
Abstract
Growing energy demand, the need to reduce greenhouse gas (GHG) emissions and the move towards a low carbon economy are driving the development of non-food lignocellulosic crops to provide an alternative to fossil fuels and to support bioenergy with carbon capture and storage (CCS). Trees offer significant potential in this role. Poplar, willow and eucalyptus are suggested here as three target tree crops however, a significant yield gap (the difference between potential and observed yield) exists that may be as much as 10 tonnes ha−1y−1. New technologies offer great potential to accelerate the breeding pipeline and provide the bioeconomy with fast growing, stress tolerant and low-input bioenergy trees with higher potential yields and smaller yield gaps. These technologies include both genomic selection (GS) and genome editing, where significant progress for trees has been made in recent years. The most challenging remaining bottleneck is the accurate phenotyping of large populations of trees for traits that underpin yield; more research is required on target traits for the sustainable intensification of the production of bioenergy tree crops.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
Global Drivers for Increasing Bioenergy from Trees
The recently concluded COP21 climate negotiations committed UN signatories to restrict global temperature increases to ‘well below’ 2 °C above pre-industrial levels and to aspire towards 1.5 °C (United Nations 2015). If such ambitious climate and GHG emissions targets are to be met and future energy security assured, it is essential that renewable and sustainable alternative energy sources are developed and utilised on a global scale. Bioenergy and bioenergy with CCS will be a central part of that commitment, since these technologies feature strongly in many forward scenarios for reduced and negative carbon emissions, as described in the work leading to COP21 (Fuss et al. 2014). Many outstanding issues remain on delivery however, the concept of ‘land sparing ’ (increasing agricultural yields and reducing farmland area to allow the spare land to be used for climate change mitigation and biodiversity conservation) could enable high-yield feedstocks to produce significant, low-impact lignocellulosic resources (Lamb et al. 2016). Sources of lignocellulosic biomass under active consideration over the past decade include energy grasses (David and Ragauskas 2010; Jørgensen 2011), crop residues (Gomez et al. 2008) and fast growing trees cultivated under short rotation coppice or short rotation forestry (Hinchee et al. 2009; Tullus et al. 2012).
Prioritisation of the Sustainable Intensifi cation of Biomass-Tree Cultivation
Bioenergy trees are prime examples of second-generation (2G) bioenergy feedstocks (as defined by Manning et al. 2015) in which lignocellulosic biomass is harvested from dedicated perennial species (Somerville et al. 2010). Such crops must be appropriate to the climate and region in which they are grown and able to grow on marginal lands thus minimising competition with food crops or the destruction of high-nature-value ecosystems. They should require few inputs; both to minimise the economic and energy costs of their cultivation and management and to reduce the environmental impacts associated with the fertilisers and pesticides necessary for intensive farming. A review by Manning et al. (2015) suggested that farmland biodiversity and the provision of regulating and cultural ecosystem services could even be improved by perennial biomass crop cultivation if appropriately managed across the landscape. Benefits (in addition to provisioning services such as low carbon energy) include the provision of habitat corridors between and within intensively farmed land areas; harbouring biodiversity including pollinators and other insect populations; preventing soil erosion and buffering water sources against nutrient run-off and sedimentation (Manning et al. 2015). Land use change modelling reported by Milner et al. (2015) supports this idea and proposes that planting perennial lignocellulosics , in temperate landscapes at least, can enhance the provision of a basket of ecosystem services. A review by Don et al. (2012) reported that soils under dedicated perennial crops emit significantly less N2O than soils under conventional arable cultivation. They also have the potential to sequester more carbon, though the effect of transition from grassland to perennial biomass cropping can be neutral or even slightly negative (Don et al. 2012; Harris et al. 2015). The net greenhouse gas balance of bioenergy crops depends strongly upon good land management practices (Davis et al. 2013) and maintaining soil-based ecosystems services is an essential aspect of sustainable agricultural intensification (Schulte et al. 2014). Figure 15.1 provides an overview of the ‘more-from-less’, paradigm for such sustainable intensification ; increasing yield in a given area without degrading the land or resorting to energy intensive cultivation practices (Allwright and Taylor 2016). This essential principle drives bioenergy tree breeding and development and thus the remainder of this chapter is focussed on research in three important species: poplar (Verlinden et al. 2015), willow (Stolarski et al. 2013) and eucalyptus (Freeman et al. 2013).

More from less—sustainable intensification . The figure illustrates how breeding targets to fill the yield gap through yield intensification should be a high priority. However, this yield intensification must make full consideration of the wider inputs required to achieve high yield and that more efficient plants with respect to resources such as water and nitrogen are required. In addition, yield intensification must occur alongside assessment of the GHG balance of the crop system such that lower GHG emissions can be targeted; perhaps through management practices related to the preservation of soil carbon. Finally, a basket of ecosystem services that includes biodiversity protection must be delivered from future multi-functional landscapes
The primary goal of bioenergy tree breeding and development is the sustainable intensification of biomass production. In addition to breeding for yield traits, this also means targeting feedstock quality to increase the efficiency of conversion to liquid fuels and decrease emissions across the whole life cycle of the system. Resource use efficiency with respect to water and nutrients is also a priority to ensure a low-input crop without high irrigation or fertigation needs. Good agronomic practices are also an essential aspect of making high yielding bioenergy trees sustainable. The crop GHG balance can be improved through the preservation of soil carbon stocks and biodiversity and ecosystems services may be preserved and enhanced by good land management.
Unravelling the Yield Gap is Central to the Sustainable Intensification of Bioenergy
Bioenergy trees have the potential to achieve high yields in sustainable, low input agronomic systems and could allow significant reductions in GHG emissions compared to conventional fossil fuels (Zanchi et al. 2012). However, these species still require investment and research effort to overcome the yield gap between typical biomass harvests and their true genetic potential (Allwright and Taylor 2016). The yield gap is widely acknowledged in the breeding and development of food crops (Affholder et al. 2013; Kassie et al. 2014). It is the difference between the potential crop yield under optimal, non-limiting conditions (water, nutrients, pest control) and the average yield under typical field conditions (Mueller et al. 2012; Van Ittersum et al. 2013). A yield gap may result from one, or a combination, of genetic (G), environmental (E) or management (M) factors and failure to address these factors may result in yield stagnation (Licker et al. 2010; Ray et al. 2012). We have already outlined that a central aim for the development of bioenergy trees is sustainable intensification (Fig. 15.1) and understanding the nature of G × E × M interactions underpinning the yield gap can help drive this. For example, through enhanced water and nutrient resource management (Mueller et al. 2012; Bredemeier et al. 2015); the optimisation of soil pH (Tilman et al. 2011); protecting and increasing soil organic carbon stocks (Powlson et al. 2011) or improving soil aeration which is important to fine root development and growth (Weltecke and Gaertig 2012). Figure 15.2 (modified from Allwright and Taylor 2016) demonstrates significant yield gaps for all three tree species; although this figure generally draws on data from small research-scale yield plots since those are the only data widely available at present for these crops for such an analysis. With this caveat, poplar and eucalyptus show a greater range of values than willow with larger maximum biomass yields reported. Poplar trials range from 3 to 35 t−1ha−1y−1 (mean 16.1), eucalyptus trials from 10.5 to 34 t−1ha−1y−1 (mean 22.4) and willow trials from 11.6 to 27.5 t−1ha−1y−1 (mean 17.3). It can be seen that the highest yields are generally achieved in trials in which irrigation or fertigation are supplied, yet potential yields are rarely reached. There is however, an exception in the case of the highest yielding eucalyptus trial whose small experimental plot size means, in the words of the authors, ‘commercial yields are likely to be considerably lower’ (Sims et al. 1999). In practice therefore, the yield gap may be greater than indicated by the shaded region of the chart as commercial yields fall short of those reported in experimental plots and trials (Nonhebel 2002) where only climatic conditions may be limiting. In the remainder of this chapter, we explore the potential for high-throughput phenotyping and transcriptomics, forward genetics (association studies and genomic selection ), reverse genetics and genetic modification/genome editing as tools to help close the yield gap and drive the sustainable intensification of the cultivation of biomass trees as part of the molecular breeding pipeline.

Reported biomass yields reveal a yield gap for biomass trees . Poplar, willow and eucalyptus show wide variation in their biomass yields. Where more than one value is reported in a publication those given here are the maximum reported oven-dry biomass yields (t ha−1 y−1) for the best performing sites, genotypes and years or coppice cycles within each study. The inset bar chart displays the mean yield and standard error for all the trials shown, pooled across feedstocks for each management practice. Numerical citations adjacent to each bar correspond to a single published field trial: 1. (Sims et al. 1999) 2. (Shankhwar and Srivastava 2015) 3. (Minhas et al. 2015) 4. (Guo et al. 2006) 5. (de Andrade et al. 2013) 6. (Herrero et al. 2014) 7. (Müller et al. 2005) 8. (Scaracia-Mugnozza et al. 1997) 9. (Pontailler et al. 1999) 10. (Carmona et al. 2015) 11. (Rae et al. 2007) 12. (Labrecque and Teodorescu 2005) 13. (Fortier et al. 2010) 14. (Nassi O Di Nasso N et al. 2010) 15. (Verlinden et al. 2015) 16. (Dillen et al. 2013) 17. (Truax et al. 2012) 18. (Nielsen et al. 2014) 19. (Bungart and Hüttl 2004) 20. (Bungart 1999) 21. (Adegbidi et al. 2001) 22. (Labrecque and Teodorescu 2003) 23. (Volk et al. 2011) 24. (Stolarski et al. 2013) 25. (Kopp et al. 2001) 26. (Stolarski et al. 2011) 27. (McElroy and Dawson 1986) 28. (Serapiglia et al. 2013) 29. (Adegbidi et al. 2003). (Modified from Allwright and Taylor 2016)
Three Tree Species for Biomass Production
There is now an extensive knowledge and technology foundation for the improvement of poplar, willow and eucalyptus . These include phenotyping facilities, genetic mapping, genetic modification and advanced molecular breeding. The publication of the poplar genome in 2006 (Tuskan et al. 2006) was followed by that of eucalyptus in 2011 (Myburg et al. 2011) while the willow genome is still in progress. All three species are of commercial significance and have been subject to extensive QTL mapping over more than two decades for traits of interest including biomass yield (Rae et al. 2009), wood quality (Brereton et al. 2010) and pest resistance (Alves et al. 2012). This depended upon the development and curation of mapping populations in all three species. More recently, genotyping -by-sequencing (GBS) and association mapping for higher resolution identification of candidate genes for bioenergy traits have been conducted in poplar and eucalyptus (Porth et al. 2013a, b; Silva-Junior et al. 2015). Genetic transformation protocols are established for all three species; there have been extensive field trials of transgenic poplar (Van Acker et al. 2014) and commercial transgenic eucalyptus is now a reality in Brazil (Ledford 2014). Table 15.1 provides an overview of the state of progress in these species while a more detailed discussion of how these resources fit together in a systems biology approach to molecular tree breeding is provided below.
A Systems Biology Approach to Molecular Tree Breeding
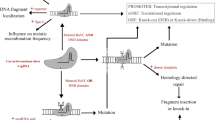
Systems biology may be broadly defined as the use of computational approaches to understand complex biological systems, using functional data from the cellular to organism perspective. As such it has much to offer tree breeding and is generally considered as the integration of ‘omics’ data, such as data from genomics, proteomics and metabolomics with data from the phenotyping. Figure 15.3 is an illustrated overview of how a systems biology approach might aid the discovery of links between genes and traits. Conducting the quantity of phenotyping required now represents a significant challenge and a bottleneck relative to the ability to obtain molecular data for genotyping . An exception to this is the ability to procure high-throughput RNA-Seq data which is now revolutionising eQTL approaches. Both reverse and forward genetics can be of value to the acceleration of the breeding pipeline. Reverse genetics seeks to elucidate a specific gene’s function through mutagenising its DNA sequence and observing the phenotypic outcome. By contrast, forward genetics seeks to map the genetic basis of a specific trait of interest by seeking a statistical relationship between genetic markers and that phenotype. In general, reverse genetics approaches are valuable for understanding the basis of traits controlled by a small number of genetic loci of large effect while forward genetics approaches are better suited to understanding polygenetic traits with multiple small effect loci. One powerful forward genetic technique is genomic selection . Here the complex nature of a polygenetic trait such as yield is explicitly recognised, with 100s or 1000s of SNP molecular markers used together to establish breeding values and obviate significance testing in association and linkage studies (Beaulieu et al. 2014). In contrast, there is also now powerful evidence that CRISPR/Cas genome editing (a cutting edge reverse genetics approach) may be deployed in tree crops such as poplar (Zhou et al. 2015) where mutation breeding could be precisely deployed, again accelerating the breeding pipeline. The following sections give more detail of progress made to date for our target bioenergy trees.

Systems biology for optimised biomass tree breeding. Phenotyping and ‘omics ’ technologies, linked to the development of both forward and reverse genetic approaches , are proposed as a mechanism to deliver the yield improvement required for sustainable intensification . (Modified from Sims et al. 2006)
Phenotyping: The Bottle Neck for Molecular Breeding
In order to make genetic gains to increase the productivity of biomass trees , it is necessary to thoroughly assess the phenotypes of large numbers of existing and emerging genotypes. Given the latest approaches use association rather than mapping populations, the number of individual genotypes and replicates can soon lead to very large and unwieldy experiments with several thousand plants (Porth et al. 2013a, b; McKown et al. 2014a, b). Phenotyping throughput is still limited and now stands as the major bottleneck for breeding programs. To this end, there is increased interest in developing high-throughput phenotyping platforms such as those which make up the International and European Plant Phenotyping Networks (http://www.plant-phenotyping.org/ and http://www.plant-phenotyping-network.eu/). These facilities include both controlled environment and field set-ups and generally increased throughput is reliant on the utilisation of imaging and remote sensing technologies (Table 15.2).
The facility at IPK Gatersleben (Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung, Germany) combines a high-throughput controlled environment phenotyping platform (IPK LemnaTec Scanlayzer) with GC-MS for metabolite profiling. This accommodates plants of small–large size with a capacity for up to 4608 plants to be grown in parallel. The system has enabled the detailed evaluation of stress-related metabolic and phenotypic traits in crops such as lentil. In this case, drought and salinity stress were the focus. Four accessions were intensively phenotyped in order to link drought and salinity tolerance to observed metabolic differences (Muscolo et al. 2015). Phenotyping platforms which utilise controlled environments allow high-precision plant phenotyping and support the study of the genetic basis of these traits. However, for traits that are subject to high GxE interaction, field phenotyping to measure plants in conditions more similar to the target commercial environment is required. For example, DIAPHEN is a field-based platform, comprising imaging tools carried by drones and phenomobiles, managed by the Institut National de la Recherche Agronomique (INRA, France). These systems are GPS-equipped and have the capacity to frequently crop physiological parameters, for example green area cover and canopy activity, in field plots of medium–large plants with a throughput of up to 100 plots h−1. The platform has been successfully used to identify genetic determinants of the drought response in an apple tree hybrid population consisting of 122 genotypes (Virlet et al. 2015).
Although substantial advances have been made in plant phenotyping in recent years (Grobkinsky et al. 2015), the gap between the relatively low-throughput of high accuracy, controlled environment platforms and the higher throughput, lower precision phenotyping which can be achieved in the field remains a challenge and several ongoing, large-scale projects are currently working on the problem of up-scaling phenotyping approaches to crop scale. High-precision, high-throughput phenotyping is necessary to support crop breeding and management .
Metabolomics , Proteomics and Transcriptomics
In addition to traditional morpho-physiological phenotyping, individuals and populations can be characterised based on their metabolome, proteome and transcriptome. Using these ‘omics’ approaches allows physiological phenotypes to be linked to the underlying metabolome, proteome and transcriptome. This is a valuable tool to elucidate the molecular and genetic basis of yield and underpin the breeding effort.
In eucalyptus , the drought stress response of two contrasting genotypes was examined at a metabolomic and physiological level (Shvaleva et al. 2006). These Eucalyptus globulus genotypes were found to differ in physiological drought avoidance mechanisms. The metabolomic analyses indicating that glutathione reductase plays a central role in response to drought. Similarly, proteomics can be applied alongside traditional phenotyping to unravel the molecular basis of traits of interest. For example, xylem development is important as it impacts donwstream bioethanol production. In Populus a proteomic method was used to identify co-expressed proteins in the secondary xylem and generate transgenic trees based on this analysis for field evaluation (Jia et al. 2011). In this way, proteomic and genomic-informed breeding strategies can be developed which utilise these rapidly advancing technologies to support breeding.
It is now possible to carry out whole-transcriptome sequencing in which expressed mRNA sequences are reverse transcribed and the cDNA (complementary DNA) sequenced to provide the entire coding region of the genome. This is a powerful and increasingly achievable tool to characterise the genetic control of traits of interest; such as yield under stress conditions. For example, a comparative transcriptomic approach has been used to identify genes with conserved expression patterns in the woody tissues of Populus trichocarpa and Eucalyptus grandis (Hefer et al. 2015). This identified conserved multi-gene orthologous gene clusters involved in secondary cell wall biosynthesis as well as species-specific gene regulation which allows xylem specialisation. Similar transcriptomic approaches have been employed in a number of plant species and many of these have been able to construct transcript correlation networks which can be linked to phenotypic traits (Porth et al. 2013a, b; Gehan et al. 2015; Vining et al. 2015). In P. balsamifera a high level of network module preservation was again present however organisation within modules and the central hub genes (highly interconnected genes at the centre of a network responsible for modulating a trait of interest) was found to vary between genotypes (Hamanishi et al. 2015). Through this transcriptome analysis , one of the six genotypes was found to have a large and distinct transcriptomic drought response while also exhibiting the smallest metabolomic response. The transcripts in this hub are likely to play a central role in regulating the drought response. This shows the power of these transcriptome-based strategies to determine critical gene hubs and gene connectivity at the genotype, organ and tissue levels. Furthermore, modelling approaches can be taken based on these identified gene hubs whereby phenotypic predictions can be made based on the alteration of genes in the hub network. The combination of transcriptomic network analysis and predictive modelling has further scope to be extended to other bioenergy tree species and to inform breeding programs for industrially-important bioenergy traits.
In the past, ‘omics’ approaches have been costly and constrained by both technologies, for example for protein identification or high-throughput sequencing, and the bioinformatics pipelines that must deal efficiently with large amounts of data. However, these are now rapidly decreasing in cost and accessibility. While it is not always straightforward; it is now possible to use these approaches as part of a powerful multi-omics strategy which can be linked with traditional phenotypic data to underpin breeding efforts.
Reverse Genetics : Proof of Concept or Direct Release of Biotechnologically Enhanced Trees?
Reverse genetic approaches such as gene knockouts or overexpression are required for proof of concept studies to confirm the function of putative candidate genes from QTL or GWAS analyses (Prado et al. 2014) but may themselves also produce new trees of valuable and distinctive phenotypes for direct commercialisation (Fig. 15.3). An example of this is seen in the successful launch of the world’s first commercial, transgenic forest tree in Brazil this year (FuturaGene 2015). FuturaGene’s GM eucalyptus has shown 20 % increases in biomass yield in field trials over a 10-year period and is ready for harvest after 5½ years instead of the usual 7 (Ledford 2014). The overexpressed protein (derived from a gene sequence identified from the model plant, Arabidopsis) accelerates growth by enhancing cell wall expansion; however, its identity remains a commercial secret. Overexpression of stress-responsive genes in eucalyptus can improve salt and cold tolerance (Navarro et al. 2011; Yu et al. 2013). The overexpression of several stress-related genes by gene stacking resulted in poplar with increased salt and pest tolerance (Polle and Chen 2015). Cold tolerance is a key breeding priority for eucalyptus to extend its growth range, sustain consistent yields and be commercially competitive in emerging bioenergy markets (Yu and Gallagher 2015). Reverse genetics can also assist in systems biology approaches to understand more complex pathways. For example, Vanholme et al. (2012) used Arabidopsis loss-of-function mutants for each of the ten genes in the lignin biosynthesis pathway to understand the responses to perturbations in this pathway. Low-lignin transgenic trees (generally knockouts or RNAi knockdowns) are of great research interest because they have the potential to yield a feedstock that is less recalcitrant to enzymatic saccharification (Studer et al. 2011). Van Acker et al. (2014) demonstrated improved saccharification and ethanol yield from field-grown GM poplar deficient in the lignin biosynthetic enzyme cinnamoyl-CoA reductase. Unfortunately, blunt reductions in lignin content can have negative consequences for yield (Van Acker et al. 2014) and pest resistance (Polle et al. 2013) and other, more novel, transgenic approaches are being investigated. One route is the heterologous expression (i.e. stimulating gene expression in cells that do not normally express the gene) of thermophilic, cell wall degrading enzymes in planta (Jung et al. 2012). These enzymes can be activated by mild temperature increases post-harvest and can decrease the energy and financial costs of the conversion of wood to ethanol. Poplar especially is known as an efficient bioreactor for the expression of foreign enzymes (Kim et al. 2012). Another exciting GM approach for reduced recalcitrance without impacting fitness has been reported by Wilkerson et al. (2014). They successfully incorporated a transferase gene into poplar which introduced ester linkages into the lignin backbone. These ester bonds can be readily hydrolysed by a mild, alkaline pre-treatment, aiding processing. In recent years, highly targeted, sequence-specific genome editing has become more feasible in eukaryotes through the development of CRISPR/Cas technology (Gaj et al. 2013). In a key development for bioenergy trees this technology has now been successfully used in poplar to target a lignin biosynthetic enzyme (4-coumarate:CoA ligase) and further innovations using this method are likely to follow (Zhou et al. 2015). Finally, tilling is a powerful and high-throughput reverse genetic approach to elucidate gene function in a mutagenised population using a mismatch endonuclease to detect the induced mutations (SNPs or indels). Ecotilling is closely related but seeks to identify polymorphisms in natural populations and evaluate their effects on genes of interest and phenotypic significance (methodology and development reviewed by Barkley and Wang 2008). Ecotilling has been successfully employed in food crops (Yu et al. 2012) and has potential for accelerating the domestication of forest trees (Harfouche et al. 2012). Marroni et al. (2011) reported the detection of rare alleles in poplar using NGS and believe that this methodology could drive next generation ecotilling in this species; allowing function to be ascribed to these low frequency variants.
From Trees to Genes: Forward Genetics
Forward genetic techniques seek to understand the genetic basis of a phenotype and identify genomic regions, markers and/or candidate genes linked with the trait of interest (Fig. 15.3). Forward genetic approaches are of particular value for elucidating quantitative, polygenic traits. Before the revolution in cost-efficient, high-throughput, next-generation sequencing (Mardis 2011), genetic marker density was generally limited and research was focussed on broad QTL mapping which can be achieved with only a few hundred SSLR or microsatellite markers. QTL have been mapped for yield in both eucalyptus (Freeman et al. 2013) and poplar (Wullschleger et al. 2005; Rae et al. 2007). Rae et al. (2009) identified five robust QTL hotspots for yield in short rotation coppice (SRC) poplar explaining 20 % of final biomass yield in the mapping population. In willow, QTL have been mapped for rust resistance; a major willow pathogen and responsible for commercial losses (Hanley et al. 2011; Samils et al. 2011). Expression QTL (eQTL) mapping is a more recent development, also known as ‘genetical genomics ’ (Joosen et al. 2009). This approach considers gene expression (quantified levels of given mRNA transcripts) as a quantitative trait and maps this expression data as QTL (Ingvarsson and Street 2010). This can permit the identification of causal genes underpinning the phenotype of interest. Genetical genomics has been widely employed for several years in a number of plant and animal species including model organisms such as Drosophila, yeast and mice (Joosen et al. 2009). In the model plant Arabidopsis the technique has been useful for understanding the genetic basis of complex responses such as genotype-by-environment interactions (Joosen et al. 2013) and genetic regulatory networks (Terpstra et al. 2010). In bioenergy research genetical genomics has been applied in the biodiesel crop jatropha for oil production traits (Liu et al. 2011) and in poplar for leaf shape variation (Drost et al. 2015). Another genetical genomics approach that has been employed in poplar is a form of bulk segregant analysis with microarray expression data. Street et al. (2006) identified extreme genotypes for drought tolerance traits in response to soil drying and used microarrays to identify differentially expressed genes between these groups. They were able to identify promising candidate genes whose differential expression co-located with traditionally mapped QTL for these drought-specific traits. More recently, with the availability of NGS approaches to provide high-throughput DNA marker data, eQTL mapping has become relatively cheap and much more tractable to elucidate the link between phenotypes and their underlying resolution at the genomic level (Majewski and Pastinen 2011). These technologies are now being applied to plant improvement and combined with QTL-Seq approaches (Takagi et al. 2013). Using NGS for RNA-Seq can offer significant new potential to resolve traits in trees in future in a more time and cost-effective manner.
Association mapping is a more powerful forward genetic approach for elucidating the genetic basis of qualitative and quantitative traits in species of interest; seeking statistical associations between SNPs and phenotypes of interest within a population (Ingvarsson and Street 2010). The finesse with which a trait can be mapped is dependent on the rate of decay of linkage disequilibrium (LD) , i.e. the non-random association of alleles at different loci. Since linkage is a major contributor to LD, LD declines with physical distance (Flint-Garcia et al. 2003). Outbreeding species (including poplar, eucalyptus and willow), which have a higher effective degree of recombination than inbreeders (Gaut and Long 2003), can achieve higher resolution association mapping but concomitantly require a higher marker frequency (Neale and Kremer 2011). The need for high marker density meant that initial association studies in bioenergy trees tended to take a candidate gene approach which are not genome-wide but useful for narrowing down genes of interest within a broader QTL region or identifying candidates within a group of genes of putatively similar function (Teare 2011). In both poplar (Wegrzyn et al. 2010; Guerra et al. 2013) and eucalyptus (Thavamanikumar et al. 2011, 2014) candidate gene approaches have been employed for wood quality traits with robust trait-marker associations identified. In poplar these have been superseded by the development of a 34,000 SNP array for P. trichocarpa with SNPs drawn from 3543 candidate genes for a variety of valuable bioenergy traits (Geraldes et al. 2013). This ‘chip’ has been employed in a number of GWAS in poplar in the past 2 years identifying hundreds of trait-marker associations for key traits including biomass yield (McKown et al. 2014a, b), wood quality (Porth et al. 2013a, b) and rust tolerance (La Mantia et al. 2013). Eucalyptus researchers are now pursuing a similar path with the recent publication of a 60,000 SNP chip that will permit GWAS in this species (Silva-Junior et al. 2015). Associations can then feed into the molecular breeding pipeline (Fig. 15.4) and marker-assisted selection (MAS), as seen in many crop plants (Miedaner and Korzun 2012), for the advanced breeding of superior bioenergy trees.

The advanced molecular breeding pipeline
Beginning with unimproved germplasm curated in a natural, wide or mapping population, advanced molecular breeding may proceed through high-throughput phenotyping for traits of interest. In parallel; GBS, GWAS , genomic selection , transcriptome sequencing and/or eQTL mapping can allow the identification of candidate genes or markers for the phenotyped traits which may also serve as high value targets for GM proof of concept studies and genome editing. Collectively these techniques feed directly into advanced, marker-assisted selection and breeding programmes for novel, high yield , low-input feedstocks.
As NGS reduces genotyping costs and marker numbers and density increase there is the potential to move towards GS in bioenergy trees. GS assigns breeding values to individuals based on genome-wide markers of sufficient density to permit the assumption or knowledge that all relevant genomic regions are in LD with some of the genotyped SNPs (single nucleotide polymorphisms or single base changes in the DNA sequence) (Grattapaglia and Resende 2010). A modelling study from Resende et al. (2012) suggests that GS could accelerate the domestication of forest trees by increasing selection efficiency resulting in a faster breeding cycle. This has huge potential for biomass poplar, willow and eucalyptus where trees take several years to reach reproductive maturity and traditional breeding can take decades. GS has recently been shown to be effective in interior spruce (Gamal El-Dien et al. 2015) using markers obtained through GBS; with ongoing research to identify the best breeding groups to deploy this technology in white spruce (Beaulieu et al. 2014).
The Breeding Pipeline
Trees are long-lived and largely out-breeding species and it is therefore difficult to make rapid improvements through breeding and selection (Harfouche et al. 2011; Allwright and Taylor 2016). In addition many tree species are dioecious (single sexed), making the selection of specific crosses difficult and genetic research complex. These lifecycle limitations have major impacts on the breeding cycles for woody plants and have been partially overcome in the past by the extensive use of vegetatively propagated or clonal material, as in the three species considered here (Liesebach and Naujoks 2004; Meilan et al. 2002; Stape et al. 2008). Recently, protoplast fusion has been introduced as a novel technique for the production of enhanced poplar germplasm (Hennig et al. 2015); however, the technique is still in its infancy.
This short review has highlighted several approaches that are combining next-generation DNA sequencing technologies with high-throughput phenotyping approaches to overcome this bottleneck in the next decade with accelerated breeding cycles possible. All pipelines begin with the collection and curation of novel germplasm material (Fig. 15.4) and future efforts to fulfil the necessity for sustainable intensification (Fig. 15.1) are likely to involve collection from extreme climate sites. The value of wild germplasm cannot be overestimated and has proved to be of central importance in recent breeding efforts in both rice (Arbelaez et al. 2015) and tomato (Blanca et al. 2015). Recent advances now mean that this material is tractable with large GWAS studies enabling the rapid development of links between traits and genes and the development of molecular markers with which to pursue MAS. The difficulty with this approach for trees is their outbreeding nature, although rare variants have been identified using a modified pooled multiplexing (the simultaneous sequencing of many DNA samples tagged for their identification, thus speeding DNA sequencing whilst reducing cost) approach that identifies rare variants of functional genes underpinning lignin production in poplar (Marroni et al. 2011). More promising are the genomic selection tools where training and validation populations are used to calculate genotypes’ breeding values from multiple markers in relation to traits of interest. Such techniques offer significant potential to reduce breeding time since selections can be made in a fraction of the time required to follow the growth and performance of a breeding population using routine harvest and assessment methods. Alongside genomic selection, genome editing has also been shown as a proven technology for poplar (Fan et al. 2015; Zhou et al. 2015) and offers a route for the rapid assessment of individual genes that might emerge from the breeding pipeline and high-throughput phenotyping . In many respects, genomic selection and genome editing offer two contrasting routes to the production of improved, high yielding biomass material for the future bioenergy landscape and both should be considered over the coming decades.
Conclusions
Tree breeding for bioenergy is important as woody lignocellulosics crops can contribute to efforts to fulfil global commitments to reduced emissions and the move towards a low carbon economy. Most future energy scenarios highlight a significant role for energy from biomass, including through co-firing; biomass burning with CCS and biomass for liquid biofuels. However, the supply of high yielding, sustainable feedstock cultivars of biomass tree species is hampered by the biology of trees. This review has highlighted the importance of high-throughput phenotyping and new molecular technologies that can be deployed to significantly accelerate the breeding pipeline, without the necessity to produce a ‘GM ’ tree; helping to address the current yield gap and increase potential yields in these important lignocellulosic crops.
References
Adegbidi HG, Volk TA, White EH et al (2001) Biomass and nutrient removal by willow clones in experimental bioenergy plantations in New York State. Biomass and Bioenergy 20:399–411
Adegbidi HG, Briggs RD, Volk TA et al (2003) Effect of organic amendments and slow-release nitrogen fertilizer on willow biomass production and soil chemical characteristics. Biomass and Bioenergy 25:389–398
Affholder F, Poeydebat C, Corbeels M et al (2013) The yield gap of major food crops in family agriculture in the tropics: assessment and analysis through field surveys and modelling. F Crop Res 143:106–118
Allwright MR, Taylor G (2016) Molecular breeding for improved second generation bioenergy crops. Trends Plant Sci 21:43–54
Alves AA, Rosado CCG, Faria DA et al (2012) Genetic mapping provides evidence for the role of additive and non-additive QTLs in the response of inter-specific hybrids of Eucalyptus to Puccinia psidii rust infection. Euphytica 183:27–38
Arbelaez JD, Moreno LT, Singh N et al (2015) Development and GBS-genotyping of introgression lines (ILs) using two wild species of rice, O. meridionalis and O. rufipogon, in a common recurrent parent, O.sativa cv. Curinga. Mol Breed 35:81
Barkley NA, Wang ML (2008) Application of TILLING and EcoTILLING as reverse genetic approaches to elucidate the function of genes in plants and animals. Curr Genomics 9:212–226
Beaulieu J, Doerksen TK, MacKay J et al (2014) Genomic selection accuracies within and between environments and small breeding groups in white spruce. BMC Genomics 15:1048
Berlin S, Lagercrantz U, von Arnold S et al (2010) High-density linkage mapping and evolution of paralogs and orthologs in Salix and Populus. BMC Genomics 11:129
Berlin S, Trybush SO, Fogelqvist J et al (2014) Genetic diversity, population structure and phenotypic variation in European Salix viminalis L. (Salicaceae). Tree Genet Genomes 10:1595–1610
Blanca J, Montero-Pau J, Sauvage C et al (2015) Genomic variation in tomato, from wild ancestors to contemporary breeding accessions. BMC Genomics 16:257
Bredemeier M, Busch G, Hartmann L et al (2015) Fast growing plantations for wood production - integration of ecological effects and economic perspectives. Front Bioeng Biotechnol 3:72
Brereton NJB, Pitre FE, Hanley SJ et al (2010) QTL mapping of enzymatic saccharification in short rotation coppice willow and its independence from biomass yield. Bioenergy Res 3:251–261
Brondani RPV, Williams ER, Brondani C, Grattapaglia D (2006) A microsatellite-based consensus linkage map for species of Eucalyptus and a novel set of 230 microsatellite markers for the genus. BMC Plant Biol 6:20
Bungart R (1999) Erzeugung von Biomasse zur energetischen Nutzung durch den Anbau schnellwachsender Baumarten auf Kippsubstraten des Lausitzer Braunkohlereviers unter besonderer Beru¨ cksichtigung der Na¨ hrelementversorgung und des Wasserhaushaltes. Cottbuser Schr Bodenschutz Rekult 7:159
Bungart R, Hüttl RF (2004) Growth dynamics and biomass accumulation of 8-year-old hybrid poplar clones in a short-rotation plantation on a clayey-sandy mining substrate with respect to plant nutrition and water budget. Eur J For Res 123:105–115
Carmona R, Nuñez T, Alonso MF (2015) Biomass yield and quality of an energy dedicated crop of poplar (Populus spp.) clones in the Mediterranean zone of Chile. Biomass and Bioenergy 74:96–102
Cervera M, Ivens B, Gusma J et al (2001) Dense genetic linkage maps of three populus species (populus deltoides, P. Nigra and P. Trichocarpa) based on AFLP and microsatellite markers. Genetics 158:787–809
David K, Ragauskas AJ (2010) Switchgrass as an energy crop for biofuel production: A review of its ligno-cellulosic chemical properties. Energy Environ Sci 3:1182
Davis SC, Boddey RM, Alves BJR et al (2013) Management swing potential for bioenergy crops. GCB Bioenergy 5:623–638
de Andrade TCGR, de Barros NF, Dias LE, Azevedo MIR (2013) Biomass yield and calorific value of six clonal stands of Eucalyptus urophylla ST Blake cultivated in Northeastern Brazil. Cern Lavras 19:467–472
Dillen SY, Djomo SN, Al Afas N et al (2013) Biomass yield and energy balance of a short-rotation poplar coppice with multiple clones on degraded land during 16 years. Biomass and Bioenergy 56:157–165
Don A, Osborne B, Hastings A et al (2012) Land-use change to bioenergy production in Europe: implications for the greenhouse gas balance and soil carbon. GCB Bioenergy 4:372–391
Drost DR, Puranik S, Novaes E et al (2015) Genetical genomics of Populus leaf shape variation. BMC Plant Biol 15:166
Fan D, Liu T, Li C et al (2015) Efficient CRISPR/Cas9-mediated targeted mutagenesis in populus in the first generation. Sci Rep 5:12217
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Fortier J, Gagnon D, Truax B, Lambert F (2010) Biomass and volume yield after 6 years in multiclonal hybrid poplar riparian buffer strips. Biomass and Bioenergy 34:1028–1040
Freeman JS, Whittock SP, Potts BM, Vaillancourt RE (2009) QTL influencing growth and wood properties in Eucalyptus globulus. Tree Genet Genomes 5:713–722
Freeman JS, Potts BM, Downes GM et al (2013) Stability of quantitative trait loci for growth and wood properties across multiple pedigrees and environments in Eucalyptus globulus. New Phytol 198:1121–1134
Fuss S, Canadell JG, Peters GP et al (2014) Betting on negative emissions. Nat Clim Chang 4:850–853
FuturaGene (2015) FuturaGene’s eucalyptus is approved for commercial use in Brazil
Gaj T, Gersbach CA, Barbas CF (2013) ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405
Gamal El-Dien O, Ratcliffe B, Klápště J et al (2015) Prediction accuracies for growth and wood attributes of interior spruce in space using genotyping-by-sequencing. BMC Genomics 16:1–16
Gaut BS, Long AD (2003) The lowdown on linkage disequilibrium. Plant Cell 15:1502–1506
Gehan MA, Greenham K, Mockler TC, McClung CR (2015) Transcriptional networks-crops, clocks, and abiotic stress. Curr Opin Plant Biol 24:39–46
Geraldes A, Pang J, Thiessen N et al (2011) SNP discovery in black cottonwood (Populus trichocarpa) by population transcriptome resequencing. Mol Ecol Resour 11:81–92
Geraldes A, Difazio SP, Slavov GT et al (2013) A 34K SNP genotyping array for populus trichocarpa: design, application to the study of natural populations and transferability to other populus species. Mol Ecol Resour 13:306–323
Gomez LD, Steele-King CG, McQueen-Mason SJ (2008) Sustainable liquid biofuels from biomass: the writing’s on the walls. New Phytol 178:473–485
Grattapaglia D, Resende MDV (2010) Genomic selection in forest tree breeding. Tree Genet Genomes 7:241–255
Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Grobkinsky DK, Pieruschka R, Svensgaard J et al (2015) Phenotyping in the fields: dissecting the genetics of quantitative traits and digital farming. New Phytol 207:950–952
Guerra FP, Wegrzyn JL, Sykes R et al (2013) Association genetics of chemical wood properties in black poplar (Populus nigra). New Phytol 197:162–176
Guo LB, Sims REH, Horne DJ (2006) Biomass production and nutrient cycling in Eucalyptus short rotation energy forests in New Zealand: II. Litter fall and nutrient return. Biomass and Bioenergy 30:393–404
Hamanishi ET, Barchet GL, Dauwe R et al (2015) Poplar trees reconfigure the transcriptome and metabolome in response to drought in a genotype- and time-of-day-dependent manner. BMC Genomics 16:329
Hanley SJ, Karp A (2014) Genetic strategies for dissecting complex traits in biomass willows (Salix spp.). Tree Physiol 34:1167–1180
Hanley SJ, Pei MH, Powers SJ et al (2011) Genetic mapping of rust resistance loci in biomass willow. Tree Genet Genomes 7:597–608
Harfouche A, Meilan R, Altman A (2011) Tree genetic engineering and applications to sustainable forestry and biomass production. Trends Biotechnol 29:9–17
Harfouche A, Meilan R, Kirst M et al (2012) Accelerating the domestication of forest trees in a changing world. Trends Plant Sci 17:64–72
Harris ZM, Spake R, Taylor G (2015) Land use change to bioenergy: a meta-analysis of soil carbon and GHG emissions. Biomass and Bioenergy 82:27–39
Hefer CA, Mizrachi E, Myburg AA et al (2015) Comparative interrogation of the developing xylem transcriptomes of two wood-forming species: Populus trichocarpa and Eucalyptus grandis. New Phytol 206:1391–1405
Hennig A, Kleinschmit JRG, Schoneberg S et al (2015) Water consumption and biomass production of protoplast fusion lines of poplar hybrids under drought stress. Front Plant Sci 6:330
Herrero C, Juez L, Tejedor C et al (2014) Importance of root system in total biomass for Eucalyptus globulus in northern Spain. Biomass and Bioenergy 67:212–222
Hinchee M, Rottmann W, Mullinax L et al (2009) Short-rotation woody crops for bioenergy and biofuels applications. In Vitro Cell Dev Biol Plant 45:619–629
Hudson CJ, Freeman JS, Kullan AR et al (2012) A reference linkage map for Eucalyptus. BMC Genomics 13:240
Ingvarsson PK, Street NR (2010) Association genetics of complex traits in plants. New Phytol 189:909–922
Jia X, Zhao M, Zhao C et al (2011) Populus biomass protein-protein interactions and their functions. BMC Proc
Joosen RVL, Ligterink W, Hilhorst HWM, Keurentjes JJB (2009) Advances in genetical genomics of plants. Curr Genomics 10:540–549
Joosen RVL, Arends D, Li Y et al (2013) Identifying genotype-by-environment interactions in the metabolism of germinating Arabidopsis seeds using generalized genetical genomics. Plant Physiol 162:553–566
Jørgensen U (2011) Benefits versus risks of growing biofuel crops: the case of Miscanthus. Curr Opin Environ Sustain 3:24–30
Jung SK, Parisutham V, Jeong SH, Lee SK (2012) Heterologous expression of plant cell wall degrading enzymes for effective production of cellulosic biofuels. J Biomed Biotechnol 2012:405842
Karp A, Hanley SJ, Trybush SO et al (2011) Genetic improvement of willow for bioenergy and biofuels. J Integr Plant Biol 53:151–165
Kassie BT, Van Ittersum MK, Hengsdijk H et al (2014) Climate-induced yield variability and yield gaps of maize (Zea mays L.) in the Central Rift Valley of Ethiopia. F Crop Res 160:41–53
Kim S, Kim Y, Ee YL et al (2012) The transgenic poplar as an efficient bioreactor system for the production of xylanase. Biosci Biotechnol Biochem 76:1140–1145
Kopp RF, Abrahamson LP, White EH et al (2001) Willow biomass production during ten successive annual harvests. Biomass and Bioenergy 20:1–7
La Mantia J, Klápště J, El-Kassaby YA et al (2013) Association analysis identifies melampsora × columbiana poplar leaf rust resistance SNPs. PLoS One 8, e78423
Labrecque M, Teodorescu TI (2003) High biomass yield achieved by Salix clones in SRIC following two 3-year coppice rotations on abandoned farmland in southern Quebec, Canada. Biomass and Bioenergy 25:135–146
Labrecque M, Teodorescu T (2005) Field performance and biomass production of 12 willow and poplar clones in short-rotation coppice in southern Quebec (Canada). Biomass and Bioenergy 29:1–9
Lamb A, Green R, Bateman I et al (2016) The potential for land sparing to offset greenhouse gas emissions from agriculture. Nat Clim Chang 1
Ledford H (2014) Brazil considers transgenic trees. Nature 512:357
Licker R, Johnston M, Foley JA et al (2010) Mind the gap: how do climate and agricultural management explain the “yield gap” of croplands around the world? Glob Ecol Biogeogr 19:769–782
Liesebach M, Naujoks G (2004) Approaches on vegetative propagation of difficult-to-root Salix caprea. Plant Cell Tissue Organ Cult 79:239–247
Liu P, Wang C, Li L et al (2011) Mapping QTLs for oil traits and eQTLs for oleosin genes in jatropha. BMC Plant Biol 11:132
Liu J, Yin T, Ye N et al (2013) Transcriptome analysis of the differentially expressed genes in the male and female shrub willows (Salix suchowensis). PLoS One 8, e60181
Majewski J, Pastinen T (2011) The study of eQTL variations by RNA-seq: From SNPs to phenotypes. Trends Genet 27:72–79
Manning P, Taylor GE, Hanley M (2015) Bioenergy, food production and biodiversity - an unlikely alliance? GCB Bioenergy 7:570–576
Mardis ER (2011) A decade’s perspective on DNA sequencing technology. Nature 470:198–203
Marroni F, Pinosio S, Di Centa E et al (2011) Large-scale detection of rare variants via pooled multiplexed next-generation sequencing: towards next-generation Ecotilling. Plant J 67:736–745
McElroy GH, Dawson WM (1986) Biomass from short-rotation coppice willow on marginal land. Biomass 10:225–240
McKown AD, Guy RD, Quamme L et al (2014a) Association genetics, geography, and ecophysiology link stomatal patterning in Populus trichocarpa with carbon gain and disease resistance trade-offs. Mol Ecol 23:5771–5790
McKown AD, Klápště J, Guy RD et al (2014b) Genome-wide association implicates numerous genes underlying ecological trait variation in natural populations of Populus trichocarpa. New Phytol 203:535–553
Meilan R, Auerbach DJ, Ma C et al (2002) Stability of herbicide resistance and GUS expression in transgenic hybrid poplars (Populus sp.) during four years of field trials and vegetative propagation. HortScience 37:277–280
Miedaner T, Korzun V (2012) Marker-assisted selection for disease resistance in wheat and barley breeding. Phytopathology 102:560–566
Milner S, Holland RA, Lovett A et al (2015) Potential impacts on ecosystem services of land use transitions to second generation bioenergy crops in GB. GCB Bioenergy 8:317–333
Minhas PS, Yadav RK, Lal K, Chaturvedi RK (2015) Effect of long-term irrigation with wastewater on growth, biomass production and water use by Eucalyptus (Eucalyptus tereticornis Sm.) planted at variable stocking density. Agric Water Manag 152:151–160
Missiaggia AA, Piacezzi AL, Grattapaglia D (2005) Genetic mapping of Eef1, a major effect QTL for early flowering in Eucalyptus grandis. Tree Genet Genomes 1:79–84
Mueller ND, Gerber JS, Johnston M et al (2012) Closing yield gaps through nutrient and water management. Nature 490:254–257
Müller MD, Filho AAT, Vale RS, do Couto L (2005) Biomass yield and energetic content in Agroforestry Systems with Eucalypt in Vazante-MG. Biomassa Energ 2:125–132
Muscolo A, Junker A, Klukas C et al (2015) Phenotypic and metabolic responses to drought and salinity of four contrasting lentil accessions. J Exp Bot 66:5467–5480
Myburg A, Grattapaglia D, Tuskan G et al (2011) The Eucalyptus grandis Genome Project: Genome and transcriptome resources for comparative analysis of woody plant biology. BMC Proc 5:I20
Nassi O Di Nasso N, Guidi W, Ragaglini G et al (2010) Biomass production and energy balance of a 12-year-old short-rotation coppice poplar stand under different cutting cycles. GCB Bioenergy 2:89–97
Navarro M, Ayax C, Martinez Y et al (2011) Two EguCBF1 genes overexpressed in Eucalyptus display a different impact on stress tolerance and plant development. Plant Biotechnol J 9:50–63
Neale DB, Kremer A (2011) Forest tree genomics: growing resources and applications. Nat Rev Genet 12:111–122
Nielsen UB, Madsen P, Hansen JK et al (2014) Production potential of 36 poplar clones grown at medium length rotation in Denmark. Biomass and Bioenergy 64:99–109
Nonhebel S (2002) Energy yields in intensive and extensive biomass production systems. Biomass and Bioenergy 22:159–167
Novaes E, Osorio L, Drost DR et al (2009) Quantitative genetic analysis of biomass and wood chemistry of Populus under different nitrogen levels. New Phytol 182:878–890
Polle A, Chen S (2015) On the salty side of life: molecular, physiological and anatomical adaptation and acclimation of trees to extreme habitats. Plant Cell Environ 38:1794–1816
Polle A, Janz D, Teichmann T, Lipka V (2013) Poplar genetic engineering: promoting desirable wood characteristics and pest resistance. Appl Microbiol Biotechnol 97:5669–5679
Pontailler JY, Ceulemans R, Guittet J (1999) Biomass yield of poplar after five 2-year coppice rotations. Forestry 72:157–163
Porth I, Klapšte J, Skyba O et al (2013a) Genome-wide association mapping for wood characteristics in Populus identifies an array of candidate single nucleotide polymorphisms. New Phytol 200:710–726
Porth I, Klápště J, Skyba O et al (2013b) Network analysis reveals the relationship among wood properties, gene expression levels and genotypes of natural Populus trichocarpa accessions. New Phytol 200:727–742
Powlson DS, Gregory PJ, Whalley WR et al (2011) Soil management in relation to sustainable agriculture and ecosystem services. Food Policy 36:S72–S87
Prado JR, Segers G, Voelker T et al (2014) Biotech crop development: from idea to product. Annu Rev Plant Biol 65:769–790
Rae AM, Pinel MPC, Bastien C et al (2007) QTL for yield in bioenergy Populus: identifying G × E interactions from growth at three contrasting sites. Tree Genet Genomes 4:97–112
Rae AM, Street NR, Robinson KM et al (2009) Five QTL hotspots for yield in short rotation coppice bioenergy poplar: the Poplar Biomass Loci. BMC Plant Biol 9:23
Ray DK, Ramankutty N, Mueller ND et al (2012) Recent patterns of crop yield growth and stagnation. Nat Commun 3:1293
Resende MFR, Muñoz P, Acosta JJ et al (2012) Accelerating the domestication of trees using genomic selection: accuracy of prediction models across ages and environments. New Phytol 193:617–624
Rocha R, Barros E, Cruz C et al (2007) Mapping of QTLS related with wood quality and developmental characteristics in hybrids (Eucalyptus Grandis X Eucalyptus mapping of QTLS related with wood quality and developmental Characteristics In Hybrids (Eucalyptus Grandis X Madeira E Crescimento EM. Rev Arvore 31:13–24
Salmon J, Ward SP, Hanley SJ et al (2014) Functional screening of willow alleles in Arabidopsis combined with QTL mapping in willow (Salix) identifies SxMAX4 as a coppicing response gene. Plant Biotechnol J 12:480–491
Samils B, Rönnberg-Wästljung AC, Stenlid J (2011) QTL mapping of resistance to leaf rust in Salix. Tree Genet Genomes 7:1219–1235
Scaracia-Mugnozza GE, Ceulemans R, Heilman PE et al (1997) Species and their hybrids grown under short rotation. II. Biomass components and harvest index of hybrid and parental species clones. Can J For Res 27:285–294
Schilling MP, Wolf PG, Duffy AM et al (2014) Genotyping-by-sequencing for populus population genomics: an assessment of genome sampling patterns and filtering approaches. PLoS One 9, e95292
Schulte RPO, Creamer RE, Donnellan T et al (2014) Functional land management: a framework for managing soil-based ecosystem services for the sustainable intensification of agriculture. Environ Sci Policy 38:45–58
Serapiglia MJ, Cameron KD, Stipanovic AJ, Smart LB (2011) Correlations of expression of cell wall biosynthesis genes with variation in biomass composition in shrub willow (Salix spp.) biomass crops. Tree Genet Genomes 8:775–788
Serapiglia MJ, Cameron KD, Stipanovic AJ et al (2013) Yield and woody biomass traits of novel shrub willow hybrids at two contrasting sites. Bioenergy Res 6:533–546
Shankhwar AK, Srivastava RK (2015) Biomass production through grey water fertigation in eucalyptus hybrid and its economic significance. Environ Prog Sustain Energy 34:222–226
Shvaleva AL, Costa E, Silva F, Breia E et al (2006) Metabolic responses to water deficit in two Eucalyptus globulus clones with contrasting drought sensitivity. Tree Physiol 26:239–248
Silva-Junior OB, Grattapaglia D (2015) Genome-wide patterns of recombination, linkage disequilibrium and nucleotide diversity from pooled resequencing and single nucleotide polymorphism genotyping unlock the evolutionary history of Eucalyptus grandis. New Phytol 208:830–845
Silva-Junior OB, Faria DA, Grattapaglia D (2015) A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol 206:1527–1540
Sims REH, Senelwa K, Maiava T, Bullock BT (1999) Eucalyptus species for biomass energy in New Zealand - part II: Coppice performance. Biomass and Bioenergy 17:333–343
Sims REH, Hastings A, Schlamadinger B et al (2006) Energy crops: current status and future prospects. Glob Chang Biol 12:2054–2076
Slavov GT, Difazio SP, Martin J et al (2012) Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa. New Phytol 196:713–725
Somerville C, Youngs H, Taylor C et al (2010) Feedstocks for lignocellulosic biofuels. Science 329:790–792
Stape JL, Binkley D, Ryan MG (2008) Production and carbon allocation in a clonal Eucalyptus plantation with water and nutrient manipulations. For Ecol Manage 255:920–930
Steane DA, Nicolle D, Sansaloni CP et al (2011) Population genetic analysis and phylogeny reconstruction in Eucalyptus (Myrtaceae) using high-throughput, genome-wide genotyping. Mol Phylogenet Evol 59:206–224
Stewart JJ, Akiyama T, Chapple C et al (2009) The effects on lignin structure of overexpression of ferulate 5-hydroxylase in hybrid poplar. Plant Physiol 150:621–635
Stolarski MJ, Szczukowski S, Tworkowski J, Klasa A (2011) Willow biomass production under conditions of low-input agriculture on marginal soils. For Ecol Manage 262:1558–1566
Stolarski MJ, Szczukowski S, Tworkowski J, Klasa A (2013) Yield, energy parameters and chemical composition of short-rotation willow biomass. Ind Crops Prod 46:60–65
Street NR, Skogström O, Sjödin A et al (2006) The genetics and genomics of the drought response in Populus. Plant J 48:321–341
Studer MH, Demartini JD, Davis MF et al (2011) Lignin content in natural Populus variants affects sugar release. Proc Natl Acad Sci U S A 108:6300–6305
Takagi H, Abe A, Yoshida K et al (2013) QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Teare MD (2011) Candidate gene association studies. In: Teare MD (ed) Genetic epidemeology. Humana Press, Totowa, NJ, pp 105–117
Terpstra IR, Snoek LB, Keurentjes JJB et al (2010) Regulatory network identification by genetical genomics: signaling downstream of the Arabidopsis receptor-like kinase ERECTA. Plant Physiol 154:1067–1078
Thavamanikumar S, Tibbits J, McManus L et al (2011) Candidate gene-based association mapping of growth and wood quality traits in Eucalyptus globulus Labill. BMC Proc 5:O15
Thavamanikumar S, McManus LJ, Ades PK et al (2014) Association mapping for wood quality and growth traits in Eucalyptus globulus ssp. globulus Labill identifies nine stable marker-trait associations for seven traits. Tree Genet Genomes 10:1661–1678
Tilman D, Balzer C, Hill J, Befort BL (2011) Global food demand and the sustainable intensification of agriculture. Proc Natl Acad Sci U S A 108:20260–20264
Truax B, Gagnon D, Fortier J, Lambert F (2012) Yield in 8 year-old hybrid poplar plantations on abandoned farmland along climatic and soil fertility gradients. For Ecol Manage 267:228–239
Tullus A, Rytter L, Tullus T et al (2012) Short-rotation forestry with hybrid aspen (Populus tremula L. × P. tremuloides Michx.) in Northern Europe. Scand J Forensic Res 27:10–29
Tuskan GA, Difazio S, Jansson S et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
United Nations (2015) Adoption of the Paris Agreement
Van Acker R, Leplé JC, Aerts D et al (2014) Improved saccharification and ethanol yield from field-grown transgenic poplar deficient in cinnamoyl-CoA reductase. Proc Natl Acad Sci U S A 111:845–850
Van Ittersum MK, Cassman KG, Grassini P et al (2013) Yield gap analysis with local to global relevance: a review. Field Crop Res 143:4–17
Vanholme R, Storme V, Vanholme B et al (2012) A systems biology view of responses to lignin biosynthesis perturbations in Arabidopsis. Plant Cell 24:3506–3529
Verlinden MS, Broeckx LS, Ceulemans R (2015) First vs. Second rotation of a poplar short rotation coppice: above-ground biomass productivity and shoot dynamics. Biomass and Bioenergy 73:174–185
Vining K, Romanel E, Jones R et al (2015) The floral transcriptome of Eucalyptus grandis. New Phytol 206:1406–1422
Virlet N, Costes E, Martinez S et al (2015) Multispectral airborne imagery in the field reveals genetic determinisms of morphological and transpiration traits of an apple tree hybrid population in response to water deficit. J Exp Bot 66:5453–5465
Volk TA, Abrahamson LP, Cameron KD et al (2011) Yields of willow biomass crops across a range of sites in North America. Asp Appl Biol 112:67–74
Wang Y, Zhang B, Sun X et al (2011) Comparative genome mapping among Populus adenopoda, P. alba, P. deltoides, P. euramericana and P. trichocarpa. Genes Genet Syst 86:257–268
Wegrzyn JL, Eckert AJ, Choi M et al (2010) Association genetics of traits controlling lignin and cellulose biosynthesis in black cottonwood (Populus trichocarpa, Salicaceae) secondary xylem. New Phytol 188:515–532
Weltecke K, Gaertig T (2012) Influence of soil aeration on rooting and growth of the Beuys-trees in Kassel, Germany. Urban For Urban Green 11:329–338
Wilkerson CG, Mansfield SD, Lu F et al (2014) Monolignol ferulate transferase introduces chemically labile linkages into the lignin backbone. Science 344(80):90–93
Wullschleger SD, Yin TM, Difazio SP et al (2005) Phenotypic variation in growth and biomass distribution for two advanced-generation pedigrees of hybrid poplar. Can J For Res 35:1779–1789
Xing Z, Maynard C (1995) Producing transgenic shining willow (Salix lucida Muhl.) shoots from stem segments via Agrobacterium tumefaciens transformation. Vitr Cell Dev Biol Plant 31:221–226
Yin T, Zhang X, Huang M et al (2002) Molecular linkage maps of the Populus genome. Genome 45:541–555
Yu A, Gallagher T (2015) Analysis on the growth rhythm and cold tolerance of five-year old eucalyptus benthamii plantation for bioenergy. Open J For 5:585–592
Yu S, Liao F, Wang F et al (2012) Identification of rice transcription factors associated with drought tolerance using the ecotilling method. PLoS One 7:1–9
Yu X, Kikuchi A, Matsunaga E et al (2013) Environmental biosafety assessment on transgenic Eucalyptus globulus harboring the choline oxidase (codA) gene in semi-confined condition. Plant Biotechnol 30:73–76
Zanchi G, Pena N, Bird N (2012) Is woody bioenergy carbon neutral? A comparative assessment of emissions from consumption of woody bioenergy and fossil fuel. GCB Bioenergy 4:761–772
Zhou X, Jacobs T, Xue L et al (2015) Exploiting SNPs for biallelic CRISPR mutations in the outcrossing woody perennial Populus reveals 4-coumarate : CoA ligase specificity and redundancy. New Phytol 208:298–301
Acknowledgement
We acknowledge support from the Seventh Framework for Research of the European Commission, for the project WATBIO (www.watbio.eu), project number, FP7-311929.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this paper
Cite this paper
Taylor, G. et al. (2016). Bioenergy Trees: Genetic and Genomic Strategies to Improve Yield. In: Barth, S., Murphy-Bokern, D., Kalinina, O., Taylor, G., Jones, M. (eds) Perennial Biomass Crops for a Resource-Constrained World. Springer, Cham. https://doi.org/10.1007/978-3-319-44530-4_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-44530-4_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-44529-8
Online ISBN: 978-3-319-44530-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)