also very frequent and probably the most common alteration recognized in the VHL syndrome being present in more than 70% of the cases by age 60 years. The ocular alterations in the anterior segment are only secondary to changes in the retina and optic nerve. Cataracts, iris neovascularization, and neovascular glaucoma are the most common [25].
The hemangiomas are usually circumscribed, round, red–orange in color and may be classified according to [25]: location (39% in the superior temporal quadrant and 27% in the lower temporal quadrant); morphology (endophytic, exophytic, and sessile); and finally, retinal effects (exudative or tractional). They can cause secondary retinal effects such as macular exudates and cause vitreous or retinal hemorrhage in about 3% of the cases, sometimes with retinal detachment [26].
The diagnosis is usually performed, on average, at 25 years of age, with the lesion presenting as a solid tumor. About one-third of the patients have multiple hemangiomas and a half have bilateral involvement. Although considered a hamartomatous abnormal formation, the capillary hemangioma does not commonly have congenital presentation; the risk of progressive growth is of 38% at 30 years and 70% at 50 years [26].
The diagnosis is performed through fundoscopic examination, in which the image of the hemangioma is typical. Differential diagnoses include the vasoproliferative tumors, Coats disease, macroaneurysms, cavernous hemangiomas, choroiditis, and choroidal neovascularization. Although the use of fluorescent angiography is not essential, the test is important in some special situations to improve treatment outcomes. Patients at risk for retinal hemangioblastoma should have routine screening by imaging with fundus photography, fluorescein angiography, and enhanced depth imaging optical coherence tomography for subclinical detection of asymptomatic tumors [27]. Echography is also important for the detection and the definition of tumor dimensions, particularly in cases associated with hemorrhage and opacity of the ocular medium. The nuclear magnetic resonance may have some advantage to detect hemangiomas starting at 2 mm in thickness [26].
In general, the decision to treat capillary hemangiomas takes into account the size, location, and associated findings, such as subretinal fluid, retinal traction, and potential threat to the vision. Among the options, laser photocoagulation (ideal for smaller lesions), cryotherapy (preferable in the larger ones), transpupillary thermal therapy, radiotherapy, photodynamic therapy, and vitreoretinal surgery can all be used. Recently, the use of anti-angiogenic molecules applied intravitreously was introduced, especially for advanced cases with more exudate and increased risk of visual loss [28].
More than 25% of the patients show some degree of permanent visual loss having visual acuity inferior to 20/40 and an additional 20% having an acuity of lower than 20/100. The severity of the loss depends on the age of the patient. 43% of the visual acuity loss occurs in the first 30 years of disease evolution. Therefore, early detection is associated with a good outcome which is a consequence of subsequent early treatment [25].
Renal tumors
Renal cell carcinomas correspond to 3% of all cancers in adults, with the highest incidence between 50 and 70 years of age. Approximately 4% of renal carcinomas are hereditary, and these cases tend to be multifocal, bilateral, and affecting younger individuals [29]. Cysts and renal cell carcinomas are present in about two-thirds of patients affected by VHL syndrome, with a cumulative incidence of 5% at 30 years of age and 69% in the sixth decade of life [2]. The cysts are benign lesions, but the coexistence of mutations in both cysts and carcinomas suggests that the cysts may be precursors of carcinomas. Corroborating this hypothesis, 1.2% of the cysts present areas of malignant transformation [30,31,32].
Renal cell carcinomas are solid lesions with malignant behavior and, when associated with VHL disease, they are always of the clear-cell subtype [13]. RCCs are the most frequent VHL-associated metastasizing tumors. Most cases, however, are asymptomatic; when present, the most common symptoms are hematuria and flank pain, drawing attention to the importance of screening protocols [33].
The radiologic diagnosis is made by ultrasound, which differentiates the solid from the cystic lesions, and CT of the abdomen which is more sensitive to detect tumors smaller than 2 cm. MRI offers no advantage over the CT [17].
Kidney lesions often grow slowly [34]. Therefore, tumors smaller than 2 cm can be clinically followed up with the purpose of avoiding multiple surgical explorations [35]. Surgery is recommended in lesions greater than 3 cm [36]; metastases are usually present when the tumor is greater than 4.5 cm. Lymph nodes, lung, liver, bone, and central nervous system are the most common sites of metastases [37].
The surgical approach seeks to preserve the kidney function as much as possible. Simple enucleation is indicated in cystic lesions [35]. In solid tumors smaller than 5 cm or multiple disease of limited extension, a partial nephrectomy can be performed [37]. In cases of larger tumors and lesions which affect the entire renal parenchyma, total nephrectomy should be performed and if it is bilateral, the patient will be started on dialysis treatment and renal transplantation may be performed 1 year after the surgery, when the chance of occurrence of metastases becomes lower [38]. The follow-up is performed by abdomen CT every six months for 2 years, and then annually [17].
Pheochromocytoma
Among the catecholamine-secreting tumors originating from chromaffin cells, pheochromocytomas, from the adrenal medulla, are the most common in the VHL syndrome, whereas paragangliomas, from sympathetic ganglia cells are rare in this syndrome. Up to one-fifth of patients with apparently sporadic pheochromocytoma may carry the VHL disease. Characteristically, the histology of these tumors shows a thick capsule and increased vascularization. In contrast to MEN-2 pheos, in VHL cases there is not adrenal medulla hyperplasia [39]. In some cases, pheochromocytomas associated with VHL syndrome express proteins related to the hypoxic response, like carbonic anhydrase IX [40]. In contrast, about 20–35% of the cases with VHL syndrome have pheochromocytomas, and these are frequently bilateral and affect patients at younger ages [41, 42]. In the retrospective analysis of patients with VHL enrolled at the National Cancer Institute, the earliest age at diagnosis was 5.5 years [43]. The presence or absence of pheochromocytoma is the basis of the classification of VHL syndrome by the National Cancer Institute [13]. Type 1 disease is related to gene deletion or nonsense mutation and patients do not suffer from pheochromocytomas while type 2 disease is characterized by the presence of pheochromocytomas and is associated with missense mutations [44].
About 40% of the families with VHL syndrome and pheochromocytomas have a mutation in codon 238 of the VHL gene [45].
Patients are often asymptomatic since in many cases there is no excessive catecholamine production by the tumor, unlike the sporadic pheochromocytoma cases [46]. When present, the most common symptoms are palpitations, sweating, headache, and paroxysmal hypertension [47] but other nonspecific signs may also be present, leading to a delay from the onset of the symptoms to the diagnosis. The lack of a pheochromocytoma diagnosis can lead to complications such as hypertensive crisis, heart failure, and stroke [48]. Furthermore, the possibility of an occult pheochromocytoma should be considered in syndrome carriers that need surgery, since there is a real risk of sympathetic hyperactivity and severe arterial hypertension during surgery with a significant increase in surgical risk and anesthetic implications [41].
Diagnosis is based on biochemical tests and radiological imaging of the abdomen by ultrasonography, CT or MRI of the abdomen, and whole body scanning with meta-iodobenzylguanidine. Biochemical evaluation consists in the determination of the catecholamines and their metabolites in the plasma or in the urine. For a blood sample, patients should be supine for 20–30 min from the time the needle is inserted and when blood is drawn, the blood sample analysis should use reference standards from supine tests, not seated tests. Urine collection should be over a 24-h period [49].
The measurement of fractionated plasma-free metanephrines is the most sensitive method for the diagnosis of pheochromocytoma, but it has lower specificity when compared with the determination of urinary catecholamines and metanephrines. Therefore, the measurement of fractionated plasma-free metanephrines should be done in patients with high level of suspicion for pheochromocytoma, since in these conditions, a highly sensitive test is fully justified. In patients with low probability of pheochromocytoma, urinary determinations of catecholamines and metanephrines are sufficient. Recently, it has been shown that increased plasma normetanephrine concentrations are usually present in the case of VHL patients with pheochromocytoma, indicating the production of the norepinephrine by the tumor [46]. Paragangliomas that have been rarely associated with VHL disease are usually parasympathetic, and do not secrete significant amounts of catecholamines [50].
Benign pheochromocytomas are commonly found, while malignant tumor with distant metastasis has been reported in about 3% of VHL patients [46]. Moreover, the risk of malignant transformation in pheochromocytomas associated with the VHL disease is not elevated. The treatment is surgical, through unilateral or bilateral adrenalectomy, depending on the form of presentation [42]. These patients should be followed up for their entire life due to the increased risk of recurrence.
Pancreatic tumors
Pancreatic lesions associated with the VHL disease occur in up to 77% of the patients [51]. They can be classified as non-secretory (cysts and cystadenomas) and secretory (islet tumors/neuroendocrine tumors—NETs). Pancreatic cysts are the most common form of presentation in the VHL syndrome. Simple cysts and serous cystadenomas are usually discovered incidentally because they are asymptomatic. They do not normally require intervention. They could be the sole manifestation of pancreatic disease in about 50% of VHL patients [52].
Cysts and cystadenomas affect young individuals, and the diagnosis is carried out between the ages of 20 and 40 years. Both are benign lesions and generally asymptomatic although epigastric pain may be referred in some cases [17]. Diagnosis is performed through imaging procedures that should be performed in every carrier of the VHL disease. Pancreatitis and pancreatic insufficiency are rare complications; it must also be stressed that there is not an increased risk of adenocarcinoma of the pancreas [52], they may coexist with pheochromocytomas [17, 51].
Pancreatic secreting or neuroendocrine tumors are constituted by cells of the Langerhans islets, and affect individuals at around 40 years of age [36]. Such tumors occur in 11–17% of the patients with the VHL disease [53]. They are usually multiple and are frequently associated with precursor lesions like nesidioblastosis or islet dysplasia [54]. These are tumors with a slow growth that are asymptomatic in most of the patients; their detection occurs at a younger age than in sporadic cases probably due to a periodic screening of family members of affected patients. Malignant behavior, however, is observed in a significant number of the cases and they can metastasize, primarily to the liver and bones.
When they become symptomatic, the symptoms are related either to the production of insulin or glucagon. Patients with neuroglycopenic symptoms should have biochemical proof of endogenous hyperinsulinemic hypoglycemia to diagnose insulinoma. On the other hand, carcinoid syndrome produces nonspecific elevations in glucagon levels. Contrary to MEN-1 pancreatic NETs, these tumors in VHL patients rarely produce gastrin [55] and in some cases produce inhibin [56].
The diagnosis is made through any abdominal imaging examination: ultrasound, CT, or MRI [17]. Somatostatin receptor scintigraphy is positive in 60% of the cases [53]. The choice of treatment is based on tumor location, size, and presence or absence of metastatic disease. Locoregional disease can be treated with tumor resection with or without lymphadenectomy. Lesions larger than 3 cm in diameter or even those larger than 2 cm, if located in the pancreas body or head should always be removed.. Minor and asymptomatic lesions must be annually evaluated [57]. Approximately 80% of the neuroendocrine pancreatic tumors express somatostatin receptors, their function is to decrease the activity of calcium channels, thereby decreasing hormone secretion. Somatostatin analogs (SSA) such as octreotide, lanreotide, and pasireotide are indicated for symptom reduction in advanced disease. The responses may vary by tumor type and some patients may become desensitized over time [58]. Although tumor size and growth rates support current indications for surgical treatment, no absolute criteria are available for selecting patients who have premalignant or malignant disease [57, 59].
Endolymphatic sac tumors of the middle ear (ELST)
Tumors of endolymphatic sac were recently recognized as belonging to this syndrome and may be more frequent than previously believed. Whenever bilateral, ELST are considered pathognomonic of the VHL disease. They are present in about 15% of the patients affected by the disease. They are usually benign cystoadenomas, but can be locally invasive, leading to hearing loss, tinnitus, dizziness, gait imbalance, and paralysis of facial muscles [60]. Three mechanisms have been described to explain the hearing loss and other symptoms [61]:
-
1.
Otic capsule invasion by the tumor, resulting in destruction of the membranous labyrinth and interruption of the endolymphatic flow;
-
2.
Labyrinthine hemorrhage causing sudden loss of hearing which may be irreversible;
-
3.
Block of the reabsorption of the endolymphatic sac fluid leading to gradual onset of hearing loss, tinnitus, and vertigo.
VHL syndrome patients should be asked annually about auditory and vestibular symptoms and evaluated with routine audiometry. Any detected abnormalities must be screened for the presence of tumors by high-resolution CT or MRI of the skull base with thin sliced sequences [60]. The treatment is surgical and may prevent deafness, but is not always mandatory in asymptomatic patients, considering the slow tumor growth and the possible complications associated with surgery [62, 63]. Stereotactic radiotherapy can be used in cases of recurrent disease [64]. The cochlear implant may be an option for patients with bilateral hearing loss [65].
Other tumoral manifestations in the VHL syndrome
Papillary cystadenoma of the epididymis is a benign palpable lesion that is present in 10–60% of patients with VHL syndrome. Unilateral cysts are common in the general population and, therefore, only the bilateral cases should be taken as an indication of a possible VHL diagnosis. They are usually asymptomatic and rarely cause pain or infertility. The diagnosis is made by ultrasound; it does not require treatment, but in symptomatic cases, it can be removed by surgery [17].
The cystadenoma of the broad ligament of the uterus is a benign and asymptomatic lesion in most of the cases; its true incidence in the VHL syndrome is unknown. Some patients may experience pain, dyspareunia, and menorrhagia [66].
Cavernous hemangiomas and hepatic hemangioblastomas, pulmonary hemangioblastomas, omentum cysts, vertebral hemangiomas, cysts and hemangiomas of the ovaries, medullary thyroid carcinomas, papillary thyroid carcinomas, dermal hemangiomas, pigmented nevus, splenic cysts and angiomas, angiomas and adenomas of the adrenal cortex, and pancreatic hemangioblastomas have also been reported [17].
Classification and diagnosis
The diagnosis is based on the assessment of three criteria: retinal or CNS hemangioma, visceral lesions, and family history. If the patient has a family history of VHL disease, only one hemangioma or visceral lesion, confirms the disease. In the cases with no family history of VHL disease, the presence of two or more hemangiomas, or a hemangioma and a visceral lesion are needed for the diagnosis [57].
The Von Hippel–Lindau syndrome is then classified according to the nature of the tumors, based on the presence or absence of pheochromocytomas (Table 1). Type 1 does not develop pheochromocytomas, but presents a high risk for other VHL lesions; it is the most common type of presentation (80%). In the type 2, there is a high risk of developing pheochromocytomas; it is further subdivided according to the presence or absence of renal cell carcinoma. The type 2A, without RCC, has the best prognosis, and the type 2B, having a high incidence of RCC, is the rarest form but it is the one that has all the manifestations of the VHL. Pheochromocytomas are the single clinical manifestation in the type 2C.
However, accurate classification can only be made in large kindred because of the variability of VHL gene mutation [67]. Genotype–phenotype correlations have been identified from analysis of the VHL mutations. Families with an increased risk of pheochromocytoma (Type 2 VHL disease) displayed missense type mutations, whereas true null VHL alleles are associated with a low risk of pheochromocytoma (Type 1 VHL disease) [5].
Several lines of evidence suggest additional pVHL functions and recognizing how VHL gene is mutated might provide new opportunities for therapeutic intervention
Genetic counseling
In the recent years, the prognosis has improved significantly, initially due to the progress in the surgical technique and in post-surgical care. This was also due to the early detection that became possible, consequent to genetic testing in asymptomatic family members (Table 2) [67]. The diagnosis in asymptomatic patients has social, psychological, and economic implications, but the benefits of its early identification outweigh the negative consequences. This has led to a clear screening protocol indication since childhood [1].
Genetic counseling is important because germline mutations in the VHL gene are virtually detected in all families with one or more affected members. Germline mutations are also identified in 4% of patients with sporadic hemangioblastomas, 1.6% of renal cell carcinomas, and 3–9% of pheochromocytomas [67].
Approximately 80% of the individuals with VHL have at least one parent affected by the syndrome. Both patients and their families should be correctly genotyped and referred for genetic counseling in conjunction with other medical consultations and performing of the appropriate tests to identify VHL clinical manifestations [68].
Affected subjects have a 50% chance of transmitting the mutation to their children, because VHL is an autosomal dominant disease. For patients with mosaicism, the risk may be as high as 50% if the germinative cells carry the mutation. The diagnosis can be made in the prenatal period through amniocentesis or chorionic villus biopsy. Parents should be informed about reproductive technologies such as egg or sperm donation. There is also the possibility of genetic diagnosis, in the pre-implantation stage of in vitro-fertilized embryos [69]. An extensive review on genetic counseling was recently published by Nielsen et al. [70]
Follow-up
There are several protocols for monitoring the VHL syndrome such as the protocol of the University of Cambridge [71] and of the American National Institute of Health [13] (Supplemental Table 1).
Overall, the ophthalmologic exam of the retina should be performed annually and started before the fifth year of age. The blood pressure monitoring should be performed routinely, at least once a year, associated with urinary catecholamine determination starting from the fifth year of age, mainly in families with a high incidence of pheochromocytoma. Abdominal ultrasound and/or CT or MRI should be performed annually, starting at 16/20 years of age [17]. It is also important that the audiological evaluation is conducted in patients with known hearing loss, followed by temporal bone resonance, if there are any changes.
In spite of its rarity, this disease is so complex that it can only be well managed in referral centers. Diagnostic delay is a very important issue as it impedes implementation of a prophylactic screening program necessary for improving median overall survival. Follow-up should be life-long and we recommend an inter-disciplinary approach (Fig. 1). Modifications of screening plan may sometimes be done with individual patients and their family history, since VHL surveillance guidelines are based on expert medical opinion, but limited evidence is available regarding the optimal initiation and frequency of surveillance regimes.

Inter-disciplinary approach for screening, treatment, and follow-up of VHL syndrome, both in the index cases and the family members carrying the same mutation
Therapeutic implications of the recent molecular understanding of VHL disease
Drugs developed to change the downstream consequences of VHL inactivation, mainly to decrease the rich vascularization observed in tumors of VHL patients, have already demonstrated significant efficacy in these patients [72]. These drugs include small molecule inhibitors of VEGFRs (sorafenib, sunitinib, valatinib, and pazopanib) and anti-VEGFR antibodies (bevacizumab) [72,73,74,75]. The effects seem to differ between the VHL types and this was suggested to be due to the differential endothelial VEGFR expression observed in the different tumors [74].
In patients with pancreatic neuroendocrine lesions, the VEGFR inhibitor, sunitinib, was able to lead to a stabilization of the disease and a phase II clinical trial was already performed, but the results were not yet released (NCT01168440) [72]. In VHL renal lesions, sunitinib led to a partial response in 33% of the patients, but contrary to this none of the hemangioblastomas responded to sunitinib [74]. Still, other anti-angiogenic drugs were tested in VHL-associated hemangioblastomas with good results. A case report reported a patient treated with pazopanib which resulted in a significant neurologic improvement and decrease of the hemangioblastoma volume [75]. Besides that, a long-term treatment with intravitreal bevacizumab, improved the visual acuity and the optical coherence tomography thickness in a patient with VHL-associated hemangioblastoma [76]. Vatalanib is actually under investigation in a phase II clinical trial for the treatment of hemangioblastomas in patients with VHL (NCT00052013).
mTOR inhibitors (everolimus and temsirolimus) have also shown favorable clinical results in advanced renal cell carcinoma and in pancreatic neuroendocrine tumors. However, studies with VHL patients are still missing despite of mTOR inhibitors being considered as promising therapies [72, 77,78,79]. Actually, a study of mutation-targeted therapy with sunitinib or everolimus in patients with neuroendocrine tumors of the gastrointestinal tract and pancreas is recruiting, and it includes VHL patients (NCT02315625).
Conclusion
In this article, we reviewed the VHL disease, a familiar syndrome of multiple benign and malignant tumors, which affects individuals since childhood and presents various forms of clinical manifestations. The discovery of any of the syndrome components, especially of hemangioblastomas of the retina and CNS, and also pheochromocytomas, should raise suspicion of this disease and other stigmas must then be investigated.
The identification of the syndrome is important due to its high risk of serious complications, which can be avoided with a proper diagnosis and early treatment. Early recognition, made possible through the genetic testing in asymptomatic members of the affected families has contributed to the improved prognosis of these patients.
For the monitoring of VHL disease, a multidisciplinary approach is needed, with professionals in the areas of endocrinology, nephrology, neurology, pathology, ophthalmology, otolaryngology, and urology, among others. The diagnosis and the follow-up of this syndrome is a challenge in the clinical practice, and the aim of this review is also to aid in the treatment of these patients.
References
Rasmussen A, Alonso E, Ochoa A, De Biase I, Familiar I, Yescas P, Sosa AL, Rodriguez Y, Chavez M, Lopez-Lopez M, Bidichandani SI (2010) Uptake of genetic testing and long-term tumor surveillance in Von Hippel–Lindau disease. BMC Med Genet 11:4. doi:10.1186/1471-2350-11-4
Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, Ferguson-Smith MA (1990) Clinical features and natural history of Von Hippel–Lindau disease. Q J Med 77(283):1151–1163
Maher ER, Neumann HP, Richard S (2011) Von Hippel–Lindau disease: a clinical and scientific review. Eur J Hum Genet 19(6):617–623. doi:10.1038/ejhg.2010.175
Friedrich CA (1999) Von Hippel–Lindau syndrome. A pleomorphic condition. Cancer 86(11 Suppl):2478–2482
Kaelin WG (2007) Von Hippel–Lindau disease. Annu Rev Pathol 2:145–173. doi:10.1146/annurev.pathol.2.010506.092049
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399(6733):271–275. doi:10.1038/20459
Semenza GL (2011) Oxygen sensing, homeostasis, and disease. N Engl J Med 365(6):537–547. doi:10.1056/NEJMra1011165
Frew IJ, Krek W (2008) pVHL: a multipurpose adaptor protein. Sci Signal 1(24):pe30. doi:10.1126/scisignal.124pe30
Gerdes JM, Davis EE, Katsanis N (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137(1):32–45. doi:10.1016/j.cell.2009.03.023
Barontini M, Dahia PL (2010) VHL disease. Best Pract Res Clin Endocrinol Metab 24(3):401–413. doi:10.1016/j.beem.2010.01.002
Musacchio A, Salmon ED (2007) The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8(5):379–393. doi:10.1038/nrm2163
Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, Hergovich A, Moch H, Meraldi P, Krek W (2009) VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol 11(8):994–1001. doi:10.1038/ncb1912
Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH (2003) Von Hippel–Lindau disease. Lancet 361(9374):2059–2067. doi:10.1016/S0140-6736(03)13643-4
Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L et al (1993) Identification of the Von Hippel–Lindau disease tumor suppressor gene. Science 260(5112):1317–1320
Couch V, Lindor NM, Karnes PS, Michels VV (2000) Von Hippel–Lindau disease. Mayo Clin Proc 75(3):265–272
Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, van Brussel A, Voest EE, Giles RH (2010) Genetic analysis of Von Hippel–Lindau disease. Hum Mutat 31(5):521–537. doi:10.1002/humu.21219
Choyke PL, Glenn GM, Walther MM, Patronas NJ, Linehan WM, Zbar B (1995) Von Hippel–Lindau disease: genetic, clinical, and imaging features. Radiology 194(3):629–642
Neumann HP, Eggert HR, Scheremet R, Schumacher M, Mohadjer M, Wakhloo AK, Volk B, Hettmannsperger U, Riegler P, Schollmeyer P et al (1992) Central nervous system lesions in Von Hippel–Lindau syndrome. J Neurol Neurosurg Psychiatry 55(10):898–901
Lonser RR, Butman JA, Huntoon K, Asthagiri AR, Wu T, Bakhtian KD, Chew EY, Zhuang Z, Linehan WM, Oldfield EH (2014) Prospective natural history study of central nervous system hemangioblastomas in Von Hippel–Lindau disease. J Neurosurg 120(5):1055–1062. doi:10.3171/2014.1.JNS131431
Wiesener MS, Seyfarth M, Warnecke C, Jurgensen JS, Rosenberger C, Morgan NV, Maher ER, Frei U, Eckardt KU (2002) Paraneoplastic erythrocytosis associated with an inactivating point mutation of the Von Hippel–Lindau gene in a renal cell carcinoma. Blood 99(10):3562–3565
Asthagiri AR, Mehta GU, Zach L, Li X, Butman JA, Camphausen KA, Lonser RR (2010) Prospective evaluation of radiosurgery for hemangioblastomas in Von Hippel–Lindau disease. Neuro Oncol 12(1):80–86. doi:10.1093/neuonc/nop018
Patrice SJ, Sneed PK, Flickinger JC, Shrieve DC, Pollock BE, Alexander E 3rd, Larson DA, Kondziolka DS, Gutin PH, Wara WM, McDermott MW, Lunsford LD, Loeffler JS (1996) Radiosurgery for hemangioblastoma: results of a multiinstitutional experience. Int J Radiat Oncol Biol Phys 35(3):493–499
Wanebo JE, Lonser RR, Glenn GM, Oldfield EH (2003) The natural history of hemangioblastomas of the central nervous system in patients with Von Hippel–Lindau disease. J Neurosurg 98(1):82–94. doi:10.3171/jns.2003.98.1.0082
Huntoon K, Wu T, Elder JB, Butman JA, Chew EY, Linehan WM, Oldfield EH, Lonser RR (2016) Biological and clinical impact of hemangioblastoma-associated peritumoral cysts in Von Hippel–Lindau disease. J Neurosurg 124(4):971–976. doi:10.3171/2015.4.JNS1533
Kreusel KM (2005) Ophthalmological manifestations in VHL and NF 1: pathological and diagnostic implications. Fam Cancer 4(1):43–47. doi:10.1007/s10689-004-1327-0
Singh AD, Nouri M, Shields CL, Shields JA, Smith AF (2001) Retinal capillary hemangioma: a comparison of sporadic cases and cases associated with Von Hippel–Lindau disease. Ophthalmology 108(10):1907–1911
Schoen MA, Shields CL, Say EA, Douglass AM, Shields JA, Jampol LM (2016) Clinically invisible retinal hemangioblastomas detected by spectral domain optical coherence tomography and fluorescein angiography in twins. Retin Cases Brief Rep. doi:10.1097/ICB.0000000000000382
Singh AD, Nouri M, Shields CL, Shields JA, Perez N (2002) Treatment of retinal capillary hemangioma. Ophthalmology 109(10):1799–1806
Linehan WM, Walther MM, Zbar B (2003) The genetic basis of cancer of the kidney. J Urol 170(6 Pt 1):2163–2172. doi:10.1097/01.ju.0000096060.92397.ed
Kragel PJ, Walther MM, Pestaner JP, Filling-Katz MR (1991) Simple renal cysts, atypical renal cysts, and renal cell carcinoma in Von Hippel–Lindau disease: a lectin and immunohistochemical study in six patients. Mod Pathol 4(2):210–214
Neumann HP, Zbar B (1997) Renal cysts, renal cancer and Von Hippel–Lindau disease. Kidney Int 51(1):16–26
Montani M, Heinimann K, von Teichman A, Rudolph T, Perren A, Moch H (2010) VHL-gene deletion in single renal tubular epithelial cells and renal tubular cysts: further evidence for a cyst-dependent progression pathway of clear cell renal carcinoma in Von Hippel–Lindau disease. Am J Surg Pathol 34(6):806–815. doi:10.1097/PAS.0b013e3181ddf54d
Malek RS, Omess PJ, Benson RC Jr, Zincke H (1987) Renal cell carcinoma in Von Hippel–Lindau syndrome. Am J Med 82(2):236–238
Choyke PL, Glenn GM, Walther MM, Zbar B, Weiss GH, Alexander RB, Hayes WS, Long JP, Thakore KN, Linehan WM (1992) The natural history of renal lesions in Von Hippel–Lindau disease: a serial CT study in 28 patients. AJR Am J Roentgenol 159(6):1229–1234
Nelson JB, Oyasu R, Dalton DP (1994) The clinical and pathological manifestations of renal tumors in Von Hippel–Lindau disease. J Urol 152(6 Pt 2):2221–2226
Karsdorp N, Elderson A, Wittebol-Post D, Hene RJ, Vos J, Feldberg MA, van Gils AP, Jansen-Schillhorn van Veen JM, Vroom TM, Hoppener JW et al (1994) Von Hippel–Lindau disease: new strategies in early detection and treatment. Am J Med 97(2):158–168
Chauveau D, Duvic C, Chretien Y, Paraf F, Droz D, Melki P, Helenon O, Richard S, Grunfeld JP (1996) Renal involvement in Von Hippel–Lindau disease. Kidney Int 50(3):944–951
Steinbach F, Novick AC, Zincke H, Miller DP, Williams RD, Lund G, Skinner DG, Esrig D, Richie JP, deKernion JB et al (1995) Treatment of renal cell carcinoma in Von Hippel–Lindau disease: a multicenter study. J Urol 153(6):1812–1816
Koch CA, Mauro D, Walther MM, Linehan WM, Vortmeyer AO, Jaffe R, Pacak K, Chrousos GP, Zhuang Z, Lubensky IA (2002) Pheochromocytoma in Von Hippel–Lindau disease: distinct histopathologic phenotype compared to pheochromocytoma in multiple endocrine neoplasia type 2. Endocr Pathol 13(1):17–27
Pinato DJ, Ramachandran R, Toussi ST, Vergine M, Ngo N, Sharma R, Lloyd T, Meeran K, Palazzo F, Martin N, Khoo B, Dina R, Tan TM (2013) Immunohistochemical markers of the hypoxic response can identify malignancy in phaeochromocytomas and paragangliomas and optimize the detection of tumours with VHL germline mutations. Br J Cancer 108(2):429–437. doi:10.1038/bjc.2012.538
Baghai M, Thompson GB, Young WF Jr, Grant CS, Michels VV, van Heerden JA (2002) Pheochromocytomas and paragangliomas in Von Hippel–Lindau disease: a role for laparoscopic and cortical-sparing surgery. Arch Surg 137(6):682–688 (discussion 688–689)
Neumann HP, Berger DP, Sigmund G, Blum U, Schmidt D, Parmer RJ, Volk B, Kirste G (1993) Pheochromocytomas, multiple endocrine neoplasia type 2, and Von Hippel–Lindau disease. N Engl J Med 329(21):1531–1538. doi:10.1056/NEJM199311183292103
Aufforth RD, Ramakant P, Sadowski SM, Mehta A, Trebska-McGowan K, Nilubol N, Pacak K, Kebebew E (2015) Pheochromocytoma screening initiation and frequency in Von Hippel–Lindau syndrome. J Clin Endocrinol Metab 100(12):4498–4504. doi:10.1210/jc.2015-3045
Cassol C, Mete O (2015) Endocrine manifestations of Von Hippel–Lindau disease. Arch Pathol Lab Med 139(2):263–268. doi:10.5858/arpa.2013-0520-RS
Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, Gnarra JR, Orcutt ML, Duh FM, Glenn G et al (1995) Germline mutations in the Von Hippel–Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat 5(1):66–75. doi:10.1002/humu.1380050109
Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, Mannelli M, Goldstein DS, Linehan WM, Lenders JW, Pacak K (2001) Pheochromocytomas in Von Hippel–Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 86(5):1999–2008
Baguet JP, Hammer L, Mazzuco TL, Chabre O, Mallion JM, Sturm N, Chaffanjon P (2004) Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol 150(5):681–686
Green JS, Bowmer MI, Johnson GJ (1986) Von Hippel–Lindau disease in a Newfoundland kindred. CMAJ 134(2):133–138, 146
Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF Jr, Endocrine S (2014) Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 99(6):1915–1942. doi:10.1210/jc.2014-1498
Gaal J, van Nederveen FH, Erlic Z, Korpershoek E, Oldenburg R, Boedeker CC, Kontny U, Neumann HP, Dinjens WN, de Krijger RR (2009) Parasympathetic paragangliomas are part of the Von Hippel–Lindau syndrome. J Clin Endocrinol Metab 94(11):4367–4371. doi:10.1210/jc.2009-1479
Hammel PR, Vilgrain V, Terris B, Penfornis A, Sauvanet A, Correas JM, Chauveau D, Balian A, Beigelman C, O’Toole D, Bernades P, Ruszniewski P, Richard S (2000) Pancreatic involvement in Von Hippel–Lindau disease. The Groupe Francophone d’Etude de la Maladie de Von Hippel–Lindau. Gastroenterology 119(4):1087–1095
Charlesworth M, Verbeke CS, Falk GA, Walsh M, Smith AM, Morris-Stiff G (2012) Pancreatic lesions in Von Hippel–Lindau disease? A systematic review and meta-synthesis of the literature. J Gastrointest Surg 16(7):1422–1428. doi:10.1007/s11605-012-1847-0
Corcos O, Couvelard A, Giraud S, Vullierme MP, Dermot OT, Rebours V, Stievenart JL, Penfornis A, Niccoli-Sire P, Baudin E, Sauvanet A, Levy P, Ruszniewski P, Richard S, Hammel P (2008) Endocrine pancreatic tumors in Von Hippel–Lindau disease: clinical, histological, and genetic features. Pancreas 37(1):85–93. doi:10.1097/MPA.0b013e31815f394a
Mete O, Asa SL (2013) Precursor lesions of endocrine system neoplasms. Pathology 45(3):316–330. doi:10.1097/PAT.0b013e32835f45c5
Lubensky IA, Pack S, Ault D, Vortmeyer AO, Libutti SK, Choyke PL, Walther MM, Linehan WM, Zhuang Z (1998) Multiple neuroendocrine tumors of the pancreas in Von Hippel–Lindau disease patients: histopathological and molecular genetic analysis. Am J Pathol 153(1):223–231. doi:10.1016/S0002-9440(10)65563-0
Gucer H, Szentgyorgyi E, Ezzat S, Asa SL, Mete O (2013) Inhibin-expressing clear cell neuroendocrine tumor of the ampulla: an unusual presentation of Von Hippel–Lindau disease. Virchows Arch 463(4):593–597. doi:10.1007/s00428-013-1465-6
Keutgen XM, Hammel P, Choyke PL, Libutti SK, Jonasch E, Kebebew E (2016) Evaluation and management of pancreatic lesions in patients with Von Hippel–Lindau disease. Nat Rev Clin Oncol 13(9):537–549. doi:10.1038/nrclinonc.2016.37
Vinik AI, Raymond E (2013) Pancreatic neuroendocrine tumors: approach to treatment with focus on sunitinib. Therap Adv Gastroenterol 6(5):396–411. doi:10.1177/1756283X13493878
Blansfield JA, Choyke L, Morita SY, Choyke PL, Pingpank JF, Alexander HR, Seidel G, Shutack Y, Yuldasheva N, Eugeni M, Bartlett DL, Glenn GM, Middelton L, Linehan WM, Libutti SK (2007) Clinical, genetic and radiographic analysis of 108 patients with Von Hippel–Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 142(6):814–818. doi:10.1016/j.surg.2007.09.012 (discussion 818 e811–e812)
Manski TJ, Heffner DK, Glenn GM, Patronas NJ, Pikus AT, Katz D, Lebovics R, Sledjeski K, Choyke PL, Zbar B, Linehan WM, Oldfield EH (1997) Endolymphatic sac tumors. A source of morbid hearing loss in Von Hippel–Lindau disease. JAMA 277(18):1461–1466
Butman JA, Kim HJ, Baggenstos M, Ammerman JM, Dambrosia J, Patsalides A, Patronas NJ, Oldfield EH, Lonser RR (2007) Mechanisms of morbid hearing loss associated with tumors of the endolymphatic sac in Von Hippel–Lindau disease. JAMA 298(1):41–48. doi:10.1001/jama.298.1.41
Hansen MR, Luxford WM (2004) Surgical outcomes in patients with endolymphatic sac tumors. Laryngoscope 114(8):1470–1474. doi:10.1097/00005537-200408000-00028
Kim HJ, Butman JA, Brewer C, Zalewski C, Vortmeyer AO, Glenn G, Oldfield EH, Lonser RR (2005) Tumors of the endolymphatic sac in patients with Von Hippel–Lindau disease: implications for their natural history, diagnosis, and treatment. J Neurosurg 102(3):503–512. doi:10.3171/jns.2005.102.3.0503
Safatle PP, Farage L, Sampaio A, Ferreira FA, Safatle HP, Oliveira CA, Ferrari I (2009) Endolymphatic sac tumor and Von Hippel–Lindau disease in a single family. Arq Neuropsiquiatr 67(4):1097–1099
Jagannathan J, Lonser RR, Stanger RA, Butman JA, Vortmeyer AO, Zalewski CK, Brewer C, Surowicz C, Kim HJ (2007) Cochlear implantation for hearing loss associated with bilateral endolymphatic sac tumors in Von Hippel–Lindau disease. Otol Neurotol 28(7):927–930. doi:10.1097/MAO.0b013e31805c7506
Gersell DJ, King TC (1988) Papillary cystadenoma of the mesosalpinx in Von Hippel–Lindau disease. Am J Surg Pathol 12(2):145–149
Hes FJ, Hoppener JW, Luijt RB, Lips CJ (2005) Von Hippel–Lindau disease. Hered Cancer Clin Pract 3(4):171–178. doi:10.1186/1897-4287-3-4-171
Schmid S, Gillessen S, Binet I, Brandle M, Engeler D, Greiner J, Hader C, Heinimann K, Kloos P, Krek W, Krull I, Stoeckli SJ, Sulz MC, van Leyen K, Weber J, Rothermundt C, Hundsberger T (2014) Management of Von Hippel–Lindau disease: an interdisciplinary review. Oncol Res Treat 37(12):761–771. doi:10.1159/000369362
Obradors A, Fernandez E, Rius M, Oliver-Bonet M, Martinez-Fresno M, Benet J, Navarro J (2009) Outcome of twin babies free of Von Hippel–Lindau disease after a double-factor preimplantation genetic diagnosis: monogenetic mutation analysis and comprehensive aneuploidy screening. Fertil Steril 91(3):933, e931–937. doi:10.1016/j.fertnstert.2008.11.013
Nielsen SM, Rhodes L, Blanco I, Chung WK, Eng C, Maher ER, Richard S, Giles RH (2016) Von Hippel–Lindau disease: genetics and role of genetic counseling in a multiple neoplasia syndrome. J Clin Oncol 34(18):2172–2181. doi:10.1200/JCO.2015.65.6140
Maher ER, Bentley E, Payne SJ, Latif F, Richards FM, Chiano M, Hosoe S, Yates JR, Linehan M, Barton DE et al (1992) Presymptomatic diagnosis of Von Hippel–Lindau disease with flanking DNA markers. J Med Genet 29(12):902–905
Agarwal R, Liebe S, Turski ML, Vidwans SJ, Janku F, Garrido-Laguna I, Munoz J, Schwab R, Rodon J, Kurzrock R, Subbiah V, Pan-Cancer Working G (2015) Targeted therapy for genetic cancer syndromes: Von Hippel–Lindau disease, Cowden syndrome, and Proteus syndrome. Discov Med 19(103):109–116
Escudier B, Bellmunt J, Negrier S, Bajetta E, Melichar B, Bracarda S, Ravaud A, Golding S, Jethwa S, Sneller V (2010) Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 28(13):2144–2150. doi:10.1200/JCO.2009.26.7849
Jonasch E, McCutcheon IE, Waguespack SG, Wen S, Davis DW, Smith LA, Tannir NM, Gombos DS, Fuller GN, Matin SF (2011) Pilot trial of sunitinib therapy in patients with Von Hippel–Lindau disease. Ann Oncol 22(12):2661–2666. doi:10.1093/annonc/mdr011
Kim BY, Jonasch E, McCutcheon IE (2012) Pazopanib therapy for cerebellar hemangioblastomas in Von Hippel–Lindau disease: case report. Target Oncol 7(2):145–149. doi:10.1007/s11523-012-0214-0
Hrisomalos FN, Maturi RK, Pata V (2010) Long-term use of intravitreal bevacizumab (avastin) for the treatment of Von Hippel–Lindau associated retinal hemangioblastomas. Open Ophthalmol J 4:66–69. doi:10.2174/1874364101004010066
Battelli C, Cho DC (2011) mTOR inhibitors in renal cell carcinoma. Therapy 8(4):359–367. doi:10.2217/thy.11.32
Faivre S, Kroemer G, Raymond E (2006) Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov 5(8):671–688. doi:10.1038/nrd2062
Yim KL (2012) Everolimus and mTOR inhibition in pancreatic neuroendocrine tumors. Cancer Manag Res 4:207–214. doi:10.2147/CMAR.S25979
Acknowledgements
We are grateful to Milaydis Sosa for proofreading.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human subjects performed by any of the authors
Informed consent
No informed consent.
Grants
This study was supported by the Portuguese Foundation for Science and Technology (FCT) through a PhD grant to Sofia S. Pereira (SFRH/BD/89308/2012).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Crespigio, J., Berbel, L.C.L., Dias, M.A. et al. Von Hippel–Lindau disease: a single gene, several hereditary tumors. J Endocrinol Invest 41, 21–31 (2018). https://doi.org/10.1007/s40618-017-0683-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-017-0683-1