
Abstract
The growing prevalence within community and healthcare settings of antibiotic resistant Gram-positive and Gram-negative bacterial pathogens is alarming. Particularly concerning are reports of bacteria that are resistant to last-resort antibiotics such as carbapenems and vancomycin. Thus, novel concepts are needed to face the serious challenge posed by multidrug-resistant bacterial infections. A promising alternative antimicrobial approach to conventional antibiotics involves the use of bacteriophage-derived protein(s), generically known as “enzybiotics.” Endolysins, one type of enzybiotic, are cell-wall enzymes, i.e., peptidoglycan hydrolases that act on the host bacterium late in the phage replication cycle. These enzymes hydrolyze critical covalent bonds essential for maintaining cell wall structural integrity. Due to the absence of an outer membrane, extrinsically applied recombinant endolysins have direct access to the bacterial cell wall to lyse susceptible Gram-positive pathogens. Highlighting their therapeutic potential, the efficacy of endolysins has been validated in vitro and/or in vivo against a variety of Gram-positive pathogens, and in less than 15 years since their first documented use as an antimicrobial in 2001, endolysins are now being commercially developed and undergoing clinical trials. Alternatively, phage-like or particulate bacteriocins comprise a second class of enzybiotics that can be used therapeutically. These multiprotein structures resemble bacteriophage tail-like assemblies and are produced by both Gram-negative and Gram-positive bacteria. Unlike fully functional bacteriophages, bacteriocins are incapable of replicating, though they nonetheless possess a pseudo-injection mechanism that results in loss of bacterial membrane integrity and subsequent bacterial death.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Endolysins, the Model Enzybiotic
Bacteriophages have been studied and used therapeutically to kill bacterial pathogens for ~100 years. Phage-encoded lysins (i.e., endolysins) are cell wall hydrolases that work in concert with holins to release progeny phage during a lytic infection cycle (chapter “Phage Infection and Lysis”) and have been studied for over 90 years (Twort 1925). While the lytic efficiency of endolysins to act on bacterial cell walls has been extensively investigated over the years, it was not until 2001 when Vincent Fischetti’s group first used an endolysin to treat a streptococcal infection in a mouse that these enzymes have been viewed for their therapeutic potential in the absence of phage (Nelson et al. 2001). In this report, the authors coined the term “enzybiotic” to describe both the ENZYmatic and antiBIOTIC properties displayed by the endolysin. Today, publically accessible databases are available that collate all current enzybiotic studies (i.e., EnzyBase (Wu et al. 2012)) as well as all genetically modified enzybiotics (i.e., GMEnzy (Wu et al. 2014)).
In addition to endolysins, virion-associated peptidoglycan hydrolases (VAPGHs) may be considered as enzybiotics (Rodriguez-Rubio et al. 2015). Recently, it has been suggested that the term “enzybiotic” should apply to any phage-derived protein that kills bacteria, which would include particulate bacteriocins, although not all bacteriocins act enzymatically. Others have suggested that enzybiotics should include any phage-derived enzyme that can be used for treatment of bacterial infections. This definition would encompass phage tailspike proteins, which may degrade the bacterial capsule and/or biofilms, but are not inherently bactericidal by themselves (Bales et al. 2013). Finally, there are some who favor a broad definition of enzybiotic that includes any enzyme that displays general antibacterial or antifungal properties, whether it is phage-encoded or not (Veiga-Crespo et al. 2007). While none of these definitions are universally agreed upon, for the purposes of this chapter, we will focus on phage-derived antimicrobial proteins (i.e., endolysins and bacteriocins), with emphasis on endolysins as the model enzybiotic.
Lysis from Without
Contrary to Gram-negative bacterial species, the absence of a protective outer membrane for Gram-positive bacteria makes their cell wall structures externally accessible to peptidoglycan-acting antimicrobial agents. While endolysins require a phage particle for DNA delivery as well as holins for substrate accessibility to achieve lysis from within the host bacterium, the extrinsic application of an endolysin to a Gram-positive organism would remove those two restrictions. Accordingly, endolysins have been expressed and purified as recombinant proteins and then extraneously applied to susceptible Gram-positive pathogens to induce lysis upon direct contact with the cell wall. An alternative concept of lysis from without, as effected by whole phage virions, is discussed elsewhere in this volume (chapter “Phage Infection and Lysis”).
Endolysin Structure
The protein structure of Gram-positive and Gram-negative endolysins varies due to differences in cell wall architecture between these major bacterial groups. Endolysins that target Gram-positive bacterial species are typically 25–40 kDa and employ a modular structure, with an N-terminal enzymatically active domain (EAD) linked to a C-terminal cell wall binding domain (CBD) (Loessner 2005; Loessner et al. 1995; Borysowski et al. 2006; Fischetti 2010; Lopez and Garcia 2004) (Fig. 1a, b). The EAD structure is evolutionarily conserved and responsible for the hydrolytic activity of the enzyme, i.e., cleaving particular covalent bonds within the bacterial peptidoglycan. The CBD structure is evolutionarily variable and accountable for guiding the endolysin to its substrate through the reversible binding of a highly specific cell wall-associated epitope, typically a carbohydrate. Efficient cleavage requires that the CBD binds to its unique cell wall ligand, thereby conferring species, strain, or serotype endolysin specificity (Fischetti 2010). For some endolysins, it has been shown that the CBD binds tightly to cell wall debris after cell lysis, and it is hypothesized that tight binding may prevent the enzyme diffusing and killing other uninfected bacterial cells (Loessner et al. 2002). There is structural evidence that the N-terminal EAD and C-terminal CBD of Gram-positive endolysins interact with each other prior to the CBD binding to the bacterial cell wall to render the EAD inactive. Along these lines, when the CBD binds to its cell wall epitope, there is a structural alteration that activates the EAD. To illustrate this phenomenon, the pneumococcal endolysin Cpl-1 was crystallized in free and choline-bound states (Hermoso et al. 2003). In the absence of choline, the tertiary structure of Cpl-1 was oriented into a hairpin conformation with the two domains directly interacting. Conversely, choline recognition by the CBD altered the structure of Cpl-1 to allow the EAD to be properly oriented for hydrolysis of the bacterial cell wall.

Endolysin Structure . Endolysins from bacteriophage that infect Gram-positive hosts have a modular structure that usually comprise one or more N-terminal globular EADs (blue), a flexible “linker” sequence (green), and a C-terminal CBD (red). These include (a) the listerial endolysin PlyPSA (PDB: 1XOV) and (b) the pneumococcal endolysin Cpl-1 (PDB: 2IXU). (c) However, endolysins can also contain a second CBD (magenta) produced from a translational start within the endolysin gene, as is the case for the clostridial CTP1L endolysin (PDB: 5A6S). (d) One endolysin, PlyC, is a holoenzyme composed of two EADs on one subunit and eight identical CBD subunits (PDB: 4F88)
Most endolysins that are active against Gram-positive organisms adopt the canonical EAD/CBD structure (Fig. 1a, b); however, some endolysins are known to possess multiple EADs. Likewise, some endolysins, including CTP1L, that targets Clostridium tyrobutyricum, CD27L, that targets Clostridium difficile, CS74L, that targets Clostridium sporogenes, and lys70, that targets Enterococcus faecalis, produce a second CBD as a separate gene product via an internal translation start site within the endolysin gene (Dunne et al. 2016; Proenca et al. 2015) (Fig. 1c). Additionally, at least one endolysin, PlyC, is a nine subunit holoenzyme that contains two EADs on one polypeptide called PlyCA and eight identical CBDs that form an octamer called PlyCB (McGowan et al. 2012) (Fig. 1d).
Endolysins derived from phage that infect Gram-negative hosts are generally 15–20 kDa and comprised of a single globular EAD . With a few noted exceptions (below), these endolysins lack a CBD. While the reasons are not completely understood, it is speculated that the lack of carbohydrates/wall teichoic acids associated with the Gram-negative peptidoglycan may explain the absence of CBDs, thereby allowing the EADs to directly bind the peptidoglycan (Stark et al. 2010). Two exceptions can be observed from endolysins encoded by the Pseudomonas aeruginosa phages φKZ and EL (Briers et al. 2007, 2009). These endolysins, KZ144 and EL188, consist of an N-terminal CBD and a C-terminal EAD. The KZ144 endolysin binds to P. aeruginosa with high affinity and can additionally interact with the peptidoglycan from other Gram-negative bacteria.
Bacteriolytic Mechanism
Peptidoglycan hydrolases, including endolysins, autolysins, and exolysins, cleave critical covalent bonds in the cell wall structure to induce lysis or cell wall remodeling. Peptidoglycan is essential for maintaining cell shape and counteracting the high intracellular osmotic pressure (Royet and Dziarski 2007). Its structure consists of a linear polysaccharide backbone of alternating units of β-1,4-linked N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) (Fig. 2). The lactyl group of every MurNAc unit is substituted with stem peptides, typically consisting of four alternating L- and D-amino acids. For Gram-negative bacteria and Gram-positive bacilli, the four amino acids in order starting closest to MurNAc are L-alanine, D-iso-glutamic acid, meso-diaminopimelic acid (mDAP), and D-alanine. This is commonly referred to as DAP-type peptidolgycan. For Gram-positive bacteria other than bacilli, the mDAP residue is replaced by L-lysine and is known as Lys-type peptidoglycan. Adjacent stem peptides are then cross-linked, either directly by mDAP to D-alanine via an interpeptide bond (Gram-negative and Gram-positive bacilli) or through an interpeptide bridge of amino acids (Gram-positive cocci). The composition of the interpeptide bridge varies. For example, the cell wall of Staphylococcus aureus contains a pentaglycine interpeptide bridge, whereas the stem peptides in the cell wall of Streptococcus pyogenes are interconnected by a dialanine interpeptide bridge.

Cleavage site specificity by the major classes of endolysins. Depending on the particular covalent bond that is disrupted in the cell wall structure, endolysins can be organized into one of five groups: (1) N-acetylmuramidases (i.e., lysozymes) and lytic transglycosylases, (2) N-acetyl-β-D-glucosaminidases, (3) N-acetylmuramoyl-L-alanine amidases, (4) stem peptide endopeptidase, and (5) interpeptide bridge endopeptidase
Depending on the particular bond that is cleaved within the cell wall structure, endolysins can be organized into at least five different groups: N-acetylmuramidases, lytic transglycosylases, N-acetyl-β-D-glucosaminidases, N-acetylmuramoyl-L-alanine amidases, and endopeptidases (Borysowski et al. 2006). The first two lytic activities are that of N-acetylmuramidases (i.e., lysozymes) and lytic transglycosylases. They both cleave the N-acetylmuramoyl-β-1,4-N-acetylglucosamine bond, which is one of the two alternating glycosidic bonds of the glycan moiety (Fig. 2, label 1). The other glycosidic bond in the glycan strand is cut by N-acetyl-β-D-glucosaminidases, which cleaves the linkage between N-acetylglucosaminyl-β-1,4-N-acetylmuramic acid (Fig. 2, label 2). N-acetylmuramoyl-L-alanine amidases, employing the most commonly observed lytic activity of endolysins, hydrolytically process the amide bond formed between MurNAc and L-alanine, which is the linkage responsible for conjoining the sugar and stem peptide moieties of peptidoglycan (Fig. 2, label 3). The last group of endolysins, termed endopeptidases, cleaves between two adjacent amino acids that constitute either the stem peptide (Fig. 2, label 4) or interpeptide bridge (Fig. 2, label 5).
According to the International Union of Biochemistry and Molecular Biology (IUBMB) enzyme nomenclature, glucosaminidases, muramidases, L-alanine-amidases, and endopeptidases are classified as hydrolases. Additionally, glucosaminidases and muramidases are further termed glycosidases or glycosyl hydrolases. Lytic transglycosylases are not hydrolases and instead use an intramolecular interaction to break the glycosidic bond between N-acetylmuramoyl-β-1,4-N-acetylgluocosamine by promoting the formation of a concomitant 1,6-anhydromuramoyl product (Holtje et al. 1975; Thunnissen et al. 1994). Due to the conservation in the EAD sequence and structure of endolysins, databases have grouped these domains into specific families. These subgroups relate to the hydrolytic mechanism for which the cleavage occurs rather than the specific bond cleaved. For example, the cysteine/histidine-dependent amidohydrolase/peptidase domains, known as CHAP domains , are a commonly observed endolysin family that utilizes a cleavage mechanism where a cysteine is deprotonated by a nearby histidine promoting a nucleophilic attack by the cysteine on the scissile bond. Some CHAP domains have been reported to display amidase activity (Nelson et al. 2006), whereas others have shown endopeptidase specificity, cleaving between the terminal D-alanine of the stem peptide and the adjacent amino acid of the interpeptide bridge (Pritchard et al. 2004; Becker et al. 2009).
Antimicrobial Development of Endolysins
Specificity and Resistance
One major concern with the application of an antibacterial agent is the eventual development of resistance by the bacterial microorganism targeted. With respect to the modern-day global health and economic implications of antibiotic resistance, there is an urgent need for novel antimicrobials that are not susceptible to resistance evolution. The misuse and overuse of broad-range antibiotics has accelerated the development and dissemination of antibiotic-resistant genes among the bacterial community by placing selective pressure not only on the targeted pathogen but also on susceptible commensal bacteria that are nearby (Nelson et al. 2012). In this regard, the high specificity of most endolysins is advantageous over classic broad-range antibiotics. Endolysins have the innate ability to selectively bind to and then destroy pathogenic bacterial species while avoiding the desirable commensal microflora that are often harmed by antibiotics or chemical preservatives. Additionally, the coevolution of phages with their explicit bacterial hosts over millions of years has resulted in Gram-positive endolysins binding to and cleaving conserved cell wall targets that are required for bacterial viability (Fischetti 2008; Loessner 2005). This is exemplified by the receptor epitopes targeted by pneumococcal and streptococcal endolysins. Pneumococcal endolysins bind to choline on the pneumococcal surface, which is essential for cell viability (Lopez and Garcia 2004; Garcia et al. 1983). Similarly, streptococcal endolysins have been shown to interact with polyrhamnose of S. pyogenes, which has been shown to be important for cell survival (Fischetti 2003; Yamashita et al. 1999). By virtue of the cell wall of Gram-positive bacteria being the outermost structure, the extrinsic application of endolysins can avoid common resistance mechanisms exploited by bacteria, including neutralization or degradation by antimicrobial-specific intracellular proteins and enzymes, expulsion by efflux pumps, and decreased membrane permeability.
Several endolysin-resistance studies have been performed by the Fischetti laboratory using both native and engineered endolysins. The pneumococcal endolysin Pal was repeatedly added to actively growing Streptococcus pneumoniae either in low concentrations on agar plates or using increasing concentrations in liquid culture. In both scenarios, no resistant pneumococcal strains were encountered (Loeffler et al. 2001). In a related study, the Bacillus anthracis endolysin, PlyG, was repeatedly added to Bacillus cells in the presence of the mutagenic chemical, methane-sulphonic acid ethyl ester, in an effort to expedite the evolution of resistant strains to the endolysin (Schuch et al. 2002). Similar to the Pal resistance studies, there were no PlyG-resistant strains of Bacillus recovered, whereas the same experimental method generated 1,000–10,000-fold increases in novobiocin and streptomycin resistance. Engineered endolysins have also been used in resistance studies. Increasing concentrations of the chimeric S. aureus endolysin ClyS were added to methicillin-resistant S. aureus (MRSA) cells in parallel with the antibiotic mupirocin over a total of 8 days (Pastagia et al. 2011). At the conclusion of the experiment, the minimum inhibitory concentration (MIC) for ClyS remained unchanged, while the MIC for mupirocin increased tenfold. It is important to note that, although there have been no documented cases of endolysin resistance to date, resistance to other peptidoglycan cell wall hydrolases has been observed. For example, bacterial strains have become resistant to human lysozyme by various secondary cell wall modifications, such as O-acetylation and N-deacetylation of the peptidoglycan and D-alanylation of teichoic acids (Vollmer 2008; Guariglia-Oropeza and Helmann 2011). Likewise, certain S. aureus strains have developed resistance against the Staphylococcus simulans bacteriocin, lysostaphin, which targets the pentaglycine interpeptide bridge of S. aureus as a glycyl-glycine endopeptidase (DeHart et al. 1995; Sugai et al. 1997; Grundling et al. 2006). S. aureus accomplishes this by altering the interpeptide bridge length (e.g., downsizing from pentaglycine to a single glycine (Sugai et al. 1997)) or amino acid composition (e.g., incorporating serine residues (DeHart et al. 1995)). Nonetheless, resistance to endolysins should be a rarer event compared to resistance development against traditional antibiotic classes.
Toxicity
One safety concern with endolysin therapy, or any bacteriolytic therapy, involves the release of proinflammatory cellular components during the treatment of systemic infections. Due to the bacteriolytic mechanism exploited by these enzymes, release of such components as teichoic acids, lipotechoic acids, and peptidoglycan could ultimately promote serious complications including septic shock and multiple organ failure (Nau and Eiffert 2002). There is in vivo evidence, however, that suggests this concern could be alleviated by varying the endolysin concentration during treatment. While one murine sepsis study showed that the continuous intravenous application of the pneumococcal Cpl-1 endolysin enhanced the concentration of proinflammatory cytokines (Entenza et al. 2005), results from a similar study showed that using lower concentrations of Cpl-1 actually reduced cytokine concentrations relative to untreated, bacteria-infected mice (Witzenrath et al. 2009). To explain this observation, it was hypothesized that using higher concentrations of endolysin stimulates a more complete digestion of the bacterial cell wall, thereby generating higher levels of proinflammatory cell wall fragments that continuously circulate in the serum. In addition to altering endolysin concentration, the concurrent use of anti-inflammatory agents, including steroids and nonsteroidal anti-inflammatory drugs (NSAIDs), can help combat inflammation.
Immunogenicity
Due to the proteinaceous nature of endolysins, immunogenicity studies relating to the systemic and mucosal therapeutic application of these antimicrobials in animals and humans have been conducted in vitro and in vivo. When rabbit hyperimmune serum was raised against the Cpl-1 endolysin, it was found that the in vitro activity of Cpl-1 against pneumococcal cells was slightly decreased but never fully inactivated (Loeffler et al. 2003). In the same study, it was elucidated that mouse IgG antibodies were raised against Cpl-1 when several doses of the endolysin were administered intravenously. Endolysin efficacy was also investigated in vivo using immunized and naïve mice. Following Cpl-1 treatment of immunized and naïve mice infected with S. pneumoniae, it was determined that there was no significant difference between the two groups regarding the reduction in pneumococcal cells by the enzyme, suggesting that the antibodies raised against Cpl-1 were unable to inhibit the enzyme in vivo. Similar results were obtained when using different endolysins and pathogens (S. pyogenes and B. anthracis). In each of these experiments, antibodies were successfully developed against these various endolysins (Fischetti 2010) but the antibodies were incapable of neutralizing their bacteriolytic efficacy (Jado et al. 2003; Rashel et al. 2007). In an attempt to explain how endolysins are able to avoid antibody inactivation, it has been theorized that the reported high binding affinity (e.g., Listeria monocytogenes endolysins Ply118 and Ply500 have nanomolar binding affinities) (Schmelcher et al. 2010; Loessner et al. 2002) and rapid lytic activity of endolysins are able to outcompete the immune system of the host.
Results from additional immunogenicity studies using the Cpl-1 endolysin in the treatment of systemic infections demonstrated that this enzyme had a half-life of approximately 20 min, which is comparable to other therapeutic proteins such as streptokinase, a thrombolytic enzyme used for treatment of acute myocardial infarction (Brucato and Pizzo 1990; Loeffler et al. 2003). Beyond delivering endolysins repeatedly or by intravenous diffusion to achieve systemic concentrations during treatment, chemically modifying or bioengineering endolysins have proven to prolong half-life. PEGylation is a commonly used chemical modification for successfully slowing the systemic clearance rate of molecules by reducing antibody binding affinity (Ramon et al. 2005; Walsh et al. 2003; Kim et al. 2008). Unfortunately, it has been reported that the cysteine-specific PEGylation of Cpl-1 ultimately abolished lytic activity (Resch et al. 2011a). However, PEGylating other peptidoglycan hydrolases, such as the bacteriocin lysostaphin, significantly improved the pharmacokinetics while rendering the enzyme still bacteriolytically active (Walsh et al. 2003). Bioengineering improved endolysin pharmacokinetic properties has proven to be more successful than introducing chemical modifications. For example, dimerization of Cpl-1 by engineering a C-terminal intermolecular disulfide bond increased activity and promoted a tenfold deceleration of plasma clearance (Resch et al. 2011b). Since several endolysins have been implicated to undergo dimerization, an engineering strategy of constructing stable disulfide-based endolysin dimers may be successful at expanding systemic half-life.
Synergy
In combination with other peptidoglycan hydrolases or antibiotics, endolysins have demonstrated synergistic therapeutic efficacy when analyzed in vitro and in vivo. Achieving synergism between two or more antimicrobials translates to lower dosage requirements and improved efficacy while reducing the chances of resistance formation by concurrently acting on multiple unique targets associated with individual bacteria. Synergy between two endolysins has been observed when using enzymes with different peptidoglycan cut sites, as the simultaneous cleavage of two distinct bonds in the cell wall would conceivably have enhanced destabilizing effects (Schmelcher et al. 2012a). Improved lytic activity could also be due to the cleavage of a particular site by the first catalytic domain that improves substrate accessibility for the catalytic domain of the second endolysin (Schmelcher et al. 2012b; Loeffler and Fischetti 2003). An example of endolysin synergism was exemplified by the pneumococcal endolysins Cpl-1 and Pal. The combinational use of Cpl-1 (muramidase) and Pal (L-alanine amidase) provoked in vitro and in vivo synergistic efficacy (Loeffler and Fischetti 2003; Jado et al. 2003). The bacteremic titer when the two pneumococcal endolysins were used together in a murine sepsis challenge was reduced to a greater extent compared to when either endolysin was used alone.
Synergistic effects were also observed in vitro when using the S. aureus endolysin LysK (L-alanine amidase/D-alanyl-glycine endopeptidase) with lysostaphin (glycyl-glycine endopeptidase) (Becker et al. 2008). Similarly, combining lysostaphin with either of the chimeric streptococcal/staphylococcal endolysins λSA2-E-Lyso-SH3b and λSA2-E-Lyso-SH3b resulted in synergistic ramifications in vitro and in vivo using a mouse model of bovine mastitis (Schmelcher et al. 2012b). Synergy has also been validated when combining endolysins with antibiotics. For example, antipneumococcal synergy was established when combining Cpl-1 with penicillin or gentamicin (Djurkovic et al. 2005). Synergistic efficacy was also seen in vitro when the staphylococcal MV-L endolysin was added to either glycopeptide antibiotics vancomycin or teicoplanin to kill vancomycin-intermediate-resistant S. aureus (Rashel et al. 2007), as well as in vivo when the chimeric staphylococcal ClyS endolysin was combined with oxacillin to cure mice from MRSA induced septicemia (Daniel et al. 2010). Recently, synergy has been noted between the antibiotic, daptomycin, and the pneumococcal Cpl-1 endolysin in a pneumococcal bacteremia model (Vouillamoz et al. 2013).
Endolysin Applications
Medicine
The molecular cloning, protein purification, and subsequent biochemical characterization of endolysins has been a research concentration in laboratories for decades. The medicinal application of these enzymes was not tested in vivo until 2001 when a streptococcal endolysin (later called PlyC) was shown to prevent and treat bacterial infections (Nelson et al. 2001). In this study, mice were first pretreated with a single 250 U (2.5 ng) dose of the endolysin in the oral cavity followed by the addition of 107 colony forming units (CFU) of live S. pyogenes. The pretreated mice were protected from bacterial colonization (28.5% infected, n = 21) compared to the untreated control mice (70.5% infected, n = 17) (P < 0.03). In the same study, S. pyogenes was introduced to the oral cavity of nine mice which were subsequently found to be heavily colonized for four consecutive days. On day 4, the infected mice were then orally treated with 500 U (5 ng) of PlyC. Two hours post-endolysin treatment, the nasopharyngeal cavity of all nine mice was found to be sterilized.
Among the most studied endolysins in vivo are Pal and Cpl-1, both of which are derived from phage that infect S. pneumoniae. The in vitro application of Pal at 100 U/ml (100 μg/ml) was able to induce a ~4-log decrease in viability after 30 s when applied to 15 different clinical strains of S. pneumoniae, including several strains that were penicillin-resistant (Loeffler et al. 2001). In an in vivo murine model for nasopharyngeal S. pneumoniae colonization, 18 mice were first colonized intranasally with 108 CFU of S. pneumoniae. Forty-two hours postinfection, half of the mice (n = 9) were then treated with 1400 U (1.4 mg) of Pal. This single dose of Pal was able to eliminate S. pneumoniae nasopharyngeal colonization within 5 h. Another study involved using a murine bacteremia model, where 20 mice were intravenously infected with 3 × 108 CFU of S. pneumoniae (Loeffler et al. 2003). One hour later, ten mice were intravenously treated with a single bolus containing 2,000 μg of Cpl-1. All endolysin-treated mice survived throughout the duration of the 48-h experiment, whereas only 20% of the untreated mice survived, with a median survival time of 25.6 h. Both Cpl-1 and Pal were also shown to protect mice from pneumococcal bacteremia induced by intraperitoneal (i.p.) injection in a synergistic manner in vivo (Jado et al. 2003). While intraperitoneally administering 200 μg of either Cpl-1 or Pal 1 h postinfection was found to be the required dose to protect mice intraperitoneally infected with 5 × 107 CFU of S. pneumoniae, only 2.5 μg of each endolysin was required to achieve the same effect when both Cpl-1 and Pal were used simultaneously. More recent in vivo studies using Cpl-1 include: continuous 250 mg/kg endolysin infusion treatment of S. pneumoniae-induced endocarditis in rats, resulting in sterilizing 105 CFU/ml pneumococci in the blood after 30 min and a > 4-log CFU/g reduction in bacterial titers on heart valve vegetations (i.e., infected masses) (Entenza et al. 2005); prevention of otitis media secondary problems as a result of primary influenza infections through Cpl-1 stimulated pneumococcal decolonization (McCullers et al. 2007); treatment of pneumococcal meningitis in rats by either intracisternal (20 mg/kg) or i.p. (200 mg/kg) injection of Cpl-1 decreased pneumococcal titers in cerebrospinal fluid (CSF) by 3- or 2-logs, respectively (Grandgirard et al. 2008); aerosolized Cpl-1 reduced mortality by 80% in a model of fatal pneumococcal pneumonia (Doehn et al. 2013); and for treatment of severe pneumonia in mice (Witzenrath et al. 2009).
Another streptococcal species that has been subjected to in vivo endolysin treatment is Streptococcus agalactiae (Group B streptococcus, GBS), the most common causative agent of neonatal sepsis and meningitis, using murine vaginal and oropharynx infection models (Cheng et al. 2005). Thirty mice were initially inoculated vaginally with 106 streptomycin-resistant GBS. Twenty-four hours later, the vaginal cavities were swabbed to determine the initial colonization rate of GBS. The mice were then treated with 10 U (~500 μg) of the GBS endolysin PlyGBS (n = 15) or enzyme buffer only (n = 15) and the vaginal cavities were swabbed at 2- and 4-h time points posttreatment to determine bacterial titers. Mice treated with PlyGBS displayed a significant ~3-log decrease in GBS CFU at the 2- and 4-h time points when compared to the buffer only controls (P < 0.0001). To determine if PlyGBS could be used for postpartum treatment in newborns, 38 mice were inoculated with 108 CFU of streptomycin-resistant GBS in the oropharynx. The next morning, mice were oropharyngeally swabbed in order to calculate baseline GBS colonization. Mice were then treated orally and nasally with 10 U (~500 μg) PlyGBS (n = 20) or enzyme buffer only (n = 18). GBS titers elucidated by oropharyngeal swabs at 2- and 24-h time points showed the endolysin-treated mice had a significant reduction in GBS colonization at both time points compared to the bacterial levels in the buffer control group (P < 0.0001).
Besides the treatment of streptococcal infections, the therapeutic efficacy of endolysins has been validated against B. anthracis, an infectious agent commonly associated with biowarfare. PlyG, an endolysin from the B. anthracis γ phage, displayed the ability to kill both vegetative cells and germinating spores (Schuch et al. 2002). Mice i.p. infected with 106 CFU of Bacillus cells died within 5 h when treated with enzyme buffer only. Mice i.p. treated with 50 U (50 μg) of PlyG 15 min postinfection led to 13 of the 19 mice (68.4%) surviving the duration of the 72 h experiment and extended the life of the remaining mice several fold over the untreated controls (P < 0.0001). Furthermore, a 5-min treatment with 10 U (10 μg) of PlyG was able to generate a 7,500-fold reduction in heat-activated Bacillus spore viability. Another Bacillus endolysin, PlyPH, was biochemically characterized to display lytic activity over a broad pH range of 4.0–10.5 and exhibited the ability to rescue mice from i.p. challenge of Bacillus (Yoong et al. 2006). Mice (n = 26) were injected with 2.5 × 106 CFU of Bacillus cells into the peritoneal cavity and then treated 10 min later with 1,200 μg of PlyPH (n = 13) or enzyme buffer only (n = 13). At the conclusion of the 38-h study, ~40% of the PlyPH-treated mice survived, whereas 100% of the buffer-treated mice were unable to be rescued (P < 0.02).
There are several studies that report an ability to control S. aureus infections in animal models through the use of endolysin therapy. The MV-L endolysin, encoded by the S. aureus phage ΦMR11, displayed in vitro and in vivo bacteriolytic activity against multidrug-resistant S. aureus strains, including MRSA and vancomycin-resistant S. aureus (VRSA) clinical isolates (Rashel et al. 2007). In a murine i.p. infection model, the peritoneal cavity of mice was injected with 5 × 109 CFU of MRSA cells, followed by treatment at different time points postinfection with 500 U (50 μg) of MV-L or enzyme buffer only. Endolysin treatment at 0, 30, and 60 min postinfection resulted in respective mouse survival rates of 100%, 100%, and 60% at the conclusion of the 60 day study. Conversely, 100% of the mice were killed within 24 h postinfection when treated with enzyme buffer only at each time point. In another study, the chimeric staphylococcal endolysin ClyS was shown to be more effective than the standard topical antimicrobial mupirocin for treating murine epidermal infections caused by methicillin-susceptible S. aureus (MSSA) and MRSA (Pastagia et al. 2011). At 24 h postcolonization with MSSA or MRSA, the infected epidermal tissue of mice was treated with either 10% (wt/wt) of ClyS in Aquaphor or 2% mupirocin. With respect to the untreated controls, ClyS treatment caused a 3-log reduction in bacterial colonization while mupirocin achieved a 2-log decrease (P = 0.001).
In a mouse nasal decolonization model, a 2-log reduction in MRSA viability was observed 1 h after nasal and oral treatment with 960 μg of ClyS (n = 20) when compared to buffer-treated mice (n = 20) (P < 0.002) (Daniel et al. 2010). Additionally, over the course of a 10 day experiment, a single i.p. injection of 1000 μg of ClyS promoted the survival of 14 out of 16 mice (87.5%) that were subjected to a murine septicemia model for MRSA infection. As a basis of comparison, mice treated with enzyme buffer (n = 14) were dead from bacterial sepsis within 24 h. Using the aforementioned murine septicemia model, therapeutic synergism was attained when using ClyS in combination with oxacillin. Mice were i.p. injected with ~5 × 105 CFU of MRSA and then treated i.p. 3 h postinfection with 166 μg ClyS, 100 μg oxacillin, or 166 μg Clys combined with 50 or 100 μg of oxacillin. At the conclusion of the 10-day study, mice treated with ClyS or oxacillin individually resulted in 30% (n = 20) and 35% (n = 23) survival rates, respectively. Contrarily, ClyS simultaneously applied with 50 or 100 μg of oxacillin significantly increased mouse survival to a respective 80% (n = 10) and 82% (n = 22).
In studies with other antistaphylococcal enzymes, a successful 2-log decrease in S. aureus colonization was reported when applying the isolated catalytic CHAP domain of the staphylococcal endolysin LysK (CHAPk) in a murine intranasal infection model (Fenton et al. 2010). S. aureus endolysins LysGH15 (50 μg per mouse, n = 3) and P-27/HP (250 μg per mouse, n = 15) were able to induce a respective ~7- and 2-log decrease in staphylococcal colonization in murine spleens and subsequently prevent bacteremia lethality (Gu et al. 2011a, b; Gupta and Prasad 2011). Finally, CF-301 (i.e. PlySs2), an enzyme being developed by the ContraFect Corp., increased survival by reducing MRSA bacteremia 100-fold within 1 h (Schuch et al. 2014). Combination therapy of CF-301 with either vancomycin or daptomycin increased mouse survival of MRSA bacteremia compared to antibiotics alone (P < 0.0001) and this endolysin/antibiotic synergy was confirmed in 26 independent MRSA and 29 MSSA strains. The U.S. Food and Drug Administration (FDA) granted a Fast Track designation to CF-301, which completed Phase 1 human clinical trials in December 2015, and is expected to began Phase 2 clinical trials in 2017.
Thus, in the past 10 years, there exists significant in vivo data supporting the therapeutic use of multiple endolysins.
Food Safety
Foodborne illness is a costly public health problem that is both prevalent and preventable. The Centers for Disease Control (CDC) estimates that 48-million people in the United States each year are infected by foodborne pathogens by consuming contaminated foods or beverages, resulting in over 120,000 hospitalizations and 3,000 deaths. There have been over 250 different foodborne diseases elucidated, for many of which the causative agent is a bacterial species. The FDA’s Center for Food Safety and Applied Nutrition (CFSAN) has constructed a list of foodborne pathogenic microorganisms that cause human disease in the United States. Some examples of Gram-positive pathogens on this list include L. monocytogenes, Clostridium perfringens, Clostridium botulinum, Bacillus cereus, and S. aureus. Significantly, lytic phages have been identified for each of the aforementioned bacterial pathogens, and in many cases, the endolysins they encode have been cloned, biochemically characterized to various extents, and experimentally validated to be effective for both detecting and eradicating foodborne pathogens.
Considering that many foodborne illnesses caused by Gram-positive pathogens develop rapidly and often require supportive care for treatment, preventing the initial infection is critical. Current bacterial diagnostic techniques, such as immunoassays and DNA-based methods, require up to 107 cells in order to accurately identify the causative bacterial pathogen. The infectious dose of many foodborne bacterial pathogens, however, is often orders of magnitude less. Additionally, these methodologies often take several days to complete. To this end, the development of new rapid diagnostics with increased sensitivity is required for effectively lowering the number of foodborne illnesses.
Endolysin CBDs have high specificity and nanomolar equilibrium association constants for their particular cell wall receptor and retain their functional binding abilities without the presence of the N-terminal EAD (Loessner et al. 2002). This mechanistic understanding outlines the potential of these domains to be utilized for biotechnological purposes as an alternative diagnostic tool for foodborne bacterial pathogens. CBDs can be chemically cross-linked with an organic dye or genetically fused to a reporter protein (e.g., GFP, CFP, DsRed) for fluorescent detection by spectrophotometric or microscopic analysis. Furthermore, CBDs can be conjugated to magnetic nanoparticles for selective enrichment of target bacteria to a detectable level for diagnostics.
Listeriosis is an infection caused by the major foodborne pathogen, L. monocytogenes, that can cause meningitis, meningoencephalitis, septicemia, and generalized infection of the fetus resulting in abortion (Vazquez-Boland et al. 2001). Depending on the risk group studied, the fatality rate associated with listeriosis can be as high as 50% for newborns and 20% for the elderly (Bortolussi 2008). The CDC estimates that approximately 1,600 illnesses and 260 deaths are due to listeriosis in the United States annually. L. monocytogenes present in contaminated food products is often difficult to detect due to the typically small L. monocytogenes cell populations coupled with the low sensitivity of current Listeria diagnostics.
Presently, growing Listeria from contaminated food products in selective enrichment media is an absolute requirement in order to increase the number of total viable Listeria cells to detectable levels. The efficiency of the standard selective enrichment protocol is hindered by the total length of the 96-h protocol. As an alternative detection methodology, paramagnetic beads coated with purified recombinant Listeria-specific endolysin CBDs were used for the detection of Listeria in food samples, a diagnostic approach known as CBD-based magnetic separation (CBD-MS). In this approach, the CBD-coated beads function to capture and concentrate Listeria cells prior to growth on standard plates. Detection rates for a variety of Listeria species and serovars (L. monocytogenes, L. ivanovii, and L. innocua) in foods naturally and artificially exceeded 90% in heterogeneous microbial communities (Kretzer et al. 2007). A total of 275 potentially naturally contaminated food samples were tested for Listeria contamination using either standard selective enrichment media or CBD-MS. In totality, Listeria was detected in 42 samples using standard selective enrichment media, compared to the 45 samples that were found to be Listeria-positive using CBD-MS. Besides the enhancement in sensitivity, the CBD-MS method is more rapid and only requires half the time (48 h) compared to the standard selective enrichment media method. The CBD-MS method was also shown to be applicable to other bacterial species, such as B. cereus and C. perfringens. In addition, the CBD-MS separation method was combined with real-time quantitative PCR to yield a highly sensitive detection limit for L. monocytogenes in raw milk ranging from 102 to 103 CFU/ml (Walcher et al. 2010).
Another endolysin-related methodology being utilized for the detection of bacteria involves fusing phage endolysin CBDs to various fluorescent markers. The fluorescent CBDs can then be used for the detection and differentiation of bacterial species and strains. For example, eight different Listeria CBDs, each of which had unique cell wall specificity, were fused to a collection of differently colored reporter proteins (Schmelcher et al. 2010). When added to a mixed population of L. monocytogenes, the fluorescent CBDs made it possible to rapidly detect and distinguish individual cells with serovar-to-strain specificity using fluorescence microscopy. This technique was further used in combination with CBD-MS for the recovery and identification of different Listeria serovars and strains from artificially contaminated milk or cheese. CBD-based detection methods have also been described for B. anthracis (Fujinami et al. 2007; Sainathrao et al. 2009). The latter study was able to employ a bioengineered CBD derived from the PlyG endolysin that was coupled to fluorescent Qdot® nanocrystals for strain-specific detection of B. anthracis. Although the authors do not discuss how this approach compares to antibody detection methods, as stated earlier in this section, CBDs have been shown to possess nanomolar affinities for the bacterial surface, which is on par to the affinities seen for diagnostic antibodies.
Supplementary to using isolated CBDs for bacterial diagnostics, the antimicrobial efficacy of full-length endolysins has been validated as a means to control a series of foodborne pathogens, including S. aureus, S. agalactiae, Streptococcus uberis, L. monocytogenes, Clostridium butyricum, and Erwinia amylovora. Purified endolysins were shown to successfully control foodborne pathogens when utilized as biopreservatives for food products. For example, the S. aureus ΦH5 phage endolysin, LysH5, rapidly reduced staphylococcal cell populations growing in pasteurized milk to undetectable levels after 4-h incubation at 37 °C (Obeso et al. 2008). A number of other endolysins, including the streptococcal endolysins B30 (Donovan et al. 2006) and Ply700 (Celia et al. 2008), as well as the chimeric streptococcal-staphylococcal constructs λSA2E-Lyso-SH3b and λSA2E-LysK-SH3b (Schmelcher et al. 2012b), have been shown to display bacteriolytic activity in milk or milk products. With the ability to retain significant lytic activity after incubating at 90 °C for 30 min, the kinetically stable L. monocytogenes endolysins Ply511, Ply118, and Ply500 could be applicable as a disinfectant for food products that undergo elevated heat treatment, such as pasteurized milk (Schmelcher et al. 2012c). These three Listeria endolysins were also effective for decreasing L. monocytogenes viability on iceberg lettuce by up to 2.4-log units after storage for 6 days. Another example of using phage endolysins to prevent the growth of foodborne pathogens involved pretreating pear slices with E. coli lysates containing overexpressed E. amylovora phage ΦEa1h lysozyme. After inoculating the fruit with bacteria and incubating the samples at 26 °C for 5 days, lysate pretreatment was able to prevent E. amylovora colonization on the surface of the pear slices (Kim et al. 2004).
Endolysins can also be produced by starter microorganisms such as Lactococccus lactis, as used for fermentation processes in order to prevent contamination and consequent economic loss for manufacturers. Contamination during fermentation can decrease food quality as well as possibly presenting future health risks if no further pasteurization occurs. Whereas broad spectrum antimicrobials cannot be used due to the nonspecific killing of the starter microorganisms, the high specificity of endolysins makes them ideal antimicrobial candidates for controlling contamination. One validated strategy involves genetically inserting endolysin genes into starter microorganisms, followed by the expression and secretion of the enzyme into the environment for the eventual eradication of unwanted bacteria. Genetically modifying fermentation bacteria for the expression and secretion of endolysins derived from both Listeria (Gaeng et al. 2000; Turner et al. 2007; Stentz et al. 2010) and Clostridium phages (Mayer et al. 2010), as well as the bacteriocin lysostaphin (Turner et al. 2007), has been reported, although the application of these microorganisms to food products has yet to be shown.
Agriculture
Several agricultural applications for phage endolysins have been explored. For instance, endolysin-producing transgenic plants are able to avoid phytopathogenic bacterial infections. Transgenic potatoes producing T4 lysozyme were resistant against Erwinia carotovora, a plant pathogen known to cause black leg and soft rot (de Vries et al. 1999; Düring et al. 1993). Lysozyme, which continuously accumulates within the transgenic plant tissue, is simultaneously released as a result of plant cell damage caused by E. carotovora pectinases. As a result, the intracellular lysozyme is liberated and has the ability to lyse the phytopathogens upon direct contact. Besides the implications affiliated with phytopathogen resistance, transgenic plants programmed to express endolysins can serve as bioreactors capable of large-scale, inexpensive production of these antimicrobials. To illustrate this point, the S. pneumoniae endolysins Cpl-1 and Pal (Oey et al. 2009b) and the S. agalactiae endolysin PlyGBS (Oey et al. 2009a) were integrated into the plastid genome of the tobacco plant Nicotiana tabacum. The total protein expression of Cpl-1 and Pal was respectively estimated to represent ~10% (i.e., 0.5 g protein per kg of fresh weight) and ~30% (i.e., 2 g protein per kg of fresh weight) of the total soluble protein levels of the plant, while expression of PlyGBS was calculated to reach more than 70% of the total plant soluble protein. These immense protein expression yields were rationalized to be caused by the highly stable intracellular environment provided by the chloroplasts, as indicated by the absence of endolysin turnover during plant development.
In the case of animal agriculture, endolysins have been investigated as a disinfectant as well as prophylactic or therapeutic antimicrobial agents as alternative means of preventing or combating animal disease. Equine strangles is a highly contagious infection of the lymph nodes of horses caused by Streptococcus equi. This disease is characterized by the expeditious onset of fever followed by upper respiratory tract catarrh, causing nasal discharge and acute swelling with subsequent abscess formation in the throat area (Sweeney et al. 2005). If left untreated, infected horses can suffocate due to enlarged lymph nodes that obstruct the airway. Serious complications associated with equine strangles occur in approximately 20% of infected horses, resulting in mortality rates as high as 8% on farms where the infection is endemic (Sweeney et al. 1987). The efficacy of the streptococcal endolysin, PlyC, was benchmarked as a narrow-spectrum disinfectant for eliminating S. equi on horse stable surfaces and equipment (Hoopes et al. 2009). PlyC was shown to be 1,000× more active per weight than Virkon-S, a common disinfecting agent, with 1 μg of endolysin possessing the ability to sterilize a 108 CFU/ml culture of S. equi in 30 min. Furthermore, PlyC was efficacious when applied as an aerosol to common stable and horse-related equipment contaminated with S. equi and capable of retaining lytic activity in conditions that mimic a horse stable, including the presence of nonionic detergents, hard water, or organic materials.
Use of peptidoglycan hydrolases for prophylactic and therapeutic applications specific to animal disease have also been tested and validated. Bovine mastitis, a microbial intramammary infection that induces inflammation of the mammary gland in cattle, affects one third of all dairy cows (Sordillo and Streicher 2002). This specific disease causes reduced milk production in addition to increased replacement costs, discarded milk, treatment costs, veterinary fees, and labor costs. The National Mastitis Council estimates economic losses are approximately $200 per cow annually in the United States, equating to a total exceeding $2 billion. Treatment of bovine mastitis with classical antibiotics is frequently ineffective and thus alternative antimicrobial approaches are needed (Brouillette and Malouin 2005). While an assortment of bacteria cause bovine mastitis (e.g., P. aeruginosa, S. agalactiae, E. coli, Mycoplasma, and Klebsiella pneumoniae), staphylococci are the most prevalent pathogen associated with the disease and, as such, have been targeted in early studies using peptidoglycan hydrolases to prevent or treat infection. Lysostaphin at a concentration of 10 μg/ml in milk was able to generate a 5- to 6-log reduction in S. aureus (Bramley and Foster 1990). After a 108 CFU inoculation of S. aureus, infusion of the lactating murine mammary gland with 10 μg of enzyme stimulated a 2- to 3-log reduction in bacteria within 30 min. Pretreating the murine mammary gland with 10 μg of lysostaphin either immediately before or 1 h prior to staphylococcal challenge caused a 6-log decrease in S. aureus viability. In an independent study, S. aureus was introduced to the teat cistern of cattle and infections were established for a minimum of 3 weeks (Oldham and Daley 1991). A bactericidal concentration of lysostaphin against 104 CFU S. aureus was maintained in the milk extracted from the mammary gland of infected cows for up to 36–48 h following treatment with a single 100 mg teat infusion of enzyme. The cure rate of infected cows administered with 100 mg of lysostaphin over three consecutive p.m. milkings was 20%, which is comparable to the 29% cure rate obtained using the antibiotic sodium cephapirin. Moreover, transgenic mice (Kerr et al. 2001) and cows (Wall et al. 2005) producing lysostaphin in the mammary glands were resistant to S. aureus-induced mastitis. Considering the application of a single antimicrobial may ultimately promote resistance formation, using chemotherapeutic agents in combination may prove to be a more efficient strategy. To this end, using lysostaphin together with the chimeric endolysin λSA2E-LysK-SH3b prompted the synergistic killing of S. aureus both in vitro and in a murine model of bovine mastitis (Schmelcher et al. 2012b).
Endolysin Engineering Approaches
The coevolution of phage and bacteria over millions, if not billions, of years has resulted in the extensive optimization of endolysins that efficiently and rapidly induce bacterial lysis starting from within the bacterial intracellular environment. Considering these enzymes have not evolved for exogenous applications, it is speculated that they are not optimized for these functions and there exists an engineering potential for endolysins to alter their functionality in, and tolerance of, complex extracellular environments, including food matrices, blood, and the surface of mucous membranes (Schmelcher et al. 2012a). Currently, a variety of protein engineering approaches can be exploited towards improved protein catalysis, specificity, folding, and stability. Engineering desirable properties to enzymes can be achieved using either rationale-based (e.g., chimeragenesis, structure guided site-directed mutagenesis, or in-silico computational modeling) or random techniques (e.g., random mutagenesis, gene shuffling, or directed evolution) (Fig. 3).

Engineering approaches for endolysins. Multiple engineering approaches can be employed to bioengineer endolysins including rational and random methods
Chimeragenesis
The modular protein architecture of endolysins derived from phages that infect Gram-positive hosts makes them ideal candidates for constructing chimeras with modified catalytic and/or binding properties. This engineering approach has been successfully exploited by nature itself, where several chimeric phage endolysins have been identified to have been formed as a result of horizontal gene transfer and intragenic recombination (Hendrix 2002), such as the L. monocytogenes endolysin PlyPSA (Zimmer et al. 2003) and the pneumococcal endolysin Pal (Sheehan et al. 1997). The structure of PlyPSA consists of a CBD that resembles that of other Listeria endolysins but consists of an evolutionary divergent EAD that is homologous to amidase domains from Bacillus and Clostridium phages.
Crystal structures of endolysins as well as detailed bioinformatic analysis of their protein sequences have provided the necessary framework to devise and construct functional chimeras consisting of domains from different origins. The earliest studies involving domain shuffling of peptidoglycan hydrolases were conducted in the 1990s. Swapping domains between the pneumococcal autolysin LytA (amidase) and endolysin Cpl-1 (muramidase) culminated in chimeric enzymes with exchanged lytic activities and regulatory properties (Diaz et al. 1990). Furthermore, cell wall specificity was altered by interchanging the choline-dependent CBD from LytA with the choline-independent CBD from the pneumococcal Cpl-7 endolysin (Diaz et al. 1991). While the previous two examples proved exchanging modules between peptidoglycan hydrolases that target the same bacterial species can result in a functional chimeric enzyme with unique properties, it was still unknown if it was feasible to synthesize chimeras using two peptidoglycan hydrolases specific to bacteria from different genus classifications. This was answered in 1993, when the EAD of the Clostridium acetobutylicum autolysin Lyc was fused to the choline-dependent CBD of Cpl-1. The resulting chimeric enzyme displayed lytic activity against choline-containing pneumococcal cell walls but was devoid of activity against C. acetobutylicum cell walls (Croux et al. 1993a). In an independent study, fusing the CBD of Lyc to the EAD of LytA improved activity against C. acetobutylicum 250-fold (Croux et al. 1993b).
Constructing chimeras has also been shown to be useful for broadening specificity. An example is the fusion of the full-length or truncated S. agalactiae B30 endolysin to lysostaphin, which yielded chimeric enzymes that displayed antibacterial efficacy towards both streptococcal and staphylococcal cells (Donovan et al. 2006). In experiments where Listeria endolysin CBDs were labeled with GFP and subsequently added to cells for bacterial diagnostic purposes, fusing the Ply500 CBD, which is specific for serovars 4, 5, and 6 L. monocytogenes strains, to the PlyP35 CBD, which targets the majority of serovar 1/2 and 3 strains, produced heterologous GFP constructs that had broad binding specificity for Listeria cells from all species and serovars (Schmelcher et al. 2011). It was also shown that combining two Ply500 CBDs together resulted in a 50-fold increase in affinity to the cell wall when compared to a construct consisting of a single Ply500 CBD. Interestingly, this higher affinity to the cell wall led to a decrease in lytic activity at physiological salt concentrations. It is speculated that this is due to slower release of the endolysin after it kills a cell, slowing its diffusion to the next target cell. While naturally advantageous for phage propagation, the high binding affinity to cell wall epitopes displayed by endolysin CBDs could decrease the antibacterial killing kinetics when extrinsically applied as a therapeutic. At high salt concentrations, however, having two CBDs increased activity over the construct with a single CBD. In this case, the binding was inhibited by the high salt which was then compensated for by having two CBDs. Therefore, depending on the application that is desired, the affinity can be engineered to favor therapeutic or instead high salt industrial applications.
One aspect of this chimeragenesis that will need further addressing is the specifics of linkage and assembly order of the EADs and CBDs. In the study of the Ply500 and Ply118 CBDs fused to GFP, changes in the order of the domains and the linkers used to join them altered the ability of the CBDs to bind the different species and serovars of Listeria (Loessner et al. 2002; Schmelcher et al. 2010). Domain linkage and assembly order in chimeric endolysins has effects not just on lytic activity but also on basic recombinant protein expression and solubility.
Chimeragenesis can also reduce the possibility of bacteria developing resistance to an endolysin. No resistance has been reported to any phage endolysin as of yet (Loeffler et al. 2001; Schuch et al. 2002; Singh et al. 2014). However, bacteria have developed resistance to the bacteriocin lysostaphin, which does resemble an endolysin. Fusing lysostaphin to the endolysin HydH5 expanded the strain specificities of lysostaphin to cover those strains hit by HydH5 (Rodriguez-Rubio et al. 2012) while, at the same time, eliminating the development of resistance by the targeted bacteria (Rodriguez-Rubio et al. 2013). Having two different EADs acting on the cell wall requires the bacteria to simultaneously develop resistance at two different cell surface targets to achieve an overall resistance, greatly reducing any chance of acquiring resistance to the chimeric endolysin.
It is envisioned that similar approaches to making chimeras of one or more EADs and/or CBDs will lead to next-generation endolysins with expanded host range and/or increased activity. These next generation endolysins could be targeted against two or three common bacterial pathogens that cause a specific illness, be it food poisoning or skin infection. This will mean fewer endolysin types will be needed per treatment, with fewer to produce, store, or to go through clinical trials. Additionally, these chimeric endolysins may see similar use in food processing, lessening the need for antibiotics in animal feed, or instead in other applications such as controlling bacterial contamination during biofuel production (Roach et al. 2013). Identification of new and divergent EADs and CBDs from phages, bacterial genomes, or metagenomic sequences will lead to opportunities to improve activity and targeting of chimeric endolysins. Other aspects of engineered endolysins such as solubility, long-term storage, and in vivo half-life may be addressed by domain swapping, or they may be addressed by other means such as directed evolution or structure guided site-directed mutagenesis.
Directed Evolution
Random mutagenesis studies have been performed with endolysins to enhance desirable properties. Specifically, directed evolution, which cultivates Darwinian evolution in a laboratory setting to engineer desirable macromolecular properties, was applied to the S. agalactiae endolysin PlyGBS, also known in the literature as B30, to improve its bacteriolytic activity (Cheng and Fischetti 2007). Random mutations were introduced into PlyGBS by either E. coli mutator strains or error-prone polymerase chain reaction (epPCR) and subsequently screened for improved lytic potency using soft agar overlay assays. These assays consist of first replica plating the original transformation plate containing PlyGBS mutant colonies while simultaneously inducing protein expression, followed by exposing the plate to chloroform vapors to liberate the overexpressed endolysins and then finally overlaying the plate with soft agar containing S. agalactiae target cells. Activity was assessed based on the qualitative evaluation of lytic clearing zones. After multiple rounds of random mutagenesis and screening, the lead PlyGBS mutant candidate exhibited a 28-fold increase in bacteriolytic activity compared to wild-type. This evolved construct consisted of a frameshift mutation that resulted in the deletion of the C-terminal CBD, leaving only the N-terminal endopeptidase EAD as well as 13 out-of-frame amino acids at the C-terminus. Aside from improving the EAD activity, directed evolution can be used for increasing CBD binding affinity, augmenting thermal stability or modifying pH and salt dependence for activity.
Directed evolution was used to improve the thermostability of the PlyC endolysin, which targets S. pyogenes. PlyC is unusual in that it consists of two subunits, i.e., separate polypeptides, PlyCA and PlyCB, which provide lytic activity and cell wall binding, respectively. Eight PlyCB subunits form a ring-like structure that binds a single PlyCA subunit. The PlyC enzyme loses activity at 45 °C. Examination of the subunits revealed that the instability is due to PlyCA, as PlyCB was stable up to ~90 °C by differential scanning calorimetry, a thermoanalytical technique that measures phase transitions. Therefore, random mutagenesis was used on PlyCA to improve its thermostability (Heselpoth and Nelson 2012). Error-prone PCR was performed on a PlyCA expressing plasmid, which was transformed into E.coli expressing PlyCB. Colonies were transferred into duplicate 96-well plates; one plate was stored at 4 °C and the second plate was grown, expressed, solubilized, and screened for lytic activity against S. pyogenes after incubation at elevated temperatures. Lead candidates were recovered from the stored duplicate plate and subject to further mutagenesis. After several rounds of mutagenesis, screening ~18,000 colonies, the leading candidate PlyC(PlyCA-N211H) was found to increase thermokinetic stability by 4.1 °C and had an 18.8-fold increase in half-life at 45 °C over wild-type PlyC (Heselpoth 2014).
While the above techniques provide promising results, both are random techniques that are limited by the number of individual mutants/clones that can be manually screened. To fully exploit the power of directed evolution, techniques must be devised that allow hundreds of thousands or millions of potential mutants to be generated under evolutionary pressure and screened rapidly. Toward this end, Bull and colleagues proposed several models for directed evolution of bacteriophage endolysins that take advantage of bacterial allelopathy (Bull et al. 2015). Allelopathy is the phenomenon by which a bacterial cell produces a “toxin” (i.e., endolysin) that influences the growth or survival of another organism. Under this premise, the goal is to evolve improved killing of a bacterial species C. An endolysin A is capable of killing C but not very efficiently. Evolutionary theory suggests that if selection is used to improve endolysin A, it must provide benefit to mutant A genes that kill C above a baseline rate demonstrated by the nonevolved A. Therefore, A must be expressed by host species B, with the following caveats: B must not be harmed by endolysin A, and C is a competitor of B such that B’s fitness is reduced in a coculture with C. If appropriate candidates for endolysin A and bacterial species B and C can be identified, then expression of A by B in a coculture with C will generate the required pressure to evolve A to be more efficient at killing C. Only B organisms that possess mutant A with enhanced killing of C will survive the coculture conditions. Several mathematical models were developed where endolysin A is encoded by a phage B or by a plasmid with bacterial host B. Additionally, coculture conditions in liquid, emulsion, or solid media were modeled. The results suggested that directed evolution in a plasmid-based system is feasible and that such a system may enable rapid improvement of an endolysin.
Structure-Guided Site-Directed Mutagenesis
As high-resolution structures of endolysins are solved by X-ray crystallography or Nuclear Magnetic Resonance (NMR), structure-guided approaches such as point mutagenesis or chimeragenesis will dramatically expand bioengineering efforts for these enzymes. Structure-guided site-directed mutagenesis (SDM) has already been effectively harnessed for modifying endolysin properties (Diez-Martinez et al. 2013; Low et al. 2011). Taking into consideration that the cell wall surface of Gram-positive bacterial species generally is negatively charged, a 2011 study showed the activity of an endolysin EAD correlated with its net (positive) charge in the absence of the CBD (Low et al. 2011). This conceptual understanding was then incorporated into endolysin bioengineering studies for increasing EAD activity and CBD-independence. For example, the endogenous XlyA EAD, which is an amidase derived from a B. subtilis prophage and consists of a net charge of Z = −3 at pH 7.5, demonstrated lytic activity in a CBD-dependent manner. By mutating five noncationic surface residues to lysines, thereby shifting the net charge from Z = −3 to Z = +3, the resulting XlyA EAD mutant lysed B. subtilis cells at a rate nearly identical to that of full-length XlyA. A reverse loss-of-function engineering approach was then conducted using the PlyBa04 EAD, which is a muramidase derived from the B. anthracis Ba04 phage and carries a net charge of Z = +1 at pH 7.5. The native PlyBa04 EAD was able to lyse B. cereus cells as efficiently as the full-length endolysin. However, when four neutral residues on the surface of the PlyBa04 EAD were mutated to aspartic acid to change the net charge from Z = +1 to Z = −3, the resulting PlyBa04 mutant showed little or no lytic activity towards Bacillus. The same negatively charged EAD mutant was made in the context of the full-length protein, and it was discovered that a significant fraction of wild-type activity was preserved. Therefore, changing the net charge of the PlyBa04 EAD from positive to negative correspondingly transitioned the lytic activity of the module from a CBD-independent to a CBD-dependent state. An alternative study established a direct correlation between the net charge of the CBD of an endolysin and its bacteriolytic activity (Diez-Martinez et al. 2013). Introducing 15 amino acid substitutions to reverse the charge of the CBD belonging to the pneumococcal endolysin Cpl-7 from Z = −14.93 to Z = +3.0 at neutral pH significantly increased the activity of the endolysin against S. pneumoniae, S. pyogenes, and other pathogens.
Crystal structures can be used to find other important differences between two similar endolysins beyond surface charges. CD27L, an endolysin that targets C. difficile, and its CD27L1–179 truncation mutant are capable of lysing certain Listeria and Bacillus species (Mayer et al. 2011). By comparing the sequences and crystal structures of the Listeria targeting endolysin PlyPSA with that of the EAD of CD27L, Mayer and colleagues discovered a tryptophan in PlyPSA in the space above the active site, pointing out towards the surface, that is represented by a leucine in CD27L. Based on its position, they posited that this tryptophan is involved in substrate binding by the EAD. Site-directed mutagenesis was used to create the L98W mutation in CD27L. This mutation conferred greater activity for CD27L and CD27L1–179 against certain serovars of L. monocytogenes. This structure-to-structure comparison further demonstrated the utility of structural knowledge in expanding the capabilities of an endolysin by site-directed mutagenesis.
With high-resolution atomic coordinates in hand, computational modeling can be used to predict mutations that can affect catalytic activity, binding to substrate, interactions with the solvent interface, or stabilizing effects of intra- or inter-domain interactions. With regard to the latter category, computational screening methods have been developed to measure the effect that amino acid mutations have on protein stability through the estimation of Gibbs free energy (ΔG) values. These algorithms use either biophysical models of amino acid interactions (Kellogg et al. 2011; Yin et al. 2007; Seeliger and de Groot 2010; Worth et al. 2011), statistical analyses of available proteins and corresponding thermodynamic properties (Guerois et al. 2002; Johnston et al. 2011; Dehouck et al. 2011), machine learning methods (Capriotti et al. 2008; Tian et al. 2010), or a combination thereof (Masso and Vaisman 2010; Li et al. 2012) to predict the effect a mutation has on stability. Some examples of algorithms that have been successfully exploited to increase protein stability are Rosetta (Kellogg et al. 2011; Dantas et al. 2007; Leaver-Fay et al. 2011) and FoldX (Guerois et al. 2002). Accordingly, these programs have been used to randomly mutate individual amino acids in silico to each of the other 19 naturally occurring amino acids and determine stabilizing mutants. Such an approach was used on PlyC, a streptococcal endolysin. SDM of the top ten mutations resulted in identification of one mutant, PlyC(PlyCA-T406R), that was shown experimentally to increase the thermal denaturation temperature by ~2.2 °C and thermokinetic stability 16-fold over wild-type (Heselpoth et al. 2015). This mutation is expected to introduce a hydrogen bond between the Q106 side-chain and the R406 side-chain of PlyCA, stabilizing the intra-molecular interaction between its N- and C-terminal domains.
Gram-Negative Engineering Approaches
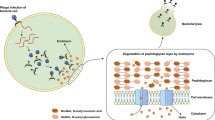
The current therapeutic strategy involving the exogenous application of endolysins has been limited to Gram-positive pathogens due to the existence of the outer membrane structure of Gram-negative bacteria. To overcome this, engineering strategies are being explored in an attempt to allow endolysins to penetrate this protective barrier in Gram-negative pathogens to access the underlying peptidoglycan layer. One strategy involves fusing peptidoglycan hydrolases to various cationic, polycationic, and other membrane-disrupting peptides. For example, hydrophobic and amphipathic peptides were fused to human lysozyme, and the resulting proteins had considerable killing activity against E. coli (Arima et al. 1997; Ibrahim et al. 1994). In another example, human lysozyme was fused to cecropin, which is an antimicrobial peptide that forms pores in the outer membrane, and the resulting construct had considerable antimicrobial efficacy against several Gram-negative bacterial species (Lu et al. 2010).
A subset of endolysins reportedly encompasses intrinsic charge and/or hydrophobic motifs that interact with the outer membrane to increase permeability. For example, the extrinsic application of the Bacillus amyloliquefaciens endolysin, Lys1521, established antibacterial efficacy towards not only Gram-positive bacteria, but E. coli and P. aeruginosa as well (Morita et al. 2001a, b; Orito et al. 2004). Likewise, in addition to Gram-positive species, the Acinetobacter baumannii endolysin LysAB2 displays activity against a variety of Gram-negative bacteria, including A. baumannii, E. coli, Citrobacter freundii, Salmonella enterica, and Enterobacter aerogenes (Lai et al. 2011). In their salient 2014 paper, Briers and colleagues focused on peptides that have cationic, hydrophobic, or amphipathic properties predicted to destabilize the lipopolysaccharide (LPS) layer and subsequently fused these peptides to endolysins at the amino- or carboxy-terminus of the proteins (Briers et al. 2014). They call their artificially engineered endolysins “Artilysins™” to distinguish them from native endolysins. The authors were able to show that by attaching these LPS-destabilizing peptides to their proteins, they improved antibacterial activity as determined by reduction of viable cells in the order of one to one-and-a-half logs, without the need for EDTA or other membrane-destabilizing chemicals that are commonly used to observe activity of endolysins on Gram-negative cells. More recently, a fusion of the KZ144 endolysin and sheep myeloid antibacterial peptide 29 (SMAP-29) produced an artilysin (ART-175) with potent antibacterial activity against P. aeruginosa and Acinetobacter baumannii (Defraine et al. 2016). Finally, there is evidence that “artilysation” of endolysins not only imparts activity against Gram-negative organisms, but it can increase activity against Gram-positive organisms as shown for a fusion of λSa2lys and the polycationic peptide PCNP, which resulted in a twofold increase against streptococci compared to the λSa2lys parental endolysin (Rodriguez-Rubio et al. 2016).
While the use of an endolysin that possesses the ability to permeabilize or disrupt outer membrane structures of Gram-negative bacteria is desirable in vitro, their therapeutic use in vivo may be limited due to toxicity issues stemming from interactions with eukaryotic cell membranes. One way to bypass these toxicity issues could be to use bacterial transport proteins to translocate the endolysin across the outer membrane. Lukacik and colleagues were able to target the pathogenicity-associated FyuA transport protein of E. coli and Yersinia pestis by fusing a FyuA-binding domain from the bacteriocin pesticin to the T4 lysozyme (Lukacik et al. 2012). This chimeric endolysin was able to lyse cells that expressed FyuA. Conversely, FyuA null-strains were not lysed. One can predict that resistance to this chimeric lysin can be attained by mutation of FyuA, but this in turn may also reduce the pathogenicity of the bacteria.
Targeting Intracellular Pathogens
Intracellular pathogens are problematic because they are able to evade the host immune system as well as antimicrobials that cannot penetrate the eukaryotic cell surface. To address this concern, peptidoglycan hydrolases are being fused to cationic or amphipathic peptides, termed protein transduction domains (PTDs) (i.e., cell-penetrating peptides, membrane-permeable peptides, or Trojan horse peptides), that naturally interact with the eukaryotic membrane to promote internalization (Dietz and Bahr 2004; Dietz 2010; Splith and Neundorf 2011). Engineering macromolecule translocation from the extracellular to intracellular environment of eukaryotic cells has been successfully accomplished through PTD fusions. Therefore, fusing peptidoglycan hydrolases to PTDs could be an effective antimicrobial strategy for targeting and killing Gram-positive pathogens that invade, persist, and replicate within eukaryotic cells, such as S. aureus, L. monocytogenes, S. pyogenes, and B. anthracis (Borysowski and Gorski 2010). Preliminary studies using lysostaphin (Donovan 2011) or staphylococcal endolysins (Becker et al. 2016) fused to PTDs have generated promising results. Both constructs were capable of reducing intracellular drug-sensitive or -resistant S. aureus cells in bovine mammary gland epithelial cells, murine osteoblasts, and human brain microvasculature epithelial cells.
In a recent development, Shen and coworkers fused PTDs to the streptococcal endolysin PlyC (Shen et al. 2016). Although the fusion construct was able to kill intracellular S. pyogenes, it was noted that the wild-type enzyme was also capable of killing intracellular streptococci. Subsequent studies showed that the PlyC holoenzyme, mediated by its PlyCB subunit, crossed epithelial cell membranes and cleared intracellular streptococci in a dose-dependent manner. It was determined that a binding pocket on PlyCB, the CBD of PlyC, interacted strongly with phosphatidylserine of the plasma membrane and this interaction allowed PlyC to translocate epithelial membranes. While the intracellular transport/trafficking mechanisms that eventually lead to the colocalization of PlyC with S. pyogenes is unknown, this is the first report of a native endolysin that can target intracellular bacteria. It is anticipated that the PlyCB binding pocket can be utilized for bioengineering efforts to extend this capability to other endolysins.
Regulatory Aspects of Endolysins
Therapeutic endolysins are classified as biologics by the FDA in the United States. Section 351 of the Public Health Service (PHS) Act defines a biological product as a “virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product, applicable to the prevention, treatment, or cure of a disease or condition of human beings.” Regulatory responsibility for therapeutic biological products is held by the Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER) at the FDA. Through an intercenter agreement reached in 2003, CDER took responsibility for “protein, peptide or carbohydrate products produced by cell culture” except for those that are blood products or components of vaccines. Because biologics are considered a subset of drugs, they are not only regulated by the aforementioned PHS Act but also by the Federal Food, Drug, and Cosmetic (FDC) Act. The primary goals of these acts are to ensure the safety and legality of all aspects of public health.
After initial laboratory testing and animal experiments, a candidate endolysin therapeutic would progress to clinical trials in humans via filing of an investigational new drug application (IND) in accordance with the regulations stated in 21 CFR 312. If the biologic was found to be effective and safe, data (pharmacological and manufacturing) collected after three rounds of clinical testing would be submitted by means of a biologics license application (BLA), which has strict regulations concerning biological therapeutics denoted in 21 CFR 600, 601, and 610. The Division of Anti-Infective Products would process BLAs concerning biologic agents against bacterial infectious disease. The license, which allows for marketing of the biologic, would then be issued upon determination that the biologic and the manufacturing process and facilities meet the benchmarks that have been set for safety, purity, and potency. The license application includes the stipulation that the facilities must be routinely inspected and that each package must bear the US license number.
After the BLA is approved, the drug can go to market, which means that it is available for public use. This is not the end of the road in regulatory surveillance, however. Typically known as Phase 4, these postmarketing studies are essential in determining the long-term effects of a drug on a large population, outside of a controlled environment. The licensee must report on distribution, any changes made to the product or manufacturing process, and any adverse effects that were not previously encountered. If the FDA feels that these unexpected side effects are severe enough to outweigh the potential benefits, they have the power to withdraw the product from the market.
One advantage of using an isolated phage protein instead of whole phage as an antimicrobial agent is that some of the complexity of the final product is eliminated. Because a protein is still considered a complex and heterogeneous substance, however, a potency assay is required. It is also likely the final endolysin product may consist of multiple elements/proteins, each of which must undergo testing individually and in conjunction with each other as the FDA law mandates. Indeed, the FDA opposition to cocktail therapies and the agency’s subsequently enacted laws that make it harder for approval of multicomponent drugs would seem to hinder endolysin development as this makes the drug-to market-process much more complicated, time-consuming, and costly. The FDA is currently overhauling these policies, however, and in 2013 released new guidelines detailing future approaches to approval of combinatorial treatment. (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM236669.pdf).
There are no endolysins that are presently on the market in the United States, though there are some endolysins and phage-based technologies that are in various stages of the approval process. The only approved products are whole phage preps that are for use as food processing aids but none are approved for use during human infection. As stated above, ContraFect Corp. has completed Phase 1 clinical trials on CF-301, an antistaphylococcal endolysin. Furthermore, this enzyme has been granted Fast Track Designation status by the FDA, which means all reviews for this drug will be expedited as it treats a serious or life-threatening condition and fills an unmet medical need. Similarly, iNtRON Biotechnology, Inc. recently published the results of a Phase 1 clinical trial on SAL200, a pharmaceutical formulation containing the staphylococcal endolysin SAL-1 (Jun et al. 2017). Other companies have endolysins in various stages of development for both human and animal use. For example, Micreos, a Dutch biotechnology company, is developing Staphefekt™, a staphylococcal endolysin, and Vetmedica, the animal health division of Boehringer Ingelheim, has acquired Artilysin™ technology from a biotech company, named Lysando AG, for pharmaceuticals, food, and hygiene applications.
Other Phage-Based Enzybiotics
Particulate Bacteriocins
Bacteriocins are another type of “enzybiotics.” The term “bacteriocin” has evolved over the years to become a catch phrase for several classes of bacterial-produced antimicrobials that do not act as traditional antibiotics. The first class includes low molecular weight, ribosomally synthesized antimicrobial peptides, such as nisin, that have been actively pursued in the food industry for their broad-spectrum killing activity against phylogenetically dissimilar bacterial species. A second class of bacteriocin is composed of enzymes that possess a narrow spectrum of killing activity directed against species that are phylogenetically similar to the producer. The classic example of this type of bacteriocin is lysostaphin, a peptidoglycan hydrolase encoded by S. simulans that rapidly lyses S. aureus. Due to the peptidoglycan hydrolase activity, lysostaphin is often mistakenly called an endolysin, but the distinction lies in the source of the enzyme. A cell wall hydrolase produced by a phage as part of the lytic system for progeny release is an endolysin, whereas those produced by bacteria are bacteriocins. Nonetheless, similar to endolysins, lysostaphin has drawn interest for its therapeutic value with the native enzyme advancing to Phase 2 clinical trials (Quickel et al. 1971) and a recombinant version of the enzyme also advancing to Phase 2 trials (Kokai-Kun 2012). The third and least studied class of bacteriocin is the “particulate bacteriocin,” also known as “high-molecular weight bacteriocin,” “killer particle,” “tailocin,” and “defective phage” since these bacteriocins can be sedimented by ultracentrifugation and resemble whole phage or headless phage tail-like assemblies when viewed by electron microscopy. The killing activity of particulate bacteriocins occurs by forming pores in the cell membrane, leading to a rapid loss of membrane potential and subsequent bacterial death (Nakayama et al. 2000; Strauch et al. 2001, 2003). Since the focus of this book is bacteriophage, we will limit our discussion here to just the particulate bacteriocins.
Particulate bacteriocins are multiprotein structures, often containing 20 or more subunits, and are informally divided into three subclasses based solely on their structural characteristics: the more-or-less complete phage containing either (i) random host-derived DNA sequences or (ii) phage-derived DNA and (iii) the headless tail-like particles (Garro and Marmur 1970). The PBSX-like bacteriocins of Bacillus are the most studied particulate bacteriocins that are found in the first subclass. These complexes appear to be complete phage-like particles, but they package random pieces of genomic DNA into heads that are too small to package a full phage genome, they do not inject their contents into sensitive cells upon adsorbing to the surface, and they subsequently do not display phage replication (Okamoto et al. 1968). Instead, these noninfectious particles are spontaneously produced at low levels resulting in cell lysis and can be induced by chemical agents or ultraviolet light similar to temperate phage.
A small number of particulate bacteriocins show a complete phage morphology under the electron microscope, package phage-derived DNA, and exhibit a killing activity but are unable to form plaques on indicator strains. For obvious reasons, it has been speculated that these particulate bacteriocins are either temperate bacteriophage that currently lack a laboratory host for propagation or instead are defective temperate bacteriophages. The prototypical member of this second subclass of particulate bacteriocin is colicin 15, produced by E. coli strain 15. Colicin 15 was discovered when it was observed that ultraviolet-irradiated E. coli strain 15 cultures lysed after several hours, releasing a sedimentable colicin activity, but not a plaque-forming temperate bacteriophage (Mukai 1960; Ryan et al. 1955). The defective phages were unable to replicate on any of the indicator strains tested and the bacteriocin was particularly unusual only for showing bactericidal action against the producer strain.
The archetypical headless phage tail-like members of the third subclass of particulate bacteriocin are the F- and R-type pyocins, produced by P. aeruginosa (originally P. pyocyanea) (Fyfe et al. 1984; Nakayama et al. 2000). R-type pyocins appear as hollow cylinders, consisting of an extended sheath and a core similar to tail assemblies of phage in the Myoviridae family (Gillor et al. 2005). In contrast, F-type pyocins are flexuous, noncontractile rods with a square-like structure at one end and a fiber-like structure at the other end resembling tail assemblies of phage in the Siphoviridae family. The –ocin nomenclature has permeated: pyocin-like bacteriocins produced by L. monocytogenes are known as monocins; those from Clostridia species are clostocins; those from C. difficile are diffocins; and those from Vibrio species are virbrocins.
The genetic, serological, morphological, and sometimes biological similarity of particulate bacteriocins and conventional phages has raised obvious questions as to their origin. Are they phage relics, phage precursors, or both (Reanney and Ackermann 1982)? It has been suggested that particulate bacteriocins are poorly evolved phages or the ancestors of temperate phages. Garro and Marmur speculated that defective phages either arose through the progressive addition of specific genetic components or through the loss of structural and replication genes (Garro and Marmur 1970). For headless phage tails, Bradley considered a devolutionary origin most probable, since it seemed to him that a functional episome was unlikely to code for an injection system before it was surrounded by a capsid (Bradley 1967; Bradley and Hoeniger 1971). Bradley’s hypothesis was strengthened by the evidence of normal phage retaining bacteriocidal activity after the loss of structural features (Uratani 1982). Bradley’s theory can be challenged, however, by recent molecular and structural elucidation of the phage tail-like Type Six Secretion System (TSSS) and examples in the literature of phage-like or phage tail-like particles apparently serving as vehicles for the delivery of proteinaceous toxins.
Applications in Crop Health
In an orchard study conducted by Jabrane and colleagues, the antibacterial action of a bacteriocin produced by “Enterobacteria strain 17” (later revealed to be serracin P, produced by Serratia plymithicum J7) was compared to streptomycin and a commercial formulation of flumequine for efficacy against Erwinia amylovora, the causal agent of fire blight (Jabrane et al. 1996). In tests, the bacteriocin decreased the total number of infections compared to the control and was comparable to that of streptomycin. Based on additional studies detailing the bactericidal activity of serracin P against strains of E. amylovora, it was proposed that this bacteriocin would make a good biopesticide candidate (Jabrane et al. 2002). In another example, the bacteriocin produced by Pseudomonas syringae pv. ciccaronei inhibited the multiplication of P. syringae subsp. savastanoi, the causal agent of olive knot disease, and decreased survival of the pathogen on the leaves and twigs of treated olive plants (Lavermicocca et al. 2002).
Many bacteria associated with plants produce bacteriocins, oftentimes under stressful conditions, to rapidly eliminate neighboring cells that are not immune or resistant to their effects. Production of these inhibitory substances can dramatically affect population dynamics on the plant surface. Thus, exploitation of these bacteriocin-producing organisms is an attractive strategy to control bacterial diseases in plants. For example, Pectobacterium carotovora subsp. carotovora strain CGE234-M403 has been used as a biological control agent of soft-rot diseases in Japan under the trade name “Biokeeper” (Chuang et al. 1999). This strain was subsequently shown to produce a phage-tail-like particulate bacteriocin that is responsible for the observed inhibitory effect (Yamada et al. 2006).
Applications in Medicine
Similar to whole phage therapy, most particulate bacteriocins have a narrow-spectrum killing activity, do not disturb the endogenous microflora, and have advantages of stability and low toxicity to humans (Gillor et al. 2005). Therapeutic efficacy of particulate bacteriocins has been demonstrated in several in vivo models, a few of which are reviewed here. Enterocoliticin, a phage tail-like bacteriocin produced by pathogenic Yersinia enterocolitica serotype O3, was shown to kill extracellular Y. enterocolitica in coculture with eukaryotic cells and reduced Y. enterocolitica titers in BALB/c mice when administered orally (Damasko et al. 2005).
Natural R-type pyocins have also shown therapeutic potential in vivo. In a mouse model of P. aeruginosa bacteremia, three strains of P. aeruginosa were intravenously (i.v.) injected into mice (Scholl et al. 2012). The mice were treated with pyocin C2, an R-type pyocin prepared from mitomycin C-induced cultures of P. aeruginosa strain 78 under various conditions: i.v. immediately after challenge with or without a booster 24 h later; i.v. 6 or 24 h after infection; or i.p. immediately after infection. Intravenous postexposure treatment soon after infection provided some protective effect against two of the P. aeruginosa strains, but not the third, despite in vitro sensitivity of this strain to the pyocin.
In a similar study, the R2 pyocin produced by P. aeruginosa PA01 was used as an antibacterial towards P. aeruginosa infection in a mouse peritonitis model. The study employed a quantitative pyocin assay to determine exact doses and explored dose response, time of administration, and multiple routes of administration (i.p. versus i.v.) to assess the potential of natural R-type pyocins as human therapeutics and prophylactics after i.p. infection (Scholl and Martin 2008). Intraperitoneal administration of 3 × 1011 pyocin particles (~18 μg total protein) 1–4 h after infection protected 90–100% of the animals at 24 and 48 h postchallenge, but further delay in treatment significantly increased mortality. At the minimally effective dose, where 80% of the animals were protected, it was estimated that 750 pyocin molecules were administered per inoculated bacterium. For i.v. administration, the effective treatment window was narrower and required a slightly higher administered pyocin dose. Intraveneous infected mice treated 1 or 8 h after challenge by i.v. administration of 3 × 1011 pyocin molecules had undetectable levels of bacteria in their blood and spleen samples up to 24 h later. Mice treated 8 h after infection showed a temporary 4-log decrease in bacteria in their blood and spleen samples, but subsequent increases in the bacterial load resulted in death. Bacteria isolated from succumbing animals remained sensitive to the R2 pyocin, which suggested that the failure to rescue mice was due to an inadequate dosing regimen rather than resistance development (Scholl and Martin 2008).
Engineering Particulate Bacteriocins
Similar to how EADs and CBDs can be swapped in endolysins to change or expand host range, parallel bioengineering/chimeragenesis approaches can be used with particulate bacteriocins. Building upon the knowledge of natural pyocins detailed above, the AvidBiotics Corp. has taken the lead in engineering particulate bacteriocins for altered host ranges. In their first study, an R2 pyocin tail fiber was engineered to display tail fibers from phage that infect E. coli or Y. pestis, thereby changing the lytic activity of the chimeric pyocin (Williams et al. 2008). Next, they fused the O-antigen tail spike of phage phiV10, specific for E. coli O157:H7 (Ritchie et al. 2011), to the pyocin chassis. The chimeric pyocin, named AvR2-V10, killed 100% of a diverse set of E. coli O157:H7 isolates but no other E. coli serotypes or Pseudomonas strains. AvR2-V10 was able to kill E. coli O157:H7 when sprayed on beef surfaces, suggesting potential disinfectant use in the food products/packaging industries (Scholl et al. 2009). AvR2-V10-resistant mutants were isolated, but all mutants lost the ability to produce the O157 antigen and had decreased virulence.
Using the proof-of-principle demonstrated for O157 above, the P. aeruginosa R-type pyocin was further engineered to target shiga-toxin producing strains of enteroaggregative E. coli O104:H4 (Scholl et al. 2012). A synthetic gene encoding a putative O-antigen specific P22-like tail spike protein identified in the draft genome of O104:H4 strain LB226692 was used as a template for PCR-mediated fusion to the 3′ end of the R2 pyocin tail fiber gene, prf15. The recombinant fusion protein expressed was composed of the first 164 amino acids of R2 fused to amino acids 114–659 or 118–659 of the putative P22-like tail spike protein, and the constructs were called AvR2–104.1 and AvR2–104.2, respectively. These engineered pyocins were shown to be highly sensitive and specific to E. coli strains that produce the full-length O104 O-antigen. Bacteria expressing other O-antigen serotypes and O-antigen truncation mutants were insensitive to the killing activity of the chimeric pyocins and survived treatment to form colonies. Most recently, a similar technology was used to engineer an expanded host range of a C. difficile R-type bacteriocin by replacing the receptor binding protein (RBP) with RBPs from phage with broad host ranges (Gebhart et al. 2015). These chimeric diffocins, known as Avidocin-CDs, survived passage through the mouse gastrointestinal tract when administered in drinking water. The Avidocins did not alter the gut microflora and prevented antibiotic-induced colonization of mice by epidemic lineages of C. difficile.
Concluding Remarks
The threat of antibiotic-resistant pathogens to public health has forced us to consider alternative antimicrobial therapies, including bacteriophage therapy or the use of enzybiotics, i.e., bacteriophage-derived proteins. Analogous to phage therapy, the use of enzybiotics has benefited by coevolution of phages with their bacterial hosts. As a result, the killing phenotype is specific to pathogenic bacteria, with little to no deleterious effects on commensal organisms, a benefit not offered by traditional antibiotic therapy. Because most enzybiotics lack DNA, RNA, or mechanisms for horizontal transfer of genes to host genomes, the particulate bacteriocins being the exception, the regulatory pathways are much more straightforward than for phage therapy. Essentially, from a regulatory perspective, enzybiotics would be treated as therapeutic proteins, no different than any other biologic therapy (e.g., antibodies, etc.). Finally, both endolysins and particulate bacteriocins employ a modular design, which can be exploited by bioengineers to expand the host range or enhance the catalytic properties through chimeragenesis, directed evolution, or structure-based design. Thus, enzybiotics offer an attractive alternative to traditional antibiotics for treatment of bacterial pathogens, including multidrug-resistant pathogens.
Cross-References
References
Arima H, Ibrahim HR, Kinoshita T, Kato A (1997) Bactericidal action of lysozymes attached with various sizes of hydrophobic peptides to the C-terminal using genetic modification. FEBS Lett 415(1):114–118
Bales PM, Renke EM, May SL, Shen Y, Nelson DC (2013) Purification and characterization of biofilm-associated EPS exopolysaccharides from ESKAPE organisms and other pathogens. PLoS One 8(6):e67950. https://doi.org/10.1371/journal.pone.0067950
Becker SC, Foster-Frey J, Donovan DM (2008) The phage K lytic enzyme LysK and lysostaphin act synergistically to kill MRSA. FEMS Microbiol Lett 287(2):185–191. https://doi.org/10.1111/j.1574-6968.2008.01308.x
Becker SC, Dong S, Baker JR, Foster-Frey J, Pritchard DG, Donovan DM (2009) LysK CHAP endopeptidase domain is required for lysis of live staphylococcal cells. FEMS Microbiol Lett 294(1):52–60. https://doi.org/10.1111/j.1574-6968.2009.01541.x
Becker SC, Roach DR, Chauhan VS, Shen Y, Foster-Frey J, Powell AM, Bauchan G, Lease RA, Mohammadi H, Harty WJ, Simmons C, Schmelcher M, Camp M, Dong S, Baker JR, Sheen TR, Doran KS, Pritchard DG, Almeida RA, Nelson DC, Marriott I, Lee JC, Donovan DM (2016) Triple-acting lytic enzyme treatment of drug-resistant and intracellular Staphylococcus aureus. Sci Rep 6:25063. https://doi.org/10.1038/srep25063
Bortolussi R (2008) Listeriosis: a primer. CMAJ 179(8):795–797. https://doi.org/10.1503/cmaj.081377
Borysowski J, Gorski A (2010) Fusion to cell-penetrating peptides will enable lytic enzymes to kill intracellular bacteria. Med Hypotheses 74(1):164–166. https://doi.org/10.1016/j.mehy.2009.07.006
Borysowski J, Weber-Dabrowska B, Gorski A (2006) Bacteriophage endolysins as a novel class of antibacterial agents. Exp Biol Med 231(4):366–377
Bradley DE (1967) Ultrastructure of bacteriophage and bacteriocins. Bacteriol Rev 31(4):230–314
Bradley DE, Hoeniger JF (1971) Structural changes in cells of Clostridium perfringens infected with a short-tailed bacteriophage. Can J Microbiol 17(3):397–402
Bramley AJ, Foster R (1990) Effects of lysostaphin on Staphylococcus aureus infections of the mouse mammary gland. Res Vet Sci 49(1):120–121
Briers Y, Volckaert G, Cornelissen A, Lagaert S, Michiels CW, Hertveldt K, Lavigne R (2007) Muralytic activity and modular structure of the endolysins of Pseudomonas aeruginosa bacteriophages phiKZ and EL. Mol Microbiol 65(5):1334–1344. https://doi.org/10.1111/j.1365-2958.2007.05870.x
Briers Y, Schmelcher M, Loessner MJ, Hendrix J, Engelborghs Y, Volckaert G, Lavigne R (2009) The high-affinity peptidoglycan binding domain of Pseudomonas phage endolysin KZ144. Biochem Biophys Res Commun 383(2):187–191. https://doi.org/10.1016/j.bbrc.2009.03.161
Briers Y, Walmagh M, Van Puyenbroeck V, Cornelissen A, Cenens W, Aertsen A, Oliveira H, Azeredo J, Verween G, Pirnay JP, Miller S, Volckaert G, Lavigne R (2014) Engineered endolysin-based “Artilysins” to combat multidrug-resistant gram-negative pathogens. MBio 5(4):e01379-01314. https://doi.org/10.1128/mBio.01379-14
Brouillette E, Malouin F (2005) The pathogenesis and control of Staphylococcus aureus-induced mastitis: study models in the mouse. Microbes and infection/Institut Pasteur 7(3):560–568. https://doi.org/10.1016/j.micinf.2004.11.008
Brucato FH, Pizzo SV (1990) Catabolism of streptokinase and polyethylene glycol-streptokinase: evidence for transport of intact forms through the biliary system in the mouse. Blood 76(1):73–79
Bull JJ, Crandall C, Rodriguez A, Krone SM (2015) Models for the directed evolution of bacterial allelopathy: bacteriophage lysins. PeerJ 3:e879. https://doi.org/10.7717/peerj.879
Capriotti E, Fariselli P, Rossi I, Casadio R (2008) A three-state prediction of single point mutations on protein stability changes. BMC Bioinformatics 9(Suppl 2):S6. https://doi.org/10.1186/1471-2105-9-S2-S6
Celia LK, Nelson D, Kerr DE (2008) Characterization of a bacteriophage lysin (Ply700) from Streptococcus uberis. Vet Microbiol 130(1–2):107–117. https://doi.org/10.1016/j.vetmic.2007.12.004
Cheng Q, Fischetti VA (2007) Mutagenesis of a bacteriophage lytic enzyme PlyGBS significantly increases its antibacterial activity against group B streptococci. Appl Microbiol Biotechnol 74(6):1284–1291. https://doi.org/10.1007/s00253-006-0771-1
Cheng Q, Nelson D, Zhu S, Fischetti VA (2005) Removal of group B streptococci colonizing the vagina and oropharynx of mice with a bacteriophage lytic enzyme. Antimicrob Agents Chemother 49(1):111–117. https://doi.org/10.1128/AAC.49.1.111-117.2005
Chuang DY, Kyeremeh AG, Gunji Y, Takahara Y, Ehara Y, Kikumoto T (1999) Identification and cloning of an Erwinia carotovora subsp. carotovora bacteriocin regulator gene by insertional mutagenesis. J Bacteriol 181(6):1953–1957
Croux C, Ronda C, Lopez R, Garcia JL (1993a) Interchange of functional domains switches enzyme specificity: construction of a chimeric pneumococcal-clostridial cell wall lytic enzyme. Mol Microbiol 9(5):1019–1025
Croux C, Ronda C, Lopez R, Garcia JL (1993b) Role of the C-terminal domain of the lysozyme of Clostridium acetobutylicum ATCC 824 in a chimeric pneumococcal-clostridial cell wall lytic enzyme. FEBS Lett 336(1):111–114
Damasko C, Konietzny A, Kaspar H, Appel B, Dersch P, Strauch E (2005) Studies of the efficacy of Enterocoliticin, a phage-tail like bacteriocin, as antimicrobial agent against Yersinia enterocolitica serotype O3 in a cell culture system and in mice. J Vet Med B Infect Dis Vet Public Health 52(4):171–179. https://doi.org/10.1111/j.1439-0450.2005.00841.x
Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA (2010) Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 54(4):1603–1612. https://doi.org/10.1128/AAC.01625-09
Dantas G, Corrent C, Reichow SL, Havranek JJ, Eletr ZM, Isern NG, Kuhlman B, Varani G, Merritt EA, Baker D (2007) High-resolution structural and thermodynamic analysis of extreme stabilization of human procarboxypeptidase by computational protein design. J Mol Biol 366(4):1209–1221. https://doi.org/10.1016/j.jmb.2006.11.080
de Vries J, Harms K, Broer I, Kriete G, Mahn A, Düring K, Wackernagel W (1999) The bacteriolytic activity in transgenic potatoes expressing the chimeric T4 lysozyme gene and the effect of T4 lysozyme on soil- and phytopathogenic bacteria. Syst Appl Microbiol 22(2):280–286
Defraine V, Schuermans J, Grymonprez B, Govers SK, Aertsen A, Fauvart M, Michiels J, Lavigne R, Briers Y (2016) Efficacy of artilysin Art-175 against resistant and persistent Acinetobacter baumannii. Antimicrob Agents Chemother 60(6):3480–3488. https://doi.org/10.1128/AAC.00285-16
DeHart HP, Heath HE, Heath LS, LeBlanc PA, Sloan GL (1995) The lysostaphin endopeptidase resistance gene (epr) specifies modification of peptidoglycan cross bridges in Staphylococcus simulans and Staphylococcus aureus. Appl Environ Microbiol 61(4):1475–1479
Dehouck Y, Kwasigroch JM, Gilis D, Rooman M (2011) PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics 12:151. https://doi.org/10.1186/1471-2105-12-151
Diaz E, Lopez R, Garcia JL (1990) Chimeric phage-bacterial enzymes: a clue to the modular evolution of genes. Proc Natl Acad Sci U S A 87(20):8125–8129
Diaz E, Lopez R, Garcia JL (1991) Chimeric pneumococcal cell wall lytic enzymes reveal important physiological and evolutionary traits. J Biol Chem 266(9):5464–5471
Dietz GP (2010) Cell-penetrating peptide technology to deliver chaperones and associated factors in diseases and basic research. Curr Pharm Biotechnol 11(2):167–174
Dietz GP, Bahr M (2004) Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci 27(2):85–131. https://doi.org/10.1016/j.mcn.2004.03.005
Diez-Martinez R, de Paz H, Bustamante N, Garcia E, Menendez M, Garcia P (2013) Improving the lethal effect of cpl-7, a pneumococcal phage lysozyme with broad bactericidal activity, by inverting the net charge of its cell wall-binding module. Antimicrob Agents Chemother 57(11):5355–5365. https://doi.org/10.1128/AAC.01372-13
Djurkovic S, Loeffler JM, Fischetti VA (2005) Synergistic killing of Streptococcus pneumoniae with the bacteriophage lytic enzyme Cpl-1 and penicillin or gentamicin depends on the level of penicillin resistance. Antimicrob Agents Chemother 49(3):1225–1228. https://doi.org/10.1128/AAC.49.3.1225-1228.2005
Doehn JM, Fischer K, Reppe K, Gutbier B, Tschernig T, Hocke AC, Fischetti VA, Loffler J, Suttorp N, Hippenstiel S, Witzenrath M (2013) Delivery of the endolysin Cpl-1 by inhalation rescues mice with fatal pneumococcal pneumonia. J Antimicrob Chemother 68(9):2111–2117. https://doi.org/10.1093/jac/dkt131
Donovan DM (2011) Fusion of peptidoglycan hydrolase enzymes to a protein transduction domain allows eradication of both extracellular and intracellular gram positive pathogens USA Patent US 12/784,675
Donovan DM, Dong S, Garrett W, Rousseau GM, Moineau S, Pritchard DG (2006) Peptidoglycan hydrolase fusions maintain their parental specificities. Appl Environ Microbiol 72(4):2988–2996. https://doi.org/10.1128/AEM.72.4.2988-2996.2006
Dunne M, Leicht S, Krichel B, Mertens HD, Thompson A, Krijgsveld J, Svergun DI, Gomez-Torres N, Garde S, Uetrecht C, Narbad A, Mayer MJ, Meijers R (2016) Crystal structure of the ctpl-1 endolysin reveals how its activity is regulated by a secondary translation product. J Biol Chem 291(10):4882–4893. https://doi.org/10.1074/jbc.M115.671172
Düring K, Porsch P, Fladung M, Lorz H (1993) Transgenic potato plants resistant to the phytopathogenic bacterium Erwinia carotovora. Plant J 3(4):587–598
Entenza JM, Loeffler JM, Grandgirard D, Fischetti VA, Moreillon P (2005) Therapeutic effects of bacteriophage Cpl-1 lysin against Streptococcus pneumoniae endocarditis in rats. Antimicrob Agents Chemother 49(11):4789–4792. https://doi.org/10.1128/AAC.49.11.4789-4792.2005
Fenton M, Casey PG, Hill C, Gahan CG, Ross RP, McAuliffe O, O’Mahony J, Maher F, Coffey A (2010) The truncated phage lysin CHAP(k) eliminates Staphylococcus aureus in the nares of mice. Bioeng Bugs 1(6):404–407. https://doi.org/10.4161/bbug.1.6.13422
Fischetti VA (2003) Novel method to control pathogenic bacteria on human mucous membranes. Ann N Y Acad Sci 987:207–214
Fischetti VA (2008) Bacteriophage lysins as effective antibacterials. Curr Opin Microbiol 11(5):393–400. https://doi.org/10.1016/j.mib.2008.09.012
Fischetti VA (2010) Bacteriophage endolysins: a novel anti-infective to control gram-positive pathogens. Int J Med Microbiol 300(6):357–362. https://doi.org/10.1016/j.ijmm.2010.04.002
Fujinami Y, Hirai Y, Sakai I, Yoshino M, Yasuda J (2007) Sensitive detection of Bacillus anthracis using a binding protein originating from gamma-phage. Microbiol Immunol 51(2):163–169
Fyfe JA, Harris G, Govan JR (1984) Revised pyocin typing method for Pseudomonas aeruginosa. J Clin Microbiol 20(1):47–50
Gaeng S, Scherer S, Neve H, Loessner MJ (2000) Gene cloning and expression and secretion of Listeria monocytogenes bacteriophage-lytic enzymes in Lactococcus lactis. Appl Environ Microbiol 66(7):2951–2958
Garcia P, Lopez R, Ronda C, Garcia E, Tomasz A (1983) Mechanism of phage-induced lysis in pneumococci. J Gen Microbiol 129(2):479–487
Garro AJ, Marmur J (1970) Defective bacteriophages. J Cell Physiol 76(3):253–263. https://doi.org/10.1002/jcp.1040760305
Gebhart D, Lok S, Clare S, Tomas M, Stares M, Scholl D, Donskey CJ, Lawley TD, Govoni GR (2015) A modified R-type bacteriocin specifically targeting Clostridium difficile prevents colonization of mice without affecting gut microbiota diversity. MBio 6(2):e02368-14. https://doi.org/10.1128/mBio.02368-14
Gillor O, Nigro LM, Riley MA (2005) Genetically engineered bacteriocins and their potential as the next generation of antimicrobials. Curr Pharm Des 11(8):1067–1075
Grandgirard D, Loeffler JM, Fischetti VA, Leib SL (2008) Phage lytic enzyme Cpl-1 for antibacterial therapy in experimental pneumococcal meningitis. J Infect Dis 197(11):1519–1522. https://doi.org/10.1086/587942
Grundling A, Missiakas DM, Schneewind O (2006) Staphylococcus aureus mutants with increased lysostaphin resistance. J Bacteriol 188(17):6286–6297. https://doi.org/10.1128/JB.00457-06
Gu J, Xu W, Lei L, Huang J, Feng X, Sun C, Du C, Zuo J, Li Y, Du T, Li L, Han W (2011a) LysGH15, a novel bacteriophage lysin, protects a murine bacteremia model efficiently against lethal methicillin-resistant Staphylococcus aureus infection. J Clin Microbiol 49(1):111–117. https://doi.org/10.1128/JCM.01144-10
Gu J, Zuo J, Lei L, Zhao H, Sun C, Feng X, Du C, Li X, Yang Y, Han W (2011b) LysGH15 reduces the inflammation caused by lethal methicillin-resistant Staphylococcus aureus infection in mice. Bioeng Bugs 2(2):96–99. https://doi.org/10.4161/bbug.2.2.14883
Guariglia-Oropeza V, Helmann JD (2011) Bacillus subtilis sigma(V) confers lysozyme resistance by activation of two cell wall modification pathways, peptidoglycan O-acetylation and D-alanylation of teichoic acids. J Bacteriol 193(22):6223–6232. https://doi.org/10.1128/JB.06023-11
Guerois R, Nielsen JE, Serrano L (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol 320(2):369–387. https://doi.org/10.1016/S0022-2836(02)00442-4
Gupta R, Prasad Y (2011) P-27/HP endolysin as antibacterial agent for antibiotic resistant Staphylococcus aureus of human infections. Curr Microbiol 63(1):39–45. https://doi.org/10.1007/s00284-011-9939-8
Hendrix RW (2002) Bacteriophages: evolution of the majority. Theor Popul Biol 61(4):471–480
Hermoso JA, Monterroso B, Albert A, Galan B, Ahrazem O, Garcia P, Martinez-Ripoll M, Garcia JL, Menendez M (2003) Structural basis for selective recognition of pneumococcal cell wall by modular endolysin from phage Cp-1. Structure 11(10):1239–1249
Heselpoth RD (2014) Engineering enhanced structural stability to the streptococcal bacteriophage endolysin PlyC. University of Maryland, College Park
Heselpoth RD, Nelson DC (2012) A new screening method for the directed evolution of thermostable bacteriolytic enzymes. J Vis Exp (69). doi:https://doi.org/10.3791/4216
Heselpoth RD, Yin Y, Moult J, Nelson DC (2015) Increasing the stability of the bacteriophage endolysin PlyC using rationale-based FoldX computational modeling. Protein Eng Des Sel 28(4):85–92. https://doi.org/10.1093/protein/gzv004
Holtje JV, Mirelman D, Sharon N, Schwarz U (1975) Novel type of murein transglycosylase in Escherichia coli. J Bacteriol 124(3):1067–1076
Hoopes JT, Stark CJ, Kim HA, Sussman DJ, Donovan DM, Nelson DC (2009) Use of a bacteriophage lysin, PlyC, as an enzyme disinfectant against Streptococcus equi. Appl Environ Microbiol 75(5):1388–1394. https://doi.org/10.1128/AEM.02195-08
Ibrahim HR, Yamada M, Matsushita K, Kobayashi K, Kato A (1994) Enhanced bactericidal action of lysozyme to Escherichia coli by inserting a hydrophobic pentapeptide into its C terminus. J Biol Chem 269(7):5059–5063
Jabrane A, Ledoux L, Thonart P, Deckers T, Lepoivre P (1996) The efficacy in vitro and in vivo of bacteriocin against Erwinia amylovora: comparison of biological and chemical control of fire blight. Acta Hortic 411:355–359
Jabrane A, Sabri A, Compere P, Jacques P, Vandenberghe I, Van Beeumen J, Thonart P (2002) Characterization of serracin P, a phage-tail-like bacteriocin, and its activity against Erwinia amylovora, the fire blight pathogen. Appl Environ Microbiol 68(11):5704–5710
Jado I, Lopez R, Garcia E, Fenoll A, Casal J, Garcia P, Spanish Pneumococcal Infection Study N (2003) Phage lytic enzymes as therapy for antibiotic-resistant Streptococcus pneumoniae infection in a murine sepsis model. J Antimicrob Chemother 52(6):967–973. https://doi.org/10.1093/jac/dkg485
Johnston MA, Sondergaard CR, Nielsen JE (2011) Integrated prediction of the effect of mutations on multiple protein characteristics. Proteins 79(1):165–178. https://doi.org/10.1002/prot.22870
Jun SY, Jang IJ, Yoon S, Jang K, KS Y, Cho JY, Seong MW, Jung GM, Yoon SJ, Kang SH (2017) Pharmacokinetics and tolerance of the phage endolysin-based candidate drug SAL200 after a single intravenous administration among healthy volunteers. Antimicrob Agents Chemother 61:e02629. https://doi.org/10.1128/AAC.02629-16
Kellogg EH, Leaver-Fay A, Baker D (2011) Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 79(3):830–838. https://doi.org/10.1002/prot.22921
Kerr DE, Plaut K, Bramley AJ, Williamson CM, Lax AJ, Moore K, Wells KD, Wall RJ (2001) Lysostaphin expression in mammary glands confers protection against staphylococcal infection in transgenic mice. Nat Biotechnol 19(1):66–70. https://doi.org/10.1038/83540
Kim WS, Salm H, Geider K (2004) Expression of bacteriophage phiEa1h lysozyme in Escherichia coli and its activity in growth inhibition of Erwinia amylovora. Microbiology 150(Pt 8):2707–2714. https://doi.org/10.1099/mic.0.27224-0
Kim KP, Cha JD, Jang EH, Klumpp J, Hagens S, Hardt WD, Lee KY, Loessner MJ (2008) PEGylation of bacteriophages increases blood circulation time and reduces T-helper type 1 immune response. Microb Biotechnol 1(3):247–257. https://doi.org/10.1111/j.1751-7915.2008.00028.x
Kokai-Kun JF (2012) Lysostaphin: a silver bullet for staph. In: Tegos A, Mylonakis E (eds) Antimicrobial drug discovery: emerging strategies. CABI, Oxfordshire, pp 147–165
Kretzer JW, Lehmann R, Schmelcher M, Banz M, Kim KP, Korn C, Loessner MJ (2007) Use of high-affinity cell wall-binding domains of bacteriophage endolysins for immobilization and separation of bacterial cells. Appl Environ Microbiol 73(6):1992–2000. https://doi.org/10.1128/AEM.02402-06
Lai MJ, Lin NT, Hu A, Soo PC, Chen LK, Chen LH, Chang KC (2011) Antibacterial activity of Acinetobacter baumannii phage varphiAB2 endolysin (LysAB2) against both gram-positive and gram-negative bacteria. Appl Microbiol Biotechnol 90(2):529–539. https://doi.org/10.1007/s00253-011-3104-y
Lavermicocca P, Lonigro SL, Valerio F, Evidente A, Visconti A (2002) Reduction of olive knot disease by a bacteriocin from Pseudomonas syringae pv. ciccaronei. Appl Environ Microbiol 68(3):1403–1407
Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popovic Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487:545–574. https://doi.org/10.1016/B978-0-12-381270-4.00019-6
Li Y, Zhang J, Tai D, Middaugh CR, Zhang Y, Fang J (2012) PROTS: a fragment based protein thermo-stability potential. Proteins 80(1):81–92. https://doi.org/10.1002/prot.23163
Loeffler JM, Fischetti VA (2003) Synergistic lethal effect of a combination of phage lytic enzymes with different activities on penicillin-sensitive and -resistant Streptococcus pneumoniae strains. Antimicrob Agents Chemother 47(1):375–377
Loeffler JM, Nelson D, Fischetti VA (2001) Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science 294(5549):2170–2172. https://doi.org/10.1126/science.1066869
Loeffler JM, Djurkovic S, Fischetti VA (2003) Phage lytic enzyme Cpl-1 as a novel antimicrobial for pneumococcal bacteremia. Infect Immun 71(11):6199–6204
Loessner MJ (2005) Bacteriophage endolysins–current state of research and applications. Curr Opin Microbiol 8(4):480–487. https://doi.org/10.1016/j.mib.2005.06.002
Loessner MJ, Wendlinger G, Scherer S (1995) Heterogeneous endolysins in Listeria monocytogenes bacteriophages: a new class of enzymes and evidence for conserved holin genes within the siphoviral lysis cassettes. Mol Microbiol 16(6):1231–1241
Loessner MJ, Kramer K, Ebel F, Scherer S (2002) C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol Microbiol 44(2):335–349
Lopez R, Garcia E (2004) Recent trends on the molecular biology of pneumococcal capsules, lytic enzymes, and bacteriophage. FEMS Microbiol Rev 28(5):553–580. https://doi.org/10.1016/j.femsre.2004.05.002
Low LY, Yang C, Perego M, Osterman A, Liddington R (2011) Role of net charge on catalytic domain and influence of cell wall binding domain on bactericidal activity, specificity, and host range of phage lysins. J Biol Chem 286(39):34391–34403. https://doi.org/10.1074/jbc.M111.244160
Lu XM, Jin XB, Zhu JY, Mei HF, Ma Y, Chu FJ, Wang Y, Li XB (2010) Expression of the antimicrobial peptide cecropin fused with human lysozyme in Escherichia coli. Appl Microbiol Biotechnol 87(6):2169–2176. https://doi.org/10.1007/s00253-010-2606-3
Lukacik P, Barnard TJ, Keller PW, Chaturvedi KS, Seddiki N, Fairman JW, Noinaj N, Kirby TL, Henderson JP, Steven AC, Hinnebusch BJ, Buchanan SK (2012) Structural engineering of a phage lysin that targets gram-negative pathogens. Proc Natl Acad Sci U S A 109(25):9857–9862. https://doi.org/10.1073/pnas.1203472109
Masso M, Vaisman II (2010) AUTO-MUTE: web-based tools for predicting stability changes in proteins due to single amino acid replacements. Protein Eng Des Sel 23(8):683–687. https://doi.org/10.1093/protein/gzq042
Mayer MJ, Payne J, Gasson MJ, Narbad A (2010) Genomic sequence and characterization of the virulent bacteriophage phiCTP1 from Clostridium tyrobutyricum and heterologous expression of its endolysin. Appl Environ Microbiol 76(16):5415–5422. https://doi.org/10.1128/AEM.00989-10
Mayer MJ, Garefalaki V, Spoerl R, Narbad A, Meijers R (2011) Structure-based modification of a Clostridium difficile-targeting endolysin affects activity and host range. J Bacteriol 193(19):5477–5486. https://doi.org/10.1128/JB.00439-11
McCullers JA, Karlstrom A, Iverson AR, Loeffler JM, Fischetti VA (2007) Novel strategy to prevent otitis media caused by colonizing Streptococcus pneumoniae. PLoS Pathog 3(3):e28. https://doi.org/10.1371/journal.ppat.0030028
McGowan S, Buckle AM, Mitchell MS, Hoopes JT, Gallagher DT, Heselpoth RD, Shen Y, Reboul CF, Law RH, Fischetti VA, Whisstock JC, Nelson DC (2012) X-ray crystal structure of the streptococcal specific phage lysin PlyC. Proc Natl Acad Sci U S A 109(31):12752–12757. https://doi.org/10.1073/pnas.1208424109
Morita M, Tanji Y, Mizoguchi K, Soejima A, Orito Y, Unno H (2001a) Antibacterial activity of Bacillus amyloliquefaciens phage endolysin without holin conjugation. J Biosci Bioeng 91(5):469–473
Morita M, Tanji Y, Orito Y, Mizoguchi K, Soejima A, Unno H (2001b) Functional analysis of antibacterial activity of Bacillus amyloliquefaciens phage endolysin against Gram-negative bacteria. FEBS Lett 500(1–2):56–59
Mukai FH (1960) Interrelationship between colicin sensitivity and phage resistance. J Gen Microbiol 23:539–551. https://doi.org/10.1099/00221287-23-3-539
Nakayama K, Takashima K, Ishihara H, Shinomiya T, Kageyama M, Kanaya S, Ohnishi M, Murata T, Mori H, Hayashi T (2000) The R-type pyocin of Pseudomonas aeruginosa is related to P2 phage, and the F-type is related to lambda phage. Mol Microbiol 38(2):213–231
Nau R, Eiffert H (2002) Modulation of release of proinflammatory bacterial compounds by antibacterials: potential impact on course of inflammation and outcome in sepsis and meningitis. Clin Microbiol Rev 15(1):95–110
Nelson D, Loomis L, Fischetti VA (2001) Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proc Natl Acad Sci U S A 98(7):4107–4112. https://doi.org/10.1073/pnas.061038398
Nelson D, Schuch R, Chahales P, Zhu S, Fischetti VA (2006) PlyC: a multimeric bacteriophage lysin. Proc Natl Acad Sci U S A 103(28):10765–10770. https://doi.org/10.1073/pnas.0604521103
Nelson DC, Schmelcher M, Rodriguez-Rubio L, Klumpp J, Pritchard DG, Dong S, Donovan DM (2012) Endolysins as antimicrobials. Adv Virus Res 83:299–365. https://doi.org/10.1016/B978-0-12-394438-2.00007-4
Obeso JM, Martinez B, Rodriguez A, Garcia P (2008) Lytic activity of the recombinant staphylococcal bacteriophage PhiH5 endolysin active against Staphylococcus aureus in milk. Int J Food Microbiol 128(2):212–218. https://doi.org/10.1016/j.ijfoodmicro.2008.08.010
Oey M, Lohse M, Kreikemeyer B, Bock R (2009a) Exhaustion of the chloroplast protein synthesis capacity by massive expression of a highly stable protein antibiotic. Plant J 57(3):436–445. https://doi.org/10.1111/j.1365-313X.2008.03702.x
Oey M, Lohse M, Scharff LB, Kreikemeyer B, Bock R (2009b) Plastid production of protein antibiotics against pneumonia via a new strategy for high-level expression of antimicrobial proteins. Proc Natl Acad Sci U S A 106(16):6579–6584. https://doi.org/10.1073/pnas.0813146106
Okamoto K, Mudd JA, Mangan J, Huang WM, Subbaiah TV, Marmur J (1968) Properties of the defective phage of Bacillus subtilis. J Mol Biol 34(3):413–428
Oldham ER, Daley MJ (1991) Lysostaphin: use of a recombinant bactericidal enzyme as a mastitis therapeutic. J Dairy Sci 74(12):4175–4182. https://doi.org/10.3168/jds.S0022-0302(91)78612-8
Orito Y, Morita M, Hori K, Unno H, Tanji Y (2004) Bacillus amyloliquefaciens phage endolysin can enhance permeability of Pseudomonas aeruginosa outer membrane and induce cell lysis. Appl Microbiol Biotechnol 65(1):105–109. https://doi.org/10.1007/s00253-003-1522-1
Pastagia M, Euler C, Chahales P, Fuentes-Duculan J, Krueger JG, Fischetti VA (2011) A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive Staphylococcus aureus strains. Antimicrob Agents Chemother 55(2):738–744. https://doi.org/10.1128/AAC.00890-10
Pritchard DG, Dong S, Baker JR, Engler JA (2004) The bifunctional peptidoglycan lysin of Streptococcus agalactiae bacteriophage B30. Microbiology 150(Pt 7):2079–2087. https://doi.org/10.1099/mic.0.27063-0
Proenca D, Velours C, Leandro C, Garcia M, Pimentel M, Sao-Jose C (2015) A two-component, multimeric endolysin encoded by a single gene. Mol Microbiol 95(5):739–753. https://doi.org/10.1111/mmi.12857
Quickel KE, Selden R, Caldwell JR, Nora NF, Schaffner W (1971) Efficacy and safety of topical lysostaphin treatment of persistent nasal carriage of Staphylococcus aureus. Appl Microbiol 22:446–450
Ramon J, Saez V, Baez R, Aldana R, Hardy E (2005) PEGylated interferon-alpha2b: a branched 40K polyethylene glycol derivative. Pharm Res 22(8):1374–1386. https://doi.org/10.1007/s11095-005-5278-4
Rashel M, Uchiyama J, Ujihara T, Uehara Y, Kuramoto S, Sugihara S, Yagyu K, Muraoka A, Sugai M, Hiramatsu K, Honke K, Matsuzaki S (2007) Efficient elimination of multidrug-resistant Staphylococcus aureus by cloned lysin derived from bacteriophage phi MR11. J Infect Dis 196(8):1237–1247. https://doi.org/10.1086/521305
Reanney DC, Ackermann H-W (1982) Comparative biology and evolution of bacteriophages. In: Lauffer MA, Bang FB, Maramorosch K, Smith KM (eds) Advances in virus research, vol 27. Academic Press, New York, pp 205–280
Resch G, Moreillon P, Fischetti VA (2011a) PEGylating a bacteriophage endolysin inhibits its bactericidal activity. AMB Express 1:29. https://doi.org/10.1186/2191-0855-1-29
Resch G, Moreillon P, Fischetti VA (2011b) A stable phage lysin (Cpl-1) dimer with increased antipneumococcal activity and decreased plasma clearance. Int J Antimicrob Agents 38(6):516–521. https://doi.org/10.1016/j.ijantimicag.2011.08.009
Ritchie JM, Greenwich JL, Davis BM, Bronson RT, Gebhart D, Williams SR, Martin D, Scholl D, Waldor MK (2011) An Escherichia coli O157-specific engineered pyocin prevents and ameliorates infection by E. coli O157:H7 in an animal model of diarrheal disease. Antimicrob Agents Chemother 55(12):5469–5474. https://doi.org/10.1128/AAC.05031-11
Roach DR, Khatibi PA, Bischoff KM, Hughes SR, Donovan DM (2013) Bacteriophage-encoded lytic enzymes control growth of contaminating Lactobacillus found in fuel ethanol fermentations. Biotechnol Biofuels 6(1):20. https://doi.org/10.1186/1754-6834-6-20
Rodriguez-Rubio L, Martinez B, Rodriguez A, Donovan DM, Garcia P (2012) Enhanced staphylolytic activity of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA88 HydH5 virion-associated peptidoglycan hydrolase: fusions, deletions, and synergy with LysH5. Appl Environ Microbiol 78(7):2241–2248. https://doi.org/10.1128/AEM.07621-11
Rodriguez-Rubio L, Martinez B, Donovan DM, Garcia P, Rodriguez A (2013) Potential of the virion-associated peptidoglycan hydrolase HydH5 and its derivative fusion proteins in milk biopreservation. PLoS One 8(1):e54828. https://doi.org/10.1371/journal.pone.0054828
Rodriguez-Rubio L, Gutierrez D, Donovan DM, Martinez B, Rodriguez A, Garcia P (2015) Phage lytic proteins: biotechnological applications beyond clinical antimicrobials. Crit Rev Biotechnol:1–11. https://doi.org/10.3109/07388551.2014.993587
Rodriguez-Rubio L, Chang WL, Gutierrez D, Lavigne R, Martinez B, Rodriguez A, Govers SK, Aertsen A, Hirl C, Biebl M, Briers Y, Garcia P (2016) ‘Artilysation’ of endolysin lambdaSa2lys strongly improves its enzymatic and antibacterial activity against streptococci. Sci Rep 6:35382. https://doi.org/10.1038/srep35382
Royet J, Dziarski R (2007) Peptidoglycan recognition proteins: pleiotropic sensors and effectors of antimicrobial defences. Nat Rev Microbiol 5(4):264–277. https://doi.org/10.1038/nrmicro1620
Ryan FJ, Fried P, Mukai F (1955) A colicin produced by cells that are sensitive to it. Biochim Biophys Acta 18(1):131
Sainathrao S, Mohan KV, Atreya C (2009) Gamma-phage lysin PlyG sequence-based synthetic peptides coupled with Qdot-nanocrystals are useful for developing detection methods for Bacillus anthracis by using its surrogates, B. anthracis-Sterne and B. cereus-4342. BMC Biotechnol 9:67. https://doi.org/10.1186/1472-6750-9-67
Schmelcher M, Shabarova T, Eugster MR, Eichenseher F, Tchang VS, Banz M, Loessner MJ (2010) Rapid multiplex detection and differentiation of Listeria cells by use of fluorescent phage endolysin cell wall binding domains. Appl Environ Microbiol 76(17):5745–5756. https://doi.org/10.1128/AEM.00801-10
Schmelcher M, Tchang VS, Loessner MJ (2011) Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb Biotechnol 4(5):651–662. https://doi.org/10.1111/j.1751-7915.2011.00263.x
Schmelcher M, Donovan DM, Loessner MJ (2012a) Bacteriophage endolysins as novel antimicrobials. Future Microbiol 7(10):1147–1171. https://doi.org/10.2217/fmb.12.97
Schmelcher M, Powell AM, Becker SC, Camp MJ, Donovan DM (2012b) Chimeric phage lysins act synergistically with lysostaphin to kill mastitis-causing Staphylococcus aureus in murine mammary glands. Appl Environ Microbiol 78(7):2297–2305. https://doi.org/10.1128/AEM.07050-11
Schmelcher M, Waldherr F, Loessner MJ (2012c) Listeria bacteriophage peptidoglycan hydrolases feature high thermoresistance and reveal increased activity after divalent metal cation substitution. Appl Microbiol Biotechnol 93(2):633–643. https://doi.org/10.1007/s00253-011-3372-6
Scholl D, Martin DW Jr (2008) Antibacterial efficacy of R-type pyocins towards Pseudomonas aeruginosa in a murine peritonitis model. Antimicrobial agents and chemotherapy 52(5):1647–1652. https://doi.org/10.1128/AAC.01479-07
Scholl D, Cooley M, Williams SR, Gebhart D, Martin D, Bates A, Mandrell R (2009) An engineered R-type pyocin is a highly specific and sensitive bactericidal agent for the food-borne pathogen Escherichia coli O157:H7. Antimicrob Agents Chemother 53(7):3074–3080. https://doi.org/10.1128/AAC.01660-08
Scholl D, Gebhart D, Williams SR, Bates A, Mandrell R (2012) Genome sequence of E. coli O104:H4 leads to rapid development of a targeted antimicrobial agent against this emerging pathogen. PLoS One 7(3):e33637. https://doi.org/10.1371/journal.pone.0033637
Schuch R, Nelson D, Fischetti VA (2002) A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 418(6900):884–889. https://doi.org/10.1038/nature01026
Schuch R, Lee HM, Schneider BC, Sauve KL, Law C, Khan BK, Rotolo JA, Horiuchi Y, Couto DE, Raz A, Fischetti VA, Huang DB, Nowinski RC, Wittekind M (2014) Combination therapy with lysin CF-301 and antibiotic is superior to antibiotic alone for treating methicillin-resistant Staphylococcus aureus-induced murine bacteremia. J Infect Dis 209(9):1469–1478. https://doi.org/10.1093/infdis/jit637
Seeliger D, de Groot BL (2010) Protein thermostability calculations using alchemical free energy simulations. Biophys J 98(10):2309–2316. https://doi.org/10.1016/j.bpj.2010.01.051
Sheehan MM, Garcia JL, Lopez R, Garcia P (1997) The lytic enzyme of the pneumococcal phage Dp-1: a chimeric lysin of intergeneric origin. Mol Microbiol 25(4):717–725
Shen Y, Barros M, Vennemann T, Gallagher DT, Yin Y, Linden SB, Heselpoth RD, Spencer DJ, Donovan DM, Moult J, Fischetti VA, Heinrich F, Losche M, Nelson DC (2016) A bacteriophage endolysin that eliminates intracellular streptococci. Elife 5. doi:https://doi.org/10.7554/eLife.13152
Singh PK, Donovan DM, Kumar A (2014) Intravitreal injection of the chimeric phage endolysin Ply187 protects mice from Staphylococcus aureus endophthalmitis. Antimicrob Agents Chemother 58(8):4621–4629. https://doi.org/10.1128/AAC.00126-14
Sordillo LM, Streicher KL (2002) Mammary gland immunity and mastitis susceptibility. J Mammary Gland Biol Neoplasia 7(2):135–146
Splith K, Neundorf I (2011) Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur Biophys J 40(4):387–397. https://doi.org/10.1007/s00249-011-0682-7
Stark CJ, Hoopes JT, Bonocora RP, Nelson DC (eds) (2010) Bacteriophage lytic enzymes as antimicrobials. Bacteriophage in the detection and control of foodborne pathogens. ASM Press, Washington, D.C.
Stentz R, Bongaerts RJ, Gunning AP, Gasson M, Shearman C (2010) Controlled release of protein from viable Lactococcus lactis cells. Appl Environ Microbiol 76(9):3026–3031. https://doi.org/10.1128/AEM.00021-10
Strauch E, Kaspar H, Schaudinn C, Dersch P, Madela K, Gewinner C, Hertwig S, Wecke J, Appel B (2001) Characterization of enterocoliticin, a phage tail-like bacteriocin, and its effect on pathogenic Yersinia enterocolitica strains. Appl Environ Microbiol 67(12):5634–5642. https://doi.org/10.1128/AEM.67.12.5634-5642.2001
Strauch E, Kaspar H, Schaudinn C, Damasko C, Konietzny A, Dersch P, Skurnik M, Appel B (2003) Analysis of enterocoliticin, a phage tail-like bacteriocin. Adv Exp Med Biol 529:249–251. https://doi.org/10.1007/0-306-48416-1_48
Sugai M, Fujiwara T, Ohta K, Komatsuzawa H, Ohara M, Suginaka H (1997) epr, which encodes glycylglycine endopeptidase resistance, is homologous to femAB and affects serine content of peptidoglycan cross bridges in Staphylococcus capitis and Staphylococcus aureus. J Bacteriol 179(13):4311–4318
Sweeney CR, Whitlock RH, Meirs DA, Whitehead SC, Barningham SO (1987) Complications associated with Streptococcus equi infection on a horse farm. J Am Vet Med Assoc 191(11):1446–1448
Sweeney CR, Timoney JF, Newton JR, Hines MT (2005) Streptococcus equi infections in horses: guidelines for treatment, control, and prevention of strangles. J Vet Intern Med 19(1):123–134
Thunnissen AM, Dijkstra AJ, Kalk KH, Rozeboom HJ, Engel H, Keck W, Dijkstra BW (1994) Doughnut-shaped structure of a bacterial muramidase revealed by X-ray crystallography. Nature 367(6465):750–753. https://doi.org/10.1038/367750a0
Tian J, Wu N, Chu X, Fan Y (2010) Predicting changes in protein thermostability brought about by single- or multi-site mutations. BMC Bioinformatics 11:370. https://doi.org/10.1186/1471-2105-11-370
Turner MS, Waldherr F, Loessner MJ, Giffard PM (2007) Antimicrobial activity of lysostaphin and a Listeria monocytogenes bacteriophage endolysin produced and secreted by lactic acid bacteria. Syst Appl Microbiol 30(1):58–67. https://doi.org/10.1016/j.syapm.2006.01.013
Twort FW (1925) The transmissible bacterial lysin and its action on dead bacteria. Lancet 206:642–644
Uratani Y (1982) A circular dichroism study of sheath contraction in pyocin R1. Biochim Biophys Acta 703(2):196–203
Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J (2001) Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev 14(3):584–640. https://doi.org/10.1128/CMR.14.3.584-640.2001
Veiga-Crespo P, Ageitos JM, Poza M, Villa TG (2007) Enzybiotics: a look to the future, recalling the past. J Pharm Sci 96(8):1917–1924. https://doi.org/10.1002/jps.20853
Vollmer W (2008) Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol Rev 32(2):287–306. https://doi.org/10.1111/j.1574-6976.2007.00088.x
Vouillamoz J, Entenza JM, Giddey M, Fischetti VA, Moreillon P, Resch G (2013) Bactericidal synergism between daptomycin and the phage lysin Cpl-1 in a mouse model of pneumococcal bacteraemia. Int J Antimicrob Agents 42(5):416–421. https://doi.org/10.1016/j.ijantimicag.2013.06.020
Walcher G, Stessl B, Wagner M, Eichenseher F, Loessner MJ, Hein I (2010) Evaluation of paramagnetic beads coated with recombinant Listeria phage endolysin-derived cell-wall-binding domain proteins for separation of Listeria monocytogenes from raw milk in combination with culture-based and real-time polymerase chain reaction-based quantification. Foodborne Pathog Dis 7(9):1019–1024. https://doi.org/10.1089/fpd.2009.0475
Wall RJ, Powell AM, Paape MJ, Kerr DE, Bannerman DD, Pursel VG, Wells KD, Talbot N, Hawk HW (2005) Genetically enhanced cows resist intramammary Staphylococcus aureus infection. Nat Biotechnol 23(4):445–451. https://doi.org/10.1038/nbt1078
Walsh S, Shah A, Mond J (2003) Improved pharmacokinetics and reduced antibody reactivity of lysostaphin conjugated to polyethylene glycol. Antimicrob Agents Chemother 47(2):554–558
Williams SR, Gebhart D, Martin DW, Scholl D (2008) Retargeting R-type pyocins to generate novel bactericidal protein complexes. Appl Environ Microbiol 74(12):3868–3876. https://doi.org/10.1128/AEM.00141-08
Witzenrath M, Schmeck B, Doehn JM, Tschernig T, Zahlten J, Loeffler JM, Zemlin M, Muller H, Gutbier B, Schutte H, Hippenstiel S, Fischetti VA, Suttorp N, Rosseau S (2009) Systemic use of the endolysin Cpl-1 rescues mice with fatal pneumococcal pneumonia. Crit Care Med 37(2):642–649. https://doi.org/10.1097/CCM.0b013e31819586a6
Worth CL, Preissner R, Blundell TL (2011) SDM – a server for predicting effects of mutations on protein stability and malfunction. Nucleic acids research 39 (Web Server issue):W215–222. doi:https://doi.org/10.1093/nar/gkr363
Wu H, Lu H, Huang J, Li G, Huang Q (2012) EnzyBase: a novel database for enzybiotic studies. BMC Microbiol 12:54. https://doi.org/10.1186/1471-2180-12-54
Wu H, Huang J, Lu H, Li G, Huang Q (2014) GMEnzy: a genetically modified enzybiotic database. PLoS One 9(8):e103687. https://doi.org/10.1371/journal.pone.0103687
Yamada K, Hirota M, Niimi Y, Nguyen HA, Takahara Y, Kamio Y, Kaneko J (2006) Nucleotide sequences and organization of the genes for carotovoricin (Ctv) from Erwinia carotovora indicate that Ctv evolved from the same ancestor as Salmonella typhi prophage. Biosci Biotechnol Biochem 70(9):2236–2247. https://doi.org/10.1271/bbb.60177
Yamashita Y, Shibata Y, Nakano Y, Tsuda H, Kido N, Ohta M, Koga T (1999) A novel gene required for rhamnose-glucose polysaccharide synthesis in Streptococcus mutans. J Bacteriol 181(20):6556–6559
Yin S, Ding F, Dokholyan NV (2007) Eris: an automated estimator of protein stability. Nat Methods 4(6):466–467. https://doi.org/10.1038/nmeth0607-466
Yoong P, Schuch R, Nelson D, Fischetti VA (2006) PlyPH, a bacteriolytic enzyme with a broad pH range of activity and lytic action against Bacillus anthracis. J Bacteriol 188(7):2711–2714. https://doi.org/10.1128/JB.188.7.2711-2714.2006
Zimmer M, Sattelberger E, Inman RB, Calendar R, Loessner MJ (2003) Genome and proteome of Listeria monocytogenes phage PSA: an unusual case for programmed + 1 translational frameshifting in structural protein synthesis. Mol Microbiol 50(1):303–317
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this entry

Cite this entry
Heselpoth, R.D., Swift, S.M., Linden, S.B., Mitchell, M.S., Nelson, D.C. (2021). Enzybiotics: Endolysins and Bacteriocins. In: Harper, D.R., Abedon, S.T., Burrowes, B.H., McConville, M.L. (eds) Bacteriophages. Springer, Cham. https://doi.org/10.1007/978-3-319-41986-2_34
Download citation
DOI: https://doi.org/10.1007/978-3-319-41986-2_34
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41985-5
Online ISBN: 978-3-319-41986-2
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences