
Abstract
Obesity is characterized by the chronic low-grade activation of the innate immune system. In this respect, macrophage-elicited metabolic inflammation and adipocyte-macrophage interaction has a primary importance in obesity. Large amounts of macrophages are accumulated by different mechanisms in obese adipose tissue. Hypertrophic adipocyte-derived chemotactic monocyte chemoattractant protein-1 (MCP-1)/C-C chemokine receptor 2 (CCR2) pathway also promotes more macrophage accumulation into the obese adipose tissue. However, increased local extracellular lipid concentrations is a final mechanism for adipose tissue macrophage accumulation. A paracrine loop involving free fatty acids and tumor necrosis factor-alpha (TNF-alpha) between adipocytes and macrophages establishes a vicious cycle that aggravates inflammatory changes in the adipose tissue. Adipocyte-specific caspase-1 and production of interleukin-1beta (IL-1beta) by macrophages; both adipocyte and macrophage induction by toll like receptor-4 (TLR4) through nuclear factor-kappaB (NF-kappaB) activation; free fatty acid-induced and TLR-mediated activation of c-Jun N-terminal kinase (JNK)-related pro-inflammatory pathways in CD11c+ immune cells; are effective in macrophage accumulation and in the development of adipose tissue inflammation. Old adipocytes are removed by macrophages through trogocytosis or sending an “eat me” signal. The obesity-induced changes in adipose tissue macrophage numbers are mainly due to increases in the triple-positive CD11b+ F4/80+ CD11c+ adipose tissue macrophage subpopulation. The ratio of M1-to-M2 macrophages is increased in obesity. Furthermore, hypoxia along with higher concentrations of free fatty acids exacerbates macrophage-mediated inflammation in obesity. The metabolic status of adipocytes is a major determinant of macrophage inflammatory output. Macrophage/adipocyte fatty-acid-binding proteins act at the interface of metabolic and inflammatory pathways. Both macrophages and adipocytes are the sites for active lipid metabolism and signaling.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Obesity
- Monocyte chemoattractant protein-1 (MCP-1)
- M1 macrophages
- M2 macrophages
- Visceral adipose tissue
- Free fatty acids
- Interleukin-6 (IL-6)
- Tumor necrosis factor-alpha (TNF-alpha)
- NOD-like receptor (NLR) family protein (NLRP3)
- C-C chemokine receptor 2 (CCR2)
- Toll like receptor 4 (TLR4)
- Chemokine (C-C motif) ligand 2 (CCL2)
- Insulin-like growth factor-1 (IGF1)
- Hypoxia-inducible factor-1 alpha (HIF-1alpha)
1 Introduction
The chronic low-grade activation of the innate immune system is evident even in childhood obesity . Thus, inflammatory markers are elevated in obese children as young as 3 years old. Furthermore, individuals are at risk for lifelong meta-inflammation (Skinner et al. 2010). Adipose tissue macrophage accumulation is directly proportional to adiposity in humans. Furthermore, adipocyte size is a strong predictor of the percentage of CD68-expressing macrophages in human subcutaneous adipose tissue . (Weisberg et al. 2003). The ratio of the macrophages is 5% in lean adipose tissue, whereas, during obesity this ratio rises up to 50% (Cinti et al. 2005). Although macrophages comprise 10–15% of stromal vascular cells (SVCs) in visceral adipose tissue of lean subjects, their numbers are increased to 40–50% of the SVCs of visceral adipose tissue in obese humans (Weisberg et al. 2003). During obesity immune cell population differs, not only in number, but also in inflammatory phenotypes . In this context, macrophage-elicited metabolic inflammation and adipocyte-macrophage interaction has the key importance in obesity (McNelis and Olefsky 2014).
2 Macrophage Infiltration into Adipose Tissue
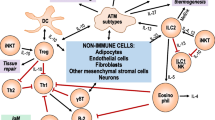
Actually the initiation of macrophage infiltration into adipose tissue occurs with four different mechanisms (Sun et al. 2011). First of all, necrosis of adipocytes driven by hypertrophy is a prominent phagocytic stimulus that regulates adipose tissue macrophage infiltration which is gradually increased by obesity (Cinti et al. 2005). In the late stages of adipose tissue obesity, macrophages form crown-like structures surrounding necrotic adipocytes (Nishimura et al. 2007; Strissel et al. 2007). In this way, white adipose tissue macrophages are selectively localized to dead adipocytes. Clearance of free lipid appears to be an important function of galectin-3 (MAC-2)-expressing macrophages in white adipose tissue (Cinti et al. 2005). Galectin-3 is one of the major factors involved in the influx of macrophages to inflammatory sites (Sano et al. 2000). At first, galectin-3 expression at sites of adipocyte necrosis may be functionally linked to macrophage aggregation and crown-like structures formation (Cinti et al. 2005). Furthermore, galectin-3 possesses anti-apoptotic activity intracellularly and that is implicated in macrophage survival at sites of inflammation (Hsu and Liu 2004). Secondly, hypertrophic adipocyte-derived chemotactic monocyte chemoattractant protein-1 (MCP-1) /CC chemokine receptor 2 (CCR2) pathway also promotes more macrophage accumulation into the obese adipose tissue. Secretion of MCP-1 from adipocytes directly triggers the recruitment of macrophages to adipose tissue. The infiltrated macrophages may in turn secrete a variety of chemokines and other cytokines that further promote a local inflammatory response and affect gene expression in adipocytes, resulting in systemic insulin resistance (Kanda et al. 2006). Adiponectin expression may occur indirectly through interactions of adipocytes with CCR2-expressing adipose tissue macrophages and the alterations of obesity-induced inflammatory gene expression (Weisberg et al. 2006) (Fig. 14.1).

Macrophage-elicited metabolic inflammation and adipocyte-macrophage interaction has a primary importance in obesity . Large amounts of macrophages are accumulated by different mechanisms in obese adipose tissue (TLR4 Toll-like receptor 4, Akt Protein kinase-B, FOXO1 Forkhead box protein O1, NF-kappaB Nuclear factor-kappa B, JNK c-Jun N-terminal kinase, IKK Inhibitor kappa B kinase, PPAR-ɣ Peroxisome proliferator-activated receptor-gamma, IRS Insulin receptor substrate, MCP-1 Monocyte chemoattractant protein-1, MCSF Macrophage colony stimulating factor, CCL2: CCR2 Chemokine (C-C motif) receptor ligand 2, IRF5 Interferon regulatory factor-5, TNF-α Tumor necrosis factor-alpha, IL-1β Interleukin-1 beta, IL-6Interleukin-6, NLRP3, NLRP NOD-like receptor (NLR) family protein; PI3K Phosphatidylinositol 3-kinase, mtROS Mitochondrial reactive oxygen species, iNOS Inducible nitric oxide synthase, Mincle mRNA Macrophage-inducible C-type lectin mRNA, HIF-1 hypoxia-inducible factor-1, RAGE Receptor for advanced glycation end product, AGE Advanced glycation end product, TNFR Tumor necrosis factor receptor)
The inhibitor of differentiation or inhibitor of DNA binding (Id) proteins are negative regulators of cell differentiation and play key roles in the regulation of cell fate decisions, and in the timing of differentiation (Norton 2000). There are several type of Id proteins. In particular, Id3 attenuates visceral fat expansion by inhibiting high fat diet -induced visceral fat vascular endothelial growth factor (VEGF), and regulated upon activation normal T-cell expressed and secreted (RANTES) expression. Eventually, increase in capillary density is restricted (Cutchins et al. 2012). The helix-loop-helix transcription regulator Id3 promotes high fat diet -induced adipocyte progenitor cell (AdPC) accumulation in visceral adipose tissue . Id3-dependent AdPC expansion is responsible for high fat diet-induced MCP-1 production, M1 macrophage accumulation, and metabolic dysfunction during obesity . Adipose depot-specific differences in inflammation may be due to depot-specific differences in AdPCs (Kaplan et al. 2015). Hence MCP-1 expression is up-regulated in white adipose tissue of high fat diet-induced obesity. The 7.2-fold increase in MCP-1 as compared with normal subjects may alter adipocyte function by decreasing insulin-stimulated glucose uptake and the expression of lipoprotein lipase , adipsin, glucose transporter type 4 (GLUT-4), fatty-acid-binding protein (aP2), beta3-adrenergic receptor, and peroxisome proliferator-activated receptor-gamma (PPAR-gamma) (Sartipy and Loskutoff 2003; Takahashi et al. 2003). Furthermore, adipocyte hypertrophy creates poorly oxygenated areas in the human obese adipose tissue. As the third mechanism, hypoxic cells secrete chemokines, which attract macrophages, presumably to clear out necrotic cells (Murdoch et al. 2004). Eventually, decreased adipose tissue partial oxygen concentration is paralleled by an increase in the expression and secretion of the chemokine and markers of macrophage infiltration (Pasarica et al. 2009). Hypoxia -induced fibrosis in adipose tissue is a key factor that ultimately stimulates the local inflammatory responses (Halberg et al. 2009). Moreover, increased local extracellular lipid concentrations, which drive adipose tissue macrophage accumulation occurs as a final mechanism for macrophage infiltration. Thus the majority of free fatty acids released from necrotic adipocytes are transported across the plasma membrane of adjacent adipocytes and locally re-esterified (Thompson et al. 2010). In this context, free fatty acids are esterified with glycerol 3-phosphate. Stimulation of glyceroneogenesis and re-esterification of free fatty acids by increasing in phosphoenolpyruvate carboxykinase (PEPCK) activity leads to ultimate rise in adipocyte size and body weight (Franckhauser et al. 2002). The close relationship between adipocyte size and the abundance of macrophages in adipose tissue indicates that the relationship of adipocyte size with adipocyte function may be regulated through a paracrine pathway involving adipose tissue macrophages (Weisberg et al. 2003).
A paracrine loop involving free fatty acids and tumor necrosis factor-alpha (TNF-alpha) between adipocytes and macrophages establishes a vicious cycle that aggravates inflammatory changes in the adipose tissue. Initially TNF-alpha increases the release of free fatty acids from adipocytes. By contrast, high amount of saturated fatty acids which are released from adipocytes can induce inflammatory changes via increasing TNF-alpha production (Suganami et al. 2005). TNF-alpha-induced activation of caspase-1 is accompanied by induction of NOD-like receptor (NLR) family protein-3 (NLRP3) in adipocytes. But caspase-1 activation does not depend on the NLRP3 inflammasome (Furuoka et al. 2016). Caspase-1 activation in adipose tissue of obese animals is partly independent of macrophage infiltration. However, caspase-1 induces the release of IL-1beta, and finally leads to the development of insulin resistance . These results indicate a crosstalk between adipocyte-specific caspase-1 and IL-1beta produced by macrophages in adipose tissue (Stienstra et al. 2010). Additionally, saturated fatty acid also serves as a ligand for toll like receptor-4 (TLR4) . Thereby the inflammatory changes in both adipocytes and macrophages are induced by TLR4 through nuclear factor-kappaB (NF-kappaB) activation (Suganami et al. 2007). Free fatty acid-induced and TLR-mediated activation of c-Jun N-terminal kinase (JNK) -related pro-inflammatory pathways in CD11c+ immune cells plays a central role in the development of adipose tissue inflammation and insulin resistance (Nguyen et al. 2007). Although the adipocyte is the key player controlling local changes in the microenvironment, macrophages have pivotal role in remodeling events. Nevertheless, resident macrophages display remarkable heterogeneity in their activities and functions (Gordon and Taylor 2005) (Fig. 14.1).
On the other hand, adipocyte-derived microparticles (MPs) are critical "find-me" signals for recruitment of monocytes and macrophages. Adipocytes exposed to saturated fatty acids show marked release of MPs, which are enriched in perilipin A. The release of MPs is highly dependent on caspase 3 and Rho-associated kinase. Thus hypertrophied and stressed adipocytes generate chemotactic signals that induce macrophage migration in a caspase 3 dependent manner (Eguchi et al. 2015). White adipose tissue is characterized by a continuous turnover of the adipocytes with approximately 10% of annual renewal (Spalding et al. 2008). In this context, old adipocytes undergoing programmed cell death are removed by macrophages (Duvall et al. 1985; Keuper et al. 2011). Removal of apoptotic adipocytes by macrophages is a process in which one cell takes bites out of another (trogocytosis). Therefore, adipocyte-derived DNA is not detected in the phagocytes (Sárvári et al. 2015). Apoptotic cells send an “eat me” signal to macrophages, triggering their own engulfment. Among the various molecules proposed to be involved in this process, in particular phosphatidylserine (PtdSer) is a strong candidate for the “eat me” signal (Krahling et al. 1999). PtdSer is transferred caspase-dependently from the inner leaflet to the outer leaflet of the plasma membrane (Martin et al. 1996). The specific signals of apoptotic cells are well recognized by the phagocytes. Thus milk fat globule-EGF-factor 8 (MFG-E8)-secreted active macrophages recognize aminophospholipids and specifically bind to apoptotic cells for engulfment (Hanayama et al. 2002). Actually the glycerophospholipid, PtdSer is perhaps the best-characterized “eat me” signal. In healthy cells, PtdSer is abundant in the inner leaflet of the plasmalemma, but during apoptosis it is quickly redistributed and exposed on the exofacial leaflet. It is recognized by specific phagocytic receptors such as T-cell immunoglobulin mucin receptor 4 (TIM4), which enhances engulfment (Miyanishi et al. 2007). Actually, MFG-E8 binds to the integrin alphavbeta3 complex in macrophages via the Rho and Rab family guaosine triphosphatases (GTPases) in MFG-E8’s EGF domain, hence bridging apoptotic cells and macrophages. TIM4 binds to PtdSer via the immunoglobulin-like domain in its extracellular region. The engulfment of apoptotic cells proceeds in two steps: TIM4 tethers apoptotic cells, and the integrin alphavbeta3 complex mediates engulfment in coordination with MFG-E8 (Toda et al. 2012). Thus, the expression of MFGE-8 and the alphavbeta5 integrin subunits are increased in adipose tissue of obese humans (Henegar et al. 2008).
3 Macrophage Phenotype in Adipose Tissue
When the visceral adipose tissue is excess, compared to subcutaneous adipose tissue , the absolute number of CD14+ adipose tissue macrophage is increased in visceral adipose tissue relative to subcutaneous adipose tissue. The absolute number of CD3+ T cells is increased in visceral adipose tissue relative to subcutaneous adipose tissue. An increase in adipose tissue macrophage predisposes to an M1 phenotype in visceral adipose tissue relative to subcutaneous adipose tissue in human obesity . Furthermore, the CD14-enriched cell population has a macrophage phenotype and is a dominant source of inflammatory cytokines in stromo-vascular cells (O’Rourke et al. 2009).
CD11c+ adipose tissue macrophages significantly increase in obesity . A novel F4/80+ CD11c+ population of macrophages in adipose tissue of obese mice is not found in lean mice (Lumeng et al. 2007a). CD11c is considered to be a classical cell-surface marker of dendritic cells (DCs) and F4/80 as substitute for function, have led to confusion on the exact contribution of DCs versus macrophages to tissue immunity in obesity. Indeed, the CD11c integrin can be expressed on macrophages. Therefore, the role of CD11c+ cells does not always reflect the role of DCs. Similarly, F4/80 is also expressed by eosinophils and neutrophils and is not a specific marker of macrophages (Hashimoto et al. 2011). However, in obese adipose tissue, the CD11c+ adipose tissue macrophages also express CD11b and F4/80 and are very different from the adipose tissue DCs, which express CD11c, but not CD11b. The obesity-induced changes in adipose tissue macrophage numbers are mainly due to increases in the triple-positive CD11b+ F4/80+ CD11c+ adipose tissue macrophage subpopulation (Lee and Lee 2014).
As mentioned above, two different populations of F4/80+CD11b+ macrophages are defined in adipose tissue; one of which also express CD11c. Obesity largely increases CD11c+ macrophage number in adipose tissue. F4/80+CD11b+CD11c+ cells are a specific population of adipose tissue macrophages recruited to adipose tissue upon high fat diet exposure (Patsouris et al. 2008). Moreover, F4/80 and CD11c are primarily regarded as cell surface markers of macrophages and dendritic cells, which are immune-phenotypically defined as F4/80+CD11c− and F4/80−CD11c+, respectively. In obese conditions, all these cells have not simultaneously completed their process of transitioning from a F4/80+CD11c− to a F4/80−CD11c+ state or cells that have transitioned from a CD11c− to CD11c+ phenotype. Some of them may show their original phenotypical characteristics and may have gradually acquired a CD11c+ phenotype (Nguyen et al. 2007). Adipose tissue macrophages from lean mice express many genes, which display characteristic of M2 or “alternatively activated” macrophages, including Ym1, arginase 1, and interleukin-10 (IL-10). Diet-induced obesity decreases expression of these genes in adipose tissue macrophages while increasing expression of genes such as those encoding TNF-alpha and inducible nitric oxide synthase (iNOS) that are characteristic of M1 or “classically activated” macrophages (Lumeng et al. 2007a). Indeed, TNF-alpha is a major macrophage-derived mediator of inflammation in adipocytes, whereas free fatty acids is an important adipocyte-derived mediators of inflammation in macrophages. TNF-alpha acts on TNF-alpha receptor of hypertrophied adipocytes. Briefly, a paracrine loop involving free fatty acids and TNF-alpha between adipocytes and macrophages establishes a vicious cycle that aggravates inflammatory changes in the adipose tissue (Suganami et al. 2005). Macrophages express some adipocyte-specific gene products such as ap2, meanwhile adipocytes secrete macrophage-specific gene products such as IL-6 or TNF-alpha. Lipid accumulation by macrophages in atherosclerotic lesions or by phagocytic capacities exhibited by adipocytes reveal an apparent coordinated activity between these two cell-types during the course of an innate immune response (Wolowczuk et al. 2008). An additional similarity between adipocytes and macrophages is further observed in the expression of TLR4 (Lin et al. 2000). The presence of functional TLR2 and TLR4 is demonstrated on human adipocytes isolated from subcutaneous fat tissue (Bès-Houtmann et al. 2007). “Adipocyte-macrophage-TLR4” pathway might be involved in the inflammatory process occurring in obesity (Wolowczuk et al. 2008). As mentioned above, diet-induced obesity is associated with the loss of tissue homeostasis and development of type 1 inflammatory responses in visceral adipose tissue . A key event is a shift of adipose tissue macrophages toward an M1 phenotype. In fact, obesity-induced adipocyte hypertrophy results in upregulated surface expression of stress markers. Adipose stress is detected by the phenotype of natural killer (NK) cells and CD8+ T cells, which produce interferon-gamma (IFN-gamma) , driving M1 macrophage polarization (Wensveen et al. 2015). The nuclear hormone receptor, PPAR-gamma is the first adipocyte-specific transcription factor, which is expressed in a highest level in adipose tissue (Chawla et al. 1994; Tontonoz et al. 1994). Further, PPAR-gamma is a critical signaling molecule in determining macrophage phenotype in adipose tissue (Charo 2007). The deficiency of PPAR gamma in immune cells favors expression of M1 and impairs M2 macrophage marker expression in adipose tissue (Bassaganya-Riera et al. 2009). Contrarily, PPAR-gamma activation induces monocyte differentiation into M2 macrophages (Bouhlel et al. 2007). The decreased PPAR-gamma protein level is associated with increased macrophage infiltration in visceral adipose tissue of obese subjects. In this case, pro-inflammatory macrophages suppress PPAR-gamma activity in adipocytes via S-nitrosylation (Yin et al. 2015). Actually, adipocytes are the source of Th2 cytokines. In response to adipocyte-derived Th2 cytokines , the expression of macrophage PPAR-beta/delta is up-regulated. Adipose tissue macrophage polarization ultimately could be modulated by adipocyte derived Th2 cytokines in a paracrine manner. PPAR-delta is induced by Th2 cytokines to control the transcriptional program of alternative activation in the macrophages. PPAR-delta may act as a factor for Th2 cytokine-induced M2 gene expression (Kang et al. 2008). In fact, obesity is accompanied by a transformation in the polarized states of macrophages from an anti-inflammatory “alternatively activated” M2 form (Kosteli et al. 2010), to a more pro-inflammatory “classically activated” M1 form (Lumeng et al. 2007a). The primary trigger for the recruitment of M1 macrophages is thought to be the secretion of TNF-alpha from hypertrophied adipocytes (Wellen and Hotamisligil 2003). The obesity-driven phenotypic adipose tissue macrophages can be characterized in these two broad classes based on the expression of particular antigens. Thus, diet-induced obesity leads to a shift in the activation state of adipose tissue macrophages from an M2-polarized state that may protect adipocytes from inflammation to an M1 pro-inflammatory state (Lumeng et al. 2007a).
TLR-dependent polarization mediators of M1 macrophages include different transcription factors such as NF-kappaB, activator protein-1 (AP-1), transcription factor PU.1 (PU.1), CCAAT/enhancer-binding protein alpha (C/EBP-alpha), signal transducer and activator of transcription 1 (STAT1) as well as interferon regulatory factor-5 (IRF5). AP-1 together with NF-kappaB in signal-dependent gene expression is necessary for innate immunity (Juhas et al. 2015). Among these transcription factors, IRF5 expression in macrophages is reversibly induced by inflammatory stimuli and contributes to the macrophage polarization . In this context, high expression of IRF5 is characteristic of M1 macrophages (Krausgruber et al. 2011). Thereby, IRF5 has been implicated in polarizing macrophages towards an inflammatory phenotype. In obese individuals, IRF5 expression is negatively associated with insulin sensitivity and collagen deposition in visceral adipose tissue (Dalmas et al. 2015), whereas another interferon regulatory factor , IRF4 is a key transcription factor that controls M2 macrophage polarization (Satoh et al. 2010). Consequently, differentiation of M1 macrophages is connected with the upregulation of IRF5 levels. The overexpression of IRF5 in M2 polarized macrophages leads to their phenotypic switch. It makes the M2 macrophages functionally similar to M1 (Juhas et al. 2015). However, Prieur et al. showed that early stages of adipose tissue expansion are characterized by M2-polarized adipose tissue macrophages and progressive lipid accumulation within adipose tissue macrophages (Prieur et al. 2011). The microenvironment in a lean adipose tissue is composed of a 4:1; M2:M1 ratio (Lumeng et al. 2008). Actually, a delicate balance of polarized populations of macrophages is necessary to maintain adequate adipocyte function. Inflammatory signals in adipose tissue of obese individuals is eliminated by retaining M2 polarization of adipose tissue macrophages or by triggering the phenotypic switch from M1 to M2 (Sun et al. 2011). Diet-induced obesity increases the number of M1 macrophages by 65-fold, whereas the number of M2 macrophages per weight basis is also increased by six-fold. Eventually, the ratio of M1-to-M2 macrophages is increased in obesity (Fujisaka et al. 2009). However, Fjeldborg et al. found a relatively higher expression of the anti-inflammatory markers, CD163 and IL-10, and a relative reduction of the pro-inflammatory markers, TNF-alpha and IL-6 , in adipose tissue from obese subjects compared to lean individuals. Thus, human adipose tissue macrophages change polarization to a more anti-inflammatory profile in obesity than towards a pro-inflammatory profile (Fjeldborg et al. 2014). There is a shift towards a M2 phenotype in non-crown like structure macrophages in adipose tissue from obese subjects compared with lean ones. Macrophages in crown like structure are predominantly M1, but most other macrophages, particularly those in fibrotic areas, are M2 (Spencer et al. 2010). At initial stage the expression of IL-10 by M2 macrophages is approximately 100-fold higher than that in adipocytes. Hence M2 macrophage-derived IL-10 constitutes the major portion of the IL-10 in adipose tissue. In this context, the phenotypic shift of adipose tissue macrophages toward an M1-dominant state cannot be explained simply by the conversion of M2 macrophages to M1. De novo accumulation of circulating monocytes to adipose tissue followed by their differentiation into M1 and M2 macrophages (Fujisaka et al. 2009). This situation can be explained as follows; first of all, transient accumulation of M2 macrophages could be essential for the control of tissue fatty acid levels during activation of lipolysis . M2 macrophage-borne signaling molecules could inhibit lipolysis and re-esterification of lipolyzed fatty acids form triacylglycerols (triacylglycerols/fatty acid cycle). Thus, M2 macrophages initially support the metabolically flexible adipocytes, which have a high capacity of both triacylglycerols/fatty acid cycling and oxidative phosphorylation in obese white adipose tissue (Masoodi et al. 2015). The M1 adipose tissue macrophage polarization in obesity is regarded as the optimization of the fat deposition and repartitioning toward adipocytes. It prevents adipose tissue macrophage lipotoxicity (Prieur et al. 2011). However, the expression of carnitine palmitoyltransferase 1A (CPT1A), the rate-limiting enzyme in mitochondrial fatty acid oxidation, is higher in human adipose tissue macrophages than in adipocytes. It is differentially expressed in visceral versus subcutaneous adipose tissue in obese individuals. Enhanced fatty acid oxidation in saturated fatty acid -incubated adipocytes and macrophages reduces triglyceride content and inflammation . While insulin sensitivity increases in adipocytes, endoplasmic reticulum (ER) stress and reactive oxygen species (ROS) damage decreases in macrophages (Malandrino et al. 2015). M2 macrophages sustain insulin sensitivity by secreting IL-4 and IL-10, while M1 macrophages induce insulin resistance through the secretion of pro-inflammatory cytokines, such as TNF-alpha (Tateya et al. 2013). M2 macrophages secrete anti-inflammatory cytokines and utilize oxidative metabolism to maintain adipose tissue homeostasis (Castoldi et al. 2015). M1 macrophages provide their energy from aerobic glycolysis. In this condition, glucose uptake as well as the conversion of pyruvate to lactate is increased. Meanwhile, the activities of the respiratory chain are attenuated, allowing for ROS production. However, M2 macrophages obtain much of their energy from fatty acid oxidation and oxidative metabolism (Galván-Peña and O’Neill 2014). In response to IL-4, STAT6 and PPARgamma-coactivator-1beta (PGC-1beta) induce the fatty acid oxidation and mitochondrial biogenesis in macrophages (Vats et al. 2006). IL-4 activates the transcriptional responses of STAT6. Active STAT6 can induce the coactivator PGC-1beta. PGC-1beta can induce mitochondrial respiration as well as mitochondrial biogenesis (Galván-Peña and O’Neill 2014; St-Pierre et al. 2003). In fact, PPAR-gamma is required for IL-4-induced increase in beta-oxidation of fatty acids. Despite IL-4 stimulation, the rate of fatty acid oxidation is reduced by approximately 70% in PPAR-gamma null macrophages. By contrast, increase in PPAR-gamma expression doubles the rate of beta-oxidation (Odegaard et al. 2007).
Progression of obesity increasingly induces a phenotypic switch from the M2 macrophages, to the M1 macrophages . Hence pro-inflammatory CD11c+ M1 macrophages is markers of insulin resistance in human obesity (Wentworth et al. 2010). Thus the enhanced macrophage-adipocyte crosstalk in obesity disrupts insulin action in adipocytes. Macrophage-secreted factors block insulin action in adipocytes via downregulation of GLUT4 and insulin receptor substrate-1 (IRS -1) , leading to a decrease in Akt phosphorylation and impaired insulin-stimulated GLUT4 translocation to the plasma membrane (Lumeng et al. 2007c). The secretion of inflammatory cytokines and chemokines by adipose tissue macrophages includes TNF-alpha , IL-6 , IL-1beta, MCP-1 , CCL2, and macrophage inhibitory factor (MIF) (Olefsky and Glass 2010). In particular IL-1beta is produced largely by macrophages. It provokes the development of obesity-associated insulin resistance by inhibition of insulin signal transduction in adipocytes. Contrarily, blocking the activity of IL-1beta improves insulin signaling in human adipocytes. This is in parallel with a reduction in macrophage-stimulated pro-inflammatory profile and lipolysis (Bing 2015). Thus the reduction in protein expression of IRS -1 , phosphatidylinositol 3-kinase (PI3K) p85alpha, and GLUT4 by macrophage-conditioned-medium is reversed when macrophage-derived IL-1beta production is inhibited. In contrast, macrophage-stimulated release of IL-6, IL-8, and chemokine (C-C motif) ligand 5 (RANTES) by adipocytes is also significantly reduced by blocking IL-1beta production (Gao et al. 2014). PGC-1beta plays a key role in saturated fatty acids metabolism and in the regulation of inflammatory signaling. PGC-1beta expression is significantly decreased in response to saturated fatty acid in macrophages in a dose dependent manner. PGC-1beta inhibits saturated fatty acid-induced TNF-alpha , MCP-1 , and IL-1beta mRNA and protein expressions. Furthermore, PGC-1beta significantly antagonizes saturated fatty acid-induced macrophage NF-kappaB-p65 and JNK activation. PGC-1beta overexpression in saturated fatty acid treated macrophages improves adipocytes PI3K-Akt insulin signaling in a paracrine fashion (Chen et al. 2016). In response to high-fat diet the tumor necrosis factor receptor (TNFR) -associated factor-3 (TRAF-3) , binds to and activates TGF-beta-kinase 1 (TAK1). Thereby the activation of downstream inhibitor of nuclear factor kappa-B kinase subunit beta (IKKbeta)–NF-κB and mitogen-activated protein kinase kinase (MAPKK)–JNK–IRS1 signaling cascades are enhanced, while disrupting AKT–glycogen synthase kinase 3 beta (GSK3beta)/forkhead box protein O1 (FOXO1) phosphorylation cascade. Eventually insulin resistance and inflammatory response is facilitated (Wang et al. 2016) (Fig. 14.1).
The release of obesity -related danger signals such as ROS , lysosomes, and other obesity-induced danger signals result in the oligomerization of NLRP3 in adipose tissue (Gurung et al. 2015; Zhou et al. 2011). Activation of inflammasomes by damaged mitochondria results in caspase-1-dependent secretion of the inflammatory cytokines IL-1beta and IL-18, and leads to an inflammatory form of cell death. In addition, regulation of mitochondria-induced inflammasome activation centrally contributes to the inflammatory process that is responsible for obesity (Gurung et al. 2015). Actually NLRP3 with macrophage marker F4/80 co-localizes in crown-like structures. Hence, obesity is associated with significant increase in serum IL-18 concentrations which is blocked upon ablation of NLRP3. Indeed, NLRP3 inflammasome is activated in response to high fat diet and controls the production of IL-1beta in adipose tissue and IL-18 in obesity. Induction of high-fat diet induced obesity causes marked caspase-1 activation in adipose tissue. Ceramides are lipid metabolites that accumulates in tissues in response to obesity, and induce caspase-1 activation in an NLRP3-dependent mechanism. In contrast, elimination of NLRP3 inflammasome reduces M1-like macrophage gene expression and increases M2-like expressed cytokines (Youm et al. 2011). Partial depletion of macrophages from adipose tissue of obese subjects decreases the expression of the macrophage marker CD68, with no significant alteration in the expression of caspase-1. This indicates that the effects of the NLRP3-apoptosis-associated speck-like protein (ASC)-caspase-1 protein complex on adipose tissue are not exerted though infiltrating macrophages (Benetti et al. 2013; Stienstra et al. 2011). Actually, innate pattern recognition receptors inflammasomes (NLRs) are cytosolic sensors that detect endogenous metabolic stress and activates caspase enzymes. Activated caspase-1 processes the cytosolic precursors of the related cytokines IL-1beta and IL-18 (Li et al. 2014; Stienstra et al. 2011).
One important molecule regulating glycolysis and macrophage activation is hypoxia -inducible factor 1-alpha (HIF-1alpha). However, hypoxia induces the inflammatory phenotypes of macrophages via HIF-1alpha-dependent and -independent mechanisms (Fujisaka et al. 2013). PI3K/Akt contributes hypoxic stress-induced TLR4 expression through the regulation of HIF-1 activation (Kim et al. 2012). Moreover, hypoxia increases the activation of JNK and p38 mitogen-activated protein kinase signaling in saturated fatty acid-treated macrophages. Inhibition of JNK blocks the hypoxic induction of pro-inflammatory cytokine expression. In any case, inhibition of hypoxia-induced transcription factors fails to reduce the expression of cytokines by saturated fatty acid -treated hypoxic macrophages. Enhanced pro-inflammatory cytokine production and JNK activity under hypoxia are prevented by inhibiting ROS generation. It is well known that hypoxia along with higher concentrations of free fatty acids exacerbates macrophage-mediated inflammation in obesity (Snodgrass et al. 2016). In white adipose tissue , increased oxidative stress due to the mRNA over-expression of NADPH oxidase subunits in ectopic fat leads to enhanced plasminogen activator inhibitor-1 (PAI-1) , IL-6 , and MCP-1 mRNA expression. Furthermore, the selective increase in ROS production in obesity eventually leads to elevation of systemic oxidative stress (Furukawa et al. 2004). In M2 macrophages, overexpression of HIF-2alpha decreases nitric oxide production and suppresses expression of pro-inflammatory cytokines through induction of arginase-1. HIF-2alpha-overexpressing macrophages alleviate pro-inflammatory responses and improve the insulin resistance in adipocytes (Choe et al. 2014). Additionally, recruited adipose tissue macrophages overexpress IL-6, iNOS , and CCR2 (Lumeng et al. 2007b). In particular, CCR2 is expressed by obese adipose tissue macrophages, which are derived from bone marrow cells (Ito et al. 2008). A high level of de novo IL-6 secretion by macrophages is a result of engulfment of the lipid content of adipocytes by macrophages. Thus, IL-6 secretion during interaction of adipocytes and macrophages might have an anti-inflammatory role in the inflamed adipose tissue by downregulating the induction and release of pro-inflammatory cytokines (Sárvári et al. 2015). In total, production of these inflammatory factors in obesity is under the transcriptional control of two key intracellular inflammatory pathways; JNK-activator protein 1 (AP1) and IKKbeta. Extracellular free fatty acids activate JNK and IKKbeta via TLR signaling pathway. Elevation of the pro-inflammatory cytokines in obesity activates JNK and IKKbeta by a specific signaling pathway (Solinas and Karin 2010). Endogenous fatty acids, which are released from adipocytes via the beta3-adrenergic stimulation, result in the activation of the TLR4 /NF-kappaB pathway. In this process, saturated fatty acids , which are released in large quantities from hypertrophied adipocytes via the macrophage-induced adipocyte lipolysis, serve as a natural ligand for TLR4. Thereby the inflammatory changes in both adipocytes and macrophages are induced through NF-kappaB activation (Suganami et al. 2007). Macrophage-inducible C-type lectin (Mincle ; also called Clec4e and Clecsf9), is a type II transmembrane C-type lectin. It is induced selectively in macrophages during the interaction between adipocytes and macrophages. Saturated fatty acid released from adipocytes induces Mincle mRNA expression in macrophages through the TLR4/NF-kappaB pathway. Macrophage-induced adipocyte lipolysis aggravates obesity-induced adipose tissue inflammation by this way (Ichioka et al. 2011). Engagement of either TLR4 or TLR2 with long-chain saturated fatty acids leads to recruitment of the myeloid differentiation primary response gene 88 (MyD88) adapter protein and formation of signaling complexes containing the IL-1 receptor associated serine/threonine kinase (IRAK1) and the TRAF family members, TRAF3 and TRAF6. The latter involves the activation of the MAP kinase kinase kinase (MAP3K) TAK1, which is required for JNK and IKKbeta activation (Kawai and Akira 2007). In addition to these pathways, IRAK-1 is also a tissue marker of meta-inflammation in obesity and a key component of the TLR2/IL-1receptor/MyD88 signaling pathway. Enhanced IRAK-1 gene expression correlates with adipose tissue infiltration by macrophages in obesity-associated chronic low-grade metabolic inflammation (Ahmad et al. 2015) (Fig. 14.1).
Inflammation leads to an increased number of microRNAs (miRNAs) detectable in both adipocytes and M1 macrophages . Indeed, under inflammatory conditions, adipocytes and M1 macrophages share the expression of 147 miRNAs, and 100 common miRNAs . Additionally, miR-221 by 2-fold, miR-222 by 2.5-fold, and miR-155 by 5-fold increased in inflamed adipocytes (Ortega et al. 2015). Although there is no significant change in the level of chemokine (C-C motif) ligand 2 (CCL2) either in adipose tissue or in circulation, the cellularity of adipose tissue macrophages (ATMs) is dramatically elevated at the early stage of obesity . Thus alternatively activated macrophages at the early stages of obesity are the major population of resident macrophages, with more specialized lipid catabolism than that of immigrant macrophages (Zheng et al. 2016). In subcutaneous white adipose tissue obtained from 56 subjects, 11 miRNAs are present in all subjects and downregulated in obesity. Of these, ten affect adipocyte CCL2 secretion for miR-126 and miR-193b. The levels of miR-193b in subcutaneous white adipose tissue are significantly associated with CCL2 secretion, whereas expression of integrin-alpha-X, an inflammatory macrophage marker, is associated with miR-193b and miR-126. In this context, miRNAs may be important regulators of adipose inflammation through their effects on CCL2 release from human adipocytes and macrophages (Arner et al. 2012). Overexpression of miR-126/-193b/-92a in different pairwise combinations reduce CCL2 secretion more efficiently than either miRNA alone. However, although effects on CCL2 secretion by co-overexpression of miR-92a/-193b and miR-92a/-126 are additive in adipocytes, the combination of miR-126/-193b is primarily additive in macrophages. Furthermore signals for miR-92a and -193b converge on the NFkappaB pathway (Kulyté et al. 2014).
The expression of insulin-like growth factor-1 (IGF1) by adipose tissue is derived from adipocytes and macrophages. In lean animals, adipocytes are the primary source of IGF1, but in obesity expression by adipocytes is reduced and by macrophages increased, as to maintain overall adipose tissue IGF1 expression (Chang et al. 2016). IGF1 reduces free fatty acid -induced JNK1 activation and TNF-alpha expression in human subcutaneous but not omental preadipocytes. Impaired anti-inflammatory action of IGF1 in omental preadipocytes is a result of the chronic inflammation in visceral adipose tissue (Neacsu et al. 2013). Actually, preadipocytes could function as macrophage-like cells and raise the possibility of a potential direct involvement of adipose tissue in inflammatory processes (Cousin et al. 1999). Moreover, preadipocyte and macrophage phenotypes are very similar and that preadipocytes have the potential to be very efficiently and rapidly converted into macrophages (Charrière et al. 2003). In this respect, both macrophages and adipocytes are the sites for active lipid metabolism and signaling. Macrophage/adipocyte aP2 expression is controlled with the same promoter or enhancer elements that confer expression in adipocytes. Hence, there is a striking overlap between the biology of adipocytes and macrophages (Makowski et al. 2001). The macrophage/adipocyte aP2 has also been expressed exclusively by differentiated adipocytes. aP2 plays a vital role in the local macrophage responses and binds a number of hydrophobic ligands which are known to influence macrophage function (Makowski et al. 2005). As lipid chaperones, fatty acid-binding proteins (FABPs) may actively facilitate the transport of lipids to specific compartments in the cell. Adipocyte/macrophage aP2 and FABP5, act at the interface of metabolic and inflammatory pathways. Although being the minor isoform in adipocytes, FABP5 protein levels are comparable to aP2 in normal macrophages (Makowski and Hotamisligil 2005). PPAR-gamma induces the expression of the adipocyte/macrophage aP2 and increases aP2 mRNA of primary human monocytes in time- and dose-dependent manner (Pelton et al. 1999). FABP -mediated lipid metabolism is closely linked to both metabolic and inflammatory processes through modulating critical lipid-sensitive pathways in adipocytes and macrophages (Furuhashi and Hotamisligil 2008). Deletion of FABPs in adipocytes results in reduced expression of inflammatory cytokines in macrophages, whereas the same deletion in macrophages led to enhanced insulin signaling and glucose uptake in adipocytes. The metabolic status of adipocytes is a major determinant of macrophage inflammatory output. Neither macrophages nor adipocytes individually could account for the total impact of FABPs on systemic metabolism (Furuhashi et al. 2008).
In adipose tissue, eosinophils are responsible for 90% of IL-4 expression and accelerate M2 macrophage polarization by secreting Th2 type cytokines such as IL-4 and IL-13. Hence, eosinophils might act as anti-inflammatory immune cells in obesity -induced adipose tissue inflammation (Huh et al. 2014). Th1 cells primarily secret IFN-gamma which stimulates monocyte differentiation into M1 type macrophages. IFN-gamma exacerbates adipose tissue inflammation in obesity (Rocha et al. 2008). IFN-gamma might also play an important role in growing adipose tissue, where adipocytes respond to inflammatory products derived from infiltrating macrophages (Wellen and Hotamisligil 2003).
Adipose tissue synthesizes complement proteins and is a target of complement activation. C3a-desArg/acylation-stimulating protein induces lipogenesis and affects lipid metabolism . The C3a receptor and C5a receptor are involved in the development of insulin resistance in adipocytes through macrophage infiltration and the activation of adipose tissue (Vlaicu et al. 2016). C5a receptor-like receptor 2 (C5L2) has been identified as a receptor for acylation-stimulating protein (ASP) and the inflammatory factor C5a. While adipocyte-conditioned medium increases C5L2-C5a receptor co-localization in macrophages, this is blocked by C5a. Induction of ASP increases Akt phosphorylation in both cell types. However, C5a induces Akt phosphorylation in adipocytes with less effect in macrophages (Poursharifi et al. 2013). ASP may induce specific inflammatory cytokines in adipocytes through PI3K- and NF-kappaB-dependent pathways, thus further promoting macrophage infiltration and local inflammation in obese adipose tissue (Tom et al. 2013).
4 Conclusion
Adipocyte death defines macrophage localization and function in adipose tissue of obese humans. However, mechanistic details of the macrophage accumulation surrounding the adipocytes and other parts of the adipose tissue is still vague. In addition, the source of TNF-alpha and the other cytokines requires a detailed analysis by taking the inflammatory alterations in obesity into account. The triggering mechanisms of lipid mediators in adipose tissue has been described by indirect evidences. Therefore, further investigations are necessary in order to clarify the molecular nature of the macrophage-adipocytes interactions in obesity-related diseases.
References
Ahmad, R., P.K. Shihab, R. Thomas, M. Alghanim, A. Hasan, S. Sindhu, and K. Behbehani. 2015. Increased expression of the interleukin-1 receptor-associated kinase (IRAK)-1 is associated with adipose tissue inflammatory state in obesity. Diabetology and Metabolic Syndrome 7: 71. doi:10.1186/s13098-015-0067-7.
Arner, E., N. Mejhert, A. Kulyté, P.J. Balwierz, M. Pachkov, M. Cormont, S. Lorente-Cebrián, A. Ehrlund, J. Laurencikiene, P. Hedén, K. Dahlman-Wright, J.-F. Tanti, Y. Hayashizaki, M. Rydén, I. Dahlman, E. van Nimwegen, C.O. Daub, and P. Arner. 2012. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes 61: 1986–1993. doi:10.2337/db11-1508.
Bassaganya-Riera, J., S. Misyak, A.J. Guri, and R. Hontecillas. 2009. PPAR gamma is highly expressed in F4/80(hi) adipose tissue macrophages and dampens adipose-tissue inflammation. Cellular Immunology 258: 138–146. doi:10.1016/j.cellimm.2009.04.003.
Benetti, E., F. Chiazza, N.S.A. Patel, and M. Collino. 2013. The NLRP3 Inflammasome as a novel player of the intercellular crosstalk in metabolic disorders. Mediators of Inflammation 2013: 678627. doi:10.1155/2013/678627.
Bès-Houtmann, S., R. Roche, L. Hoareau, M.-P. Gonthier, F. Festy, H. Caillens, P. Gasque, C. Lefebvre d’Hellencourt, and M. Cesari. 2007. Presence of functional TLR2 and TLR4 on human adipocytes. Histochemistry and Cell Biology 127: 131–137. doi:10.1007/s00418-006-0230-1.
Bing, C. 2015. Is interleukin-1β a culprit in macrophage-adipocyte crosstalk in obesity? Adipocytes 4: 149–152. doi:10.4161/21623945.2014.979661.
Bouhlel, M.A., B. Derudas, E. Rigamonti, R. Dièvart, J. Brozek, S. Haulon, C. Zawadzki, B. Jude, G. Torpier, N. Marx, B. Staels, and G. Chinetti-Gbaguidi. 2007. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metabolism 6: 137–143. doi:10.1016/j.cmet.2007.06.010.
Castoldi, A., C. Naffah de Souza, N.O.S. Câmara, and P.M. Moraes-Vieira. 2015. The macrophage switch in obesity development. Frontiers in Immunology 6: 637. doi:10.3389/fimmu.2015.00637.
Chang, H.R., H.J. Kim, X. Xu, and A.W. Ferrante. 2016. Macrophage and adipocyte IGF1 maintain adipose tissue homeostasis during metabolic stresses. Obesity (Silver Spring, Md.) 24: 172–183. doi:10.1002/oby.21354.
Charo, I.F. 2007. Macrophage polarization and insulin resistance: PPARgamma in control. Cell Metabolism 6: 96–98. doi:10.1016/j.cmet.2007.07.006.
Charrière, G., B. Cousin, E. Arnaud, M. André, F. Bacou, L. Penicaud, and L. Casteilla. 2003. Preadipocyte conversion to macrophage. Evidence of plasticity. Journal of Biological Chemistry 278: 9850–9855. doi:10.1074/jbc.M210811200.
Chawla, A., E.J. Schwarz, D.D. Dimaculangan, and M.A. Lazar. 1994. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 135: 798–800. doi:10.1210/endo.135.2.8033830.
Chen, H., Y. Liu, D. Li, J. Song, and M. Xia. 2016. PGC-1β suppresses saturated fatty acid-induced macrophage inflammation by inhibiting TAK1 activation. IUBMB Life 68: 145–155. doi:10.1002/iub.1470.
Choe, S.S., K.C. Shin, S. Ka, Y.K. Lee, J.-S. Chun, and J.B. Kim. 2014. Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes 63: 3359–3371. doi:10.2337/db13-1965.
Cinti, S., G. Mitchell, G. Barbatelli, I. Murano, E. Ceresi, E. Faloia, S. Wang, M. Fortier, A.S. Greenberg, and M.S. Obin. 2005. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. Journal of Lipid Research 46: 2347–2355. doi:10.1194/jlr.M500294-JLR200.
Cousin, B., O. Munoz, M. Andre, A.M. Fontanilles, C. Dani, J.L. Cousin, P. Laharrague, L. Casteilla, and L. Pénicaud. 1999. A role for preadipocytes as macrophage-like cells. The FASEB Journal 13: 305–312.
Cutchins, A., D.B. Harmon, J.L. Kirby, A.C. Doran, S.N. Oldham, M. Skaflen, A.L. Klibanov, N. Meller, S.R. Keller, J. Garmey, and C.A. McNamara. 2012. Inhibitor of differentiation-3 mediates high fat diet-induced visceral fat expansion. Arteriosclerosis, Thrombosis, and Vascular Biology 32: 317–324. doi:10.1161/ATVBAHA.111.234856.
Dalmas, E., A. Toubal, F. Alzaid, K. Blazek, H.L. Eames, K. Lebozec, M. Pini, I. Hainault, E. Montastier, R.G.P. Denis, P. Ancel, A. Lacombe, Y. Ling, O. Allatif, C. Cruciani-Guglielmacci, S. André, N. Viguerie, C. Poitou, V. Stich, A. Torcivia, F. Foufelle, S. Luquet, J. Aron-Wisnewsky, D. Langin, K. Clément, I.A. Udalova, and N. Venteclef. 2015. Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nature Medicine 21: 610–618. doi:10.1038/nm.3829.
Duvall, E., A.H. Wyllie, and R.G. Morris. 1985. Macrophage recognition of cells undergoing programmed cell death (apoptosis). Immunology 56: 351–358.
Eguchi, A., A. Mulya, M. Lazic, D. Radhakrishnan, M.P. Berk, D. Povero, A. Gornicka, and A.E. Feldstein. 2015. Microparticles release by adipocytes act as “find-me” signals to promote macrophage migration. PLoS One 10: e0123110. doi:10.1371/journal.pone.0123110.
Fjeldborg, K., S.B. Pedersen, H.J. Møller, T. Christiansen, M. Bennetzen, and B. Richelsen. 2014. Human adipose tissue macrophages are enhanced but changed to an anti-inflammatory profile in obesity. Journal of Immunology Research 2014: 309548. doi:10.1155/2014/309548.
Franckhauser, S., S. Muñoz, A. Pujol, A. Casellas, E. Riu, P. Otaegui, B. Su, and F. Bosch. 2002. Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes 51: 624–630.
Fujisaka, S., I. Usui, A. Bukhari, M. Ikutani, T. Oya, Y. Kanatani, K. Tsuneyama, Y. Nagai, K. Takatsu, M. Urakaze, M. Kobayashi, and K. Tobe. 2009. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes 58: 2574–2582. doi:10.2337/db08-1475.
Fujisaka, S., I. Usui, M. Ikutani, A. Aminuddin, A. Takikawa, K. Tsuneyama, A. Mahmood, N. Goda, Y. Nagai, K. Takatsu, and K. Tobe. 2013. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1α-dependent and HIF-1α-independent manner in obese mice. Diabetologia 56: 1403–1412. doi:10.1007/s00125-013-2885-1.
Furuhashi, M., and G.S. Hotamisligil. 2008. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nature Reviews Drug Discovery 7: 489–503. doi:10.1038/nrd2589.
Furuhashi, M., R. Fucho, C.Z. Görgün, G. Tuncman, H. Cao, and G.S. Hotamisligil. 2008. Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. The Journal of Clinical Investigation 118: 2640–2650. doi:10.1172/JCI34750.
Furukawa, S., T. Fujita, M. Shimabukuro, M. Iwaki, Y. Yamada, Y. Nakajima, O. Nakayama, M. Makishima, M. Matsuda, and I. Shimomura. 2004. Increased oxidative stress in obesity and its impact on metabolic syndrome. The Journal of Clinical Investigation 114: 1752–1761. doi:10.1172/JCI21625.
Furuoka, M., K.-I. Ozaki, D. Sadatomi, S. Mamiya, T. Yonezawa, S. Tanimura, and K. Takeda. 2016. TNF-α induces caspase-1 activation independently of simultaneously induced NLRP3 in 3T3-L1 cells. Journal of Cellular Physiology 231: 2761–2767. doi:10.1002/jcp.25385.
Galván-Peña, S., and L.A.J. O’Neill. 2014. Metabolic reprograming in macrophage polarization. Frontiers in Immunology 5: 420. doi:10.3389/fimmu.2014.00420.
Gao, D., M. Madi, C. Ding, M. Fok, T. Steele, C. Ford, L. Hunter, and C. Bing. 2014. Interleukin-1β mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. American Journal of Physiology: Endocrinology and Metabolism 307: E289–E304. doi:10.1152/ajpendo.00430.2013.
Gordon, S., and P.R. Taylor. 2005. Monocyte and macrophage heterogeneity. Nature Reviews Immunology 5: 953–964. doi:10.1038/nri1733.
Gurung, P., J.R. Lukens, and T.-D. Kanneganti. 2015. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends in Molecular Medicine 21: 193–201. doi:10.1016/j.molmed.2014.11.008.
Halberg, N., T. Khan, M.E. Trujillo, I. Wernstedt-Asterholm, A.D. Attie, S. Sherwani, Z.V. Wang, S. Landskroner-Eiger, S. Dineen, U.J. Magalang, R.A. Brekken, and P.E. Scherer. 2009. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Molecular and Cellular Biology 29: 4467–4483. doi:10.1128/MCB.00192-09.
Hanayama, R., M. Tanaka, K. Miwa, A. Shinohara, A. Iwamatsu, and S. Nagata. 2002. Identification of a factor that links apoptotic cells to phagocytes. Nature 417: 182–187. doi:10.1038/417182a.
Hashimoto, D., J. Miller, and M. Merad. 2011. Dendritic cell and macrophage heterogeneity in vivo. Immunity 35: 323–335. doi:10.1016/j.immuni.2011.09.007.
Henegar, C., J. Tordjman, V. Achard, D. Lacasa, I. Cremer, M. Guerre-Millo, C. Poitou, A. Basdevant, V. Stich, N. Viguerie, D. Langin, P. Bedossa, J.-D. Zucker, and K. Clement. 2008. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biology 9: R14. doi:10.1186/gb-2008-9-1-r14.
Hsu, D.K., and F.-T. Liu. 2004. Regulation of cellular homeostasis by galectins. Glycoconjugate Journal 19: 507–515. doi:10.1023/B:GLYC.0000014080.95829.52.
Huh, J.Y., Y.J. Park, M. Ham, and J.B. Kim. 2014. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Molecules and Cells 37: 365–371. doi:10.14348/molcells.2014.0074.
Ichioka, M., T. Suganami, N. Tsuda, I. Shirakawa, Y. Hirata, N. Satoh-Asahara, Y. Shimoda, M. Tanaka, M. Kim-Saijo, Y. Miyamoto, Y. Kamei, M. Sata, and Y. Ogawa. 2011. Increased expression of macrophage-inducible C-type lectin in adipose tissue of obese mice and humans. Diabetes 60: 819–826. doi:10.2337/db10-0864.
Ito, A., T. Suganami, A. Yamauchi, M. Degawa-Yamauchi, M. Tanaka, R. Kouyama, Y. Kobayashi, N. Nitta, K. Yasuda, Y. Hirata, W.A. Kuziel, M. Takeya, S. Kanegasaki, Y. Kamei, and Y. Ogawa. 2008. Role of CC chemokine receptor 2 in bone marrow cells in the recruitment of macrophages into obese adipose tissue. The Journal of Biological Chemistry 283: 35715–35723. doi:10.1074/jbc.M804220200.
Juhas, U., M. Ryba-Stanisławowska, P. Szargiej, and J. Myśliwska. 2015. Different pathways of macrophage activation and polarization. Postepy Higieny i Medycyny Doswiadczalnej (Online) 69: 496–502. doi:10.5604/17322693.1150133.
Kanda, H., S. Tateya, Y. Tamori, K. Kotani, K. Hiasa, R. Kitazawa, S. Kitazawa, H. Miyachi, S. Maeda, K. Egashira, and M. Kasuga. 2006. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. The Journal of Clinical Investigation 116: 1494–1505. doi:10.1172/JCI26498.
Kang, K., S.M. Reilly, V. Karabacak, M.R. Gangl, K. Fitzgerald, B. Hatano, and C.-H. Lee. 2008. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metabolism 7: 485–495. doi:10.1016/j.cmet.2008.04.002.
Kaplan, J.L., M.A. Marshall, C.C. McSkimming, D.B. Harmon, J.C. Garmey, S.N. Oldham, P. Hallowell, and C.A. McNamara. 2015. Adipocyte progenitor cells initiate monocyte chemoattractant protein-1-mediated macrophage accumulation in visceral adipose tissue. Molecular Metabolism 4: 779–794. doi:10.1016/j.molmet.2015.07.010.
Kawai, T., and S. Akira. 2007. TLR signaling. Seminars in Immunology 19: 24–32. doi:10.1016/j.smim.2006.12.004.
Keuper, M., M. Blüher, M.R. Schön, P. Möller, A. Dzyakanchuk, K. Amrein, K.-M. Debatin, M. Wabitsch, and P. Fischer-Posovszky. 2011. An inflammatory micro-environment promotes human adipocyte apoptosis. Molecular and Cellular Endocrinology 339: 105–113. doi:10.1016/j.mce.2011.04.004.
Kim, S.Y., E. Jeong, S.M. Joung, and J.Y. Lee. 2012. PI3K/Akt contributes to increased expression of Toll-like receptor 4 in macrophages exposed to hypoxic stress. Biochemical and Biophysical Research Communications 419: 466–471. doi:10.1016/j.bbrc.2012.02.015.
Kosteli, A., E. Sugaru, G. Haemmerle, J.F. Martin, J. Lei, R. Zechner, and A.W. Ferrante. 2010. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. The Journal of Clinical Investigation 120: 3466–3479. doi:10.1172/JCI42845.
Krahling, S., M.K. Callahan, P. Williamson, and R.A. Schlegel. 1999. Exposure of phosphatidylserine is a general feature in the phagocytosis of apoptotic lymphocytes by macrophages. Cell Death and Differentiation 6: 183–189. doi:10.1038/sj.cdd.4400473.
Krausgruber, T., K. Blazek, T. Smallie, S. Alzabin, H. Lockstone, N. Sahgal, T. Hussell, M. Feldmann, and I.A. Udalova. 2011. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nature Immunology 12: 231–238. doi:10.1038/ni.1990.
Kulyté, A., Y. Belarbi, S. Lorente-Cebrián, C. Bambace, E. Arner, C.O. Daub, P. Hedén, M. Rydén, N. Mejhert, and P. Arner. 2014. Additive effects of microRNAs and transcription factors on CCL2 production in human white adipose tissue. Diabetes 63: 1248–1258. doi:10.2337/db13-0702.
Lee, B.-C., and J. Lee. 2014. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochimica et Biophysica Acta 1842: 446–462. doi:10.1016/j.bbadis.2013.05.017.
Li, H.-B., C. Jin, Y. Chen, and R.A. Flavell. 2014. Inflammasome activation and metabolic disease progression. Cytokine & Growth Factor Reviews 25: 699–706. doi:10.1016/j.cytogfr.2014.07.020.
Lin, Y., H. Lee, A.H. Berg, M.P. Lisanti, L. Shapiro, and P.E. Scherer. 2000. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. The Journal of Biological Chemistry 275: 24255–24263. doi:10.1074/jbc.M002137200.
Lumeng, C.N., J.L. Bodzin, and A.R. Saltiel. 2007a. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of Clinical Investigation 117: 175–184. doi:10.1172/JCI29881.
Lumeng, C.N., S.M. Deyoung, J.L. Bodzin, and A.R. Saltiel. 2007b. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 56: 16–23. doi:10.2337/db06-1076.
Lumeng, C.N., S.M. Deyoung, and A.R. Saltiel. 2007c. Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. American Journal of Physiology: Endocrinology and Metabolism 292: E166–E174. doi:10.1152/ajpendo.00284.2006.
Lumeng, C.N., J.B. DelProposto, D.J. Westcott, and A.R. Saltiel. 2008. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57: 3239–3246. doi:10.2337/db08-0872.
Makowski, L., and G.S. Hotamisligil. 2005. The role of fatty acid binding proteins in metabolic syndrome and atherosclerosis. Current Opinion in Lipidology 16: 543–548.
Makowski, L., J.B. Boord, K. Maeda, V.R. Babaev, K.T. Uysal, M.A. Morgan, R.A. Parker, J. Suttles, S. Fazio, G.S. Hotamisligil, and M.F. Linton. 2001. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nature Medicine 7: 699–705. doi:10.1038/89076.
Makowski, L., K.C. Brittingham, J.M. Reynolds, J. Suttles, and G.S. Hotamisligil. 2005. The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor gamma and IkappaB kinase activities. The Journal of Biological Chemistry 280: 12888–12895. doi:10.1074/jbc.M413788200.
Malandrino, M.I., R. Fucho, M. Weber, M. Calderon-Dominguez, J.F. Mir, L. Valcarcel, X. Escoté, M. Gómez-Serrano, B. Peral, L. Salvadó, S. Fernández-Veledo, N. Casals, M. Vázquez-Carrera, F. Villarroya, J.J. Vendrell, D. Serra, and L. Herrero. 2015. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. American Journal of Physiology: Endocrinology and Metabolism 308: E756–E769. doi:10.1152/ajpendo.00362.2014.
Martin, S.J., D.M. Finucane, G.P. Amarante-Mendes, G.A. O’Brien, and D.R. Green. 1996. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. The Journal of Biological Chemistry 271: 28753–28756.
Masoodi, M., O. Kuda, M. Rossmeisl, P. Flachs, and J. Kopecky. 2015. Lipid signaling in adipose tissue: Connecting inflammation & metabolism. Biochimica et Biophysica Acta 1851: 503–518. doi:10.1016/j.bbalip.2014.09.023.
McNelis, J.C., and J.M. Olefsky. 2014. Macrophages, immunity, and metabolic disease. Immunity 41: 36–48. doi:10.1016/j.immuni.2014.05.010.
Miyanishi, M., K. Tada, M. Koike, Y. Uchiyama, T. Kitamura, and S. Nagata. 2007. Identification of Tim4 as a phosphatidylserine receptor. Nature 450: 435–439. doi:10.1038/nature06307.
Murdoch, C., A. Giannoudis, and C.E. Lewis. 2004. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 104: 2224–2234. doi:10.1182/blood-2004-03-1109.
Neacsu, O., K. Cleveland, H. Xu, T.T. Tchkonia, J.L. Kirkland, and C.M. Boney. 2013. IGF-I attenuates FFA-induced activation of JNK1 phosphorylation and TNFα expression in human subcutaneous preadipocytes. Obesity (Silver Spring, Md.) 21: 1843–1849. doi:10.1002/oby.20329.
Nguyen, M.T.A., S. Favelyukis, A.-K. Nguyen, D. Reichart, P.A. Scott, A. Jenn, R. Liu-Bryan, C.K. Glass, J.G. Neels, and J.M. Olefsky. 2007. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. The Journal of Biological Chemistry 282: 35279–35292. doi:10.1074/jbc.M706762200.
Nishimura, S., I. Manabe, M. Nagasaki, Y. Hosoya, H. Yamashita, H. Fujita, M. Ohsugi, K. Tobe, T. Kadowaki, R. Nagai, and S. Sugiura. 2007. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes 56: 1517–1526. doi:10.2337/db06-1749.
Norton, J.D. 2000. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. Journal of Cell Science 113(Pt 22): 3897–3905.
O’Rourke, R.W., M.D. Metcalf, A.E. White, A. Madala, B.R. Winters, I.I. Maizlin, B.A. Jobe, C.T. Roberts, M.K. Slifka, and D.L. Marks. 2009. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. International Journal of Obesity 2005(33): 978–990. doi:10.1038/ijo.2009.133.
Odegaard, J.I., R.R. Ricardo-Gonzalez, M.H. Goforth, C.R. Morel, V. Subramanian, L. Mukundan, A. Red Eagle, D. Vats, F. Brombacher, A.W. Ferrante, and A. Chawla. 2007. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447: 1116–1120. doi:10.1038/nature05894.
Olefsky, J.M., and C.K. Glass. 2010. Macrophages, inflammation, and insulin resistance. Annual Review of Physiology 72: 219–246. doi:10.1146/annurev-physiol-021909-135846.
Ortega, F.J., M. Moreno, J.M. Mercader, J.M. Moreno-Navarrete, N. Fuentes-Batllevell, M. Sabater, W. Ricart, and J.M. Fernández-Real. 2015. Inflammation triggers specific microRNA profiles in human adipocytes and macrophages and in their supernatants. Clinical Epigenetics 7: 49. doi:10.1186/s13148-015-0083-3.
Pasarica, M., O.R. Sereda, L.M. Redman, D.C. Albarado, D.T. Hymel, L.E. Roan, J.C. Rood, D.H. Burk, and S.R. Smith. 2009. Reduced adipose tissue oxygenation in human obesity: Evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 58: 718–725. doi:10.2337/db08-1098.
Patsouris, D., P.-P. Li, D. Thapar, J. Chapman, J.M. Olefsky, and J.G. Neels. 2008. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metabolism 8: 301–309. doi:10.1016/j.cmet.2008.08.015.
Pelton, P.D., L. Zhou, K.T. Demarest, and T.P. Burris. 1999. PPARgamma activation induces the expression of the adipocyte fatty acid binding protein gene in human monocytes. Biochemical and Biophysical Research Communications 261: 456–458. doi:10.1006/bbrc.1999.1071.
Poursharifi, P., M. Lapointe, D. Pétrin, D. Devost, D. Gauvreau, T.E. Hébert, and K. Cianflone. 2013. C5L2 and C5aR interaction in adipocytes and macrophages: Insights into adipoimmunology. Cellular Signalling 25: 910–918. doi:10.1016/j.cellsig.2012.12.010.
Prieur, X., C.Y.L. Mok, V.R. Velagapudi, V. Núñez, L. Fuentes, D. Montaner, K. Ishikawa, A. Camacho, N. Barbarroja, S. O’Rahilly, J.K. Sethi, J. Dopazo, M. Orešič, M. Ricote, and A. Vidal-Puig. 2011. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes 60: 797–809. doi:10.2337/db10-0705.
Rocha, V.Z., E.J. Folco, G. Sukhova, K. Shimizu, I. Gotsman, A.H. Vernon, and P. Libby. 2008. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: A role for adaptive immunity in obesity. Circulation Research 103: 467–476. doi:10.1161/CIRCRESAHA.108.177105.
Sano, H., D.K. Hsu, L. Yu, J.R. Apgar, I. Kuwabara, T. Yamanaka, M. Hirashima, and F.T. Liu. 2000. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. Journal of Immunology (Baltimore, Md.) 1950(165): 2156–2164.
Sartipy, P., and D.J. Loskutoff. 2003. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America 100: 7265–7270. doi:10.1073/pnas.1133870100.
Sárvári, A.K., Q.-M. Doan-Xuan, Z. Bacsó, I. Csomós, Z. Balajthy, and L. Fésüs. 2015. Interaction of differentiated human adipocytes with macrophages leads to trogocytosis and selective IL-6 secretion. Cell Death & Disease 6: e1613. doi:10.1038/cddis.2014.579.
Satoh, T., O. Takeuchi, A. Vandenbon, K. Yasuda, Y. Tanaka, Y. Kumagai, T. Miyake, K. Matsushita, T. Okazaki, T. Saitoh, K. Honma, T. Matsuyama, K. Yui, T. Tsujimura, D.M. Standley, K. Nakanishi, K. Nakai, and S. Akira. 2010. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nature Immunology 11: 936–944. doi:10.1038/ni.1920.
Skinner, A.C., M.J. Steiner, F.W. Henderson, and E.M. Perrin. 2010. Multiple markers of inflammation and weight status: Cross-sectional analyses throughout childhood. Pediatrics 125: e801–e809. doi:10.1542/peds.2009-2182.
Snodgrass, R.G., M. Boß, E. Zezina, A. Weigert, N. Dehne, I. Fleming, B. Brüne, and D. Namgaladze. 2016. Hypoxia potentiates palmitate-induced pro-inflammatory activation of primary human macrophages. The Journal of Biological Chemistry 291: 413–424. doi:10.1074/jbc.M115.686709.
Solinas, G., and M. Karin. 2010. JNK1 and IKKbeta: Molecular links between obesity and metabolic dysfunction. The FASEB Journal 24: 2596–2611. doi:10.1096/fj.09-151340.
Spalding, K.L., E. Arner, P.O. Westermark, S. Bernard, B.A. Buchholz, O. Bergmann, L. Blomqvist, J. Hoffstedt, E. Näslund, T. Britton, H. Concha, M. Hassan, M. Rydén, J. Frisén, and P. Arner. 2008. Dynamics of fat cell turnover in humans. Nature 453: 783–787. doi:10.1038/nature06902.
Spencer, M., A. Yao-Borengasser, R. Unal, N. Rasouli, C.M. Gurley, B. Zhu, C.A. Peterson, and P.A. Kern. 2010. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. American Journal of Physiology: Endocrinology and Metabolism 299: E1016–E1027. doi:10.1152/ajpendo.00329.2010.
Stienstra, R., L.A.B. Joosten, T. Koenen, B. van Tits, J.A. van Diepen, S.A.A. van den Berg, P.C.N. Rensen, P.J. Voshol, G. Fantuzzi, A. Hijmans, S. Kersten, M. Müller, W.B. van den Berg, N. van Rooijen, M. Wabitsch, B.-J. Kullberg, J.W.M. van der Meer, T. Kanneganti, C.J. Tack, and M.G. Netea. 2010. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metabolism 12: 593–605. doi:10.1016/j.cmet.2010.11.011.
Stienstra, R., J.A. van Diepen, C.J. Tack, M.H. Zaki, F.L. van de Veerdonk, D. Perera, G.A. Neale, G.J. Hooiveld, A. Hijmans, I. Vroegrijk, S. van den Berg, J. Romijn, P.C.N. Rensen, L.A.B. Joosten, M.G. Netea, and T.-D. Kanneganti. 2011. Inflammasome is a central player in the induction of obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America 108: 15324–15329. doi:10.1073/pnas.1100255108.
St-Pierre, J., J. Lin, S. Krauss, P.T. Tarr, R. Yang, C.B. Newgard, and B.M. Spiegelman. 2003. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. The Journal of Biological Chemistry 278: 26597–26603. doi:10.1074/jbc.M301850200.
Strissel, K.J., Z. Stancheva, H. Miyoshi, J.W. Perfield, J. DeFuria, Z. Jick, A.S. Greenberg, and M.S. Obin. 2007. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56: 2910–2918. doi:10.2337/db07-0767.
Suganami, T., J. Nishida, and Y. Ogawa. 2005. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arteriosclerosis, Thrombosis, and Vascular Biology 25: 2062–2068. doi:10.1161/01.ATV.0000183883.72263.13.
Suganami, T., K. Tanimoto-Koyama, J. Nishida, M. Itoh, X. Yuan, S. Mizuarai, H. Kotani, S. Yamaoka, K. Miyake, S. Aoe, Y. Kamei, and Y. Ogawa. 2007. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology 27: 84–91. doi:10.1161/01.ATV.0000251608.09329.9a.
Sun, K., C.M. Kusminski, and P.E. Scherer. 2011. Adipose tissue remodeling and obesity. The Journal of Clinical Investigation 121: 2094–2101. doi:10.1172/JCI45887.
Takahashi, K., S. Mizuarai, H. Araki, S. Mashiko, A. Ishihara, A. Kanatani, H. Itadani, and H. Kotani. 2003. Adiposity elevates plasma MCP-1 levels leading to the increased CD11b-positive monocytes in mice. The Journal of Biological Chemistry 278: 46654–46660. doi:10.1074/jbc.M309895200.
Tateya, S., F. Kim, and Y. Tamori. 2013. Recent advances in obesity-induced inflammation and insulin resistance. Frontiers in Endocrinology 4: 93. doi:10.3389/fendo.2013.00093.
Thompson, B.R., S. Lobo, and D.A. Bernlohr. 2010. Fatty acid flux in adipocytes: The in’s and out’s of fat cell lipid trafficking. Molecular and Cellular Endocrinology 318: 24–33. doi:10.1016/j.mce.2009.08.015.
Toda, S., R. Hanayama, and S. Nagata. 2012. Two-step engulfment of apoptotic cells. Molecular and Cellular Biology 32: 118–125. doi:10.1128/MCB.05993-11.
Tom, F.-Q., D. Gauvreau, M. Lapointe, H. Lu, P. Poursharifi, X.-P. Luo, and K. Cianflone. 2013. Differential chemoattractant response in adipocytes and macrophages to the action of acylation stimulating protein. European Journal of Cell Biology 92: 61–69. doi:10.1016/j.ejcb.2012.10.005.
Tontonoz, P., E. Hu, R.A. Graves, A.I. Budavari, and B.M. Spiegelman. 1994. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes & Development 8: 1224–1234.
Vandanmagsar, B., Y.-H. Youm, A. Ravussin, J.E. Galgani, K. Stadler, R.L. Mynatt, E. Ravussin, J.M. Stephens, and V.D. Dixit. 2011. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature Medicine 17: 179–188. doi:10.1038/nm.2279.
Vats, D., L. Mukundan, J.I. Odegaard, L. Zhang, K.L. Smith, C.R. Morel, R.A. Wagner, D.R. Greaves, P.J. Murray, and A. Chawla. 2006. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metabolism 4: 13–24. doi:10.1016/j.cmet.2006.05.011.
Vlaicu, S.I., A. Tatomir, D. Boodhoo, S. Vesa, P.A. Mircea, and H. Rus. 2016. The role of complement system in adipose tissue-related inflammation. Immunologic Research 64: 653–664. doi:10.1007/s12026-015-8783-5.
Wang, P.-X., X.-J. Zhang, P. Luo, X. Jiang, P. Zhang, J. Guo, G.-N. Zhao, X. Zhu, Y. Zhang, S. Yang, and H. Li. 2016. Hepatocyte TRAF3 promotes liver steatosis and systemic insulin resistance through targeting TAK1-dependent signalling. Nature Communications 7: 10592. doi:10.1038/ncomms10592.
Weisberg, S.P., D. McCann, M. Desai, M. Rosenbaum, R.L. Leibel, and A.W. Ferrante. 2003. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation 112: 1796–1808. doi:10.1172/JCI19246.
Weisberg, S.P., D. Hunter, R. Huber, J. Lemieux, S. Slaymaker, K. Vaddi, I. Charo, R.L. Leibel, and A.W. Ferrante. 2006. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. The Journal of Clinical Investigation 116: 115–124. doi:10.1172/JCI24335.
Wellen, K.E., and G.S. Hotamisligil. 2003. Obesity-induced inflammatory changes in adipose tissue. The Journal of Clinical Investigation 112: 1785–1788. doi:10.1172/JCI20514.
Wensveen, F.M., S. Valentić, M. Šestan, T. Turk Wensveen, and B. Polić. 2015. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation. European Journal of Immunology 45: 2446–2456. doi:10.1002/eji.201545502.
Wentworth, J.M., G. Naselli, W.A. Brown, L. Doyle, B. Phipson, G.K. Smyth, M. Wabitsch, P.E. O’Brien, and L.C. Harrison. 2010. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes 59: 1648–1656. doi:10.2337/db09-0287.
Wolowczuk, I., C. Verwaerde, O. Viltart, A. Delanoye, M. Delacre, B. Pot, and C. Grangette. 2008. Feeding our immune system: Impact on metabolism. Clinical & Developmental Immunology 2008: 639803. doi:10.1155/2008/639803.
Yin, R., L. Fang, Y. Li, P. Xue, Y. Li, Y. Guan, Y. Chang, C. Chen, and N. Wang. 2015. Pro-inflammatory Macrophages suppress PPARγ activity in Adipocytes via S-nitrosylation. Free Radical Biology & Medicine 89: 895–905. doi:10.1016/j.freeradbiomed.2015.10.406.
Zheng, C., Q. Yang, J. Cao, N. Xie, K. Liu, P. Shou, F. Qian, Y. Wang, and Y. Shi. 2016. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death & Disease 7: e2167. doi:10.1038/cddis.2016.54.
Zhou, R., A.S. Yazdi, P. Menu, and J. Tschopp. 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225. doi:10.1038/nature09663.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Engin, A.B. (2017). Adipocyte-Macrophage Cross-Talk in Obesity. In: Engin, A., Engin, A. (eds) Obesity and Lipotoxicity. Advances in Experimental Medicine and Biology, vol 960. Springer, Cham. https://doi.org/10.1007/978-3-319-48382-5_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-48382-5_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48380-1
Online ISBN: 978-3-319-48382-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)