
Abstract
The decrease in adiponectin levels are negatively correlated with chronic subclinical inflammation markers in obesity. The hypertrophic adipocytes cause obesity-linked insulin resistance and metabolic syndrome. Furthermore, macrophage polarization is a key determinant regulating adiponectin receptor (AdipoR1/R2) expression and differential adiponectin-mediated macrophage inflammatory responses in obese individuals. In addition to decrease in adiponectin concentrations, the decline in AdipoR1/R2 mRNA expression leads to a decrement in adiponectin binding to cell membrane, and this turns into attenuation in the adiponectin effects. Within the receptor complex, adaptor protein-containing pleckstrin homology domain, phosphotyrosine-binding domain, and leucine zipper motif 1 (APPL1) is the intracellular binding partner of AdipoR1 and AdipoR2. The expression levels of APPL1 or APPL2 lead to an altered adiponectin activity. Despite normal or high adiponectin levels, an impaired post receptor signaling due to APPL1/APPL2 may alter adiponectin efficiency and activity. However, APPL2 blocks adiponectin signaling through AdipoR1 and AdipoR2 by competitive inhibition of APPL1. APPL1 is also an important mediator of adiponectin dependent insulin sensitization. In this context, adiponectin resistance is associated with insulin resistance and is thought to be partly due to the down-regulation of the AdipoRs in high-fat diet fed subjects. Actually, adiponectin resistance occurs very rapidly after saturated fatty acid feeding, this metabolic disturbance is not due to a decrease in AdipoR1 protein content. Intra-abdominal adipose tissue-AdipoR2 expression is reduced in obesity, whereas AdipoR1 expression is not changed. Adiponectin resistance together with insulin resistance forms a vicious cycle. The elevated adiponectin levels with adiponectin resistance is a compensatory response in the condition of an unusual discordance between insulin resistance and adiponectin unresponsiveness.
Additionally, different mechanisms are involved in vascular adiponectin resistance at different stages of obesity. Nevertheless, diet-induced hyperlipidemia is the leading cause of vascular adiponectin resistance. Leptin/adiponectin imbalance may also be an important marker of the elevated risk of developing abdominal obesity-associated cardiovascular diseases.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Obesity
- Adiponectin
- High molecular weight (HMW) adiponectin
- Globular adiponectin
- Insulin resistance
- Hyperadiponectinemia
- Adiponectin receptor R1/R2 (AdipoR1/R2)
- Adaptor protein-containing pleckstrin homology domain, phosphotyrosine-binding domain, and leucine zipper motif 1 (APPL1)
- APPL1/APPL2
- Adenosine monophosphate (AMP)-activated protein kinase (AMPK)
- Tumour necrosis factor-alpha (TNF-alpha)
- Endothelial nitric oxide synthase (eNOS)
1 Introduction
Scherer et al. described a novel 30-kDa adipocyte complement-related protein (Acrp30), that is made exclusively in adipocytes. This molecule is called adiponectin and structurally similar to complement factor C1q. Secretion of adiponectin is enhanced by insulin. Actually its mRNA is induced over 100-fold during adipocyte differentiation (Scherer et al. 1995). Adiponectin is present in the circulation of healthy individuals at very high concentrations accounting for approximately 0.05% of total plasma proteins, with levels ranging from 3 to 30 μg/mL (Arita et al. 1999; Shibata et al. 2009). The median concentrations of plasma adiponectin are 5.5 μg/mL in men and 8.7 μg /mL in women. The subjects are divided into two groups according to their adiponectin concentrations using a cut-off point of 4.0 μg /mL. In men with an adiponectin concentration less than 4.0 μg /mL showed a higher prevalence of abdominal obesity . The prevalence of the metabolic syndrome is higher in subjects with an adiponectin concentration less than 4.0 μg /mL than in those with a concentration more than 4.0 μg /mL. In low concentration group, metabolic syndrome occurs at a rate of 52.3% and 37.5% in men and in women, respectively. Furthermore, the relative risk of adverse cardiovascular events is 1.56-fold higher among patients with the lower adiponectin levels than in those with the higher levels (Ryo et al. 2004). Mean preoperative fasting adiponectin concentration is significantly increased in response to weight loss after bariatric surgery . The plasma adiponectin levels correlate negatively with fat mass. Each kilogram of fat mass lost corresponds to an increase in plasma adiponectin concentration by approximately 6%. The subjects in obese state show no variations in adiponectin plasma concentration fluctuations during the evening and night, whereas in the post-obese state diurnal variation of adiponectin is restored again (Calvani et al. 2004). In obese patients, short-term weight loss does not change total adiponectin levels and insulin resistance , while the distribution pattern of adiponectin oligomers changes due to significant increment of high molecular weight adiponectin and reduction of medium molecular weight adiponectin. Especially, high molecular weight adiponectin is promptly responsive to short-term weight loss prior to changes in insulin resistance (Mai et al. 2014). According to adiponectin hypothesis reduced adiponectin levels can be caused by genetic factors or high-fat diet. In this context, decreased adiponectin signaling or disruption of adiponectin receptors may serve as an upstream pathway of increased oxidative stress , monocyte chemoattractant protein-1 (MCP-1) expression and inflammation in white adipose tissue (Yamauchi et al. 2007). In obese subjects, the hypertrophic adipocytes cause obesity-linked insulin resistance and metabolic syndrome . The decrease in adiponectin receptors (AdipoR1/R2) mRNA expression leads to a decrement in adiponectin binding, and this turns into attenuation in the adiponectin effects. Consequently, adiponectin resistance together with insulin resistance forms a vicious cycle (Yamauchi and Kadowaki 2008). Recently, new treatment strategies are being developed for the improvement of adiponectin resistance . The up-regulation of the expression and stimulation of adiponectin receptors by using adiponectin receptor agonists would be an effective method to treat obesity-related conditions. However, current drug development based on adiponectin and adiponectin receptors for clinical applications is scarce, and there is a lack of clinical trial data availability (Hossain et al. 2015). Dual activation of peroxisome proliferator-activated receptor-alpha (PPAR-alpha) and peroxisome proliferator-activated receptor-gamma (PPAR-gamma) enhances the action of adiponectin by increasing both adiponectin and AdipoRs, which can result in the amelioration of obesity-induced inflammation and insulin resistance (Tsuchida et al. 2005). Despite the conflicting evidences, leptin administration within the subphysiological to physiological range increases circulating levels and adipose tissue mRNA expression of adiponectin (Hoffmann et al. 2016). Different insights and existing contradictions about adiponectin resistance could be reconciled by explaining the details of related metabolic pathways. In this respect, this chapter will attempt to clarify the mechanisms and contributing factors of adiponectin resistance in obesity.
2 Regulation of Adiponectin Expression
Adiponectin is found in serum in number of complexes which include trimers and hexamers. These are collectively defined as low-molecular weight (LMW) trimer, medium molecular weight (MMW) hexamer and high-molecular weight (HMW) multimers (Waki et al. 2003; Whitehead et al. 2006).Trimeric and HMW/hexameric adiponectin activate different signal transduction pathways. The hexamer consists of two adjacent trimeric globular domains and a single stalk composed of collagen domains from two trimers. Although they are not necessary for trimer formation or stability, two of the three monomers in an adiponectin trimer are covalently linked by disulfide bonds between cysteine residues (Tsao et al. 2003). The concentrations of total adiponectin and HMW adiponectin are 25% and 45% lower, respectively, in obese children compared to controls. Thereby, HMW to total-adiponectin ratio is lower in the obese children than in the controls, whereas LMW adiponectin/total adiponectin ratio is higher in the obese as compared to the normal-weight children. These ratios are reversed with weight loss in obese patients after weight-reduction programme (Gajewska et al. 2011). HMW complexes circulating in serum represent a precursor pool. Activation of HMW complex is triggered by metabolic stimuli, which may subsequently trigger the induction or activation of a serum reductase. Trimeric adiponectin has a significantly shortened half-life when compared with the endogenous oligomeric complexes. Active adiponectin may be subjected to proteolysis by membrane-bound proteases that are found on the cell surface of target cells. This process leads to the formation of final active ligand that is rapidly cleared (Pajvani et al. 2003).
The adiponectin cDNA encodes a polypeptide of 247 amino acids with a secretory signal sequence at the amino terminus, a collagenous region and a globular domain. The expression of adiponectin is observed exclusively in mature fat cells. By contrast, the stromal-vascular fraction of fat tissue does not contain adiponectin mRNA. Hormone-induced differentiation of pre-adipocytes dramatically increases the level of adiponectin expression, however, the expression of adiponectin mRNA is significantly reduced in the adipose tissues from obese humans (Hu et al. 1996). Acute hyperinsulinaemia results in a significant reduction of total adiponectin. In this case, HMW adiponectin does not change, whereas MMW adiponectin and LMW adiponectin decrease. In the state of obesity , both hyperinsulinaemia and hyperlipidaemia contribute to low adiponectin levels (Bobbert et al. 2009). Regulation of adiponectin expression in non-adipose tissues is different from that in adipose tissue. Thus, adiponectin is up-regulated in human skeletal muscle in response to inflammatory stimuli. The underlying mechanisms may involve the inducible nitric oxide synthase (iNOS) -dependent pathway (Delaigle et al. 2004).
Inhibition of mitogen-activated protein kinase kinase (MAPKK)/extracellular signal-regulated kinase (ERK) 1/2 pathway decreases intracellular and secretory adiponectin levels in adipocytes, whereas adiponectin gene expression is increased. Thereby, MAPKK/ERK1/2 pathway activity plays a critical role in controlling adiponectin protein turnover in adipocytes via the ubiquitin-proteasome pathway. Thus, in adipose tissues of high-fat diet fed mice, ubiquitinated adiponectin protein levels are significantly higher (Gu et al. 2013).
The decrease in adiponectin levels are negatively correlated with chronic subclinical inflammation markers in obesity . Weight loss in morbidly obese patients induces a significant rise in adiponectin, while decreases the chronic inflammatory markers (Kopp et al. 2005). In these cases, profound reducing effects of interleukin-6 (IL-6 ) plus IL-6 receptor (IL-6R) and tumour necrosis factor-alpha (TNF-alpha) are shown on adiponectin mRNA levels. The inverse relationship between the plasma adiponectin and cytokines in vivo and the cytokine-induced reduction in adiponectin mRNA in vitro suggest that endogenous cytokines may inhibit adiponectin expression (Bruun et al. 2003). While TNF-alpha, IL-6, and IL-18 downregulate adiponectin production, transcription factors such as PPAR-gamma, CCAAT-enhancer-binding protein (C/EBP) alpha, and forkhead box transcription factor 1 (FOXO1) upregulate the adiponectin expression (Hino and Nagata 2012). In this respect, Vilarrasa et al. showed that massive weight loss induced by gastric bypass in a series of 65 morbidly obese patients had reduced IL-18, TNF-alpha receptors, and C-reactive protein (CRP). However, there was no relationship between either adiponectin and IL-18 or TNF-alpha receptors and CRP . This result suggests that IL-18 and TNF-alpha receptors do not play a remarkable role in the inhibition of the adiponectin production (Vilarrasa et al. 2007).
Leptin secretion rises acutely during feeding or in response to over-nutrition as adipocytes expand in size and number (Unger 2002), whereas adiponectin secretion is increased during under-nutrition and exercise. In the latter case, adipocytes tend to diminish in size because lipolysis is most active in this period (Miyazaki et al. 2010). Actually, leptin and adiponectin have similar lipo-oxidative actions (Unger 2002). In contrast to leptin, adiponectin levels are significantly reduced among obese subjects in comparison with lean controls. Although the majority of adiponectin is secreted from adipose tissue, the mean plasma adiponectin levels are found significantly lower in obese patients (Arita et al. 1999). In this context, leptin and adiponectin are inversely correlated in obesity . However, leptin can not participate directly in adiponectin synthesis. Nontheless, the long-term regulation of adiponectin expression in white adipose tissue appears to be the opposite of that of leptin. Additionally, adiponectin levels are more sensitive to changes in adiposity or insulin sensitivity (Zhang et al. 2002).
3 Adiponectin Receptor Complex and Adiponectin Resistance
Adiponectin exerts its effects by binding to adiponectin receptors, two of which are AdipoR1 and AdipoR2 (Yamauchi et al. 2014). Adiponectin receptors have a suitable structure related to their functions. The seven transmembrane helices of AdipoR1 form a helix bundle, which are arranged circularly in a clockwise manner, from helix-1 to helix-7. The structure of AdipoR2 is quite similar to that of AdipoR1. In both, the AdipoR1 and AdipoR2 structures surround a large internal cavity. This cavity is formed between the four- and three-helix subdomains. Adiponectin may broadly interact with the extracellular face of receptor, rather than the carboxy-terminal tail of the receptors. Moreover, the adiponectin-binding sites seem to be shared by AdipoR1 and AdipoR2 (Tanabe et al. 2015). In addition to AdipoR1 and AdipoR2, T-cadherin has been identified as a receptor for the HMW form of adiponectin, but not for trimeric or globular adiponectin (gAD). Although lacking in the known cellular functions , T-cadherin is expressed in endothelial and smooth muscle cells, where it is positioned to interact with adiponectin (Hug et al. 2004). Circulating levels of adiponectin, particularly HMW form of adiponectin are elevated in T-cadherin-deficient mice (Denzel et al. 2010).
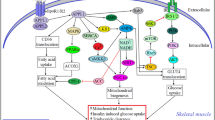
Actually, adiponectin receptors bind to globular and full-length adiponectin and mediate increased adenosine monophosphate (AMP)-activated protein kinase and PPAR-alpha ligand activities. Thereupon, adiponectin also enhances glucose uptake and fatty-acid oxidation (Kadowaki and Yamauchi 2005). Basically, the adiponectin receptors are transmembrane receptors that undergo conformational changes and couple the intracellular domain with other signaling molecules upon extracellular adiponectin binding. Adaptor protein [adaptor protein containing pleckstrin homology (PH) domain, phosphotyrosine binding (PTB) domain and leucine zipper motif (APPL)] directly binds to the intracellular domains of AdipoR1 and AdipoR2 via its C-terminal PTB and CC domains (Ruan and Dong 2016). The first identified adaptor proteins that interact directly with adiponectin receptors are APPL1 and APPL2 (Mao et al. 2006). Within the receptor complex, APPL1 is the intracellular binding partner of AdipoR1 and AdipoR2 (Mao et al. 2006). Furthermore, the general function of PTB domain is to act as an adaptor or scaffold for the binding of proteins, particularly those in signaling pathways. The PTB domain of APPL1 is located near the -COOH terminus, away from BIN-Amphiphysin-Rvs (BAR)-PH domain. Therefore, PTB is an easily accessible structure for its binding partners (Deepa and Dong 2009). APPL1 mediates the actions of adiponectin in the regulation of energy metabolism and insulin sensitivity (Ruan and Dong 2016). Thus, insulin-stimulated activation of protein kinase B (Akt) and suppression of gluconeogenesis in hepatocytes are enhanced by APPL1 overexpression, but are attenuated by APPL1 deficiency. In this context, APPL1 is an important mediator of adiponectin dependent insulin sensitization in skeletal muscle. Decreased APPL1 expression reduces insulin-dependent Akt activation. Moreover, while adiponectin treatment itself does not affect Akt phosphorylation, co-treatment of myotubes with both adiponectin and insulin leads to a synergistic increase in Akt phosphorylation (Hosch et al. 2006). APPL1 also plays an important role in insulin-stimulated insulin-responsive glucose transporter-4 (GLUT4) translocation in muscle and adipose tissues and its N-terminal portion may be critical for APPL1 function. Thereby, deficiency of APPL1 attenuates insulin-mediated Akt phosphorylation, glucose uptake and GLUT4 translocation in adipocytes. APPL1 is localized mainly in the cytosol, and it shows a small degree of re-localization to the microsomes and nucleus in response to insulin (Saito et al. 2007b) (Fig. 18.1).

Adiponectin resistance together with insulin resistance forms a vicious cycle. Insulin-adiponectin resistance cross talk causes intracellular signal transduction disturbances. Adiponectin resistance is a compensatory response in the condition of an unusual discordance between insulin resistance and adiponectin unresponsiveness (PKC protein kinase C, AMPK 5′ adenosine monophosphate-activated protein kinase, GLUT4 insulin-responsive glucose transporter-4, NADP nicotinamide adenine dinucleotide phosphate, iNOS inducible nitric oxide synthase, NO nitric oxide, ROS reactive oxygen radicals, AdipoR1/R2 adiponectin receptors, TLR4 Toll-like receptor-4, NF-kappaB nuclear factor-kappa B, JNK c-Jun N-terminal kinase, IKK inhibitor kappa B kinase, IRS -1 insulin receptor substrate-1, PI3-K Phosphatidylinositol 3-kinase, Akt protein kinase B, eNOS endothelial nitric oxide synthase, MAPK mitogen-activated protein-kinase, ET-1 endothelin-1, TNF-alpha Tumor necrosis factor-alpha, GSK-3 glycogen synthase kinase-3, FL full-length human adiponectin, G globular adiponectin, IL-6 interleukin-6, IL-8 interleukin-8, PPAR-ɣ peroxisome proliferator-activated receptor-gamma, PPAR-α peroxisome proliferator-activated receptor-alpha, FAO fatty acid oxidation, FOXO1 forkhead box transcription factor 1, APPL2 adaptor protein-containing pleckstrin homology domain, phosphotyrosine-binding domain, and leucine zipper motif 2)
Eventually, the expression levels of APPL1 or APPL2 lead to an altered adiponectin activity. Decreases in AMP-activated protein kinase (AMPK) activation, hepatic glucose uptake and free fatty acid oxidation increase the de novo lipogenesis and gluconeogenesis in liver. Final stage of this process involves intrahepatic lipid accumulation and fatty liver disease. Genetically, C-APPL1/A-APPL2 allele combination is associated with an increase in nonalcoholic fatty liver disease (NAFLD) occurrence, with a more severe degree of hepatic steatosis and with reduced cytoprotective effects of adiponectin on hepatocytes. NAFLD risk alleles have been found to be associated with hepatocellular injury and a more severe hepatic steatosis. Actually, these effects may be independent of the plasma adiponectin levels. Despite normal or high adiponectin levels, an impaired post receptor signalling due to APPL1/APPL2 single nucleotide polimorphisms may alter adiponectin efficiency and activity (Barbieri et al. 2013). Thus, APPL2 blocks adiponectin signaling through AdipoR1 and AdipoR2 by competitive inhibition of APPL1 (Ruan and Dong 2016).
On the other hand, DNA methylation in the obesity decreases adiponectin expression. The AdipoR2 is highly methylated in adipocytes from high fat diet -fed obese mice as compared with lean mice. Moreover, AdipoR2 methylation levels are inversely correlated with the amounts of adiponectin mRNA. By contrast, the AdipoR1 methylation is unaltered regardless of obesity. In human adipocytes, the AdipoR2 methylation is not only positively correlated with body mass index but also negatively associated with adiponectin transcripts. DNA methylation involves the particular region of adiponectin promoter. Thus, the AdipoR2 methylation is mediated by DNA methyltransferase, and induces the subsequent formation of heterochromatin structure to suppress adiponectin gene expression in obesity (Kim et al. 2015). While the AdipoR1 activates the AMPK pathways, AdipoR2 induces the PPAR-alpha pathways such as increased expression of uncoupling protein 2 (UCP2). In this respect, disruption of AdipoR1 results in the abrogation of adiponectin-induced AMPK activation, whereas that of AdipoR2 results in decreased activity of PPAR-alpha signaling pathway. Furthermore, simultaneous disruption of both AdipoR1 and R2 abolishes adiponectin binding and actions. Collective elimination of the adiponectin effects results in increased tissue triglyceride content, inflammation and oxidative stress , and thus leads to insulin resistance (Yamauchi et al. 2007). Conversely, UCP2 could exert anti-steatotic and anti-inflammatory activities through promoting mitochondrial respiration, attenuating non-esterified mitochondrial fatty acid accumulation and reactive oxygen species (ROS) production, and activating AMPK (Baffy 2005; Zhou et al. 2010). Acute adiponectin loading fails to lower glycemia, which is consistent with systemic adiponectin resistance. The mechanism for this failure appears to correlate with a blunted response of AMPK activation but not with changes in AdipoR1 and AdipoR2 mRNA expression. Actually, adiponectin resistance correlates with impaired hepatic AMPK response to adiponectin. The reduced AMPK response is associated with increased basal AMPK phosphorylation and AMPK resistance (Lin et al. 2007). Tsuchida et al. have previously shown that the expression of AdipoR1/R2 is inversely correlated with plasma insulin levels in vivo. Moreover, the expressions of AdipoR1/R2 in obese mice are significantly decreased in skeletal muscle and adipose tissue, which is correlated with decreased adiponectin binding to membrane fractions of skeletal muscle and decreased AMPK activation by adiponectin (Tsuchida et al. 2004). AdipoR1 mRNA expression in skeletal muscle myotubes derived from lean healthy individuals is stimulated 1.8- and 2.5-fold with gAD and leptin, respectively. No increase in AdipoR1 gene expression is measured in myotubes derived from obese subjects. AdipoR2 mRNA expression is unaltered after gAD and leptin exposure in myotubes (McAinch et al. 2006). Evidences of adiponectin resistance have been shown in peripheral tissues of obese humans. In this manner, decrease in the expressions of AdipoR1/R2 and adiponectin concentration reduces the adiponectin binding to cell membrane (Tsuchida et al. 2004). Furthermore, AdipoR1 expression in human adipose tissue is also reduced in obese subjects. Thus, it suggests that adiponectin might have reduced biological effects in adipose tissue due to low levels of adiponectin receptors in omental adipocytes of obese individuals (Rasmussen et al. 2006). Receptor endocytosis is a key event in regulation of signaling transduction. Enhanced adiponectin-stimulated AMPK phosphorylation involves the downregulation of adiponectin signaling through the endocytosis of AdipoR1. Indeed, AdipoR1 is internalized through a clathrin- and Rab5-dependent pathway and that endocytosis may play a role in the regulation of adiponectin signaling. Thus, blocking clathrin-mediated endocytosis abolishes adiponectin internalization (Ding et al. 2009).
4 Adiponectin and Insulin Resistance
Overexpression of glycogen synthase kinase (GSK)-3 in skeletal muscle of obese type 2 diabetic humans are associated with an impaired ability of insulin to activate glucose disposal and glycogen synthase. Inhibition of GSK-3 causes improvements in insulin-stimulated glucose transport activity by enhancing post-insulin receptor insulin signaling and GLUT4 glucose transporter translocation (Henriksen and Dokken 2006). Chronic treatment of insulin-resistant, prediabetic obese rats with a highly selective GSK-3 inhibitor improves insulin receptor substrate-1 (IRS -1)-dependent insulin signaling in skeletal muscle and enhances whole body insulin sensitivity (Dokken and Henriksen 2006). Thus, GSK-3 overactivity in obesity is associated with enhanced IRS -1 serine phosphorylation and defective IRS -1-dependent signaling, ultimately resulting in reduced GLUT4 translocation and glucose transport activity in skeletal muscle (Henriksen 2010). Actually, insulin-induced signaling involves in activation of phosphatidylinositol 3-kinase (PI3K) and Akt. Akt inactivates GSK-3. PI3K-dependent GSK3 inactivation causes the inhibition of adiponectin formation in diet-induced obesity. The expression of PI3K-resistant-GSK3 stimulates the production of adiponectin and protects from diet-induced obesity (Chen et al. 2016). In obesity-linked insulin resistance , both adiponectin and adiponectin receptors are downregulated. Up-regulation of adiponectin/adiponectin receptors or enhancing adiponectin receptor function has protective effect against obesity-linked insulin resistance (Caselli 2014). AdipoR1 overexpression increases glucose uptake and glycogen accumulation in the high-fat diet-fed rats, and it locally ameliorates muscle insulin resistance . These effects are associated with increased phosphorylation of IRS -1, Akt, and GSK-3. A direct role for adiponectin action via AdipoR1 in the enhancement of insulin sensitivity involves activation of the PI3K and AMPK signaling pathways (Patel et al. 2012). APPL1 has been shown to interact directly with Akt (Saito et al. 2007b). However, IRS -1 phosphorylation is also increased by either APPL1 or AdipoR1 overexpression (Patel et al. 2012). APPL1 promotes glucose disposal into skeletal muscle in vivo through activation of the PI3K pathway (Cleasby et al. 2011). Plasma concentrations of adiponectin in obese subjects are significantly lower than those in non-obese subjects (Arita et al. 1999). Adiponectin resistance is initially described in obesity. This condition is associated with insulin resistance and is thought to be partly due to the down-regulation of the AdipoR1 due to high-fat diet (Mullen et al. 2009). Additionally, the severe degree of insulin resistance occurs in patients with genetic defect of the insulin receptors. These cases have the combination of high plasma adiponectin with low leptin (Semple et al. 2006). This discordance between high plasma adiponectin and extreme insulin resistance may be accounted for their either direct effects in adipocytes leading to the loss of insulin receptor function, or their effects on the developed adipose tissue that has severely impaired insulin receptor function (Semple et al. 2006). The hyperadiponectinaemia in states of insulin receptor dysfunction is explained by a superiority of HMW multimers to the other counterparts. HMW adiponectin shows a significant correlation with the dissociation between plasma adiponectin and insulin sensitivity when compared to total plasma adiponectin (Semple et al. 2007). Furthermore, deficiency of both IRS -1 and 2 in adipocytes reduces adiponectin secretion. Lack of insulin receptor or IRSs in adipocytes increases adiponectin mRNA expression, but reduces adiponectin secretion. Adiponectin mRNA expression shows no significant response to insulin receptor deficiency, but is modestly increased by IRS -1 and 2 deficiency. Loss of insulin receptor function may affect adiponectin levels indirectly through alteration of adipocyte turnover (Groeneveld et al. 2016). The expressions of both AdipoR1 and AdipoR2 are significantly decreased in most of the insulin-sensitive tissues of genetically obese mice as compared with the controls. FOXO1 increases the AdipoR1/R2 expressions. In these animals, insulin can reduce the AdipoR1/R2 expressions via down-regulation of FOXO1 activity. Actually, the expression levels of AdipoR1/R2 might regulate adiponectin binding and AMPK activation by adiponectin. This process is referred as adiponectin sensitivity. Chronic overfeeding and attending elevation of insulin results in the decreased expression of AdipoR1/R2. The decrease in AdipoR1/R2 mRNA leads to a decrease in adiponectin binding, and this turns into a decrease in the adiponectin effects. This is defined as adiponectin resistance . Adiponectin resistance together with insulin resistance forms a vicious cycle (Tsuchida et al. 2004). The changes in AdipoR2 levels may reflect decreased FOXO1-dependent transcription. In contrast, AdipoR1 mRNA expression is not changed in liver and tend to be reduced in skeletal muscle of both normoglycemic and hyperglycemic conditions. This reflects the differential regulation of AdipoR1 and AdipoR2 under insulin-resistant state. The reduced AdipoR1 expression in muscle also contributes to adiponectin resistance in addition to insulin resistance . In any way possible, insulin resistance can result in hyperadiponectinemia and adiponectin resistance. In contrast, Lin et al. showed that stepwise decreases in adiponectin and AdipoR1/AdipoR2 expressions are associated with the progression from insulin resistance to overt diabetes (Lin et al. 2007). During the prolonged hyperglycemia-associated oxidative stress , acetylated FOXO1 is retained in the nucleus, where it engages sirtuin 1 (SIRT1). Deacetylation of FOXO1 by SIRT1 promotes FOXO1-dependent transcription and accelerates FOXO1 degradation (Kitamura et al. 2005). During prolonged incubation with insulin, insulin-stimulated glucose uptake is significantly reduced. Insulin resistance and adiponectin mRNA expression develops in these adipocytes. In insulin-resistant adipocyte, adiponectin deficiency does not change insulin-stimulated glucose uptake, whereas in insulin-sensitive adipocytes, adiponectin deficiency suppresses insulin signaling , expression of IRS -1 and GLUT4, and GLUT4 translocation to the membrane (Chang et al. 2015) (Fig. 18.1).
5 Discordance Between Adiponectin Response and Insulin Resistance
The variation in fat cell size that occurs in obesity may have an important impact on adipose tissue function. Adipose tissue morphology correlates with insulin levels and is linked to the total adipocyte number independent of sex and body fat level (Arner et al. 2010). However, in visceral fat cells, the lipolytic activity is higher than in subcutaneous fat cells owing in part to less marked insulin action and lower alpha 2-adrenergic receptor mediated antilipolytic effect of catecholamines (Engfeldt and Arner 1988). Adipose tissue displays a variable balance between increases in fat cell size and increases in fat cell number in obesity. Thus, the total adipocyte number is greatest in pronounced hyperplasia and smallest in pronounced hypertrophy (Arner et al. 2010). Adipose tissue expands via the combination of hypertrophy and hyperplasia in diet-induced obesity. Subcutaneous adipose tissue has a primary role in supporting circulating adiponectin levels in lean subjects. Fat cell diameter and percentage of hypertrophic fat cells are greatest in subcutaneous adipose tissue and correlate negatively with both serum and secreted adiponectin levels in obese subjects, because increases in fat cell size have been shown to impair fat cell function. In contrast, hyperplastic expansion appears predominant in visceral adipose tissue, which contained a significantly greater percentage of small fat cells and a significantly smaller mean fat cell diameter. Visceral adipose tissue correlated positively with both serum HMW and secreted total adiponectin. Eventually, reductions in the percentage of small fat cells in subcutaneous adipose tissue are associated with the reductions in both secreted and circulating levels of total and HMW adiponectin (Meyer et al. 2013). Actually, AMPK activity is lower in visceral than in subcutaneous abdominal adipose tissue of insulin sensitive obese or insulin-resistant obese patients. Furthermore, AMPK activity is lower in all adipose tissues of obese patients who are insulin resistant than in body mass index-matched insulin sensitive subjects. These evidences indicate the close links between reduced AMPK activity, increased inflammation in white adipose tissue and whole-body insulin resistance in obese humans (Gauthier et al. 2011). In fact, AMPK is a heterotrimeric enzyme complex consisting of a catalytic subunit alpha and two regulatory subunits beta and gamma. AMPK is activated by the rising of AMP and decreasing of ATP. AMPK system is a regulator of energy balance that once activated by low energy status, switches on ATP-producing catabolic pathways such as fatty acid oxidation and glycolysis, and switches off ATP-consuming anabolic pathways such as lipogenesis. The AMPK system also regulates food intake and energy expenditure by mediating the insulin sensitizing effect of adiponectin (Viollet et al. 2007). Elevated adiponectin protein expression in adipocytes and elevated adiponectin serum concentrations could help to compensate the adipocyte-specific insulin resistance in fat-specific insulin receptor deficient mice. Thus, hyperadiponectinemia results from a lack of insulin signalling in adipose tissue (Blüher et al. 2002). Furthermore, Kim et al. showed that the elevated adiponectin levels with adiponectin resistance is a compensatory response in the condition of an unusual discordance between insulin resistance and adiponectin responsiveness. However, this type of adiponectin resistance could not be explained by decreased adiponectin receptor gene expression or AMPK phosphorylation in skeletal muscle and liver. These mice are resistant or unresponsive to metabolic actions of both insulin and adiponectin (Kim et al. 2006). Serum adiponectin is significantly associated with AMPK phosphorylation in skeletal muscles of men but not in women. Serum adiponectin is significantly and negatively associated with skeletal muscle ceramide content in men. Furthermore, ceramide content is negatively associated with AdipoR1 expression in skeletal muscles of men. These associations suggest that the insulin-sensitizing effect of adiponectin on human male skeletal muscles may be mediated via AdipoR1 leading to lowering of ceramide content (Høeg et al. 2013). For women only, there is a negative correlation between C16-ceramide and plasma adiponectin and a positive correlation between total ceramide content and insulin resistance (Blachnio-Zabielska et al. 2012). Evidence of adiponectin resistance has been shown in peripheral tissues of obese humans. Thus, decrease in the expressions of AdipoR1/R2 or adiponectin concentration in obese mice reduces the adiponectin binding to membrane fractions of skeletal muscle (Tsuchida et al. 2004).
Actually, adiponectin stimulates fatty acid oxidation and improves insulin sensitivity in humans, due in part to the activation of AMPK and subsequent deactivation of acetyl coenzyme A carboxylase (ACC). Although both high fat diets result in the loss of ability to stimulate fatty acid oxidation of adiponectin, high saturated fat diet shows reduced rates of maximal insulin-stimulated glucose transport compared with high unsaturated fat diet. The lack of stimulation of fatty acid oxidation in response to adiponectin is supported by the lack of ACC phosphorylation in high fat diets. The development of adiponectin resistance does not necessarily coincide with the development of impaired glucose transport (Mullen et al. 2007). gAD increases glucose uptake in skeletal muscle cells via GLUT4 translocation and subsequently reduces the rate of glycogen synthesis and shifts glucose metabolism toward lactate production. These effects are consistent with the increased phosphorylation of AMPK and ACC and oxidation of fatty acids, which is induced by gAD (Ceddia et al. 2005). Thus, gAD increases AMPK activity and ACC phosphorylation and decreased malonyl CoA concentration in muscle. In fact, adiponectin is known to increase fatty acid oxidation in skeletal muscle by the inactivation of ACC, and reducing inhibition of carnitine palmitoyl transferase 1 (CPT1) by malonyl-CoA, leading to increased fatty acid uptake in the mitochondria (Tomas et al. 2002). In contrast, the stimulatory effect of gAD on fatty acid oxidation in skeletal muscle is blunted in obese humans. Combined exposure of insulin and globular head group of adiponectin exhibits an additive effect on glucose uptake in lean and obese individuals but this effect is reduced by 50% in obese muscle. In accordance with glucose, fatty acid oxidation is significantly increased with adiponectin in both lean and obese subjects. The absolute change in fatty acid oxidation in response to gAD is 58% lower in obese subjects. While the ratio of palmitate esterification to oxidation is significantly elevated in obese muscle, the stimulation of AMPK -alpha2 by gAD is impaired. In this case, resulting metabolic changes are attributed to reduced AMPK activity. These evidences indicate that adiponectin resistance develops during the progression of obesity (Bruce et al. 2005). Interestingly, reduced activation of AMPK signaling and fatty acid oxidation in obese and obese diabetic myotubes is not associated with reduced protein expression of AMPK-alpha and ACCbeta or the expression and activity of the upstream AMPK kinase. Moreover, obese subjects tend to have higher AdipoR1 expression. Thus, reduced activation of AMPK by gAD in obese subjects is not caused by reduced adiponectin receptor expression (Chen et al. 2005). Practically, only the animals fed with the saturated diet become insulin resistant, whereas polyunsaturated-fed animals are insulin responsive. By high fat feeding, increased fatty acid translocase (FAT/CD36) at the plasma membrane of skeletal muscle accompanies by increased total fatty acid uptake in accumulation of reactive diacylglycerol (DAG) and ceramide lipid species and impaired insulin response. Virtually, a 60% saturated-fat diet can induce skeletal muscle adiponectin resistance , as evidenced by a failure of gAD to increase fatty acid oxidation or phosphorylate ACC above basal levels. However, it is unclear whether adiponectin resistance precedes intramuscular lipid accumulation and the development of insulin resistance . Nevertheless, adiponectin resistance occurs very rapidly after saturated fatty acid feeding, and this is not due to a decrease in AdipoR1 protein content (Mullen et al. 2009).
On the other hand, leptin stimulates AMPK activity and increases AMPK Thr172 and ACC-beta Ser222 phosphorylation and fatty acid oxidation in lean myotubes but that in obese subject leptin-dependent AMPK signaling and fatty acid oxidation are suppressed. Reduced activation of AMPK is associated with elevated expression of IL-6 and suppressor of cytokine signaling 3 (SOCS3) mRNA in myotubes of obese subjects. SOCS3 has been shown to inhibit leptin activation of AMPK in human myotubes and contributes to leptin resistance observed in obese subject (Steinberg et al. 2006). Furthermore, leptin and adiponectin stimulates fatty acid oxidation through similar mechanisms, it is possible that SOCS3 may interfere with intracellular gAD signal transduction. Adiponectin resistance worsens insulin resistance in obese mice. The expression of AdipoR1/R2 is regulated by insulin via the insulin/ PI3K/FOXO1 pathway and is correlated with adiponectin sensitivity (Tsuchida et al. 2004). According to Cui et al. Foxo1 silencing inhibits AdipoR1 expression and the activation of AMPK . Insulin induces a decrease in skeletal muscle AdipoR1 expression in a PI3K/Akt/FOXO1-dependent fashion (Cui et al. 2012). It is well-known that Akt regulates gene transcriptions through the inactivation of FOXO1 . As such, the PI3K/Akt/FOXO1 axis has a central role in energy metabolism and signal transduction of insulin, governing insulin sensitivity (Mullen et al. 2009). Obesity decreases the expression of adiponectin receptors, thereby reducing adiponectin sensitivity finally leads to insulin resistance (Kadowaki and Yamauchi 2005). These observations confirm the important connections between insulin and AdipoR1. Eventually, elevated insulin levels may result in a decreased expression of AdipoR1, leading to diminished binding of adiponectin and a reduction in PPARgamma coactivator 1alpha (PGC-1alpha). These events ultimately trigger adiponectin resistance by simultaneously increasing the sphingolipid-ceramide levels (Sente et al. 2016). PPAR-gamma2 is a key regulator of adipogenesis , and PPAR-gamma2 transcription is activated by C/EBP-delta through the direct binding of C/EBP-delta to the PPAR-gamma2 promoter. By contrast, glucocorticoid-induced leucine-zipper protein (GILZ) binds to the PPAR-gamma2 promoter element and inhibits C/EBP-delta-mediated transcription. GILZ inhibits the transcription of the PPAR-gamma2 gene and blocks adipocyte differentiation (Shi et al. 2003). GILZ overexpression decreases leptin mRNA and protein secretion. Although GILZ silencing decreases adiponectin mRNA levels, it does not affect the amount of adiponectin secreted. While GILZ silencing increases basal ERK1/2 and c-Jun N-terminal kinase (JNK) phosphorylation, it decreases MAPK phosphatase-1 (MKP1) protein levels. Thus, adipose tissue GILZ mRNA levels are reduced in proportion to the degree of obesity (Lee et al. 2016). Surprisingly, adiponectin overexpression reduces MKP1 protein levels. High adiponectin levels enhance MAPK/PGC-1alpha signaling and mitochondrial biogenesis in skeletal muscle by suppressing MKP1 protein expression (Qiao et al. 2012). Despite the low level of adiponectin in obesity, the reason for this discrepancy is unclear.
Furthermore, a high-fat maternal diet decreases AdipoR1 expression in offspring, which could contribute to reduced sensitivity to adiponectin (Hou et al. 2015). Thus, AdipoR1 deficiency leads to diet-induced metabolic dysfunction. These mice have greater body weight and fat mass, hepatic steatosis, impaired glucose disposal rate, and elevations in serum insulin and leptin, whereas, AdipoR2-deficient mice are protected from diet-induced weight gain and metabolic perturbations (Parker-Duffen et al. 2014). In contrast, the AdipoR1 overexpressing mice resist diet-induced obesity while decreasing lipid accumulation, oxidative stress and autophagic damage (Chou et al. 2014a). On the other hand, the adiponectin promoter drives expression of lipoprotein lipase (LPL) in adipocytes and increases the amount of lipase in adipose tissue lipid storages. Thereby, adipose tissue protects against the accumulation of ectopic lipids in the peripheral tissues by storing fat as neutral lipid (triglyceride). In addition to lipid diversion, another predicted effect of increased adipose LPL is the stimulation of PPAR transcription factors by the free fatty acids , which are generated by lipoprotein hydrolysis. Consequently, increased LPL in adipocytes improves adipocyte function and protects against glucose and insulin intolerance in diet-induced obesity (Walton et al. 2015). Simultaneous disruption of both AdipoR1- and R2-adiponectin binding and actions result in increased tissue triglyceride content, inflammation and oxidative stress , and thus lead to insulin resistance and marked glucose intolerance. In this regard, AdipoR1 and R2 play important roles in the regulation of glucose and lipid metabolism (Yamauchi et al. 2007). Actually, adiponectin exerts insulin-sensitizing effects through binding to own receptors. This signaling primarily leads to activation of AMPK and PPAR-alpha. As mentioned above, in obesity-linked insulin resistance , both adiponectin and adiponectin receptors are downregulated. From this perspective, up-regulation of adiponectin or enhancing adiponectin receptor functions may be a therapeutic strategy for obesity-linked insulin resistance (Caselli 2014) (Fig. 18.1).
Fifty-four obese patients with NAFLD showed significantly lower serum adiponectin levels when compared with the normal participants; and also its level is lower in nonalcoholic steatohepatitis (NASH) patients in comparison to patients with simple steatosis. Furthermore, adiponectin receptor gene expression in liver biopsy of NASH patients is lower in comparison to non-NASH patients. Adipo R2 receptor depletion in these patients suggests that not only adiponectin deficiency has a role in progression of severity of NAFLD, but also adiponectin resistance is important in the pathogenesis of NAFLD (Salman et al. 2015). Adiponectin potently stimulates ceramidase activity through AdipoR1 and AdipoR2. Ceramide catabolism is enhanced and its antiapoptotic metabolite, sphingosine-1-phosphate (S1P) is formed independent from AMPK activity. Ceramidase activity is impaired in cells lacking both adiponectin receptor isoforms, leading to elevated ceramide levels and enhanced susceptibility to palmitate-induced cell death (Holland et al. 2011). Actually, AdipoR1-deficiency markedly reduces gAD-induced neutral ceramidase activation, whereas AdipoR2-deficiency only slightly inhibits ceramidase. More importantly, small interfering RNA-mediated neutral ceramidase-deficiency markedly blocks the effect of adiponectin on TNF-alpha -induced intercellular adhesion molecule-1 (ICAM-1) expression. Eventually, adiponectin inhibits TNF-alpha-induced inflammatory response via ceramidase recruitment and activation in an AdipoR1-dependent fashion (Wang et al. 2014). Actually, ceramide impairs insulin sensitivity in peripheral tissues by blocking the plasma membrane translocation and promoting dephosphorylation of Akt (Holland and Summers 2008). Upon binding adiponectin, the adiponectin receptors convert ceramides into sphingosines by stimulating ceramidase activity. Hence, adiponectin-related activities depend primarily on the lowering of cellular ceramides or the concomitant increase in sphingosines (Ye and Scherer 2013). AdipoRon , synthetic small-molecule agonist of the AdipoR1, exhibits very similar effects to adiponectin in muscle and liver, such as activation of AMPK and PPAR-alpha pathways by binding to both AdipoR1 and AdipoR2. Moreover, AdipoRon ameliorates insulin resistance and glucose intolerance in mice fed a high-fat diet or genetically obese mice (Okada-Iwabu et al. 2013). Consequently, treatment with AdipoRon increases AdipoR1/AdipoR2, AMPK , and PPAR-alpha mRNA expressions as well as the mRNA levels of genes involved in fatty acid beta-oxidation. Moreover, AdipoRon contributes to increased mitochondrial biogenesis through the up-regulation of PGC-1alpha expression (Sente et al. 2016).
As a dietary saturated fatty acid , palmitate not only rapidly inhibits transcription of the adiponectin gene and the release of adiponectin from adipocytes but also stimulates lysosomal degradation of newly synthesized adiponectin (Karki et al. 2011). Palmitate induces a 45% decrease in insulin-stimulated glucose uptake in adipocytes. In accordance with this, the mRNA and protein expression of adiponectin are reduced by 43% and 36%, respectively, by palmitate treatment. These changes are accompanied by a 54% increase in intracellular ROS levels (Xi et al. 2007). Palmitate also increases the expression levels of iNOS and endoplasmic reticulum (ER) stress, ER stress response markers, and decreased mitochondrial protein contents. Palmitate-induced mitochondrial dysfunction is the primary event that leads to iNOS induction, ER stress, and decreased adiponectin synthesis in adipocytes (Jeon et al. 2012). Nitric oxide exerts its action on specific amino acid residues within target proteins by increasing generation of the nitric oxide-derivide peroxynitrite . iNOS represses adipose PPAR-gamma expression. In contrast, iNOS disruption sensitizes adipose tissue to PPAR-gamma agonism, thus raising plasma high-molecular weight adiponectin levels. This leads to increased AMPK activation in the liver and improvement of hepatic insulin sensitivity and glucose tolerance in obese mice (Dallaire et al. 2008) (Fig. 18.1). Lipid peroxidation-induced protein carbonylation is a potential mechanism underlying mitochondrial dysfunction (Frohnert and Bernlohr 2013). Protein carbonylation plays a major stimulating role in cytokine-dependent mitochondrial dysfunction and may be linked to the development of insulin resistance in the adipocyte (Curtis et al. 2012). Actually, mitochondrial function is closely linked to adiponectin synthesis in adipocytes, and mitochondrial dysfunction in adipose tissue decreases plasma adiponectin levels in obesity (Koh et al. 2007). Impaired mitochondrial function increases ER stress, and reduces adiponectin transcription via activation of JNK and consequent induction of activating transcription factor (ATF)-3 (Koh et al. 2007). ATF-3 negatively regulates human AdipoR1 expression via binding to an ATF-3-responsive region in the promoter, which plays an important role in attenuation of adiponectin signaling and induction of insulin resistance (Park et al. 2010). Further, palmitate-induced ER stress induces adiponectin resistance , via AMPK phosphorylation and reducing APPL1 expression (Park et al. 2015). The induction of ER stress is also accompanied by a decrease in adiponectin multimerization in adipocytes (Mondal et al. 2012). APPL1 interacts with adiponectin receptors in mammalian cells and this interaction is stimulated by adiponectin. Overexpression of APPL1 increases adiponectin signalling and adiponectin-mediated downstream events; such as lipid oxidation, glucose uptake and the membrane translocation of GLUT4, whereas suppression of APPL1 reduces all these processes (Mao et al. 2006). Adaptor protein APPL1 as a critical molecule promotes IRS 1/2-insulin receptor interaction. APPL1 forms a complex with IRS1 /2 under basal conditions, and this complex is then recruited to the insulin receptor in response to insulin or adiponectin stimulation. The interaction between APPL1 and insulin receptor depends on insulin- or adiponectin-stimulated APPL1 phosphorylation, which is greatly reduced in insulin target tissues in obese mice (Ryu et al. 2014).
AdipoR2 is induced by both PPAR-alpha and PPAR-gamma in primary and THP-1 macrophages. Actually, AdipoR1 is more abundant than AdipoR2 in monocytes and its expression decreases upon differentiation into macrophages, whereas AdipoR2 remains constant (Chinetti et al. 2004). Decreased HMW adiponectin plays a crucial and causal role in obesity -linked insulin resistance and metabolic syndrome . Decreased adiponectin action and increased MCP-1 form a vicious adipokine network causing obesity-linked insulin resistance and metabolic syndrome. While PPAR-gamma upregulates HMW adiponectin, PPAR-alpha upregulates AdipoRs (Yamauchi and Kadowaki 2008). The abundance of MCP-1 mRNA in adipose tissue and the plasma concentration of MCP-1 are increased in white adipose tissue of both genetically obese diabetic mice and mice with obesity induced by a high-fat diet. MCP-1 expression in adipose tissue contributes to the macrophage infiltration into this tissue (Kanda et al. 2006). Initially, the C-C motif chemokine receptor-2 (CCR2) regulates monocyte and macrophage recruitment in adipose tissue. Thereby CCR2 is necessary for macrophage-dependent inflammatory responses. Adipose tissue-derived MCP1 (also known as CCL2) is a major chemoattractant, which is responsible for macrophage infiltration and activation. This cytokine is a high-affinity ligand for CCR2 and is significantly elevated in obesity. Consequently, the high amount of CCR2 influences the development of obesity-associated adipose tissue inflammation and systemic insulin resistance (Weisberg et al. 2006). Free fatty acid-stimulated expression of monocyte chemotactic factors is dose and time dependent in obesity. Inhibition of the JNK pathway partially reduces free fatty acid -induced upregulation of MCP-1 and MCP-3. JNK is not only important for mediating free fatty acid -induced insulin resistance but also involves in free fatty acid -induced expression of monocyte chemotactic factors in adipocytes. Adipocytes-derived chemokines contribute to obesity-related white adipose tissue macrophage infiltration through the stimulation of free fatty acids (Jiao et al. 2009). High mobility group box 1 (HMGB1) is a pro-inflammatory adipocytokine involved in white adipose tissue inflammation and insulin resistance in patients with obesity. HMGB1 is secreted from TNF-alpha -induced adipocytes through JNK signaling. Adiponectin protects against HMGB1-induced adipose tissue inflammation (Shimizu et al. 2016). Additionally, adiponectin also promotes macrophage polarization toward the anti-inflammatory M2 phenotype . In this respect, adiponectin stimulates the expression of M2 markers and attenuates the expression of M1 markers in human monocyte-derived macrophages and stromal vascular fraction cells isolated from human adipose tissue (Ohashi et al. 2010). Macrophage polarization controls AdipoR1 and AdipoR2 expression. Activation of classical M1 macrophages suppress AdipoRs expression, whereas alternatively activated (M2) macrophages preserve AdipoRs. Recombinant full-length human adiponectin treatment largely restores AdipoR levels of M1-polarized macrophages compared with those in nonpolarized macrophages, even in the presence of M1 cytokines . In contrast to M1 macrophages, M2 macrophages maintain higher AdipoR levels compared with nonpolarized macrophages and exhibit no AdipoR up-regulation in response to adiponectin. Adiponectin exerts a strong proinflammatory response by inducing IL-12, IL-6 , and TNF-alpha in M1 macrophages . Notably, this proinflammatory adiponectin response is absent in nonpolarized and M2 macrophages. In contrast, in M2 macrophages, adiponectin induces the anti-inflammatory cytokine IL-10 without altering AdipoR expression (van Stijn et al. 2015). It is clear that macrophage polarization is a key determinant regulating AdipoR expression and differential adiponectin-mediated macrophage inflammatory responses (van Stijn et al. 2015).
Preadipocytes have a heightened inflammatory cytokine response following acute saturated fatty acid and monounsaturated fatty acid exposure, when compared to mature adipocytes. This effect is pronounced for MCP-1 . Preadipocytes also demonstrate activation of the NF-kappaB pathway following fatty acid exposure (Dordevic et al. 2014). Adiponectin pre-treatment significantly reduces the increase in MCP-1 mRNA in stimulated adipocytes. Thus, adiponectin exerts anti-inflammatory activity by suppressing IL-6 and MCP-1 production from inflamed adipocytes. This anti-inflammatory action may be mediated through inhibition of NF-kappaB activity as well as through increased PPAR-gamma expression. Furthermore, adiponectin significantly attenuates nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IkappaB-alpha) and IkappaB kinase (IKK) gene expression (Zoico et al. 2009). Irrespective of abdominal visceral fat, low adiponectin and high free fatty acid levels are associated with insulin resistance , and that the effect of low adiponectin is stronger than that of high free fatty acid levels in circulation (Medina-Urrutia et al. 2015). An excess flux of fatty acids causes an altered adipokine production and an increase in oxidative stress in mature adipocytes. Free fatty acids exposed adipocytes demonstrate decreased FOXO1 protein levels in a dose-dependent manner. The FOXO1 downregulation correlates with an increase in the production of ROS and a proinflammatory adipokine pattern, which is characterized with a decrease in adiponectin and an increase in IL-6, plasminogen activator inhibitor-1 (PAI-1) , and MCP-1 mRNA expression (Subauste and Burant 2007). In hepatic cells, adiponectin stimulation produces a transient burst of ROS through activation of the small guanosine triphosphatase (GTPase) Rac1 and 5-lipoxygenase . Furthermore, adiponectin-induced oxidants cause the oxidation and inhibition of protein-tyrosine phosphatase 1B (PTP1B), one of the major phosphotyrosine phosphatases that involves in the control of insulin receptor phosphorylation. Adiponectin causes increased association of PTP1B to insulin receptor. ROS is a critical regulator of the cross-talk between adiponectin and insulin pathways and provides a redox-based molecular mechanism for the insulin-sensitizing function of adiponectin (Fiaschi et al. 2007).
The transcript level of ACC and fatty acid synthase is upregulated by saturated fatty acid treatment. In this case, AdipoR1 reverses the effect induced by palmitate and enhances fatty acid metabolism . Actually, AdipoR1 also increases the gene expression of cytochrome C oxidase, PPAR-alpha, and decreases the gene expression of PGC1-alpha, AMPK -alpha in palmitate-treated human hepatocytes (Chou et al. 2014b). Adiponectin alleviates the endothelial dysfunction caused by elevated free fatty acids concentration through the cross talk between cyclic AMP (cAMP) and NF-kappaB signaling pathway (Wang et al. 2012). The homeostatic model assessment (HOMA)-insulin resistance is associated positively with body mass index , serum non-esterified fatty acid , leptin, IL-6 , and TNF-alpha levels, but negatively with adiponectin. Of these, only serum level of leptin, and in a lesser degree IL-6 and adiponectin are independent determinants of the severity of insulin resistance (Peti et al. 2011). The long-chain saturated fatty acids are positively associated and delta 9–18 desaturase activity is significantly but inversely associated with adiponectin concentrations after dietary fat intake (Gallo et al. 2010).
Plasma adiponectin level correlates positively with peripheral and hepatic insulin sensitivity and negatively with fasting proinsulin and the proinsulin-to-insulin ratio. Indeed, adiponectin, but not adiposity, is the significantly independent determinant of the proinsulin-to-insulin ratio (Bacha et al. 2004). Nevertheless, compared with subcutaneous-derived adipocytes, adiponectin secretion from omental adipocytes is increased by insulin. Thereby, reduced secretion of adiponectin from the omental adipose depot may account for the decline in plasma adiponectin in obesity (Motoshima et al. 2002). Thus, Drolet et al. showed that omental adipocyte adiponectin release is reduced to a greater extent in visceral obese women. Hence, omental adiponectin release contributes to hypo-adiponectinemia on a large scale (Drolet et al. 2009). C/EBP and Nuclear Factor-Y (NF-Y) bind on the −117/−73 region of the adiponectin promoter. This region is critical for the activity of the adiponectin. The C/EBP binding increases in both re-fed animal and high glucose-treated adipocytes (Park et al. 2004). The expression of adiponectin mRNA is significantly reduced in the adipose tissues from obese humans (Hu et al. 1996), whereas mRNA levels of key adiponectin-regulatory transcription factors, including PPAR-gamma2 or C/EBP-alpha, are not significantly changed (Kim et al. 2015). The higher storage capacity of adipocytes prevents the formation of lipotoxic intermediates in adipocytes from the excessive fatty acids. Thereby, excessive lipids efficiently form triglyceride-enriched lipid droplets . If lipid influx into a cell exceeds the oxidative or storage capacity of the cell, then lipotoxic lipid intermediates are likely to accumulate (Unger et al. 2013). Both adiponectin and leptin increase fatty acid oxidation and protect the lipid-intolerant cells from the lipotoxic consequences of fatty acid spillover and aberrant ceramide accumulation (Unger et al. 2013). In particular, adiponectin increases IkappaB-alpha and PPAR-gamma levels to prevent high-fat diet-induced impairment of insulin signalling in adipose tissue. Furthermore, gAD administration is able to improve pathways of insulin signaling and lipid storage in adipose tissue of high-fat diet-fed rats (Matafome et al. 2014). In addition, chronic adipose tissue hypoxia induces dysfunction of adipocytes by triggering oxidative stress in human adipocytes and reduce the production of adiponectin in obesity (Netzer et al. 2015). In this case, antioxidants improve insulin resistance and restore adiponectin production. Recent studies have demonstrated that adiponectin protects against oxidative stress-induced damage in the vascular endothelium (Matsuda and Shimomura 2014). Intra-abdominal adipose tissue-AdipoR2 expression is reduced in obesity, whereas AdipoR1 expression is not altered by obesity. While AdipoR1 expression is directly associated with plasma free fatty acids concentration, AdipoR2 is inversely correlated with plasma levels of triglycerides (Morínigo et al. 2006).
Considering the beneficial metabolic effects, adiponectin-induced AMPK activation is associated with increased lipid oxidation. In this case, adiponectin initially enhances the association of AdipoR1 with APPL1, subsequent binding of APPL1 with AMPKalpha2, leads to phosphorylation and inhibition of ACC and increases fatty acid oxidation (Fang et al. 2010). Additionally, adiponectin also stimulates the ceramidase activity through AdipoR1 and AdipoR2, and enhances ceramide catabolism and formation of its anti-apoptotic metabolite, S1P, independent of AMPK. Overproduction of adiponectin decreases caspase-8-mediated death, whereas genetic ablation of adiponectin increases apoptosis in vivo through a sphingolipid-mediated pathway (Holland et al. 2011). Collectively, adiponectin may directly oppose lipotoxicity by targeting the degradation of ceramide, which is most commonly implicated as a mediator of lipotoxic cell death. It has been shown that adiponectin-mediated enhancements in adipose tissue expansion are highly effective at limiting lipid spillover to other tissues and diminishing formation of lipotoxic metabolites in nonadipose tissues (Unger et al. 2013). Saturated fatty acid -induced ER stress also induces adiponectin resistance , assessed via AMPK phosphorylation, via reducing APPL1 expression (Park et al. 2015). On the other hand, toxic free fatty acids can activate the intrinsic lipoapoptosis pathway in hepatocytes via JNK. Reduced adiponectin levels increase vulnerability to lipotoxicity , which promotes progression from simple steatosis to nonalcoholic steatohepatitis and even advanced hepatic fibrosis (Wree et al. 2011).
6 Adiponectin Resistance and Endothelial Dysfunction in Obesity
Adiponectin is protective against endothelial dysfunction through its pleiotropic actions in obesity . Data from human investigations demonstrate that adiponectin is an important component of the adipo-vascular axis that mediates the cross-talk between adipose tissue and vasculature (Li et al. 2011). High fat diet abolishes microvascular responses to either gAD or insulin and decreases insulin-stimulated glucose disposal by approximately 60%. In this respect, high fat diet induces vascular adiponectin and insulin resistance but gAd administration can restore vascular insulin responses and improve the metabolic action of insulin via an AMPK - and nitric oxide-dependent mechanisms (Zhao et al. 2015). Actually, adiponectin exerts anti-inflammatory and anti-atherogenic properties via its ability to stimulate vascular endothelial nitric oxide production (Chen et al. 2003). Indeed, both gAd and full-length adiponectin induce a relevant dose-dependent vasodilation in Zucker lean rats, but not in hypoadiponectinemic Zucker fatty diabetic rats. This vasodilator effect is totally nitric oxide-dependent. AdipoR1 is much more highly expressed than AdipoR 2 in both animal groups, but APPL1 is significantly decreased in obese rats. The endothelial nitric oxide synthase (eNOS) expression is not significantly different between Zucker lean and obese Zucker fatty diabetic rats. Adiponectin exerts a nitric oxide-dependent vasodilation in resistant arteries of normoglycemic Zucker lean rats, but not Zucker fatty diabetic rats. It is reasonable to claim that alterations in the expression of APPL1 may be involved in the resistance to adiponectin in obese rats (Schmid et al. 2011). Furthermore, the recombinant globular domain of human adiponectin reduces the generation of endothelial ROS , which are induced by oxidized low density lipoprotein (Motoshima et al. 2004). Additionally, adiponectin suppresses high-glucose-induced peroxide production in endothelial cells. Suppression of excess ROS production via adiponectin is mediated by a cAMP/protein kinase A (PKA) -dependent pathway, because globular domain of adiponectin increases cellular cAMP content in hyperglycemic conditions. Inhibition of PKA blocks the effect of both globular domain and full-length adiponecting to suppress ROS generation (Ouedraogo et al. 2006). On the other hand, both superoxide and peroxynitrite productions are increased in adiponectin deficient vessels. Furthermore, although the eNOS expression is normal, nitric oxide production and eNOS phosphorylation is significantly reduced in adiponectin-deficient vessels. Thus, the globular domain of adiponectin reduces aortic superoxide production, increases eNOS phosphorylation, and improves vasodilatory response to acetyl choline. Increased nitric oxide inactivation combined with decreased basal nitric oxide production contributes to endothelial dysfunction in adiponectin deficient subjects. The replacement of plasma adiponectin may improve endothelial function, and reduces cardiovascular complications (Cao et al. 2009).
As mentioned above, in hyperlipidemic state endothelial dysfunction is mainly dependent on decreased synthesis of bioavailable nitric oxide. By contrast, stimulation of nitric oxide production and inhibition of superoxide radical production would provide protection against endothelial dysfunction. However, increase in the production of nitric oxide simultaneously with superoxide radical production leads to formation of peroxynitrite and aggravates vascular injury (Li et al. 2007). In this context, membrane-associated reduced nicotinamide adenine dinucleotide phosphate (NAD(P)H)-dependent oxidases, which can be activated by PKC in the hyperlipidemic condition, is the primary source of superoxide anion. Peroxynitrite formation is a result of the swift reaction between nitric oxide and superoxide anion (Mohazzab et al. 1994). In this respect, treatment with the globular domain of adiponectin significantly enhances eNOS but reduces iNOS activity in hyperlipidemic vessels. Indeed, globular domain of adiponectin reduces superoxide production and peroxynitrite formation in hyperlipidemic vascular segments by approximately 78%. Collectively, these results indicate that adiponectin protects the endothelium against hyperlipidemic injury in obesity by multiple mechanisms, including promoting eNOS activity, inhibiting iNOS activity, preserving bioactive nitric oxide, and attenuating oxidative and nitrative stress (Li et al. 2007). Insulin-resistance decreases nitric oxide bioavailability . In accordance with the insulin resistance , adiponectin-resistance specific pathways decrease nitric oxide bioavailability by enhancing oxidative/nitrative stress. Consequently, insulin-adiponectin resistance cross talk causes intracellular signal transduction disturbances (Li et al. 2010a). In this context, the major intracellular pathway activated by adiponectin, AMPK is responsible for vascular protective and anti-ischemic properties of adiponectin. Conversely, adiponectin resistance is a serious risk factor for cardiovascular injury including atherosclerosis and endothelial dysfunction (Lau et al. 2011). Adiponectin promotes the phosphorylation of AMPK, Akt and eNOS in endothelial cells. Both AMPK and Akt signals are required for adiponectin-induced endothelial migration and differentiation. PI3K functions upstream from the Akt-eNOS regulatory axis in adiponectin-stimulated endothelial cells (Ouchi et al. 2004). Additionally, AMPK inhibition does not abrogate the reduction of high-glucose–induced ROS production by gAD in endothelial cells. This suggests that the involvement of the PKA pathway in this process is not dependent on AMPK . Furthermore, increasing cAMP levels or blocking PKA activity in human endothelial cells do not effect the ability of gAD to activate AMPK (Ouedraogo et al. 2006).
Endothelial cell is activated by various inflammatory stimuli, including TNF-alpha , and this results in the synthesis of adhesion molecules and increases the adherence of monocytes. This monocyte adhesion to the arterial endothelium is considered to be crucial for the development of vascular diseases (Ross 1993). Physiological concentrations of adiponectin dose-dependently inhibit TNF-alpha-induced THP-1 adhesion and expression of vascular cell adhesion molecule-1 (VCAM-1), E-selectin, and ICAM-1 on human aortic endothelial cells (Ouchi et al. 1999). Overexpression of AdipoR1 and 2 in endothelial cells significantly enhances the suppressive effect of a sub-effective dose of adiponectin on TNF-alpha -induced ICAM-1 expression and NF-kappaB activation. Upregulation of AdipoRs in endothelial cells potentiates the anti-inflammatory effects of adiponectin (Zhang et al. 2009) (Fig. 18.1).
Adiponectin over-expression in diet -induced obese mice lead to the inhibition of macrophage infiltration and the elimination of crown-like structures. In this context, adiponectin transgenic mice display a remarkable sensitivity to insulin, and thus the hepatic steatosis diminish. Increased circulating adiponectin levels are associated with increased adipose tissue vascularization and perfusion under conditions of long term diet-induced obesity , subsequently, metabolic functions improve despite the obese environment (Aprahamian 2013). Leptin activates cellular signaling pathways, and increases adiponectin mRNA in the adipose tissue from normal-weight individuals, but the same increase in adiponectin mRNA can not be achieved in the adipose tissue from obese participants. First of all, weight gain increases adiponectin expression in healthy humans. In this case leptin up-regulates adiponectin expression during weight gain. Actually, obese subjects have increased caveolin-1 expression, which attenuates leptin-signal. This leptin signal impairment may prevent concordant increases in adiponectin levels in obese subjects despite their high levels of leptin. In other words, increases in leptin to adiponectin ratio may suggest decreased leptin sensitivity, and altered insulin sensitivity (Singh et al. 2016). Actually, the leptin to adiponectin ratio is proposed as a good biomarker for the prevalence of metabolic syndrome in comparison to the adiponectin and leptin levels alone. However, this ratio is closely dependent on visceral fat accumulation and cardiorespiratory performance (Kumagai et al. 2005). In addition, a high serum leptin to adiponectin ratio and high levels of serum triglycerides may be indicators of “at-risk” obesity, independent of other obesity markers, especially in young severely obese individuals (Labruna et al. 2011).
Despite unchanged or elevated plasma adiponectin levels, vascular AMPK and eNOS phosphorylation levels are significantly reduced in high fat-diet animals. The disassociation between plasma adiponectin levels and vascular AMPK/eNOS phosphorylation in obese/hyperlipidemic rats suggests that high fat-fed animals have reduced vascular response to adiponectin. Although different mechanisms are involved in vascular adiponectin resistance in high fat-fed rats at different stages of obesity , diet-induced obesity/hyperlipidemia causes significant vascular adiponectin resistance. In advanced stages of high fat-diet, adiponectin inactivation, adiponectin receptor downregulation, and circulating adiponectin reduction collectively occur (Li et al. 2010b). In obesity-related cardiovascular diseases , the beneficial effects of perivascular adipose tissue on vascular functions are impaired. The contribution of perivascular adipose tissue dysfunction to obesity-related cardiovascular diseases is associated with decreased levels of adiponectin and increased levels of TNF-alpha . These changes lead to increased quantity of adipose tissue, inflammation , cell proliferation and endothelial dysfunction (Ozen et al. 2015). Furthermore, TNF-alpha is upregulated by leptin. In contrast, adiponectin downregulates the expression and release of a number of proinflammatory immune mediators. Therefore, leptin/adiponectin imbalance may be an important mediator of the elevated risk of developing abdominal obesity associated cardiovascular diseases (López-Jaramillo et al. 2014). Adiponectin and its receptor system have an important protective role against oxidative stress - or TNF-alpha -mediated myocardial insulin resistance and dysfunction after ischemic heart disease (Saito et al. 2007a). It is well-known that in the advanced stages of heart failure, there is a downregulation in fatty acid oxidation, increased glycolysis and glucose oxidation, reduced respiratory chain activity, and an impaired reserve for mitochondrial oxidative capacity (Stanley et al. 2005). Thus, the abrogation of TNF-alpha -induced suppression of AdipoR1 expression might account for the improvement of glucose uptake. Furthermore, adiponectin and its receptor system may have an important protective role against oxidative stress- or TNF-alpha-mediated myocardial insulin resistance and dysfunction after myocardial injury (Saito et al. 2007a). In the myocardium, adiponectin-mediated protection from ischemia-reperfusion injury is linked to cyclooxygenase-2 (COX-2) -mediated suppression of TNF signaling, inhibition of apoptosis by AMPK , inhibition of iNOS and NADPH-oxidase protein expression and resultant excessive peroxynitrite -induced oxidative and nitrosative stress (Goldstein et al. 2009; Tao et al. 2007).
Clinical studies have demonstrated the impaired production of eNOS in the vasculature in subjects with decreased adiponectin levels. Consequently, endothelium-dependent vasorelaxation decreases due to lack of nitric oxide availability (Adya et al. 2015). Obesity and type 2 diabetes are associated with low plasma adiponectin concentrations in different ethnic groups and indicate that the degree of hypoadiponectinemia is more closely related to the degree of insulin resistance and hyperinsulinemia than to the degree of adiposity and glucose intolerance (Weyer et al. 2001). gAd acts as a critical physiological factor which protects against fluctuating high glucose-induced endothelial damage. It may act via attenuating apoptosis and increasing synthesis of nitric oxide through both the PI3K/Akt and AMPK signaling pathway to reduce oxidative stress and cell apoptosis (Xiao et al. 2011). Globular adiponectin reverses high glucose-impaired endothelial progenitor cells functions through nitric oxide- and p38 MAPK-related mechanisms (Huang et al. 2011). Both full-length adiponectin and gAD increase endothelial COX-2 expression, with gAD -mediated upregulation of COX-2 that is dependent on AdipoR1 and NFkappaB activation. With respect to full-length adiponectin, gAD more efficiently increases activation of NFkappaB signaling pathways, resulting in COX-2 overexpression and COX-2-dependent prostacyclin 2 release. In contrast to the full-length adiponectin, gAD also increases p38 MAPK phosphorylation and VCAM-1 expression , ultimately enhancing adhesion of monocytes to endothelial cells (Addabbo et al. 2011).
7 Conclusion
In obesity , the interactions between insulin and adiponectin are extremely complex. While insulin negatively regulates the expression of AdipoRs and adiponectin sensitivity, insulin resistance may result in hyperadiponectinaemia and adiponectin resistance. Adaptor protein of AdipoRs, APPL1 also promotes the insulin receptor substrat-insulin receptor interaction. On the other hand, saturated fatty acid -induced ER stress enhances adiponectin resistance , via reducing APPL1 expression. This condition is also associated with insulin resistance and is thought to be partly due to the down-regulation of the adiponectin and AdipoR1 in high-fat diet.
Even, new treatment strategies are being developed for the improvement of adiponectin resistance, lack of clinical evidences complicates the solution of the problems associated with obesity . Further understanding of the molecular mechanisms of the AdipoRs expression or stimulation, adiponectin-AdipoRs interaction and adiponectin/insulin resistance would facilitate the treatment of obesity-related conditions and adipose tissue dysfunction.
References
Addabbo, F., C. Nacci, L. De Benedictis, V. Leo, M. Tarquinio, M.J. Quon, and M. Montagnani. 2011. Globular adiponectin counteracts VCAM-1-mediated monocyte adhesion via AdipoR1/NF-κB/COX-2 signaling in human aortic endothelial cells. American Journal of Physiology. Endocrinology and Metabolism 301: E1143–E1154. doi:10.1152/ajpendo.00208.2011.
Adya, R., B.K. Tan, and H.S. Randeva. 2015. Differential effects of leptin and adiponectin in endothelial angiogenesis. Journal of Diabetes Research 2015: 648239. doi:10.1155/2015/648239.
Aprahamian, T.R. 2013. Elevated adiponectin expression promotes adipose tissue vascularity under conditions of diet-induced obesity. Metabolism 62: 1730–1738. doi:10.1016/j.metabol.2013.07.010.
Arita, Y., S. Kihara, N. Ouchi, M. Takahashi, K. Maeda, J. Miyagawa, K. Hotta, I. Shimomura, T. Nakamura, K. Miyaoka, H. Kuriyama, M. Nishida, S. Yamashita, K. Okubo, K. Matsubara, M. Muraguchi, Y. Ohmoto, T. Funahashi, and Y. Matsuzawa. 1999. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochemical and Biophysical Research Communications 257: 79–83.
Arner, E., P.O. Westermark, K.L. Spalding, T. Britton, M. Rydén, J. Frisén, S. Bernard, and P. Arner. 2010. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 59: 105–109. doi:10.2337/db09-0942.
Bacha, F., R. Saad, N. Gungor, and S.A. Arslanian. 2004. Adiponectin in youth: Relationship to visceral adiposity, insulin sensitivity, and beta-cell function. Diabetes Care 27: 547–552.
Baffy, G. 2005. Uncoupling protein-2 and non-alcoholic fatty liver disease. Frontiers in Bioscience: A virtual library of medicine 10: 2082–2096.
Barbieri, M., A. Esposito, E. Angellotti, M.R. Rizzo, R. Marfella, and G. Paolisso. 2013. Association of genetic variation in adaptor protein APPL1/APPL2 loci with non-alcoholic fatty liver disease. PLoS One 8: e71391. doi:10.1371/journal.pone.0071391.
Blachnio-Zabielska, A.U., C. Koutsari, T. Tchkonia, and M.D. Jensen. 2012. Sphingolipid content of human adipose tissue: Relationship to adiponectin and insulin resistance. Obesity (Silver Spring) 20: 2341–2347. doi:10.1038/oby.2012.126.
Blüher, M., M.D. Michael, O.D. Peroni, K. Ueki, N. Carter, B.B. Kahn, and C.R. Kahn. 2002. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Developmental Cell 3: 25–38.
Bobbert, T., J. Weicht, K. Mai, M. Möhlig, A.F.H. Pfeiffer, and J. Spranger. 2009. Acute hyperinsulinaemia and hyperlipidaemia modify circulating adiponectin and its oligomers. Clinical Endocrinology 71: 507–511. doi:10.1111/j.1365-2265.2008.03519.x.
Bruce, C.R., V.A. Mertz, G.J.F. Heigenhauser, and D.J. Dyck. 2005. The stimulatory effect of globular adiponectin on insulin-stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes 54: 3154–3160.
Bruun, J.M., A.S. Lihn, C. Verdich, S.B. Pedersen, S. Toubro, A. Astrup, and B. Richelsen. 2003. Regulation of adiponectin by adipose tissue-derived cytokines: In vivo and in vitro investigations in humans. American Journal of Physiology. Endocrinology and Metabolism 285: E527–E533. doi:10.1152/ajpendo.00110.2003.
Calvani, M., A. Scarfone, L. Granato, E.V. Mora, G. Nanni, M. Castagneto, A.V. Greco, M. Manco, and G. Mingrone. 2004. Restoration of adiponectin pulsatility in severely obese subjects after weight loss. Diabetes 53: 939–947.
Cao, Y., L. Tao, Y. Yuan, X. Jiao, W.B. Lau, Y. Wang, T. Christopher, B. Lopez, L. Chan, B. Goldstein, and X.L. Ma. 2009. Endothelial dysfunction in adiponectin deficiency and its mechanisms involved. Journal of Molecular and Cellular Cardiology 46: 413–419. doi:10.1016/j.yjmcc.2008.10.014.
Caselli, C. 2014. Role of adiponectin system in insulin resistance. Molecular Genetics and Metabolism 113: 155–160. doi:10.1016/j.ymgme.2014.09.003.
Ceddia, R.B., R. Somwar, A. Maida, X. Fang, G. Bikopoulos, and G. Sweeney. 2005. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia 48: 132–139. doi:10.1007/s00125-004-1609-y.
Chang, E., J.M. Choi, S.E. Park, E.-J. Rhee, W.-Y. Lee, K.-W. Oh, S.-W. Park, and C.-Y. Park. 2015. Adiponectin deletion impairs insulin signaling in insulin-sensitive but not insulin-resistant 3 T3-L1 adipocytes. Life Sciences 132: 93–100. doi:10.1016/j.lfs.2015.02.013.
Chen, H., M. Montagnani, T. Funahashi, I. Shimomura, and M.J. Quon. 2003. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. The Journal of Biological Chemistry 278: 45021–45026. doi:10.1074/jbc.M307878200.
Chen, M.B., A.J. McAinch, S.L. Macaulay, L.A. Castelli, P.E. O’brien, J.B. Dixon, D. Cameron-Smith, B.E. Kemp, and G.R. Steinberg. 2005. Impaired activation of AMP-kinase and fatty acid oxidation by globular adiponectin in cultured human skeletal muscle of obese type 2 diabetics. The Journal of Clinical Endocrinology and Metabolism 90: 3665–3672. doi:10.1210/jc.2004-1980.
Chen, H., A. Fajol, M. Hoene, B. Zhang, E.D. Schleicher, Y. Lin, C. Calaminus, B.J. Pichler, C. Weigert, H.U. Häring, F. Lang, and M. Föller. 2016. PI3K-resistant GSK3 controls adiponectin formation and protects from metabolic syndrome. Proceedings of the National Academy of Sciences of the United States of America 113: 5754–5759. doi:10.1073/pnas.1601355113.
Chinetti, G., C. Zawadski, J.C. Fruchart, and B. Staels. 2004. Expression of adiponectin receptors in human macrophages and regulation by agonists of the nuclear receptors PPARalpha, PPARgamma, and LXR. Biochemical and Biophysical Research Communications 314: 151–158.
Chou, I.-P., Y.-P. Chiu, S.-T. Ding, B.-H. Liu, Y.Y. Lin, and C.-Y. Chen. 2014a. Adiponectin receptor 1 overexpression reduces lipid accumulation and hypertrophy in the heart of diet-induced obese mice—Possible involvement of oxidative stress and autophagy. Endocrine Research 39: 173–179. doi:10.3109/07435800.2013.879165.
Chou, I.-P., Y.Y. Lin, S.-T. Ding, and C.-Y. Chen. 2014b. Adiponectin receptor 1 enhances fatty acid metabolism and cell survival in palmitate-treated HepG2 cells through the PI3 K/AKT pathway. European Journal of Nutrition 53: 907–917. doi:10.1007/s00394-013-0594-7.
Cleasby, M.E., Q. Lau, E. Polkinghorne, S.A. Patel, S.J. Leslie, N. Turner, G.J. Cooney, A. Xu, and E.W. Kraegen. 2011. The adaptor protein APPL1 increases glycogen accumulation in rat skeletal muscle through activation of the PI3-kinase signalling pathway. The Journal of Endocrinology 210: 81–92. doi:10.1530/JOE-11-0039.
Cui, X.-B., C. Wang, L. Li, D. Fan, Y. Zhou, D. Wu, Q.-H. Cui, F.-Y. Fu, and L.-L. Wu. 2012. Insulin decreases myocardial adiponectin receptor 1 expression via PI3K/Akt and FoxO1 pathway. Cardiovascular Research 93: 69–78. doi:10.1093/cvr/cvr273.
Curtis, J.M., W.S. Hahn, M.D. Stone, J.J. Inda, D.J. Droullard, J.P. Kuzmicic, M.A. Donoghue, E.K. Long, A.G. Armien, S. Lavandero, E. Arriaga, T.J. Griffin, and D.A. Bernlohr. 2012. Protein carbonylation and adipocyte mitochondrial function. The Journal of Biological Chemistry 287: 32967–32980. doi:10.1074/jbc.M112.400663.
Dallaire, P., K. Bellmann, M. Laplante, S. Gélinas, C. Centeno-Baez, P. Penfornis, M.-L. Peyot, M.G. Latour, J. Lamontagne, M.E. Trujillo, P.E. Scherer, M. Prentki, Y. Deshaies, and A. Marette. 2008. Obese mice lacking inducible nitric oxide synthase are sensitized to the metabolic actions of peroxisome proliferator-activated receptor-gamma agonism. Diabetes 57: 1999–2011. doi:10.2337/db08-0540.
Deepa, S.S., and L.Q. Dong. 2009. APPL1: Role in adiponectin signaling and beyond. American Journal of Physiology. Endocrinology and Metabolism 296: E22–E36. doi:10.1152/ajpendo.90731.2008.
Delaigle, A.M., J.-C. Jonas, I.B. Bauche, O. Cornu, and S.M. Brichard. 2004. Induction of adiponectin in skeletal muscle by inflammatory cytokines: In vivo and in vitro studies. Endocrinology 145: 5589–5597. doi:10.1210/en.2004-0503.
Denzel, M.S., M.-C. Scimia, P.M. Zumstein, K. Walsh, P. Ruiz-Lozano, and B. Ranscht. 2010. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. The Journal of Clinical Investigation 120: 4342–4352. doi:10.1172/JCI43464.
Ding, Q., Z. Wang, and Y. Chen. 2009. Endocytosis of adiponectin receptor 1 through a clathrin- and Rab5-dependent pathway. Cell Research 19: 317–327. doi:10.1038/cr.2008.299.
Dokken, B.B., and E.J. Henriksen. 2006. Chronic selective glycogen synthase kinase-3 inhibition enhances glucose disposal and muscle insulin action in prediabetic obese Zucker rats. American Journal of Physiology. Endocrinology and Metabolism 291: E207–E213. doi:10.1152/ajpendo.00628.2005.
Dordevic, A.L., N. Konstantopoulos, and D. Cameron-Smith. 2014. 3T3-L1 preadipocytes exhibit heightened monocyte-chemoattractant protein-1 response to acute fatty acid exposure. PLoS One 9: e99382. doi:10.1371/journal.pone.0099382.
Drolet, R., C. Bélanger, M. Fortier, C. Huot, J. Mailloux, D. Légaré, and A. Tchernof. 2009. Fat depot-specific impact of visceral obesity on adipocyte adiponectin release in women. Obesity (Silver Spring) 17: 424–430. doi:10.1038/oby.2008.555.
Engfeldt, P., and P. Arner. 1988. Lipolysis in human adipocytes, effects of cell size, age and of regional differences. Hormone and Metabolic Research. Supplement Series 19: 26–29.
Fang X, Palanivel R, Cresser J, Schram K, Ganguly R, Thong FS, Tuinei J, Xu A, Abel ED, Sweeney G. An APPL1-AMPK signaling axis mediates beneficial metabolic effects of adiponectin in the heart. Am J Physiol Endocrinol Metab. 2010 Nov;299(5):E721–9.
Fiaschi, T., F. Buricchi, G. Cozzi, S. Matthias, M. Parri, G. Raugei, G. Ramponi, and P. Chiarugi. 2007. Redox-dependent and ligand-independent trans-activation of insulin receptor by globular adiponectin. Hepatology 46: 130–139. doi:10.1002/hep.21643.
Frohnert, B.I., and D.A. Bernlohr. 2013. Protein carbonylation, mitochondrial dysfunction, and insulin resistance. Advances in Nutrition 4: 157–163. doi:10.3945/an.112.003319.
Gajewska, J., H. Weker, J. Ambroszkiewicz, M. Chełchowska, M. Więch, and T. Laskowska-Klita. 2011. Changes in concentration of serum adiponectin multimeric forms following weight reduction programme in prepubertal obese children. Medycyna Wieku Rozwojowego 15: 298–305.
Gallo, S., G. Egeland, S. Meltzer, L. Legault, and S. Kubow. 2010. Plasma fatty acids and desaturase activity are associated with circulating adiponectin in healthy adolescent girls. The Journal of Clinical Endocrinology and Metabolism 95: 2410–2417. doi:10.1210/jc.2009-1975.
Gauthier, M.-S., E.L. O’Brien, S. Bigornia, M. Mott, J.M. Cacicedo, X.J. Xu, N. Gokce, C. Apovian, and N. Ruderman. 2011. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochemical and Biophysical Research Communications 404: 382–387. doi:10.1016/j.bbrc.2010.11.127.
Goldstein, B.J., R.G. Scalia, and X.L. Ma. 2009. Protective vascular and myocardial effects of adiponectin. Nature Clinical Practice. Cardiovascular Medicine 6: 27–35. doi:10.1038/ncpcardio1398.
Groeneveld, M.P., G.V. Brierley, N.M. Rocha, K. Siddle, and R.K. Semple. 2016. Acute knockdown of the insulin receptor or its substrates Irs1 and 2 in 3 T3-L1 adipocytes suppresses adiponectin production. Scientific Reports 6: 21105. doi:10.1038/srep21105.
Gu, D., Z. Wang, X. Dou, X. Zhang, S. Li, L. Vu, T. Yao, and Z. Song. 2013. Inhibition of ERK1/2 pathway suppresses adiponectin secretion via accelerating protein degradation by Ubiquitin-proteasome system: Relevance to obesity-related adiponectin decline. Metabolism 62: 1137–1148. doi:10.1016/j.metabol.2013.01.025.
Henriksen, E.J. 2010. Dysregulation of glycogen synthase kinase-3 in skeletal muscle and the etiology of insulin resistance and type 2 diabetes. Current Diabetes Reviews 6: 285–293.
Henriksen, E.J., and B.B. Dokken. 2006. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Current Drug Targets 7: 1435–1441.
Hino, K., and H. Nagata. 2012. Screening for adiponectin secretion regulators. Vitamins and Hormones 90: 125–141. doi:10.1016/B978-0-12-398313-8.00005-1.
Høeg, L.D., K.A. Sjøberg, A.-M. Lundsgaard, A.B. Jordy, N. Hiscock, J.F.P. Wojtaszewski, E.A. Richter, and B. Kiens. 2013. Adiponectin concentration is associated with muscle insulin sensitivity, AMPK phosphorylation, and ceramide content in skeletal muscles of men but not women. Journal of Applied Physiology (1985) 114: 592–601. doi:10.1152/japplphysiol.01046.2012.
Hoffmann, A., T. Ebert, N. Klöting, J. Dokas, F. Jeromin, B. Jessnitzer, R. Burkhardt, M. Fasshauer, and S. Kralisch. 2016. Leptin dose-dependently decreases atherosclerosis by attenuation of hypercholesterolemia and induction of adiponectin. Biochimica et Biophysica Acta 1862: 113–120. doi:10.1016/j.bbadis.2015.10.022.
Holland, W.L., and S.A. Summers. 2008. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocrine Reviews 29: 381–402. doi:10.1210/er.2007-0025.
Holland, W.L., R.A. Miller, Z.V. Wang, K. Sun, B.M. Barth, H.H. Bui, K.E. Davis, B.T. Bikman, N. Halberg, J.M. Rutkowski, M.R. Wade, V.M. Tenorio, M.-S. Kuo, J.T. Brozinick, B.B. Zhang, M.J. Birnbaum, S.A. Summers, and P.E. Scherer. 2011. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nature Medicine 17: 55–63. doi:10.1038/nm.2277.
Hosch, S.E., J.M. Olefsky, and J.J. Kim. 2006. APPLied mechanics: Uncovering how adiponectin modulates insulin action. Cell Metabolism 4: 5–6. doi:10.1016/j.cmet.2006.06.003.
Hossain, M.M., A. Mukheem, and T. Kamarul. 2015. The prevention and treatment of hypoadiponectinemia-associated human diseases by up-regulation of plasma adiponectin. Life Sciences 135: 55–67. doi:10.1016/j.lfs.2015.03.010.
Hou, M., Z. Chu, T. Liu, H. Lv, L. Sun, B. Wang, J. Huang, and W. Yan. 2015. A high-fat maternal diet decreases adiponectin receptor-1 expression in offspring. The Journal of Maternal-Fetal & Neonatal Medicine 28: 216–221. doi:10.3109/14767058.2014.914489.
Hu, E., P. Liang, and B.M. Spiegelman. 1996. AdipoQ is a novel adipose-specific gene dysregulated in obesity. The Journal of Biological Chemistry 271: 10697–10703.
Huang, P.-H., J.-S. Chen, H.-Y. Tsai, Y.-H. Chen, F.-Y. Lin, H.-B. Leu, T.-C. Wu, S.-J. Lin, and J.-W. Chen. 2011. Globular adiponectin improves high glucose-suppressed endothelial progenitor cell function through endothelial nitric oxide synthase dependent mechanisms. Journal of Molecular and Cellular Cardiology 51: 109–119. doi:10.1016/j.yjmcc.2011.03.008.
Hug, C., J. Wang, N.S. Ahmad, J.S. Bogan, T.-S. Tsao, and H.F. Lodish. 2004. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proceedings of the National Academy of Sciences of the United States of America 101: 10308–10313. doi:10.1073/pnas.0403382101.
Jeon, M.J., J. Leem, M.S. Ko, J.E. Jang, H.-S. Park, H.S. Kim, M. Kim, E.H. Kim, H.J. Yoo, C.-H. Lee, I.-S. Park, K.-U. Lee, and E.H. Koh. 2012. Mitochondrial dysfunction and activation of iNOS are responsible for the palmitate-induced decrease in adiponectin synthesis in 3T3L1 adipocytes. Experimental & Molecular Medicine 44: 562–570. doi:10.3858/emm.2012.44.9.064.
Jiao, P., Q. Chen, S. Shah, J. Du, B. Tao, I. Tzameli, W. Yan, and H. Xu. 2009. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: Involvement of nuclear factor-kappaB and c-Jun NH2-terminal kinase pathways. Diabetes 58: 104–115. doi:10.2337/db07-1344.
Kadowaki, T., and T. Yamauchi. 2005. Adiponectin and adiponectin receptors. Endocrine Reviews 26: 439–451. doi:10.1210/er.2005-0005.
Kanda, H., S. Tateya, Y. Tamori, K. Kotani, K. Hiasa, R. Kitazawa, S. Kitazawa, H. Miyachi, S. Maeda, K. Egashira, and M. Kasuga. 2006. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. The Journal of Clinical Investigation 116: 1494–1505. doi:10.1172/JCI26498.
Karki, S., P. Chakrabarti, G. Huang, H. Wang, S.R. Farmer, and K.V. Kandror. 2011. The multi-level action of fatty acids on adiponectin production by fat cells. PLoS One 6: e28146. doi:10.1371/journal.pone.0028146.
Kim, C.-H., P. Pennisi, H. Zhao, S. Yakar, J.B. Kaufman, K. Iganaki, J. Shiloach, P.E. Scherer, M.J. Quon, and D. LeRoith. 2006. MKR mice are resistant to the metabolic actions of both insulin and adiponectin: Discordance between insulin resistance and adiponectin responsiveness. American Journal of Physiology. Endocrinology and Metabolism 291: E298–E305. doi:10.1152/ajpendo.00319.2005.
Kim, A.Y., Y.J. Park, X. Pan, K.C. Shin, S.-H. Kwak, A.F. Bassas, R.M. Sallam, K.S. Park, A.A. Alfadda, A. Xu, and J.B. Kim. 2015. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nature Communications 6: 7585. doi:10.1038/ncomms8585.
Kitamura, Y.I., T. Kitamura, J.-P. Kruse, J.C. Raum, R. Stein, W. Gu, and D. Accili. 2005. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metabolism 2: 153–163. doi:10.1016/j.cmet.2005.08.004.
Koh, E.H., J.-Y. Park, H.-S. Park, M.J. Jeon, J.W. Ryu, M. Kim, S.Y. Kim, M.-S. Kim, S.-W. Kim, I.S. Park, J.H. Youn, and K.-U. Lee. 2007. Essential role of mitochondrial function in adiponectin synthesis in adipocytes. Diabetes 56: 2973–2981. doi:10.2337/db07-0510.
Kopp, H.-P., K. Krzyzanowska, M. Möhlig, J. Spranger, A.F.H. Pfeiffer, and G. Schernthaner. 2005. Effects of marked weight loss on plasma levels of adiponectin, markers of chronic subclinical inflammation and insulin resistance in morbidly obese women. International Journal of Obesity 2005(29): 766–771. doi:10.1038/sj.ijo.0802983.
Kumagai, S., H. Kishimoto, Masatakasuwa, B. Zou, and Harukasasaki. 2005. The leptin to adiponectin ratio is a good biomarker for the prevalence of metabolic syndrome, dependent on visceral fat accumulation and endurance fitness in obese patients with diabetes mellitus. Metabolic Syndrome and Related Disorders 3: 85–94. doi:10.1089/met.2005.3.85.
Labruna, G., F. Pasanisi, C. Nardelli, R. Caso, D.F. Vitale, F. Contaldo, and L. Sacchetti. 2011. High leptin/adiponectin ratio and serum triglycerides are associated with an “at-risk” phenotype in young severely obese patients. Obesity (Silver Spring) 19: 1492–1496. doi:10.1038/oby.2010.309.
Lau, W.B., L. Tao, Y. Wang, R. Li, and X.L. Ma. 2011. Systemic adiponectin malfunction as a risk factor for cardiovascular disease. Antioxidants & Redox Signaling 15: 1863–1873. doi:10.1089/ars.2010.3743.
Lee, M.-J., R.-Z. Yang, K. Karastergiou, S.R. Smith, J.R. Chang, D.-W. Gong, and S.K. Fried. 2016. Low expression of the GILZ may contribute to adipose inflammation and altered adipokine production in human obesity. Journal of Lipid Research 57: 1256–1263. doi:10.1194/jlr.M067728.
Li, R., W.-Q. Wang, H. Zhang, X. Yang, Q. Fan, T.A. Christopher, B.L. Lopez, L. Tao, B.J. Goldstein, F. Gao, and X.L. Ma. 2007. Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/iNOS activity. American Journal of Physiology. Endocrinology and Metabolism 293: E1703–E1708. doi:10.1152/ajpendo.00462.2007.
Li, R., W.B. Lau, and X.L. Ma. 2010a. Adiponectin resistance and vascular dysfunction in the hyperlipidemic state. Acta Pharmacologica Sinica 31: 1258–1266. doi:10.1038/aps.2010.95.
Li, R., M. Xu, X. Wang, Y. Wang, W.B. Lau, Y. Yuan, W. Yi, X. Wei, B.L. Lopez, T.A. Christopher, X.-M. Wang, and X.-L. Ma. 2010b. Reduced vascular responsiveness to adiponectin in hyperlipidemic rats—Mechanisms and significance. Journal of Molecular and Cellular Cardiology 49: 508–515. doi:10.1016/j.yjmcc.2010.03.002.
Li, F.Y.L., K.K.Y. Cheng, K.S.L. Lam, P.M. Vanhoutte, and A. Xu. 2011. Cross-talk between adipose tissue and vasculature: Role of adiponectin. Acta Physiologica (Oxford, England) 203: 167–180. doi:10.1111/j.1748-1716.2010.02216.x.
Lin, H.V., J.-Y. Kim, A. Pocai, L. Rossetti, L. Shapiro, P.E. Scherer, and D. Accili. 2007. Adiponectin resistance exacerbates insulin resistance in insulin receptor transgenic/knockout mice. Diabetes 56: 1969–1976. doi:10.2337/db07-0127.
López-Jaramillo, P., D. Gómez-Arbeláez, J. López-López, C. López-López, J. Martínez-Ortega, A. Gómez-Rodríguez, and S. Triana-Cubillos. 2014. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Hormone Molecular Biology and Clinical Investigation 18: 37–45. doi:10.1515/hmbci-2013-0053.
Mai, S., G.E. Walker, A. Brunani, G. Guzzaloni, G. Grossi, A. Oldani, G. Aimaretti, M. Scacchi, and P. Marzullo. 2014. Inherent insulin sensitivity is a major determinant of multimeric adiponectin responsiveness to short-term weight loss in extreme obesity. Scientific Reports 4: 5803. doi:10.1038/srep05803.
Mao, X., C.K. Kikani, R.A. Riojas, P. Langlais, L. Wang, F.J. Ramos, Q. Fang, C.Y. Christ-Roberts, J.Y. Hong, R.-Y. Kim, F. Liu, and L.Q. Dong. 2006. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nature Cell Biology 8: 516–523. doi:10.1038/ncb1404.
Matafome, P., T. Rodrigues, A. Pereira, L. Letra, H. Azevedo, A. Paixão, M. Silvério, A. Almeida, C. Sena, and R. Seiça. 2014. Long-term globular adiponectin administration improves adipose tissue dysmetabolism in high-fat diet-fed Wistar rats. Archives of Physiology and Biochemistry 120: 147–157. doi:10.3109/13813455.2014.950590.
Matsuda, M., and I. Shimomura. 2014. Roles of adiponectin and oxidative stress in obesity-associated metabolic and cardiovascular diseases. Reviews in Endocrine & Metabolic Disorders 15: 1–10. doi:10.1007/s11154-013-9271-7.
McAinch, A.J., G.R. Steinberg, J. Mollica, P.E. O’Brien, J.B. Dixon, S.L. Macaulay, B.E. Kemp, and D. Cameron-Smith. 2006. Differential regulation of adiponectin receptor gene expression by adiponectin and leptin in myotubes derived from obese and diabetic individuals. Obesity (Silver Spring) 14: 1898–1904. doi:10.1038/oby.2006.221.
Medina-Urrutia, A., C. Posadas-Romero, R. Posadas-Sánchez, E. Jorge-Galarza, T. Villarreal-Molina, M.D.C. González-Salazar, G. Cardoso-Saldaña, G. Vargas-Alarcón, M. Torres-Tamayo, and J.G. Juárez-Rojas. 2015. Role of adiponectin and free fatty acids on the association between abdominal visceral fat and insulin resistance. Cardiovascular Diabetology 14: 20. doi:10.1186/s12933-015-0184-5.
Meyer, L.K., T.P. Ciaraldi, R.R. Henry, A.C. Wittgrove, and S.A. Phillips. 2013. Adipose tissue depot and cell size dependency of adiponectin synthesis and secretion in human obesity. Adipocytes 2: 217–226. doi:10.4161/adip.24953.
Miyazaki, S., T. Izawa, J. Ogasawara, T. Sakurai, S. Nomura, T. Kizaki, H. Ohno, and T. Komabayashi. 2010. Effect of exercise training on adipocyte-size-dependent expression of leptin and adiponectin. Life Sciences 86: 691–698. doi:10.1016/j.lfs.2010.03.004.
Mohazzab, K.M., P.M. Kaminski, and M.S. Wolin. 1994. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. The American Journal of Physiology 266: H2568–H2572.
Mondal, A.K., S.K. Das, V. Varma, G.T. Nolen, R.E. McGehee, S.C. Elbein, J.Y. Wei, and G. Ranganathan. 2012. Effect of endoplasmic reticulum stress on inflammation and adiponectin regulation in human adipocytes. Metabolic Syndrome and Related Disorders 10: 297–306. doi:10.1089/met.2012.0002.
Morínigo, R., M. Musri, J. Vidal, R. Casamitjana, S. Delgado, A.M. Lacy, C. Ayuso, R. Gomis, and H. Corominola. 2006. Intra-abdominal fat adiponectin receptors expression and cardiovascular metabolic risk factors in obesity and diabetes. Obesity Surgery 16: 745–751. doi:10.1381/096089206777346736.
Motoshima, H., X. Wu, M.K. Sinha, V.E. Hardy, E.L. Rosato, D.J. Barbot, F.E. Rosato, and B.J. Goldstein. 2002. Differential regulation of adiponectin secretion from cultured human omental and subcutaneous adipocytes: Effects of insulin and rosiglitazone. The Journal of Clinical Endocrinology and Metabolism 87: 5662–5667. doi:10.1210/jc.2002-020635.
Motoshima, H., X. Wu, K. Mahadev, and B.J. Goldstein. 2004. Adiponectin suppresses proliferation and superoxide generation and enhances eNOS activity in endothelial cells treated with oxidized LDL. Biochemical and Biophysical Research Communications 315: 264–271. doi:10.1016/j.bbrc.2004.01.049.
Mullen, K.L., A.C. Smith, K.A. Junkin, and D.J. Dyck. 2007. Globular adiponectin resistance develops independently of impaired insulin-stimulated glucose transport in soleus muscle from high-fat-fed rats. American Journal of Physiology. Endocrinology and Metabolism 293: E83–E90. doi:10.1152/ajpendo.00545.2006.
Mullen, K.L., J. Pritchard, I. Ritchie, L.A. Snook, A. Chabowski, A. Bonen, D. Wright, and D.J. Dyck. 2009. Adiponectin resistance precedes the accumulation of skeletal muscle lipids and insulin resistance in high-fat-fed rats. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 296: R243–R251. doi:10.1152/ajpregu.90774.2008.
Netzer, N., H. Gatterer, M. Faulhaber, M. Burtscher, S. Pramsohler, and D. Pesta. 2015. Hypoxia, oxidative stress and fat. Biomolecules 5: 1143–1150. doi:10.3390/biom5021143.
Ohashi, K., J.L. Parker, N. Ouchi, A. Higuchi, J.A. Vita, N. Gokce, A.A. Pedersen, C. Kalthoff, S. Tullin, A. Sams, R. Summer, and K. Walsh. 2010. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. The Journal of Biological Chemistry 285: 6153–6160. doi:10.1074/jbc.M109.088708.
Okada-Iwabu, M., T. Yamauchi, M. Iwabu, T. Honma, K. Hamagami, K. Matsuda, M. Yamaguchi, H. Tanabe, T. Kimura-Someya, M. Shirouzu, H. Ogata, K. Tokuyama, K. Ueki, T. Nagano, A. Tanaka, S. Yokoyama, and T. Kadowaki. 2013. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 503: 493–499. doi:10.1038/nature12656.
Ouchi, N., S. Kihara, Y. Arita, K. Maeda, H. Kuriyama, Y. Okamoto, K. Hotta, M. Nishida, M. Takahashi, T. Nakamura, S. Yamashita, T. Funahashi, and Y. Matsuzawa. 1999. Novel modulator for endothelial adhesion molecules: Adipocyte-derived plasma protein adiponectin. Circulation 100: 2473–2476.
Ouchi, N., H. Kobayashi, S. Kihara, M. Kumada, K. Sato, T. Inoue, T. Funahashi, and K. Walsh. 2004. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. The Journal of Biological Chemistry 279: 1304–1309. doi:10.1074/jbc.M310389200.
Ouedraogo, R., X. Wu, S.-Q. Xu, L. Fuchsel, H. Motoshima, K. Mahadev, K. Hough, R. Scalia, and B.J. Goldstein. 2006. Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells: Evidence for involvement of a cAMP signaling pathway. Diabetes 55: 1840–1846. doi:10.2337/db05-1174.
Ozen, G., A. Daci, X. Norel, and G. Topal. 2015. Human perivascular adipose tissue dysfunction as a cause of vascular disease: Focus on vascular tone and wall remodeling. European Journal of Pharmacology 766: 16–24. doi:10.1016/j.ejphar.2015.09.012.
Pajvani, U.B., X. Du, T.P. Combs, A.H. Berg, M.W. Rajala, T. Schulthess, J. Engel, M. Brownlee, and P.E. Scherer. 2003. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. The Journal of Biological Chemistry 278: 9073–9085. doi:10.1074/jbc.M207198200.
Park, S., S.-Y. Oh, M.-Y. Lee, S. Yoon, K.-S. Kim, and J. Kim. 2004. CCAAT/enhancer binding protein and nuclear factor-Y regulate adiponectin gene expression in adipose tissue. Diabetes 53: 2757–2766.
Park, H.J., Y.M. Kang, C.H. Kim, and M.H. Jung. 2010. ATF3 negatively regulates adiponectin receptor 1 expression. Biochemical and Biophysical Research Communications 400: 72–77. doi:10.1016/j.bbrc.2010.08.011.
Park, M., A. Sabetski, Y. Kwan Chan, S. Turdi, and G. Sweeney. 2015. Palmitate induces ER stress and autophagy in H9c2 cells: Implications for apoptosis and adiponectin resistance. Journal of Cellular Physiology 230: 630–639. doi:10.1002/jcp.24781.
Parker-Duffen, J.L., K. Nakamura, M. Silver, M.A. Zuriaga, S. MacLauchlan, T.R. Aprahamian, and K. Walsh. 2014. Divergent roles for adiponectin receptor 1 (AdipoR1) and AdipoR2 in mediating revascularization and metabolic dysfunction in vivo. The Journal of Biological Chemistry 289: 16200–16213. doi:10.1074/jbc.M114.548115.
Patel, S.A., K.L. Hoehn, R.T. Lawrence, L. Sawbridge, N.A. Talbot, J.L. Tomsig, N. Turner, G.J. Cooney, J.P. Whitehead, E.W. Kraegen, and M.E. Cleasby. 2012. Overexpression of the adiponectin receptor AdipoR1 in rat skeletal muscle amplifies local insulin sensitivity. Endocrinology 153: 5231–5246. doi:10.1210/en.2012-1368.
Peti, A., A. Juhasz, P. Kenyeres, Z. Varga, I. Seres, G.L. Kovacs, G. Paragh, and L. Bajnok. 2011. Relationship of adipokines and non-esterified fatty acid to the insulin resistance in non-diabetic individuals. Journal of Endocrinological Investigation 34: 21–25. doi:10.1007/BF03346690.
Qiao, L., B. Kinney, H.S. Yoo, B. Lee, J. Schaack, and J. Shao. 2012. Adiponectin increases skeletal muscle mitochondrial biogenesis by suppressing mitogen-activated protein kinase phosphatase-1. Diabetes 61: 1463–1470. doi:10.2337/db11-1475.
Rasmussen, M.S., A.S. Lihn, S.B. Pedersen, J.M. Bruun, M. Rasmussen, and B. Richelsen. 2006. Adiponectin receptors in human adipose tissue: Effects of obesity, weight loss, and fat depots. Obesity (Silver Spring) 14: 28–35. doi:10.1038/oby.2006.5.
Ross, R. 1993. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 362: 801–809. doi:10.1038/362801a0.
Ruan, H., and L.Q. Dong. 2016. Adiponectin signaling and function in insulin target tissues. Journal of Molecular Cell Biology 8: 101–109. doi:10.1093/jmcb/mjw014.
Ryo, M., T. Nakamura, S. Kihara, M. Kumada, S. Shibazaki, M. Takahashi, M. Nagai, Y. Matsuzawa, and T. Funahashi. 2004. Adiponectin as a biomarker of the metabolic syndrome. Circulation Journal 68: 975–981.
Ryu, J., A.K. Galan, X. Xin, F. Dong, M.A. Abdul-Ghani, L. Zhou, C. Wang, C. Li, B.M. Holmes, L.B. Sloane, S.N. Austad, S. Guo, N. Musi, R.A. DeFronzo, C. Deng, M.F. White, F. Liu, and L.Q. Dong. 2014. APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Reports 7: 1227–1238. doi:10.1016/j.celrep.2014.04.006.
Saito, Y., D. Fujioka, K. Kawabata, T. Kobayashi, T. Yano, T. Nakamura, Y. Kodama, H. Takano, Y. Kitta, J. Obata, and K. Kugiyama. 2007a. Statin reverses reduction of adiponectin receptor expression in infarcted heart and in TNF-alpha-treated cardiomyocytes in association with improved glucose uptake. American Journal of Physiology. Heart and Circulatory Physiology 293: H3490–H3497. doi:10.1152/ajpheart.00310.2007.
Saito, T., C.C. Jones, S. Huang, M.P. Czech, and P.F. Pilch. 2007b. The interaction of Akt with APPL1 is required for insulin-stimulated Glut4 translocation. The Journal of Biological Chemistry 282: 32280–32287. doi:10.1074/jbc.M704150200.
Salman, A., M. Hegazy, and S. AbdElfadl. 2015. Combined adiponectin deficiency and resistance in obese patients: Can it solve part of the puzzle in nonalcoholic steatohepatitis. Open Access Macedonian Journal of Medical Sciences 3: 298–302. doi:10.3889/oamjms.2015.057.
Scherer, P.E., S. Williams, M. Fogliano, G. Baldini, and H.F. Lodish. 1995. A novel serum protein similar to C1q, produced exclusively in adipocytes. The Journal of Biological Chemistry 270: 26746–26749.
Schmid, P.M., M. Resch, A. Steege, S. Fredersdorf-Hahn, B. Stoelcker, C. Birner, C. Schach, C. Buechler, G.A.J. Riegger, A. Luchner, and D.H. Endemann. 2011. Globular and full-length adiponectin induce NO-dependent vasodilation in resistance arteries of Zucker lean but not Zucker diabetic fatty rats. American Journal of Hypertension 24: 270–277. doi:10.1038/ajh.2010.239.
Semple, R.K., M.A. Soos, J. Luan, C.S. Mitchell, J.C. Wilson, M. Gurnell, E.K. Cochran, P. Gorden, V.K.K. Chatterjee, N.J. Wareham, and S. O’Rahilly. 2006. Elevated plasma adiponectin in humans with genetically defective insulin receptors. The Journal of Clinical Endocrinology and Metabolism 91: 3219–3223. doi:10.1210/jc.2006-0166.
Semple, R.K., N.H. Halberg, K. Burling, M.A. Soos, T. Schraw, J. Luan, E.K. Cochran, D.B. Dunger, N.J. Wareham, P.E. Scherer, P. Gorden, and S. O’Rahilly. 2007. Paradoxical elevation of high-molecular weight adiponectin in acquired extreme insulin resistance due to insulin receptor antibodies. Diabetes 56: 1712–1717. doi:10.2337/db06-1665.
Sente, T., A.M. Van Berendoncks, V.Y. Hoymans, and C.J. Vrints. 2016. Adiponectin resistance in skeletal muscle: Pathophysiological implications in chronic heart failure. Journal of Cachexia, Sarcopenia and Muscle 7: 261–274. doi:10.1002/jcsm.12086.
Shi, X., W. Shi, Q. Li, B. Song, M. Wan, S. Bai, and X. Cao. 2003. A glucocorticoid-induced leucine-zipper protein, GILZ, inhibits adipogenesis of mesenchymal cells. EMBO Reports 4: 374–380. doi:10.1038/sj.embor.embor805.
Shibata, R., N. Ouchi, and T. Murohara. 2009. Adiponectin and cardiovascular disease. Circulation Journal 73: 608–614.
Shimizu, T., M. Yamakuchi, K.K. Biswas, B. Aryal, S. Yamada, T. Hashiguchi, and I. Maruyama. 2016. HMGB1 is secreted by 3 T3-L1 adipocytes through JNK signaling and the secretion is partially inhibited by adiponectin. Obesity (Silver Spring) 24: 1913–1921. doi:10.1002/oby.21549.
Singh, P., P. Sharma, K.R. Sahakyan, D.E. Davison, F.H. Sert-Kuniyoshi, A. Romero-Corral, J.M. Swain, M.D. Jensen, F. Lopez-Jimenez, T. Kara, and V.K. Somers. 2016. Differential effects of leptin on adiponectin expression with weight gain versus obesity. International Journal of Obesity 2005(40): 266–274. doi:10.1038/ijo.2015.181.
Stanley, W.C., F.A. Recchia, and G.D. Lopaschuk. 2005. Myocardial substrate metabolism in the normal and failing heart. Physiological Reviews 85: 1093–1129. doi:10.1152/physrev.00006.2004.
Steinberg, G.R., A.J. McAinch, M.B. Chen, P.E. O’Brien, J.B. Dixon, D. Cameron-Smith, and B.E. Kemp. 2006. The suppressor of cytokine signaling 3 inhibits leptin activation of AMP-kinase in cultured skeletal muscle of obese humans. The Journal of Clinical Endocrinology and Metabolism 91: 3592–3597. doi:10.1210/jc.2006-0638.
Subauste, A.R., and C.F. Burant. 2007. Role of FoxO1 in FFA-induced oxidative stress in adipocytes. American Journal of Physiology. Endocrinology and Metabolism 293: E159–E164. doi:10.1152/ajpendo.00629.2006.
Tanabe, H., Y. Fujii, M. Okada-Iwabu, M. Iwabu, Y. Nakamura, T. Hosaka, K. Motoyama, M. Ikeda, M. Wakiyama, T. Terada, N. Ohsawa, M. Hato, S. Ogasawara, T. Hino, T. Murata, S. Iwata, K. Hirata, Y. Kawano, M. Yamamoto, T. Kimura-Someya, M. Shirouzu, T. Yamauchi, T. Kadowaki, and S. Yokoyama. 2015. Crystal structures of the human adiponectin receptors. Nature 520: 312–316. doi:10.1038/nature14301.
Tao, L., E. Gao, X. Jiao, Y. Yuan, S. Li, T.A. Christopher, B.L. Lopez, W. Koch, L. Chan, B.J. Goldstein, and X.L. Ma. 2007. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation 115: 1408–1416. doi:10.1161/CIRCULATIONAHA.106.666941.
Tomas, E., T.-S. Tsao, A.K. Saha, H.E. Murrey, Zhang Cc Cc, S.I. Itani, H.F. Lodish, and N.B. Ruderman. 2002. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: Acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proceedings of the National Academy of Sciences of the United States of America 99: 16309–16313. doi:10.1073/pnas.222657499.
Tsao, T.-S., E. Tomas, H.E. Murrey, C. Hug, D.H. Lee, N.B. Ruderman, J.E. Heuser, and H.F. Lodish. 2003. Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity. Different oligomers activate different signal transduction pathways. The Journal of Biological Chemistry 278: 50810–50817. doi:10.1074/jbc.M309469200.
Tsuchida, A., T. Yamauchi, Y. Ito, Y. Hada, T. Maki, S. Takekawa, J. Kamon, M. Kobayashi, R. Suzuki, K. Hara, N. Kubota, Y. Terauchi, P. Froguel, J. Nakae, M. Kasuga, D. Accili, K. Tobe, K. Ueki, R. Nagai, and T. Kadowaki. 2004. Insulin/Foxo1 pathway regulates expression levels of adiponectin receptors and adiponectin sensitivity. The Journal of Biological Chemistry 279: 30817–30822. doi:10.1074/jbc.M402367200.
Tsuchida, A., T. Yamauchi, S. Takekawa, Y. Hada, Y. Ito, T. Maki, and T. Kadowaki. 2005. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: Comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes 54: 3358–3370.
Unger, R.H. 2002. Lipotoxic diseases. Annual Review of Medicine 53: 319–336. doi:10.1146/annurev.med.53.082901.104057.
Unger, R.H., P.E. Scherer, and W.L. Holland. 2013. Dichotomous roles of leptin and adiponectin as enforcers against lipotoxicity during feast and famine. Molecular Biology of the Cell 24: 3011–3015. doi:10.1091/mbc.E12-10-0774.
van Stijn, C.M.W., J. Kim, A.J. Lusis, G.D. Barish, and R.K. Tangirala. 2015. Macrophage polarization phenotype regulates adiponectin receptor expression and adiponectin anti-inflammatory response. The FASEB Journal 29: 636–649. doi:10.1096/fj.14-253831.
Vilarrasa, N., J. Vendrell, R. Sánchez-Santos, M. Broch, A. Megia, C. Masdevall, N. Gomez, J. Soler, J. Pujol, C. Bettónica, H. Aranda, and J.M. Gómez. 2007. Effect of weight loss induced by gastric bypass on proinflammatory interleukin-18, soluble tumour necrosis factor-alpha receptors, C-reactive protein and adiponectin in morbidly obese patients. Clinical Endocrinology 67: 679–686. doi:10.1111/j.1365-2265.2007.02945.x.
Viollet, B., R. Mounier, J. Leclerc, A. Yazigi, M. Foretz, and F. Andreelli. 2007. Targeting AMP-activated protein kinase as a novel therapeutic approach for the treatment of metabolic disorders. Diabetes & Metabolism 33: 395–402. doi:10.1016/j.diabet.2007.10.004.
Waki, H., T. Yamauchi, J. Kamon, Y. Ito, S. Uchida, S. Kita, K. Hara, Y. Hada, F. Vasseur, P. Froguel, S. Kimura, R. Nagai, and T. Kadowaki. 2003. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. The Journal of Biological Chemistry 278: 40352–40363. doi:10.1074/jbc.M300365200.
Walton, R.G., B. Zhu, R. Unal, M. Spencer, M. Sunkara, A.J. Morris, R. Charnigo, W.S. Katz, A. Daugherty, D.A. Howatt, P.A. Kern, and B.S. Finlin. 2015. Increasing adipocyte lipoprotein lipase improves glucose metabolism in high fat diet-induced obesity. The Journal of Biological Chemistry 290: 11547–11556. doi:10.1074/jbc.M114.628487.
Wang, B., Y. Yu, and L. Han. 2012. Adiponectin improves endothelial dysfunction caused by elevated FFAs levels, partially through cAMP-dependent pathway. Diabetes Research and Clinical Practice 97: 119–124. doi:10.1016/j.diabres.2012.02.009.
Wang, Y., X. Wang, W.B. Lau, Y. Yuan, D. Booth, J.-J. Li, R. Scalia, K. Preston, E. Gao, W. Koch, and X.-L. Ma. 2014. Adiponectin inhibits tumor necrosis factor-α-induced vascular inflammatory response via caveolin-mediated ceramidase recruitment and activation. Circulation Research 114: 792–805. doi:10.1161/CIRCRESAHA.114.302439.
Weisberg, S.P., D. Hunter, R. Huber, J. Lemieux, S. Slaymaker, K. Vaddi, I. Charo, R.L. Leibel, and A.W. Ferrante. 2006. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. The Journal of Clinical Investigation 116: 115–124. doi:10.1172/JCI24335.
Weyer, C., T. Funahashi, S. Tanaka, K. Hotta, Y. Matsuzawa, R.E. Pratley, and P.A. Tataranni. 2001. Hypoadiponectinemia in obesity and type 2 diabetes: Close association with insulin resistance and hyperinsulinemia. The Journal of Clinical Endocrinology and Metabolism 86: 1930–1935. doi:10.1210/jcem.86.5.7463.
Whitehead, J.P., A.A. Richards, I.J. Hickman, G.A. Macdonald, and J.B. Prins. 2006. Adiponectin—A key adipokine in the metabolic syndrome. Diabetes, Obesity and Metabolism 8: 264–280. doi:10.1111/j.1463-1326.2005.00510.x.
Wree, A., A. Kahraman, G. Gerken, and A. Canbay. 2011. Obesity affects the liver—The link between adipocytes and hepatocytes. Digestion 83: 124–133. doi:10.1159/000318741.
Xi, L., Z. Qian, G. Xu, C. Zhou, and S. Sun. 2007. Crocetin attenuates palmitate-induced insulin insensitivity and disordered tumor necrosis factor-alpha and adiponectin expression in rat adipocytes. British Journal of Pharmacology 151: 610–617. doi:10.1038/sj.bjp.0707276.
Xiao, X., Y. Dong, J. Zhong, R. Cao, X. Zhao, G. Wen, and J. Liu. 2011. Adiponectin protects endothelial cells from the damages induced by the intermittent high level of glucose. Endocrine 40: 386–393. doi:10.1007/s12020-011-9531-9.
Yamauchi, T., and T. Kadowaki. 2008. Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. International Journal of Obesity 2005(32 Suppl 7): S13–S18. doi:10.1038/ijo.2008.233.
Yamauchi, T., Y. Nio, T. Maki, M. Kobayashi, T. Takazawa, M. Iwabu, M. Okada-Iwabu, S. Kawamoto, N. Kubota, T. Kubota, Y. Ito, J. Kamon, A. Tsuchida, K. Kumagai, H. Kozono, Y. Hada, H. Ogata, K. Tokuyama, M. Tsunoda, T. Ide, K. Murakami, M. Awazawa, I. Takamoto, P. Froguel, K. Hara, K. Tobe, R. Nagai, K. Ueki, and T. Kadowaki. 2007. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nature Medicine 13: 332–339. doi:10.1038/nm1557.
Yamauchi, T., M. Iwabu, M. Okada-Iwabu, and T. Kadowaki. 2014. Adiponectin receptors: A review of their structure, function and how they work. Best Practice & Research. Clinical Endocrinology & Metabolism 28: 15–23. doi:10.1016/j.beem.2013.09.003.
Ye, R., and P.E. Scherer. 2013. Adiponectin, driver or passenger on the road to insulin sensitivity? Molecular Metabolism 2: 133–141. doi:10.1016/j.molmet.2013.04.001.
Zhang, Y., M. Matheny, S. Zolotukhin, N. Tumer, and P.J. Scarpace. 2002. Regulation of adiponectin and leptin gene expression in white and brown adipose tissues: influence of beta3-adrenergic agonists, retinoic acid, leptin and fasting. Biochimica et Biophysica Acta 1584: 115–122.
Zhang, P., Y. Wang, Y. Fan, Z. Tang, and N. Wang. 2009. Overexpression of adiponectin receptors potentiates the antiinflammatory action of subeffective dose of globular adiponectin in vascular endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology 29: 67–74. doi:10.1161/ATVBAHA.108.178061.
Zhao, L., Z. Fu, J. Wu, K.W. Aylor, E.J. Barrett, W. Cao, and Z. Liu. 2015. Globular adiponectin ameliorates metabolic insulin resistance via AMPK-mediated restoration of microvascular insulin responses. The Journal of Physiology 593: 4067–4079. doi:10.1113/JP270371.
Zhou, M., A. Xu, K.S.L. Lam, P.K.H. Tam, C.-M. Che, L. Chan, I.-K. Lee, D. Wu, and Y. Wang. 2010. Rosiglitazone promotes fatty acyl CoA accumulation and excessive glycogen storage in livers of mice without adiponectin. Journal of Hepatology 53: 1108–1116. doi:10.1016/j.jhep.2010.05.034.
Zoico, E., U. Garbin, D. Olioso, G. Mazzali, A.M. Fratta Pasini, V. Di Francesco, A. Sepe, L. Cominacini, and M. Zamboni. 2009. The effects of adiponectin on interleukin-6 and MCP-1 secretion in lipopolysaccharide-treated 3 T3-L1 adipocytes: Role of the NF-kappaB pathway. International Journal of Molecular Medicine 24: 847–851.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Engin, A. (2017). Adiponectin-Resistance in Obesity. In: Engin, A., Engin, A. (eds) Obesity and Lipotoxicity. Advances in Experimental Medicine and Biology, vol 960. Springer, Cham. https://doi.org/10.1007/978-3-319-48382-5_18
Download citation
DOI: https://doi.org/10.1007/978-3-319-48382-5_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48380-1
Online ISBN: 978-3-319-48382-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)