
Abstract
Parkinson’s disease (PD) is a highly complex neurodegenerative disorder with a multifactorial origin. Although several cellular mechanisms and genes have been implicated in the onset and progression of the disease, the precise molecular underpinnings of the disease remain unclear. In this context, epigenetic modulation of gene expression by environmental factors is emerging as an important mechanism in PD and in other neurodegenerative disorders. Thus, epigenetic mechanisms, such as DNA methylation, histone modifications and altered microRNA expression, have been under intense investigation due to their possible involvement in PD. Epigenetic modulation is responsible for inducing differential gene expression, a phenomenon which is essential throughout life in order to regulate multiple cellular responses such as development, cellular fate commitment and adaptation to the environment. Disturbances of a balanced gene expression can, therefore, have detrimental effects. Environmental factors can challenge the establishment and maintenance of epigenetic modifications and could thereby fill the gap in our further understanding of origin and/or progression of neurodegenerative diseases. In this chapter, we focus on the role of epigenetics in PD.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
The inability of the brain to replenish certain cell types, upon their death, is associated with the development of specific conditions, known as neurodegenerative disorders. In Parkinson’s disease (PD), one of those conditions that usually manifests after 60 years of age, the demise of dopaminergic neurons from the substantia nigra, explains the typical motor symptoms of the disease. Due to the increase in life expectancy, the number of individuals affected by PD has also drastically increased, resulting in extensive socioeconomic challenges. In the USA alone, it has been estimated that the annual costs of PD rise up to $23 billion [1]. Several therapeutic options are presently available to treat some of the symptoms associated with PD. However, there is currently no cure or preventive strategy. The majority of PD cases is sporadic, with no known cause, and is thought to occur due to the interplay between susceptibility genes and the environment, in ways that are poorly understood.
The term epigenetics refers to alterations in gene expression, usually reversible, which can be inherited but are not engraved in the DNA sequence. These modifications can be implemented via methylation of the DNA, histone modifications or microRNAs (miRNAs). Chemical pollutants, nutrition, temperature changes and other environmental stresses can influence gene expression via changes in epigenetic modifications. Although no solid relationship has been yet identified, epigenetic deregulation is thought to play an important and poorly understood role in the aetiopathogenesis of various neurodegenerative disorders, including PD.
2 Parkinson’s Disease
2.1 Pathology and Clinical Features
PD, named after Dr. James Parkinson who first documented it in 1817, constitutes the second most prevalent neurodegenerative disorder today. With a prevalence of 1–2% over the age of 65 [2, 3] and of 4–5% over the age of 85 [4], it is estimated that this progressive disorder affects approximately 6.3 million individuals worldwide, with the number expected to increase to 8.3 million by 2030 [5].
The typical neuropathological hallmarks of PD are the loss of dopaminergic neurons from the substantia nigra pars compacta (SN) and the accumulation of intracellular protein inclusions termed Lewy bodies (LBs), mainly composed of alpha-synuclein (aSyn) [6, 7]. Dopaminergic neurons extend their fibres from the SN towards the striatum, where they release dopamine, the neurotransmitter responsible for the learning and execution of motor functions [8, 9]. Due to decreased levels of dopamine, PD patients present characteristic motor dysfunctions such as bradykinesia, muscle rigidity, resting tremor and postural instability [10, 11]. Nonmotor symptoms, including anxiety, depression, dementia, sleep disturbances, constipation, hyposmia and anosmia, are also apparent and limit the quality of life of patients even further [8, 12]. Motor features remain the principal criteria for the clinical diagnosis of PD, although some nonmotor impairments are now valued as predictive markers for the disorder since they tend to appear prior to the onset of motor symptoms [13, 14]. Indeed, according to the Braak staging hypothesis, Lewy body pathology is quite dispersed not only throughout the brain but also in other tissues, such as the gut. According to this hypothesis, the progression of PD is classified into six stages. Stages 1–2 are linked with the presymptomatic phase where Lewy bodies appear in the enteric and peripheral autonomic nervous system and also spread from the olfactory bulb and vagus nerve to the lower brainstem. The symptomatic period starts on stage 3, when the midbrain, including the SN, starts to be affected. Finally, pathological changes involve the mesocortex in stage 4 and the neocortex in stages 5 and 6 [15]. Although this staging system has been confirmed by other groups and applies for the majority of the cases, deviations from this model can be observed, raising questions about the overall validity of the hypothesis [16, 17].
2.2 Genetic Forms of PD
Familial forms of PD account for only about 10–15% of all the cases [18]. However, it is possible that additional cases might be associated with yet unidentified genes, as additional genetic studies are conducted [19]. Thus, the list of genes implicated in the onset of PD (PARK genes) is expanding. The PARK gene family currently comprises 20 genes (Table 19.1) which are responsible for autosomal recessive, dominant or X-linked modes of inheritance. Moreover, PD-related genes can present point mutations, duplications or triplications and account for both early- or late-onset forms PD [20, 21]. Interestingly, over 500 DNA variants have been described in only five of the PD-associated genes [22].
A mutation in gene encoding for alpha-synuclein (SNCA) was the first to be associated with familial PD. Presently, six point mutations leading to amino acid substitutions have been linked with autosomal dominant forms of PD. In addition, duplications and triplications of the SNCA locus have also been associated with autosomal dominant forms of PD [23,24,25]. Although SNCA is an extensively studied gene, the precise function of alpha-synuclein (aSyn) and how it causes disease remain elusive. aSyn is typically described as a presynaptic protein participating in the regulation of the synaptic vesicle pool and in neurotransmitter release. However, other studies reported aSyn binds mitochondria and is present in the interconnection of mitochondrial membranes and ER or in the nucleus [26,27,28,29].
LRRK2 mutations are the most common cause of autosomal dominant PD [30]. Some LRRK2 mutations are more prevalent in certain ethnic groups [22]. The majority of patients carrying LRRK2 mutations present the classical pathological features of PD, including the presence of LBs, but the age of onset of the symptoms can vary appearing either earlier or later than idiopathic forms of the disease [30].
The VPS35 gene codes for the vacuolar protein sorting 35 (VPS35). VPS35 is one of the central components of the retromer cargo-recognition complex which is involved in the trafficking and recycling of synaptic vesicles and proteins [30]. The p.D620N mutation was recognised as a novel cause of autosomal dominant, late-onset PD [31, 32], displaying a dominant negative protein sorting phenotype [33, 34].
The lysosomal enzyme β-glucocerebrosidase, encoded by GBA, plays an important role in glycolipid metabolism [35]. Mutations in this gene are known to cause Gaucher disease, one of a growing list of lysosomal storage disorders. However, GBA mutations have been described to increase the risk of developing PD and are quite common in PD patients [36,37,38,39].
On the other hand, mutations in PARK2, PINK1 and PARK7 can cause autosomal recessive forms of early-onset PD. All three genes share identical clinical phenotypes, but LB pathology appears to be more variable [35]. PARK3, PARK10 and PARK12 loci have been implicated in PD, but the genes have not yet been identified. Thus, further analyses will be necessary in order to elucidate the role these loci play in PD pathogenesis [40].
2.3 Sporadic Forms of PD
Most PD cases have no known cause, suggesting environmental and lifestyle factors play important and poorly understood roles in the disease. Although these factors are indeed valid and important, it is now estimated that genetics may explain up to 60% of PD cases, underscoring the complexity of the disorder [12]. Toxins, such as methyl-phenyl-tetrahydropyridine (MPTP) [41], 6-hydroxydopamine [42], the herbicide paraquat [43] and the pesticide rotenone [44], have been shown to cause loss of dopaminergic cells in the substantia nigra. In addition, exposure to heavy metals or electromagnetic radiation, head trauma and viral infections are also known risk factors in PD [12, 45]. On the contrary, caffeine [46], uric acid levels [47], nicotine [48] and antagonists of the A2A receptor [49] have been suggested to act as neuroprotectors.
3 Epigenetics in PD
PD, as other neurodegenerative diseases, is a complex disorder occurring from the interplay between genetic, environmental, nutritional and other factors, together with ageing. As epigenetics may be altered in response to, at least, some of these factors, it is becoming increasingly accepted; it may also play an important role in the aetiology and pathogenesis of PD.
3.1 The Role of DNA Methylation
DNA methylation involves the covalent addition of a methyl group from S-adenosyl methionine (SAM) to the 5′ position of cytosines. In this way, 5-methylcytosine is formed (5-mC), with the concomitant conversion of SAM to S-adenosylhomocysteine (SAH) [50,51,52]. Methylation is a dynamic process that is apparent in multiple genomic sites, although it is mainly described to occur in repeats of CG dinucleotides [53]. In the human genome, these dinucleotides cluster in areas known as CpG islands which are associated with promoter regions, at least for about 60% of human genes [54]. Functionally, DNA methylation is associated with transcriptional inhibition. This can be executed either directly, by hindering the association of the DNA machinery with chromatin, or indirectly, with the recruitment of methyl-CpG-binding domain proteins (MBDs) [55, 56]. MBDs, in turn, attract histone-modifying and chromatin-remodelling complexes to the methylated sites. DNA methyltransferases (DNMTs) are the enzymes responsible for mediating DNA methylation. In mammals, DNMT1 is able to maintain DNA methylation following replication, while DNMT3a and DNMT3b exert de novo methylation [50].
Genome-wide DNA methylation analysis in blood and brain samples of healthy individuals and PD patients revealed a significant dysregulation of CpG island methylation in the group of patients. Many genes were found to be either hypo- or hypermethylated, including PD risk genes [57]. Another study identified 20 genes that were differentially methylated in blood samples obtained from PD patients in comparison to controls [58].
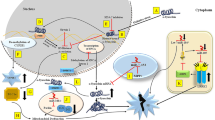
The observation that the SNCA promoter is hypermethylated in patients with alcoholism [59] or anorexia [60] suggested that epigenetics, perhaps through metabolic alterations, may also play a role in PD. Indeed, it was described that SNCA expression was upregulated upon methylation-mediated inhibition of SNCA intron 1 and that the SN, putamen and cortex of PD patients exhibited a significant hypomethylation pattern compared to healthy controls (Fig. 19.1) [61]. Another study was not able to detect methylation differences in the anterior cingulate or putamen of PD patients when examined a CpG region of the promoter of SNCA. However, substantial methylation reduction was apparent in the SN of these patients [62]. A reduction in the nuclear levels of DNMT1 was reported in postmortem brain tissue from dementia with Lewy bodies (DLB) or PD patients, as well as in brains from transgenic mice overexpressing SNCA. This alteration in the subcellular localisation of DNMT1 resulted in a global hypomethylation, including CpG islands upstream of SNCA and other genes, while aSyn was identified as the sequester of DNMT1 from the nucleus to the cytoplasm (Fig. 19.1) [63]. On the other hand, when the promoter and a CpG-rich region of SNCA intron 1 were analysed in patients with PD versus healthy individuals, hypermethylation at various positions in different brain regions was detected [64].

Epigenetic modifications in dopaminergic neurons. Certain toxins enter the neuronal cells and cause histone modifications, thereby influencing the expression of several genes. In the nucleus, aSyn interacts with H1 forming a tight complex and also with H3 inhibiting its acetylation. In turn, histones trigger the aggregation of aSyn. Several PD-associated genes, such as PARK16, GPNMB and STX1B, show altered expression as a result of aberrant DNA methylation. The promoter of SNCA is usually found hypomethylated in PD, leading to increased levels of aSyn. aSyn is able to sequester DNMT1 from the cytoplasm in the nucleus resulting in a general reduction of the methylation pattern. Ac acetylation, de-Ac deacetylation, ↑ increase, ↓ decrease, ┤ inhibition
The methylation status of SNCA intron 1 was further investigated in blood samples [65], peripheral blood mononuclear cells (PBMCs) [66] or leukocytes of PD patients [67]. In agreement with results in brain tissue, these studies reported a significant decrease in methylation of the SNCA promoter. Nevertheless, a correlation between SNCA mRNA levels and the methylation pattern of its promoter could not be firmly established [65, 66]. Another study in leukocytes from PD patients and healthy individuals revealed no alterations in the levels of methylation in any of the investigated regions [68].
Additional genes, namely, PARK16, GPNMB and STX1B, have been found to present aberrant methylation in postmortem PD brain samples (Fig. 19.1) [69]. The methylation status of the TNF promoter was significantly diminished in the SN compared to the cortex of both PD patients and healthy individuals, suggesting that a possible overexpression of TNF may trigger inflammatory reactions compromising the vulnerability of the dopaminergic neurons [70]. Postmortem samples obtained from the cortex and putamen showed decreased CpG methylation and increased mRNA levels of the CYP2E1 gene in PD patients [71]. Interestingly, a single nucleotide polymorphism (SNP) in this gene has been associated with PD [72], and its protein product, cytochrome P450 2E1, is implicated in the production of toxic metabolites that influence degeneration of dopaminergic neurons [50]. Although mutations in PARK2 have been associated with autosomal recessive juvenile parkinsonism, abnormal methylation levels of PARK2 promoter have been described in acute lymphoblastic and in chronic myeloid leukaemia [73], but not in PD cases [74]. In a similar manner, increased methylation of the UCHL1 promoter was reported in diverse types of cancer [75, 76], while no significant alterations in CpG methylation was observed in the hippocampus and frontal cortex from PD brains [77]. Similar results were obtained for ATP13A2 gene. DNA methylation of the promoter revealed an association with the progression of Kufor-Rakeb syndrome, although no such link has been made for PD so far [78].
DNA methylation in mitochondria might also be a relevant phenomenon in the context of PD. Recently, the mammalian mitochondrial DNMT (mtDNMT) was discovered [79]. Despite some controversy regarding CpG methylation in the genome of human mitochondria [80], some studies claim this can occur [81, 82]. Moreover, alterations in mitochondrial DNA methylation have been associated with cancer [83] and liver disease [84]. Finally, it was suggested that age-related changes in the DNA methylation of mitochondria may influence gene expression, alter mitochondrial metabolism and increase ROS production [85]. On the other hand, both PARK2 and PINK1 genes are essential for physiological mitochondrial function, and, when either of them is mutated, they can lead to mitochondrial impairment [12]. Considering the involvement of mitochondria in PD, further investigation will unravel possible implication of mitochondrial DNA methylation in PD pathogenesis.
3.2 Hydroxymethylation
Recently, the enzyme ten-eleven translocation1 (Tet1) was found to catalyse the oxidation of 5-mC to 5-hydroxymethylcytosine (5-hmC) [86]. Following studies have associated 5-hmC with euchromatin, indicating its relation with promoter regions and increased transcriptional levels [87, 88]. This intriguing, novel epigenetic modification is essentially unexplored in the context of neurodegeneration.
A detailed study revealed that 5-hmC levels increase in the mouse cerebellum in an age-dependent manner. In addition, an intragenic and proximal (to transcription start or termination sites regions) enrichment of 5-hmC was identified and associated with elevated gene expression. Gene ontology pathway analysis of the differentially expressed genes pointed towards pathways which are associated with neurodegenerative diseases such as Alzheimer’s disease, Huntington’s disease and PD [89], but additional studies are necessary in order to establish whether this type of DNA alteration is relevant in neurodegeneration.
3.3 Histone Modifications
The N-terminal tails of the histones are around 25–40 amino acid residues long and constitute a suitable region where chromatin-modifying enzymes can execute their function [90]. Histone modifications include methylation of lysine or arginine residues, acetylation, phosphorylation, ubiquitination, SUMOylation, ADP-ribosylation, crotonylation, hydroxylation and proline isomerisation [52, 81]. Histone modifications have been described to play pivotal roles in the development, differentiation and maintenance of dopaminergic neurons [91]. However, little is known concerning alterations in the physiological pattern histone modifications and their implications in PD pathogenesis.
In a recent study, the use of isolated dopaminergic neurons from brain tissue from PD patients revealed increased acetylation levels of histone H2A, H3 and H4 compared to age-matched control individuals. Furthermore, the levels of various histone deacetylases (HDACs) are reduced in 1-methyl-4-phenylpyridinium (MPP+)-treated cells and in MPTP-treated mouse brains and also in midbrain samples from PD patients [92]. These findings highlighted the presence of histone modifications suggesting that chromatin remodelling may be highly implicated in the pathogenesis of PD. Exposure to additional toxins also induces alterations into histones. For instance, when the pesticide dieldrin was administered in mice, elevated acetylation of histones H3 and H4 occurred in mesencephalic dopaminergic neurons due to proteasomal dysfunction (Fig. 19.1). Subsequently, the cAMP response element-binding protein, a histone acetyltransferase (HAT), was found to accumulate in the cells [93]. Another neurotoxic agent, paraquat, induces acetylation of histone H3 in dopaminergic cells in vitro (Fig. 19.1) [94].
In murine and primate models of levodopa-induced dyskinesia (LDID), dopamine depletion via MPTP administration was associated with a reduction in histone H3 trimethylation at Lys4 (Fig. 19.1). Chronic levodopa (or l-DOPA) therapy of these models was accompanied by deacetylation of striatal histone H4 at Lys5, 8, 12 and 16 (Fig. 19.1). The presence of histone modifications is evident, suggesting they may contribute to the development and maintenance of LDID in PD [95]. LDID has been associated with abnormal dopamine D1 receptor transmission. Histone H3 phosphoacetylation is blocked by D1 receptor inactivation, suggesting that inhibition of histone H3 acetylation and/or phosphorylation may be used for the prevention or reversion of dyskinesia [96]. In a mouse model of PD, it was shown that administration of l-DOPA induced phosphorylation of histone H3 on Ser28 in regions marked by trimethylation of the adjacent Lys27 (Fig. 19.1). This phenomenon was specifically observed in neurons expressing the D1 receptor and correlated with aberrant expression of genes that may be accountable for motor complications or dyskinesia [97].
Dopaminergic neurons of paraquat-treated mice displayed accumulation of aSyn in the nucleus, where it co-localises with acetylated histone H3. Further investigation revealed that aSyn binds directly to histone H1 and forms a tight 2:1 complex (Fig. 19.1). On the other hand, histone H1, together with the core histones, was able to boost the formation of aSyn fibrils (Fig. 19.1) [98]. Another study reported both in vitro and in Drosophila that nuclear aSyn associated with histone H3 reduces its acetylation (Fig. 19.1) [99]. Similar results were also described in PC12 cells expressing monoamine oxidase B. aSyn co-localised with histone H3 and once more was able to decrease its acetylation [27]. Finally, overexpression of dHDAC6 in a Drosophila model of PD ectopically expressing SNCA promoted aSyn inclusion formation and reduced aSyn oligomerisation. On the other hand, depletion of dHDAC6 enhanced the detrimental effects of aSyn overexpression, including the loss of dopaminergic neurons and locomotor dysfunction [100].
In C. elegans overexpressing human wt or A53T SNCA, nine histone genes coding for linker H1 and two core histones, H2B and H4, were downregulated [101].
3.4 miRNAs in PD
miRNAs bind to the 3′ untranslated region (UTR) of mRNA targets and modulate protein translation [102]. Thus, given their pleiotropic effects in cell biology, miRNAs are also emerging as relevant contributors to neurodegeneration in PD. Recently, an overall downregulation of miRNAs was found in tissue samples isolated from the SN of PD patients when compared to samples from healthy individuals [103].
Transgenic mice lacking Dicer in their dopaminergic neurons display neuronal cell death in the SN [104], suggesting overall miRNA processing is detrimental for dopaminergic cell function. Interestingly, studies in PD patients revealed that miR-133b, which is specifically expressed in midbrain dopaminergic neurons, is deficient in midbrain tissue. miR-133b is involved in a negative feedback circuit that contains the paired-like homeodomain transcription factor Pitx3, having a regulatory role in the maturation and function of midbrain dopaminergic neurons [104]. miR-132 has also been linked to midbrain dopaminergic neuronal differentiation. In a rat model of PD, miR-132 was significantly increased, and, in turn, the levels of its target protein, nuclear receptor-related 1 protein (Nurr1), were reduced [105, 106].
In a study using the MPTP-induced mouse model of PD, miR-124 was found to be downregulated in the SN of the mice, along with an increase in the levels of calpain/CDK5 proteins [107]. Interestingly, activation of calpains has been associated with dopaminergic cell death in the MPTP-induced mouse model and in postmortem nigral tissue from PD brains [108]. Another study reported a functional role of elevated miR-126 in SN dopaminergic neurons of PD patients through the inhibition of IGF-1/PI3K signalling pathway, contributing to neurotoxicity [109].
The levels of miR-1, miR-22* and miR-29 are reduced in blood samples of PD patients. Interestingly, the levels of miR-16-2*, miR-26a2* and miR30a enabled the distinction between treated from non-treated PD patients [110]. On the other hand, miR-1826/miR-450b-3p, miR-505 and miR-626 are upregulated in the plasma of PD patients and may be useful as PD biomarkers [111].
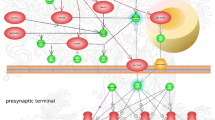
LRRK2 was found to influence the miRNA pathway, possibly by associating with Argonaute (Ago), in both human and Drosophila samples. Furthermore, in a Drosophila model of PD, it was observed that mutant LRRK2 suppresses the function of let-7 and miR-184* which normally regulate the translation of E2F1/DP complex, involved in cell cycle and survival control (Fig. 19.2) [112]. Furthermore, frontal cortex samples from PD patients contain high levels of LRRK2 and reduced levels of miR-205. It was then found that miR-205 is able to bind to the 3′ UTR of LRRK2 mRNA and suppress its expression. Further in vitro studies included the introduction of miR-205 in neurons carrying the R1441G LRRK2 mutation, which prevented outgrowth defects [113]. These findings suggested the regulatory role of miR-205 on LRRK2 expression and, therefore, a possible role in PD pathogenesis (Fig. 19.2).

The impact of miRNAs on TH+ neurons. miR-205 is able to suppress the expression of LRRK2 protein by binding to its 3′ UTR mRNA region. On the contrary, mutant LRRK2 inhibits let-7 and miR-184* which participate in cell survival. Overexpression of miR-494 reduces the levels of PARK7. Furthermore, several miRNAs bind to snca mRNA sequence and prevent its translation. On the other hand, the levels and aggregation of aSyn are indirectly increased due to increased FGF20 or decreased Hsp70 protein levels. Finally, mutant aSyn is thought to affect the production of certain miRNAs. *LRRK2 mutant LRRK2, *aSyn mutant aSyn, ↑ increase, ↓ decrease, ┤inhibition
DJ-1, the product of PARK7, is thought to be an oxidative sensor that protects cells from oxidative stress. Decreased levels of DJ-1 have been detected in the SN of sporadic PD patients suggesting a connection with PD. miR-494 was found to bind to the 3′ UTR of PARK7 mRNA and, when overexpressed, was able to significantly reduce DJ-1 protein levels in vitro and in an MPTP mouse model, while concomitantly rendering the cells more susceptible to oxidative stress and leading to dopaminergic cell death (Fig. 19.2) [114].
A global miRNA expression profiling in C. elegans showed that three members of the let-7 family (cel-miR-241, 230 and 48) were deregulated in animals mutated for PARK2. Similarly, 12 differentially regulated miRNAs from the miR-64/miR-65 and let-7 families were identified in animals overexpressing human A53T SNCA (Fig. 19.2) [115].
The levels of miR-34b and c were found significantly reduced in the amygdala, frontal cortex, cerebellum and SN of PD patients, accompanied by a decrease in the expression of PARK2 and PARK7. In addition, depletion of miR34-b and c in in vitro differentiated dopaminergic neurons caused an alteration of mitochondrial function and oxidative stress [116, 117]. In addition, both miRNAs appear to repress SNCA expression. Overexpression of miR-34b and c in SH-SY5Y cells resulted in a substantial reduction of aSyn protein levels via targeting the 3′ UTR of SNCA mRNA (Fig. 19.2), while inhibition, using anti-miRs, increased both the levels and the aggregation of the protein. Finally, a polymorphic variation in the 3′ UTR of human SNCA mRNA was associated with resistance to miR-34b binding and therefore to increased aSyn [118].
Two other abundant brain miRNAs, miR-7 and miR-153, bind to the 3′ UTR of SNCA mRNA and inhibit its translation (Fig. 19.2). More precisely, miR-7, a neuron-specific miRNA, was found to downregulate the expression of SNCA in HEK293T cells, protecting against oxidative stress. On the other hand, a specific miR-7 inhibitor caused a significant increase of aSyn protein levels in SH-SY5Y cells. Results obtained from MPTP-treated mice were in agreement with those obtained in the in vitro models, showing a substantial reduction of miR-7 levels and suggesting that elevated SNCA expression may be attributed to this downregulation [119]. Furthermore, treatment of primary cortical neurons with MPP+ followed by miR-7 overexpression resulted in neuronal protection from MPP+-induced toxicity and restored neuronal viability [120]. This protection from cell death was achieved via preservation of active mTOR signalling, possibly promoting aSyn clearance [120, 121].
miR-153 is another brain predominant miRNA that binds to the 3′ UTR of SNCA mRNA resulting in a significant decrease of its mRNA and protein levels [122]. The miR-153 binding site is predicted to be located within nucleotides 459–465. A variation identified in one male PD patient (464 C > A) was never encountered in healthy individuals or in patients with familial PD that were involved in the study and was suggested to be a rare cause of PD [123]. Interestingly, it seems that miR-7 and miR-153 have a synergistic effect on reducing aSyn levels [122].
In contrast, it was reported that SH-SY5Y cells treated with miR-106a* significantly increased their aSyn protein levels [124]. Moreover, other miRNAs such as miR-301b, miR-26b, miR-373* and miR-21 which regulate the levels of chaperone-mediated autophagy proteins were significantly increased in the SN of human PD brain tissues [124].
Administration of MPP+ or MPTP to cell or mouse models, respectively, resulted in a decline of miR-214 levels and in an increase in aSyn levels. In particular, a miR-214 inhibitor caused a reduction in the amount of TH+ cells when administered in vivo. Thus, as a result, miR-214 may contribute to the upregulation of SNCA and, therefore, to the toxic effects of aSyn in dopaminergic neurons (Fig. 19.2) [125].
Alterations in synaptosomal proteins were investigated in early symptomatic A30P SNCA transgenic mice, indicating that several proteins related to mitochondrial function were differentially expressed. Moreover, miRNA expression profiling revealed that the levels of miR-10a, 10b, 212, 132 and 495 were altered in brainstem samples when compared those from wild-type control animals [126]. In a Drosophila A30P SNCA model, high-throughput sequencing of small RNAs revealed that five miRNAs were upregulated. Among them, miR-13b, miR-133 and miR-137 are enriched in the brain and highly conserved from Drosophila to humans. miR-137 was shown to target the 3′ UTR mRNA of the dopamine D2 receptor. Therefore, it was suggested that mutant aSyn may be responsible for the dysregulation of miRNAs which are implicated in neuroactive-ligand receptor pathways (Fig. 19.2) [127].
Heat sock protein 70 (Hsp70) is capable of inhibiting cellular toxicity caused by aSyn via reduction of aSyn misfolding and aggregation [128,129,130,131,132]. Chemical blockade of Hsp70 in a cellular model (SH-SY5Y cells) overexpressing SNCA promotes aSyn aggregation. Interestingly, administration of miR-16-1 mimics those results given that miR-16-1 targets HSP70 mRNA and downregulates both its mRNA and protein levels (Fig. 19.2) [133]. Therefore, aSyn toxicity and the protective effects of Hsp70 are corroborated via this novel mechanism, opening new perspectives for intervention in PD.
A polymorphism (rs1989754) in the FGF20 gene was reported to be associated with increased risk of developing PD [134]. Another FGF20 polymorphism that was identified a few years later (rs12720208) was suggested to obstruct the binding of miR-433 to the FGF20 mRNA both in vitro and in vivo and, therefore, lead to increased FGF20 protein levels. Interestingly, elevated FGF20 protein levels have been linked to the subsequent increase of aSyn levels, observed both in SH-SY5Y cells and in human brain samples. In this way, elevated FGF20 levels may account for susceptibility towards developing PD through the increase of aSyn (Fig. 19.2) [135].
4 Epigenetic-Based Therapeutic Approaches for PD
HDAC inhibitors (HDACis) are commonly used as anticancer molecules. However, they have also emerged in the field of neurodegenerative disorders, in models of PD and AD, due to their effects on different members of the histone deacetylase family of proteins [136,137,138,139]. Valproic acid (VPA) has been shown to protect against rotenone [140], aSyn [140] and MPTP toxicity [141]. The responses triggered by VPA were mediated by decreasing the levels of pro-inflammatory factors and inducing microglia apoptosis [142, 143]. Finally, trichostatin A (TSA) has been described to increase the expression of HSP70, thereby having neuroprotective and anti-inflammatory properties [144], and to induce microglia apoptosis accompanied by increased histone H3 acetylation [143]. Nevertheless, the positive effects of these compounds conceal certain drawbacks. For example, in one study, it was shown that hyperacetylation of histone H4 via the administration of sodium butyrate, an HDACi, induces the expression of the protein kinase C δ (PKCδ) in the striatum and SN of mice. This upregulation was responsible for increasing the sensitivity of the cells to oxidative stress, rendering the dopaminergic neurons more prone to cell death and potentially contributing to PD [145]. TSA was also found to induce neuronal cell death and activate pro-apoptotic genes, likely contributing to PD pathogenesis [146, 147]. In addition, it was described that TSA potentiated pro-inflammatory responses in microglial cells, a process that is associated with several degenerative conditions [148]. The balance between HAT and HDAC activities is vital for normal cellular function, and, although many studies are evaluating the therapeutic potential of HDACis in PD, it should also be noted that they may cause undesired side effects and responses not only in neurons but also in other cell types, due to putative effects in nonhistone protein targets. Thus, despite current hopes and potential, additional work is still necessary in order to improve the applicability of these approaches.
Abbreviations
- 3′ UTR:
-
3′ untranslated region
- 5-hmC:
-
5-hydroxymethylcytosine
- 5-mC:
-
5-methylcytosine is formed
- Ago:
-
Argonaute
- aSyn:
-
Alpha-synuclein protein
- DLB:
-
dementia with Lewy bodies
- DNMT1:
-
DNA methyltransferase 1
- DNMT3a:
-
DNA methyltransferase 3a
- DNMT3b:
-
DNA methyltransferase 3b
- DNMTs:
-
DNA methyltransferases
- FGF20:
-
Fibroblast growth factor 20
- HAT:
-
Histone acetyltransferase
- HDACis:
-
Histone deacetylase inhibitors
- HDACs:
-
Histone deacetylases
- Hsp70:
-
Heat sock protein 70
- LB:
-
Lewy bodies
- LDID:
-
Levodopa-induced dyskinesia
- l-DOPA:
-
Levodopa
- LN:
-
Lewy neurites
- MBDs:
-
methyl-CpG-binding domain proteins
- mDNMT:
-
Mitochondrial DNMT
- miRNAs:
-
microRNAs
- MPP+ :
-
1-methyl-4-phenylpyridinium
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NACP-Rep1:
-
polymorphic microsatellite repeat region
- Nurr1:
-
Nuclear receptor-related 1 protein
- PBMCs:
-
Peripheral blood mononuclear cells
- PD:
-
Parkinson’s disease
- PKC δ:
-
Protein kinase C δ
- SAH:
-
S-adenosylhomocysteine
- SAM:
-
S-adenosyl methionine
- SN:
-
substantia nigra pars compacta
- SNCA :
-
alpha-synuclein gene
- SNPs:
-
Single nucleotide polymorphisms
- TSA:
-
Trichostatin
- VPA:
-
Valproic acid
- VSP35:
-
vacuolar protein sorting 35
References
Huse DM, Schulman K, Orsini L, Castelli-Haley J, Kennedy S, Lenhart G. Burden of illness in Parkinson’s disease. Mov Disord [Internet]. 2005;20(11):1449–54. http://www.ncbi.nlm.nih.gov/pubmed/16007641
de Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, et al. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology [Internet]. Department of Epidemiology & Biostatistics, Erasmus Medical Center, Rotterdam, The Netherlands.; 2000;54:S21–3. http://www.ncbi.nlm.nih.gov/pubmed/10854357
Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, et al. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol [Internet]. Division of Research, Kaiser Permanente, Oakland, CA 94612, USA. skv@dor.kaiser.org; 2003;157:1015–22. http://www.ncbi.nlm.nih.gov/pubmed/12777365
de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol [Internet]. Department of Epidemiology & Biostatistics, Erasmus Medical Centre Rotterdam, Netherlands.; 2006;5:525–35. http://www.ncbi.nlm.nih.gov/pubmed/16713924
Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68(5):384–6.
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol [Internet]. Mayo Clinic, Jacksonville, FL, USA.; 2009;8:1150–7. http://www.ncbi.nlm.nih.gov/pubmed/19909913
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature [Internet]. 1997;388:839–40. http://www.ncbi.nlm.nih.gov/pubmed/9278044
Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur J Neurosci. 2011;33(6):1139–51.
Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2(7):492–501.
Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci [Internet]. Department of Neurology, Columbia University College of Physicians Surgeons, New York, New York 10032, USA. fahn@neuro.columbia.edu; 2003;991:1–14. http://www.ncbi.nlm.nih.gov/pubmed/12846969
Lang AE, Lozano AM. Parkinson’s disease. Second of two parts. N Engl J Med [Internet]. Department of Medicine, University of Toronto and the Toronto Hospital, Canada.; 1998;339:1130–43. http://www.ncbi.nlm.nih.gov/pubmed/9770561
Mhyre TR, Boyd JT, Hamill RW, Maguire-Zeiss KA. Parkinson’s disease. Subcell Biochem [Internet]. Department of Neuroscience, Georgetown University Medical Center, 3970 Reservoir Road, NW NRB WP-24A, 20057, Washington, DC, USA, trm36@georgetown.edu.; 2012;65:389–455. http://www.ncbi.nlm.nih.gov/pubmed/23225012
Savica R, Rocca W a, Ahlskog JE. When does Parkinson disease start? Arch Neurol. 2010;67(7):798–801.
Hawkes CH. The prodromal phase of sporadic Parkinson’s disease: Does it exist and if so how long is it? Movement Disorders. 2008. p. 1799–807.
Braak H, Del Tredici K, Rüb U, De Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211.
Halliday G, Hely M, Reid W, Morris J. The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol. 2008;115(4):409–15.
Zaccai J, Brayne C, McKeith I, Matthews F, Ince PG. Patterns and stages of alpha-synucleinopathy: Relevance in a population-based cohort. Neurology. 2008;70(13):1042–8.
Verstraeten A, Theuns J, Van Broeckhoven C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet [Internet]. Elsevier Ltd; 2015;31(3):140–9. http://dx.doi.org/10.1016/j.tig.2015.01.004
Hamza TH, Payami H. The heritability of risk and age at onset of Parkinson’s disease after accounting for known genetic risk factors. J Hum Genet [Internet]. Division of Genetics, New York State Department of Health, Wadsworth Center, Albany, NY 12201–2002, USA.; 2010;55:241–3. http://www.ncbi.nlm.nih.gov/pubmed/20203693
Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet [Internet]. 2011;12:301–25. http://www.annualreviews.org/doi/abs/10.1146/annurev-genom-082410-101440
Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson’s disease and parkinsonism. Annals of Neurology. 2006. p. 389–98.
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum Mutat. 2010;31(7):763–80.
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364(9440):1167–9.
Singleton a B, Farrer M, Johnson J, Singleton a, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841.
Ibáñez P, Bonnet A-M, Débarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet (London, England) [Internet]. 2004;364(9440):1169–71. http://www.sciencedirect.com/science/article/pii/S0140673604171043
Guardia-Laguarta C, Area-Gomez E, Schon E a, Przedborski S. Novel subcellular localization for α-synuclein: possible functional consequences. Front Neuroanat [Internet]. 2015;9(February):17. http://journal.frontiersin.org.ezproxy.nihlibrary.nih.gov/article/10.3389/fnana.2015.00017/abstract
Siddiqui A, Chinta SJ, Mallajosyula JK, Rajagopolan S, Hanson I, Rane A, et al. Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson’s disease. Free Radic Biol Med [Internet]. Buck Institute for Age Research, 8001 Redwood Boulevard, Novato, CA 94945, USA.; 2012;53:993–1003. http://www.ncbi.nlm.nih.gov/pubmed/22705949
Yu S, Zuo X, Li Y, Zhang C, Zhou M, Zhang YA, et al. Inhibition of tyrosine hydroxylase expression in alpha-synuclein-transfected dopaminergic neuronal cells. Neurosci Lett [Internet]. Department of Neurobiology and the Sino-Japan Joint Laboratory on Neurodegenerative Diseases, Beijing Institute of Geriatrics, Xuanwu Hospital of the Capital University of Medical Sciences, 45 Changchun Street, Beijing 100053, China.; 2004;367:34–9. http://www.ncbi.nlm.nih.gov/pubmed/15308292
Specht CG, Tigaret CM, Rast GF, Thalhammer A, Rudhard Y, Schoepfer R. Subcellular localisation of recombinant alpha- and gamma-synuclein. Mol Cell Neurosci [Internet]. Laboratory for Molecular Pharmacology, Department of Pharmacology, University College London, UCL, London WC1E 6BT, UK.; 2005;28:326–34. http://www.ncbi.nlm.nih.gov/pubmed/15691713
Bonifati V. Genetics of Parkinson’s disease – state of the art, 2013. Parkinsonism Relat Disord [Internet]. Elsevier Ltd; 2014;20:S23–8. http://linkinghub.elsevier.com/retrieve/pii/S1353802013700099
Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset parkinson disease. Am J Hum Genet. 2011;89(1):168–75.
Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in parkinson disease. Am J Hum Genet. 2011;89(1):162–7.
Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol. 2010;20(14):1310–5.
MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, MacCabe BD, et al. RAB7L1 Interacts with LRRK2 to Modify Intraneuronal Protein Sorting and Parkinson’s Disease Risk. Neuron. 2013;77(3):425–39.
Klein C, Westenberger A. Genetics of Parkinson’s Disease. Cold Spring Harb Perspect Med [Internet]. Cold Spring Harbor Laboratory Press; 2012 Jan;2(1):a008888. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3253033/
The Glucocerebrosidase Gene and Parkinson’s Disease in Ashkenazi Jews. N Engl J Med [Internet]. Massachusetts Medical Society; 2005 Feb 17;352(7):728–31. http://dx.doi.org/10.1056/NEJM200502173520719
Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet [Internet]. 2004;41(12):937–40. http://jmg.bmj.com/content/41/12/937.full
Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab. 2004;81(1):70–3.
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med [Internet]. 2009;361(17):1651–61. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2856322&tool=pmcentrez&rendertype=abstract
Schulte C, Gasser T. Genetic basis of Parkinson’s disease: Inheritance, penetrance, and expression. Application of Clinical Genetics. 2011. p. 67–80.
Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219(4587):979–80.
Ungerstedt U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol. 1968;5:107–10.
Dauer W, Przedborski S. Parkinson’s disease: Mechanisms and models. Neuron. 2003. p. 889–909.
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov a V, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3(12):1301–6.
Shulman JM, De Jager PL, Feany MB. Parkinson’s Disease: Genetics and Pathogenesis. Annu Rev Pathol Mech Dis [Internet]. 2011;6(1):193–222. http://www.annualreviews.org/doi/abs/10.1146/annurev-pathol-011110-130242
Kalda A, Yu L, Oztas E, Chen JF. Novel neuroprotection by caffeine and adenosine A(2A) receptor antagonists in animal models of Parkinson’s disease. J Neurol Sci [Internet]. 2006;248(1–2):9–15. http://www.ncbi.nlm.nih.gov/pubmed/16806272
Wirdefeldt K, Adami H-O, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence [Internet]. Eur J Epidemiol 2011 p. 1–58. http://springerlink.bibliotecabuap.elogim.com/article/10.1007/s10654-011-9581-6/fulltext.html
Quik M. Smoking, nicotine and Parkinson’s disease. Trends in Neurosciences. 2004. p. 561–8.
Ferreira DG, Batalha VL, Vicente Miranda H, Coelho JE, Gomes R, Gonçalves FQ, et al. Adenosine A2A Receptors Modulate α-Synuclein Aggregation and Toxicity. Cereb Cortex [Internet]. 2015;bhv268. http://cercor.oxfordjournals.org/content/early/2015/11/02/cercor.bhv268.abstract
Feng Y, Jankovic J, Wu YC. Epigenetic mechanisms in Parkinson’s disease. J Neurol Sci [Internet]. Department of Neurology, Shanghai First People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200080, PR China. Parkinson's Disease Center and Movement Disorders Clinic, Department of Neurology, Baylor College of Medicine, Houston,; 2015;349:3–9. http://www.ncbi.nlm.nih.gov/pubmed/25553963
Coppede F. The potential of epigenetic therapies in neurodegenerative diseases. Front Genet [Internet]. Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa Pisa, Italy.; 2014;5:220. http://www.ncbi.nlm.nih.gov/pubmed/25071843
Urdinguio RG, Sanchez-Mut J V., Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol [Internet]. Elsevier Ltd; 2009;8(11):1056–72. http://dx.doi.org/10.1016/S1474-4422(09)70262-5
Georgel PT. The danger of epigenetics misconceptions (epigenetics and stuff…). Biochem Cell Biol [Internet]. 2015;4(August):1–4. http://www.nrcresearchpress.com/doi/abs/10.1139/bcb-2015-0091?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub=pubmed
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol [Internet]. Nature Publishing Group; 2010;28(10):1057–68. http://www.ncbi.nlm.nih.gov/pubmed/20944598
Lopez-Serra L, Esteller M. Proteins that bind methylated DNA and human cancer: reading the wrong words. Br J Cancer [Internet]. 2008;98(12):1881–5. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2441952&tool=pmcentrez&rendertype=abstract
Qureshi IA, Mehler MF. Epigenetic mechanisms governing the process of neurodegeneration. Mol Asp Med [Internet]. Roslyn and Leslie Goldstein Laboratory for Stem Cell Biology and Regenerative Medicine, Albert Einstein College of Medicine, Bronx, New York, NY 10461, USA. irfan@jhu.edu; 2013; 34:875–82. http://www.ncbi.nlm.nih.gov/pubmed/22782013
Masliah E, Dumaop W, Galasko D, Desplats P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics [Internet]. Department of Neuroscience; University of California San Diego; La Jolla, CA USA; Department of Pathology; University of California San Diego; La Jolla, CA USA. Department of Pathology; University of California San Diego; La Jolla, CA USA. Department of N; 2013;8:1030–8. http://www.ncbi.nlm.nih.gov/pubmed/23907097
Moore K, McKnight AJ, Craig D, O’Neill F. Epigenome-wide association study for Parkinson’s disease. Neuromolecular Med [Internet]. Queens University Belfast, Belfast, UK, kerry.moore@btinternet.com.; 2014;16:845–55. http://www.ncbi.nlm.nih.gov/pubmed/25304910
Bonsch D, Lenz B, Kornhuber J, Bleich S. DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport [Internet]. Department of Psychiatry and Psychotherapy, Friedrich-Alexander-University of Erlangen-Nuremberg, Schwabachanlage 6–10, D-91054 Erlangen, Germany.; 2005;16:167–70. http://www.ncbi.nlm.nih.gov/pubmed/15671870
Frieling H, Gozner A, Romer KD, Lenz B, Bonsch D, Wilhelm J, et al. Global DNA hypomethylation and DNA hypermethylation of the alpha synuclein promoter in females with anorexia nervosa. Mol Psychiatry [Internet]. 2007;12:229–30. http://www.ncbi.nlm.nih.gov/pubmed/17325715
Jowaed A, Schmitt I, Kaut O, Wullner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients' brains. J Neurosci [Internet]. Department of Neurology, Rheinische Friedrich-Wilhelms-Universitat, Universitatsklinikum Bonn, D-53105 Bonn, Germany.; 2010;30:6355–9. http://www.ncbi.nlm.nih.gov/pubmed/20445061
Matsumoto L, Takuma H, Tamaoka A, Kurisaki H, Date H, Tsuji S, et al. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One [Internet]. Division of Neuroscience, Department of Neurology, Graduate School of Medicine, The University of Tokyo, Bunkyo, Tokyo, Japan.; 2010;5:e15522. http://www.ncbi.nlm.nih.gov/pubmed/21124796
Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, et al. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem [Internet]. Department of Neurosciences, School of Medicine, University of California at San Diego, La Jolla, California 92093, USA. pdesplat@ucsd.edu; 2011;286:9031–7. http://www.ncbi.nlm.nih.gov/pubmed/21296890
de Boni L, Tierling S, Roeber S, Walter J, Giese A, Kretzschmar HA. Next-generation sequencing reveals regional differences of the alpha-synuclein methylation state independent of Lewy body disease. Neuromolecular Med [Internet]. The Center for Neuropathology and Prion Research, Ludwig-Maximilians-University Munich, Feodor-Lynen-Str. 23, 81377, Munich, Germany. Laura.de_Boni@med.uni.muenchen.de; 2011;13:310–20. http://www.ncbi.nlm.nih.gov/pubmed/22042430
Pihlstrom L, Berge V, Rengmark A, Toft M. Parkinson’s disease correlates with promoter methylation in the alpha-synuclein gene. Mov Disord [Internet]. Department of Neurology, Oslo University Hospital, Oslo, Norway.; 2015;30:577–80. http://www.ncbi.nlm.nih.gov/pubmed/25545759
Ai SX, Xu Q, Hu YC, Song CY, Guo JF, Shen L, et al. Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. J Neurol Sci [Internet]. Department of Neurology, Xiangya Hospital, Central South University, Changsha, China. Department of Geriatric Neurology, Xiangya Hospital, Central South University, Changsha, China. Department of Neurology, Xiangya Hospital, Central South University, Chan; 2014;337:123–8. http://www.ncbi.nlm.nih.gov/pubmed/24326201
Tan YY, Wu L, Zhao ZB, Wang Y, Xiao Q, Liu J, et al. Methylation of alpha-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Park Relat Disord [Internet]. Department of Neurology and Institute of Neurology, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China. Electronic address: rabbit82@gmail.com. Department of Neurology and Institute of Neurology, Ruijin; 2014;20:308–13. http://www.ncbi.nlm.nih.gov/pubmed/24398085
Song Y, Ding H, Yang J, Lin Q, Xue J, Zhang Y, et al. Pyrosequencing analysis of SNCA methylation levels in leukocytes from Parkinson’s disease patients. Neurosci Lett [Internet]. Department of Neurology, Xuanwu Hospital of Capital Medical University, Key Laboratory for Neurodegenerative Diseases of Ministry of Education, Beijing 100053, PR China. Department of Neurology, Xuanwu Hospital of Capital Medical University, Key Laborator; 2014;569:85–8. http://www.ncbi.nlm.nih.gov/pubmed/24721670
Plagnol V, Nalls MA, Bras JM, Hernandez DG, Sharma M, Sheerin U-M, et al. A Two-Stage Meta-Analysis Identifies Several New Loci for Parkinson’s Disease. Gibson G, editor. PLoS Genet [Internet]. 2011 Jun 30;7(6):e1002142. http://dx.plos.org/10.1371/journal.pgen.1002142
Pieper HC, Evert BO, Kaut O, Riederer PF, Waha A, Wüllner U. Different methylation of the TNF-alpha promoter in cortex and substantia nigra: Implications for selective neuronal vulnerability. Neurobiol Dis. 2008;32(3):521–7.
Kaut O, Schmitt I, Wüllner U. Genome-scale methylation analysis of Parkinson’s disease patients' brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics. 2012;13(1):87–91.
Shahabi HN, Westberg L, Melke J, Håkansson A, Belin AC, Sydow O, et al. Cytochrome P450 2E1 gene polymorphisms/haplotypes and Parkinson’s disease in a Swedish population. J Neural Transm. 2009;116(5):567–73.
Agirre X, Román-Gómez J, Vázquez I, Jiménez-Velasco A, Garate L, Montiel-Duarte C, et al. Abnormal methylation of the common PARK2 and PACRG promoter is associated with downregulation of gene expression in acute lymphoblastic leukemia and chronic myeloid leukemia. Int J Cancer. 2006;118(8):1945–53.
Cai M, Tian J, Zhao G, Luo W, Zhang B. Study of methylation levels of parkin gene promoter in Parkinson’s disease patients. Int J Neurosci [Internet]. 2011;121(9):497–502. http://www.ncbi.nlm.nih.gov/pubmed/21663383
Yu J, Tao Q, Cheung KF, Jin H, Poon FF, Wang X, et al. Epigenetic identification of ubiquitin carboxyl-terminal hydrolase L1 as a functional tumor suppressor and biomarker for hepatocellular carcinoma and other digestive tumors. Hepatology. 2008;48(2):508–18.
Kagara I, Enokida H, Kawakami K, Matsuda R, Toki K, Nishimura H, et al. CpG Hypermethylation of the UCHL1 Gene Promoter is Associated With Pathogenesis and Poor Prognosis in Renal Cell Carcinoma. J Urol. 2008;180(1):343–51.
Barrachina M, Ferrer I. DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J Neuropathol Exp Neurol. 2009;68(8):880–91.
Behrens MI, Brüggemann N, Chana P, Venegas P, Kägi M, Parrao T, et al. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord. 2010;25(12):1929–37.
Shock LS, Thakkar P V, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A [Internet]. 2011;108(9):3630–5. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3048134&tool=pmcentrez&rendertype=abstract
Hong EE, Okitsu CY, Smith AD, Hsieh C-L. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol Cell Biol [Internet]. 2013;33(14):2683–90. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3700126&tool=pmcentrez&rendertype=abstract
Lardenoije R, Iatrou A, Kenis G, Kompotis K, Steinbusch HW, Mastroeni D, et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol [Internet]. School for Mental Health and Neuroscience (MHeNS), Department of Psychiatry and Neuropsychology, Maastricht University, Universiteitssingel 50, 6200 MD Maastricht, The Netherlands. Center for Integrative Genomics, University of Lausanne, Genopode Building; 2015;131:21–64. http://www.ncbi.nlm.nih.gov/pubmed/26072273
Iacobazzi V, Castegna A, Infantino V, Andria G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Molecular Genetics and Metabolism. 2013. p. 25–34.
Feng S, Xiong L, Ji Z, Cheng W, Yang H. Correlation between increased ND2 expression and demethylated displacement loop of mtDNA in colorectal cancer. Mol Med Rep. 2012;6(1):125–30.
Pirola CJ, Fernandez Gianotti T, Burgueno a. L, Rey-Funes M, Loidl CF, Mallardi P, et al. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. 2012;1356–63.
Zinovkina LA, Zinovkin RA. DNA Methylation, Mitochondria, and Programmed Aging. Biochemistry [Internet]. 2015;80(12):1571–7. http://www.ncbi.nlm.nih.gov/pubmed/26638681
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science [Internet]. 2009;324(5929):930–5. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2715015&tool=pmcentrez&rendertype=abstract
Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore T a, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature [Internet]. 2011;473(7347):398–402. http://www.ncbi.nlm.nih.gov/pubmed/21460836
Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet [Internet]. 2011;13(1):7–13. http://dx.doi.org/10.1038/nrg3080
Song C-X, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29(1):68–72.
Habibi E, Masoudi-Nejad A, Abdolmaleky HM, Haggarty SJ. Emerging roles of epigenetic mechanisms in Parkinson’s disease. Funct Integr Genomics [Internet]. Laboratory of Systems Biology and Bioinformatics (LBB), Institute of Biochemistry and Biophysics and Center of Excellence in Biomathematics, University of Tehran, Tehran, Iran.; 2011;11:523–37. http://www.ncbi.nlm.nih.gov/pubmed/21892731
van Heesbeen HJ, Mesman S, Veenvliet J V, Smidt MP. Epigenetic mechanisms in the development and maintenance of dopaminergic neurons. Development [Internet]. 2013;140(6):1159–69. http://www.ncbi.nlm.nih.gov/pubmed/23444349
Park G, Tan J, Garcia G, Kang Y, Salvesen G, Zhang Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J Biol Chem [Internet]. Sanford-Burnham Medical Research Institute, United States; Central South University, China. Central South University, China zhangzhuohua@sklmg.edu.cn.; 2015; http://www.ncbi.nlm.nih.gov/pubmed/26699403
Song C, Kanthasamy A, Anantharam V, Sun F, Kanthasamy AG. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration. Mol Pharmacol [Internet]. Iowa Center for Advanced Neurotoxicology, Department of Biomedical Sciences, Iowa State University, Ames, IA 50011, USA.; 2010;77:621–32. http://www.ncbi.nlm.nih.gov/pubmed/20097775
Song C, Kanthasamy A, Jin H, Anantharam V, Kanthasamy AG. Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology [Internet]. Department of Biomedical Sciences, Iowa Center for Advanced Neurotoxicology, Iowa State University, Ames, IA 50011, USA.; 2011;32:586–95. http://www.ncbi.nlm.nih.gov/pubmed/21777615
Nicholas AP, Lubin FD, Hallett PJ, Vattem P, Ravenscroft P, Bezard E, et al. Striatal histone modifications in models of levodopa-induced dyskinesia. J Neurochem [Internet]. Center for Neurodegeneration and Experimental Therapeutics, Department of Neurology, University of Alabama at Birmingham, AL 35294–0017, USA. nicholas@uab.edu; 2008;106:486–94. http://www.ncbi.nlm.nih.gov/pubmed/18410512
Darmopil S, Martín AB, De Diego IR, Ares S, Moratalla R. Genetic Inactivation of Dopamine D1 but Not D2 Receptors Inhibits L-DOPA-Induced Dyskinesia and Histone Activation. Biol Psychiatry. 2009;66(6):603–13.
Södersten E, Feyder M, Lerdrup M, Gomes AL, Kryh H, Spigolon G, et al. Dopamine Signaling Leads to Loss of Polycomb Repression and Aberrant Gene Activation in Experimental Parkinsonism. PLoS Genet. 2014;10(9).
Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, et al. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry [Internet]. Department of Chemistry and Biochemistry, University of California, Santa Cruz, California 95064, USA.; 2003;42:8465–71. http://www.ncbi.nlm.nih.gov/pubmed/12859192
Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet [Internet]. Department of Pathology, Brigham and Women’s Hospital, Program in Neuroscience, Harvard Medical School, 77 Avenue Louis Pasteur, Boston, MA 02115, USA.; 2006;15:3012–23. http://www.ncbi.nlm.nih.gov/pubmed/16959795
Du G, Liu X, Chen X, Song M, Yan Y, Jiao R, et al. Drosophila Histone Deacetylase 6 Protects Dopaminergic Neurons against -Synuclein Toxicity by Promoting Inclusion Formation. Mol Biol Cell [Internet]. 2010;21(13):2128–37. http://www.molbiolcell.org/content/21/13/2128.abstract
Vartiainen S, Pehkonen P, Lakso M, Nass R, Wong G. Identification of gene expression changes in transgenic C. elegans overexpressing human α-synuclein. Neurobiol Dis. 2006;22(3):477–86.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009/01/27 ed. Howard Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. dbartel@wi.mit.edu; 2009;136:215–33.
Cardo LF, Coto E, Ribacoba R, Menendez M, Moris G, Suarez E, et al. MiRNA profile in the substantia nigra of Parkinson’s disease and healthy subjects. J Mol Neurosci [Internet]. Genetica Molecular-Laboratorio de Medicina, Hospital Universitario Central de Asturias, 33006, Oviedo, Spain.; 2014;54:830–6. http://www.ncbi.nlm.nih.gov/pubmed/25284245
Kim J, Inoue K, Ishii J, Vanti WB, Voronov S V, Murchison E, et al. A MicroRNA feedback circuit in midbrain dopamine neurons. Science (80- ) [Internet]. Departments of Pathology and Neurology, Center for Neurobiology and Behavior, and Taub Institute, Columbia University, College of Physicians and Surgeons 15–403, 630 West 168th Street, New York, NY 10032, USA.; 2007;317:1220–4. http://www.ncbi.nlm.nih.gov/pubmed/17761882
Jankovic J, Chen S, Le WD. The role of Nurr1 in the development of dopaminergic neurons and Parkinson’s disease. Prog Neurobiol. 2005;77(1–2):128–38.
Lungu G, Stoica G, Ambrus A. MicroRNA profiling and the role of microRNA-132 in neurodegeneration using a rat model. Neurosci Lett. 2013;553:153–8.
Kanagaraj N, Beiping H, Dheen ST, Tay SS. Downregulation of miR-124 in MPTP-treated mouse model of Parkinson’s disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience [Internet]. Department of Anatomy, Yong Loo Lin School of Medicine, National University Health System, National University of Singapore, Singapore 117597, Singapore. Department of Anatomy, Yong Loo Lin School of Medicine, National University Health System, National U; 2014;272:167–79. http://www.ncbi.nlm.nih.gov/pubmed/24792712
Crocker SJ, Smith PD, Jackson-Lewis V, Lamba WR, Hayley SP, Grimm E, et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J Neurosci. 2003;23(10):4081–91.
Kim W, Lee Y, McKenna ND, Yi M, Simunovic F, Wang Y, et al. miR-126 contributes to Parkinson’s disease by dysregulating the insulin-like growth factor/phosphoinositide 3-kinase signaling. Neurobiol Aging [Internet]. Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, MA, USA; Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, MA, USA. Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, MA, USA.; 2014;35:1712–21. http://www.ncbi.nlm.nih.gov/pubmed/24559646
Margis R, Margis R, Rieder CR. Identification of blood microRNAs associated to Parkinsonis disease. J Biotechnol [Internet]. Neurology Section, Movement Disorders Unit, Hospital de Clinicas de Porto Alegre, Rua Ramiro Barcelos 2350, 90035–00 Porto Alegre, RS, Brazil.; 2011;152:96–101. http://www.ncbi.nlm.nih.gov/pubmed/21295623
Khoo SK, Petillo D, Kang UJ, Resau JH, Berryhill B, Linder J, et al. Plasma-based circulating MicroRNA biomarkers for Parkinson’s disease. J Park Dis [Internet]. Center for Neurodegenerative Science, Van Andel Institute, Grand Rapids, MI 49503, USA. Kean.Khoo@vai.org; 2012;2:321–31. http://www.ncbi.nlm.nih.gov/pubmed/23938262
Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature [Internet]. 2010;466(7306):637–41. http://han.sub.uni-goettingen.de/han/GoogleScholar/www.nature.com/nature/journal/v466/n7306/full/nature09191.html
Cho HJ, Liu G, Jin SM, Parisiadou L, Xie C, Yu J, et al. Microrna-205 regulates the expression of parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum Mol Genet. 2013;22(3):608–20.
Xiong R, Wang Z, Zhao Z, Li H, Chen W, Zhang B, et al. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol Aging. 2014;35(3):705–14.
Asikainen S, Rudgalvyte M, Heikkinen L, Louhiranta K, Lakso M, Wong G, et al. Global microRNA expression profiling of Caenorhabditis elegans Parkinson’s disease models. J Mol Neurosci. 2010/01/22 ed. Department of Biosciences, Kuopio University, Kuopio, Finland.; 2010;41:210–8.
Minones-Moyano E, Porta S, Escaramis G, Rabionet R, Iraola S, Kagerbauer B, et al. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet. 2011/05/12 ed. Genetic Causes of Disease Group, Genes and Disease Program, Centre for Genomic Regulation, Barcelona, Catalonia, Spain.; 2011;20:3067–78.
Villar-Menendez I, Porta S, Buira SP, Pereira-Veiga T, Diaz-Sanchez S, Albasanz JL, et al. Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol Dis. 2014/06/04 ed. Institute of Neuropathology, Bellvitge University Hospital-ICS, [Bellvitge Biomedical Research Institute-] IDIBELL, L’Hospitalet de Llobregat, Spain. Departamento de Quimica Inorganica, Organica y Bioquimica, Facultad de Ciencias y Tecnologias Quimicas, C; 2014;69:206–14.
Kabaria S, Choi DC, Chaudhuri AD, Mouradian MM, Junn E. Inhibition of miR-34b and miR-34c enhances alpha-synuclein expression in Parkinson’s disease. FEBS Lett. 2014/12/30 ed. Center for Neurodegenerative and Neuroimmunologic Diseases, Department of Neurology, Rutgers - Robert Wood Johnson Medical School, Piscataway, NJ 08854, USA. Center for Neurodegenerative and Neuroimmunologic Diseases, Department of Neurology, Rutgers - Ro; 2015;589:319–25.
Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009/07/25 ed. Center for Neurodegenerative and Neuroimmunologic Diseases, Department of Neurology, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School, Piscataway, NJ 08854, USA. junneu@umdnj.edu; 2009;106:13052–7.
Fragkouli A, Doxakis E. miR-7 and miR-153 protect neurons against MPP(+)-induced cell death via upregulation of mTOR pathway. Front Cell Neurosci. 2014/07/30 ed. Lab of Molecular and Cellular Neuroscience, Center for Basic Research, Biomedical Research Foundation of the Academy of Athens Athens, Greece.; 2014;8:182.
Chen LL, Song JX, Lu JH, Yuan ZW, Liu LF, Durairajan SS, et al. Corynoxine, a natural autophagy enhancer, promotes the clearance of alpha-synuclein via Akt/mTOR pathway. J Neuroimmune Pharmacol. 2014/02/14 ed. School of Chinese Medicine, Hong Kong Baptist University, Kowloon, Hong Kong.; 2014;9:380–7.
Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010/01/29 ed. Basic Neurosciences Division, Biomedical Research Foundation of the Academy of Athens, Soranou Efesiou 4, Athens 11527, Greece. edoxakis@bioacademy.gr; 2010;285:12726–34.
Kim HJ, Park G, Jeon BS, Park WY, Kim YE. A mir-153 binding site variation in SNCA in a patient with Parkinson’s disease. Mov Disord. 2013/05/16 ed. Departments of Neurology and Movement Disorder Center, Parkinson Study Group, and Neuroscience Research Institute, Seoul National University College of Medicine, Seoul, Korea.; 2013;28:1755–6.
Alvarez-Erviti L, Seow Y, Schapira AH V, Rodriguez-Oroz MC, Obeso JA, Cooper JM. Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson’s disease. Cell Death Dis [Internet]. 2013;4:e545. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3615743&tool=pmcentrez&rendertype=abstract
Wang ZH, Zhang JL, Duan YL, Zhang QS, Li GF, Zheng DL. MicroRNA-214 participates in the neuroprotective effect of Resveratrol via inhibiting alpha-synuclein expression in MPTP-induced Parkinson’s disease mouse. Biomed Pharmacother. 2015/09/10 ed. Department of Internal Neurology, Huaihe Hospital of Henan University, Kaifeng 475000, China. Electronic address: wzhdyl0526@163.com. Department of Internal Neurology, Huaihe Hospital of Henan University, Kaifeng 475000, China. Department of Ultrasound, K; 2015;74:252–6.
Gillardon F, Mack M, Rist W, Schnack C, Lenter M, Hildebrandt T, et al. MicroRNA and proteome expression profiling in early-symptomatic α-synuclein(A30P)-transgenic mice. Proteomics - Clin Appl. 2008;2(5):697–705.
Kong Y, Liang X, Liu L, Zhang D, Wan C, Gan Z, et al. High Throughput Sequencing Identifies MicroRNAs Mediating α-Synuclein Toxicity by Targeting Neuroactive-Ligand Receptor Interaction Pathway in Early Stage of Drosophila Parkinson’s Disease Model. PLoS One [Internet]. 2015;10:e0137432. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4567341/
Outeiro TF, Klucken J, Strathearn KE, Liu F, Nguyen P, Rochet JC, et al. Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem Biophys Res Commun [Internet]. Alzheimer’s Research Unit, MassGeneral Institute for Neurodegenerative Disease, MGH, Harvard Medical School, Charlestown, MA 02129, USA. touteir@partners.org; 2006;351:631–8. http://www.ncbi.nlm.nih.gov/pubmed/17081499
Klucken J, Outeiro TF, Nguyen P, McLean PJ, Hyman BT. Detection of novel intracellular alpha-synuclein oligomeric species by fluorescence lifetime imaging. FASEB J [Internet]. MassGeneral Institute for Neurodegenerative Disease, Alzheimer’s Disease Research Unit, Massachusetts General Hospital, 114 16 St., Charlestown, MA 02129, USA.; 2006;20:2050–7. http://www.ncbi.nlm.nih.gov/pubmed/17012257
Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biol Chem [Internet]. Alzheimer’s Disease Research Laboratory, Harvard Medical School, Massachusetts General Hospital, Charlestown, Massachusetts 02129, USA.; 2004;279:25497–502. http://www.ncbi.nlm.nih.gov/pubmed/15044495
Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, et al. Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One [Internet]. Alzheimer’s Research Unit, MassGeneral Institute for Neurodegenerative Disease, MGH Harvard Medical School, Charlestown, Massachusetts, United States of America. touteiro@fm.ul.pt; 2008;3:e1867. http://www.ncbi.nlm.nih.gov/pubmed/18382657
Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem [Internet]. Alzheimer Disease Research Unit, Department of Neurology, Massachusetts General Hospital, Charlestown, Massachusetts 02129, USA.; 2005;280:23727–34. http://www.ncbi.nlm.nih.gov/pubmed/15845543
Zhang Z, Cheng Y. miR-16-1 Promotes the Aberrant α-Synuclein Accumulation in Parkinson Disease via Targeting Heat Shock Protein 70. Sci World J [Internet]. 2014;2014:8. http://dx.doi.org/10.1155/2014/938348
van der Walt JM, Noureddine MA, Kittappa R, Hauser MA, Scott WK, McKay R, et al. Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. Am J Hum Genet. 2004/05/04 ed. Department of Medicine and Center for Human Genetics, Duke University Medical Center, Durham, NC 27710, USA.; 2004;74:1121–7.
Wang G, van der Walt JM, Mayhew G, Li YJ, Zuchner S, Scott WK, et al. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am J Hum Genet. 2008/02/07 ed. Center for Human Genetics, Duke University Medical Center, Durham, NC 27710, USA.; 2008;82:283–9.
Bahari-Javan S, Sananbenesi F, Fischer A. Histone-acetylation: a link between Alzheimer’s disease and post-traumatic stress disorder? Front Neurosci [Internet]. Department of Psychiatry and Psychotherapy, University Medical Center Gottingen Gottingen, Germany ; Research Group for Epigenetics in Neurodegenerative Diseases, German Center for Neurodegenerative Diseases (DZNE) Gottingen Germany. Research Group for Ep; 2014;8:160. http://www.ncbi.nlm.nih.gov/pubmed/25009454
Benito E, Urbanke H, Ramachandran B, Barth J, Halder R, Awasthi A, et al. HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models. J Clin Invest [Internet]. 2015;125:3572–84. http://www.ncbi.nlm.nih.gov/pubmed/26280576
Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci [Internet]. Laboratory for Aging and Cognitive Diseases, European Neuroscience Institute, Grisebach Str. 5, D-37077 Goettingen, Germany. afische2@gwdg.de; 2010;31:605–17. http://www.ncbi.nlm.nih.gov/pubmed/20980063
Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science (80- ) [Internet]. Alzheimer’s Research Unit, MGH, Harvard Medical School, CNY 114, 16th Street, Charlestown, MA 02129, USA.; 2007;317:516–9. http://www.ncbi.nlm.nih.gov/pubmed/17588900
Monti B, Gatta V, Piretti F, Raffaelli SS, Virgili M, Contestabile A. Valproic acid is neuroprotective in the rotenone rat model of Parkinson’s disease: involvement of alpha-synuclein. Neurotox Res [Internet]. Department of Biology, University of Bologna, Bologna, Italy.; 2010;17:130–41. http://www.ncbi.nlm.nih.gov/pubmed/19626387
Kidd SK, Schneider JS. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience [Internet]. Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, 1020 Locust Street, JAH 521, Philadelphia, PA 19107, USA.; 2011;194:189–94. http://www.ncbi.nlm.nih.gov/pubmed/21846494
Peng GS, Li G, Tzeng NS, Chen PS, Chuang DM, Hsu YD, et al. Valproate pretreatment protects dopaminergic neurons from LPS-induced neurotoxicity in rat primary midbrain cultures: role of microglia. Brain Res Mol Brain Res [Internet]. Department of Neurology, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan.; 2005;134:162–9. http://www.ncbi.nlm.nih.gov/pubmed/15790540
Chen PS, Wang CC, Bortner CD, Peng GS, Wu X, Pang H, et al. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience [Internet]. Laboratory of Pharmacology and Chemistry, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC 27709, USA.; 2007;149:203–12. http://www.ncbi.nlm.nih.gov/pubmed/17850978
Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S, et al. Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem [Internet]. Molecular Neurobiology Section, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892–1363, USA.; 2009;111:976–87. http://www.ncbi.nlm.nih.gov/pubmed/19765194
Jin H, Kanthasamy A, Harischandra DS, Kondru N, Ghosh A, Panicker N, et al. Histone hyperacetylation up-regulates protein kinase Cdelta in dopaminergic neurons to induce cell death: relevance to epigenetic mechanisms of neurodegeneration in Parkinson disease. J Biol Chem [Internet]. From the Department of Biomedical Sciences, Iowa Center for Advanced Neurotoxicology, Iowa State University, Ames, Iowa 50011. the Department of Molecular Pharmacology and Therapeutics, Stritch School of Medicine, Loyola University Chicago, Maywood, Illin; 2014;289:34743–67. http://www.ncbi.nlm.nih.gov/pubmed/25342743
Boutillier AL, Trinh E, Loeffler JP. Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J Neurochem [Internet]. Laboratoire de Signalisations Moleculaires et Neurodegenerescence, EA no. 3433, 11 rue Humann, 67085 Strasbourg cedex, France. laurette@neurochem.u-strasbg.fr; 2003;84:814–28. http://www.ncbi.nlm.nih.gov/pubmed/12562525
Wang Y, Wang X, Liu L, Wang X. HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci Lett [Internet]. Department of Physiology and Key Laboratory of the Neurodegenerative Disorders of the Chinese Ministry of Education, Capital Medical University, Youanmen, Beijing 100069, China.; 2009;467:212–6. http://www.ncbi.nlm.nih.gov/pubmed/19835929
Suuronen T, Huuskonen J, Pihlaja R, Kyrylenko S, Salminen A. Regulation of microglial inflammatory response by histone deacetylase inhibitors. J Neurochem [Internet]. Department of Neuroscience and Neurology, University of Kuopio, Kuopio, Finland.; 2003;87:407–16. http://www.ncbi.nlm.nih.gov/pubmed/14511118
Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science. 1996;274(5290):1197–9.
Takahashi H, Ohama E, Suzuki S, Horikawa Y, Ishikawa a, Morita T, et al. Familial juvenile parkinsonism: clinical and pathologic study in a family. Neurology [Internet]. 1994;44(3 Pt 1):437–41. http://www.ncbi.nlm.nih.gov/pubmed/8145912
Matsumine H, Saito M, Shimoda-Matsubayashi S, Tanaka H, Ishikawa a, Nakagawa-Hattori Y, et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am J Hum Genet. 1997;60:588–96.
Gasser T, Müller-Myhsok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V, et al. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat Genet [Internet]. 1998;18(3):262–5. http://www.ncbi.nlm.nih.gov/pubmed/9500549, http://www.nature.com/ng/journal/v18/n3/pdf/ng0398-262.pdf
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395(6701):451–2.
Leroy E. Intron-exon Structure of Ubiquitin C-terminal Hydrolase-L1. DNA Res [Internet]. Oxford University Press; 1998 Jan 1 [cited 2016 Apr 19];5(6):397–400. http://dnaresearch.oxfordjournals.org/cgi/content/long/5/6/397
Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet [Internet]. 2001;68(4):895–900. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1275643&tool=pmcentrez&rendertype=abstract
van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, et al. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet. 2001;69(3):629–34.
Annesi G, Savettieri G, Pugliese P, D’Amelio M, Tarantino P, Ragonese P, et al. DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann Neurol. 2005;58(5):803–7.
Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW. The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann Neurol. 2003;54(3):283–6.
Hasegawa K, Kowa H. Autosomal dominant familial Parkinson disease: Older onset of age, and good response to Levodopa therapy. Eur Neurol. 1997;38(suppl1):39–43.
Wszolek ZK, Pfeiffer B, Fulgham JR, Parisi JE, Thompson BM, Uitti RJ, et al. Western Nebraska family (family D) with autosomal dominant parkinsonism. Neurology. 1995;45(3 Pt 1):502–5.
Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, Van Der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600.
Lin CH, Tan EK, Chen ML, Tan LC, Lim HQ, Chen GS, et al. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology. 2008;71(21):1727–32.
Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68(19):1557–62.
Chan AYY, Baum L, Tang NLS, Lau CYK, Ng PW, Hui KF, et al. The role of the Ala746Thr variant in the ATP13A2 gene among Chinese patients with Parkinson’s disease. J Clin Neurosci. 2013;20(5):761–2.
Li YJ, Deng J, Mayhew GM, Grimsley JW, Huo X, Vance JM. Investigation of the PARK10 gene in Parkinson disease. Ann Hum Genet. 2007;71(5):639–47.
Hicks AA, Petursson H, Jonsson T, Stefansson H, Johannsdottir HS, Sainz J, et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol. 2002;52(5):549–55.
Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C, et al. Significant linkage of Parkinson disease to chromosome 2q36-37. Am J Hum Genet. 2003;72(4):1053–7.
Pankratz N, Nichols WC, Uniacke SK, Halter C, Murrell J, Rudolph A, et al. Genome-wide linkage analysis and evidence of gene-by-gene interactions in a sample of 362 multiplex Parkinson disease families. Hum Mol Genet. 2003;12(20):2599–608.
Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C, et al. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am J Hum Genet. 2002;71:124–35.
Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, Lyons K, et al. Complete genomic screen in Parkinson disease: evidence for multiple genes. JAMA [Internet]. 2001;286(18):2239–44. http://www.ncbi.nlm.nih.gov/pubmed/11710888
Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet. 2005;14(15):2099–111.
Simón-Sánchez J, Singleton AB. Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum Mol Genet. 2008;17(13):1988–93.
Shi CH, Tang BS, Wang L, Lv ZY, Wang J, Luo LZ, et al. PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort. Neurology. 2011;77(1):75–81.
Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M, et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology. 2010;75(15):1356–61.
Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65(1):19–23.
Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology [Internet]. 2009;72(3):240–5. http://www.ncbi.nlm.nih.gov/pubmed/19038853, http://www.neurology.org/content/72/3/240.full.pdf
Shojaee S, Sina F, Banihosseini SS, Kazemi MH, Kalhor R, Shahidi GA, et al. Genome-wide Linkage Analysis of a Parkinsonian-Pyramidal Syndrome Pedigree by 500 K SNP Arrays. Am J Hum Genet. 2008;82(6):1375–84.
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet [Internet]. 2009;41(12):1303–7. http://www.ncbi.nlm.nih.gov/pubmed/19915576
Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet [Internet]. 2009;41(12):1308–12. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2787725&tool=pmcentrez&rendertype=abstract
Wider C, Skipper L, Solida A, Brown L, Farrer M, Dickson D, et al. Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Park Relat Disord. 2008;14(6):465–70.
Chartier-Harlin MC, Dachsel JC, Vilariño-Güell C, Lincoln SJ, Leprêtre F, Hulihan MM, et al. Translation initiator EIF4G1 mutations in familial parkinson disease. Am J Hum Genet. 2011;89(3):398–406.
Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet [Internet]. 2010;42(9):781–5. http://dx.doi.org/10.1038/ng.642
Nuytemans K, Bademci G, Inchausti V, Dressen A, Kinnamon DD, Mehta A, et al. Whole exome sequencing of rare variants in EIF4G1 and VPS35 in Parkinson disease. Neurology. 2013;80(11):982–9.
Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating Co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One. 2012;7(5).
Köroĝlu Ç, Baysal L, Cetinkaya M, Karasoy H, Tolun A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Park Relat Disord. 2013;19(3):320–4.
Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, et al. The sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive parkinsonism with generalized seizures. Hum Mutat. 2013;34(9):1200–7.
Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset parkinsonism. Hum Mutat. 2013;34(9):1208–15.
Acknowledgements
TFO is supported by the DFG Center for Nanoscale Microscopy and Molecular Physiology of the Brain and from BMBF Grant DecipherPD (01KU1503B).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Pavlou, M.A.S., Outeiro, T.F. (2017). Epigenetics in Parkinson’s Disease. In: Delgado-Morales, R. (eds) Neuroepigenomics in Aging and Disease. Advances in Experimental Medicine and Biology(), vol 978. Springer, Cham. https://doi.org/10.1007/978-3-319-53889-1_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-53889-1_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53888-4
Online ISBN: 978-3-319-53889-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)