
Abstract
To investigate the nature and origin of the antibacterial activity of the lytic phage ϕAB2 toward Acinetobacter baumannii, we successfully isolated and characterized a novel phage lysozyme (endolysin) from ϕAB2 and named it LysAB2. To analyze antibacterial activity of LysAB2, the complete LysAB2 and two deletion derivatives were constructed, purified and characterized. Zymographic assays showed that only the intact LysAB2 could lyse the peptidoglycan of A. baumannii and the Staphylococcus aureus cell wall. Antibacterial analysis also showed that only the intact LysAB2 retained the complete bactericidal activity. When applied exogenously, LysAB2 exhibited a broad bacteriolytic activity against a number of Gram-negative and Gram-positive bacteria. Thermostability assays indicated that LysAB2 was stable at 20∼40°C. Its optimal pH was 6.0, and it was active from pH 4 to 8. Scanning electron microscopy revealed that exposure to 500 μg ml−1 LysAB2 for up to 60 min caused a remarkable modification of the cell shape of the bacteria. Treating bacteria with LysAB2 clearly enhanced permeation of the bacterial cytoplasmic membrane. These results indicate that LysAB2 is an effective lysozyme against bacteria, and they suggest that it is a good candidate for a therapeutic/disinfectant agent to control nosocomial infections caused by multiple drug-resistant bacteria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acinetobacter baumannii is a non-fermentative, Gram-negative, non-motile, oxidase-negative coccobacillus that commonly exists in the hospital environment. This bacterium has been reported as an emerging nosocomial pathogen, with a growing prevalence worldwide (Peleg et al. 2008). In the past, A. baumannii was an easy-to-treat pathogen because it was susceptible to a wide range of antibiotic agents (Bergogne-Berezin and Towner 1996). However, it is now attracting great attention and emerging as a cause of numerous global outbreaks due to the increase in its antimicrobial resistance to virtually all available drugs (Dijkshoorn et al. 2007; Perez et al. 2007). Therefore, there is a need to find novel agents that can prevent the spread of multidrug-resistant strains of A. baumannii (MDRAB) (Wroblewska 2006). Bacteriophages (phages) are known to be active against bacteria but inactive against eukaryotic cells. Therefore, phage therapy is being considered as a possible therapeutic alternative for the treatment of infections caused by MDR strains (Barrow and Soothill 1997; Sulakvelidze et al. 2001; Stone 2002). Presently, endolysins (phage-encoded lysozymes that break down bacterial peptidoglycan at the terminal stage of the phage reproduction cycle) have been used successfully to control antibiotic-resistant pathogenic bacteria in animal models (Entenza et al. 2005). Their cell wall-binding domains target the enzymes to their substrate, helping their corresponding catalytic domains to cleave bonds in the peptidoglycan network. The ability of endolysins to digest cell walls when applied exogenously (as recombinant proteins) to bacterial cells has enabled their use as alternative antibacterials (Borysowski et al. 2006; Hermoso et al. 2007).
In previous work, the characterization of a lytic phage specifically infecting A. baumannii was performed (Lin et al. 2010). This A. baumannii phage ϕAB2 was isolated from wastewater to displayed infection specificity toward many multidrug-resistant A. baumannii isolates. In addition to the features of a short-tail, icosahedral capsid and a dsDNA genome of about 40 kb, the partial genomic sequence of ϕAB2 also indicated its close relation to the phage LKA1 (Ceyssens et al. 2006; Lavigne et al. 2003). To determine the putative endolysin gene in ϕAB2, some random DNA fragments from the ϕAB2 genome were cloned to sequenced. By running blast sequence analysis (Johnson et al. 2008), a partial open reading frame (ORF) encoding an endolysin-like protein was detected in one of the obtained DNA fragments. The full ORF was then completed by primer walking. In this study, in order to analyze antibacterial activity of LysAB2, the complete LysAB2 and two deletion derivatives were constructed and purified. Their enzymatic and antibacterial activities were tested. This is believed to be the first characterization of a cell wall-hydrolyzing enzyme encoded by an Acinetobacter phage.
Materials and methods
Bacteria, phage, vector, and growth conditions
The bacteria, plasmids, and phage used in this study are summarized in Table 1. A. baumannii strain M3237 (BCRC 80276) was used for ϕAB2 phage (DSM 23600) propagation. Bacteria were cultivated in Luria Bertani (LB) broth or LB agar (Difco Laboratories, Detroit, MI, USA) at 37°C. The vector pET-30b (+) (Novagen, USA) was used for cloning and expression of the lysAB2 endolysin gene in Escherichia coli.
DNA manipulation
Bacteriophage DNA was prepared according to Lin (Lin et al. 2010). Plasmid DNA was isolated from E. coli with a QIAprep Spin MiniPrep Kit (Qiagen). Polymerase chain reaction (PCR) amplification, using the DNA of phage ϕAB2 as a template, was performed to generate the putative endolysin gene lysAB2. To construct the LysAB2 expression vector, a 558-base pair DNA fragment of lysAB2 was amplified by PCR with lysAB2-FP and lysAB2-RP primers and cloned into a TA cloning site of pGEM-T-easy (Promega); the resulting recombinant DNA (lysAB2) was digested with EcoRI and cloned into the EcoRI sites of pET-30b (Novagen). The resulting plasmid, designated pET30b-LysAB2, was then used to transform E. coli strain BL21 (DE3) for expression. The two deletion derivative mutants were constructed in a similar manner to pET30b-LysAB2, using lysAB2-FP as a forward primer, and lysAB2-354RP (pET30b-LysAB2-D1) and lysAB2-124RP (pET30b-LysAB2-D2) as the reverse primers. The schematic map is shown in Fig. 2a. The constructed plasmids for mutant expression were sequenced to confirm the expected deletion.
Overexpression and purification of LysAB2 and its derivatives
The E. coli BL21 (DE3) strains, harboring the plasmids pET30b-LysAB2, pET30b-LysAB2-D1 or pET30b-LysAB2-D2 were grown overnight in LB medium containing kanamycin (50 μg/mL). The overnight culture of the transformed E. coli was diluted 100 times with the same medium and incubated at 37°C with shaking (150 rpm) until the optical density of the medium at OD600 nm reached 0.5. The expression of the target gene was induced by the addition of isopropyl-l-d-thiogalacto pyranoside at a final concentration of 0.1 mM. After further incubation for 3 h, the cells were harvested by centrifugation. The cell pellet was suspended in 10 mL of lysis-equilibration-wash (LEW) buffer containing 50 mM NaH2PO4/300 mM NaCl (pH 8.0), disrupted by sonication and centrifuged at 10,000 g for 15 min to remove debris. Crude supernatant was loaded onto Protino Ni-TED packed columns (MACHEREY-NAGEL, Germany) equilibrated with LEW buffer. The fractions were eluted with the elution buffer containing 50 mM NaH2PO4/300 mM NaCl/250 mM imidazole (pH 8.0). Active fractions were pooled and dialyzed against the elution buffer and concentrated by Amicon Ultra-0.5 centrifugal filter (MILLPORE). The purified proteins were mixed with sample buffer (62.5 mM Tris–HCl, pH 6.8, containing 5% 2-mercaptoethanol, 2% sodium dodecyl sulfate, 10% glycerol, and 0.01% bromophenol blue) and heated in a boiling water bath for 3 min, followed by separation of the proteins using SDS-polyacrylamide gel (12%) electrophoresis.
Zymogram
The preparation of the substrate (A. baumannii or Staphylococcus aureus cells) for the zymogram was performed according to the methods of Kakikawa (Kakikawa et al. 2002), with some modifications. Bacteria were propagated in LB broth at 37°C. The cells were collected, washed once and suspended in distilled water. After the suspension was autoclaved and mildly sonicated (four times for 5 min at 20 kHz), the cells were treated with 1 mg/ml trypsin in 50 mM potassium phosphate buffer for 4 h at 37°C. The suspension was centrifuged, and the sediment was treated with 10% tricholoroacetic acid at 25°C overnight, washed ten times with water, and used as the substrate for the zymogram. An SDS-polyacrylamide separating gel (pH 8.8) containing the substrate (3 wt.%/vol.) was used to detect the lytic activity. After SDS-PAGE, the gel was soaked for 30 min in distilled water at room temperature. The gel was then transferred into the renaturing buffer (50 mM Tris–HCl, pH 8.0, containing 1% Triton X-100) and shaken gently overnight at 37°C. The lytic activity of the renatured protein was examined by observing the clear translucent bands on the opaque background. The contrast was enhanced by staining the gel with 0.1% methylene blue–0.01% potassium hydroxide and subsequent destaining with demineralized water.
Determination of lysozyme activity
For lysozyme activity measurement, A. baumannii (BCRC 80276) treated with CHCl3 to remove the outer cell membrane was used as the substrate. LB medium (50 ml) containing exponentially growing cells (±0.6 at OD600) were centrifuged (4,000×g, 15 min, 4°C) and decanted. The cell pellet was suspended in chloroform-saturated 0.05 M Tris buffer (pH 7.7). After gently shaking for 45 min at room temperature, protoplasts were collected (4,000×g, 15 min, 4°C), washed and suspended in 10 mM phosphate buffer (pH 7) at an OD600 between 0.6 and 1.0
Protein samples of 30 μl were added to aliquots of 270 μl cell suspension in tissue culture plate wells and measured in a Multiskan Spectrum (Thermo) at room temperature. Each sample was prepared in duplicate to measure the drop in turbidity (OD600). All samples were plotted relative to the negative controls and compared with CEWL 20,000 U/mg (Amresco, Ohio, USA)
Thermostability and pH optimum of LysAB2
The thermostability of LysAB2 was evaluated at six different temperatures (20°C, 30°C, 40°C, 50°C, 60°C, and 70°C). After incubating the enzyme solutions in the absence of the substrate for 15 min at the test temperature, the residual enzyme activities were determined under the conditions of the antibacterial activity assay. The optimum pH was determined by performing the antibacterial assay at 37°C for 1 h using buffers with different pH values (4.0, 5.0, 6.0, 7.0, and 8.0).
Antibacterial assays
In vitro antibacterial assays were performed for the purified endolysin LysAB2 and its two deletion derivatives. The concentration of each purified protein was determined by the Bradford assay using bovine serum albumin as a standard. Multidrug-resistant strains of A. baumannii M3237 (BCRC 80276), methicillin-resistant S. aureus strain MR602 (BCRC 80277), and seven other reference strains (listed in Table 1) were grown at 37°C in LB broth medium until an absorbance of A600 1 was reached. The cells were centrifuged, washed and resuspended in phosphate buffer (10 mM K2HPO4 and KH2PO4, pH = 7.2). Approximately 105 cells were mixed with 50 μg of enzyme diluted in phosphate buffer to make a final volume of 100 μl (final concentration of 500 μg ml−1). The control experiment was performed with phosphate buffer. The mixture was incubated in an Eppendorf tube at 37°C for 1 h. The mixture was then overlaid on a LB agar plate. After overnight incubation at 37°C, the surviving bacteria, expressed as colony-forming units, were counted. The antibacterial rates were obtained using the following equation: \( {\hbox{antibacterial}}\,{\hbox{rate}}\,\left( \% \right) = \left( {{N_0}-{N_1}} \right)/{N_0} \times 100\% \), where N 0 and N 1 are the numbers of colonies in the control culture plates and the experimental culture plates, respectively. The assays were conducted in triplicate.
Turbidity test
To determine the exogenous lytic activity of the purified endolysin LysAB2, a suspension of viable cells (in LB medium, OD600: 1) of A. baumannii was mixed with 500 μg of purified endolysin LysAB2 in 1 ml phosphate buffer and incubated in an Eppendorf tube at 37°C with shaking. The experiment was performed using phosphate buffer as a negative control. The lytic activity was determined by measuring the turbidity at OD600 at various times. The assays were conducted in triplicate.
Scanning electron microscopy
Multidrug-resistant A. baumannii strain M3237 and methicillin-resistant S. aureus strain MR602 were grown in LB broth to a log phase, harvested by centrifugation, washed twice with deionized water and resuspended in the same water. Approximately 109 cells were incubated at 37°C for 1 h with 500 μg ml−1 LysAB2. Controls were run in the presence of deionized water. The volume was adjusted to 100 μl. The treated cells were fixed with 2.5% (w/v) glutaraldehyde in 0.1 M cacodylate buffer and 1% tannic acid, extensively washed with the phosphate buffer and dehydrated with a graded ethanol series. After critical-point drying and gold coating, the samples were observed with a HITACHI H-7500 instrument.
Bacteria cytoplasmic membrane permeated assay
The ability of LysAB2 to permeate the cytoplasmic membrane was assayed by the fluorescein isothiocyanate (FITC) staining method, as described by Mangoni (Mangoni et al. 2004). Briefly, A. baumannii was grown at 37°C in LB broth medium until an absorbance of A600 1 was reached. The cells were centrifuged, washed and resuspended in phosphate buffer. Approximately 107 cells were mixed with 50 μg of LysAB2 diluted in phosphate buffer to make a final volume of 100 μl (final concentration of 500 μg ml−1). The control experiment was performed in the absence of LysAB2 and in the presence of phosphate buffer. The mixture was incubated in an Eppendorf tube at 37°C for 1 h. The cell suspension was poured on to glass slides and kept at 37°C for 30 min to allow the adhesion of LysAB2-treated A. baumannii cells to the glass slides. The slides were then washed with sodium phosphate buffer gently, and 1 ml of FITC (Sigma) solution (6 μg/ml in sodium phosphate buffer) was added to the slides. After 30 min at 30°C, the FITC solution was removed and the slides were rinsed again with sodium phosphate buffer. The slides were then examined by bright field and fluorescence microscopy to assess the FITC influx inside the bacterial cells.
Nucleotide sequence accession number
The nucleotide sequence of lysAB2 has been deposited in GenBank (accession no. HM755898).
Results
Identification of the lysAB2 gene and sequence analysis of the LysAB2 protein
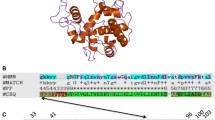
To find the possible endolysin gene existing in the lytic phage ϕAB2 infecting A. baumannii, random cloning and sequencing of the DNA fragments generated from the ϕAB2 genome were performed. One of the DNA fragments containing a partial ORF encoding possible endolysins was identified. The complete sequence of the ORF with a length of 558 bp was then obtained with primer walking. The BLAST search for the putative product deduced from the ORF showed a strong sequence similarity to the chitinases belonging to glycoside hydrolase (GH) family 19 and the broad lysozyme-like superfamily. Chitinases are degradative enzymes produced by a wide range of organisms, ranging from viruses to bacteria to plants and animals (Patil et al. 2000). In addition to using chitin as the substrate, some chitinases have been shown to hydrolyze the glycosidic bonds in arabinogalactan proteins (van Hengel et al. 2001) and bacterial peptidoglycan (Brunner et al. 1998). Previous structural analyses also revealed that the proteins in the GH19 chitinase family share several secondary structural elements with lysozymes that are mainly localized in areas around the substrate binding and catalytic sites (Brunner et al. 1998; Hart et al. 1995; Hahn et al. 2000; Ubhayasekera et al. 2009). Based on these clues, we hypothesized that the derived product from the cloned ORF in ϕAB2 was an endolysin displaying sequence similarity to the GH family 19 chitinases and named it LysAB2. The nucleotide sequence of lysAB2 was deposited in GenBank (accession no. HM755898). The result shown in Fig. 1 is the multiple sequence alignment between LysAB2 and its homologues to the catalytic cores of the GH family 19 chitinase domain. Particularly notable is the resemblance between the two groups of proteins with respect to the relative location of the predicted catalytic residues (marked with hash signs), the conserved residues presumably involved in sugar binding (marked with star signs), and the predicted secondary structure elements around the catalytic residues.

Multiple sequence alignment of LysAB2, showing its homology to the catalytic core of the GH family 19 chitinase domain. The alignment between LysAB2 and its homologues identified by BLAST (Johnson et al. 2008) was constructed using T-coffee (Notredame et al. 2000). Five pre-aligned GH family 19 chitinase sequences (boxed) were obtained from the cd00325 cluster in the Conserved Domain Database, NCBI (Marchler-Bauer et al. 2009). The alignment between the two groups were further generated by clustalw_3D program in STRAP (Gille and Frommel 2001), and adjusted manually. The predicted residues involved in catalysis and sugar binding (Brunner et al. 1998; Hart et al. 1995; Hahn et al. 2000; Ubhayasekera et al. 2009) are indicated by hash and star signs, respectively, under the alignment. Dotted line highlights the region of the putative amphipathic helices (aa 113–145) in the C terminus of LysAB2, and the basic amino acids between aa 113–145 of LysAB2 are shown in bold font. psipred SSE prediction of secondary structural elements for LysAB2 generated by PSIPRED (McGuffin et al. 2000), 2BAA SSE the known secondary structural elements of the barley chitinase 2BAA (Hart et al. 1995); ribbons indicate helices. This figure was prepared with the program Jalview v. 2.4 (Waterhouse et al. 2009)
Interestingly, in the C terminus of LysAB2, the rich distribution of the basic amino acids within and surrounding the putative amphipathic helices (aa 113–145 in Fig. 1) has been observed. In the previous studies, some endolysins, especially those from phages of Gram-negative bacteria, were demonstrated to be able to induce microbial membrane disturbance by the amphipathic helix stretches in the C terminus (During et al. 1999; Orito et al. 2004). Therefore, in the subsequent experiments, we constructed and purified the complete LysAB2 and its two deletion derivatives to characterize their enzymatic and antibacterial activities.
Cloning, overexpression and purification of endolysin LysAB2 and its two derivatives
To confirm that the lysAB2 gene encods an endolysin-like enzyme and to identify the effective antibacterial region of LysAB2, lysAB2 gene and its two deletion derivatives were cloned for further characterization. After the PCR reaction, full-length lysAB2 amplified from the phage ϕAB2 was inserted into the E. coli expression vector pET-30b, yielding pET30b-LysAB2, which encoded His-Tag-fused LysAB2 with a calculated molecular mass of 26.7 kDa. In E. coli BL21 (DE3), pET30b-LysAB2 directed the synthesis of a single individual protein with an apparent molecular mass of about 26 kDa on an SDS gel (Fig. 2b). The other two LysAB2 deletion derivatives, LysAB2-D1 and LysAB2-D1, were expressed from the plasmids pET30b-LysAB2-D1 and pET30b-LysAB2-D2, respectively. LysAB2-D1 is a His-Tag-fused protein with a calculated molecular mass of 19.1 kDa (Fig. 2b), containing the first 118 amino acids of LysAB2. The other mutant, LysAB2-D2 is a His-Tag-fused protein with a calculated molecular mass of 10.2 kDa (Fig. 2b), containing the first 41 amino acids of LysAB2.

The schematic map and SDS-PAGE of the full-length LysAB2 and its deletion derivatives, LysAB2-D1 and LysAB2-D2. a Schematic structure of LysAB2 and its deletion constructs, LysAB2-D1 and LysAB2-D2. The black bar depicts the full length of LysAB2; the predicted residues involved in catalysis and sugar binding are indicated by hash and star signs, respectively; dotted line indicates the predicted amphipathic helix domains. Gray bar depicts the deletion mutant LysAB2-D1 where the putative amphipathic helix domains in the C-terminal have been removed. White bar depicts the deletion mutant LysAB2-D2 which contains only the first 41 aa of LysAB2, with all putative functional domains being removed. Arrows indicate the oligonucleotide primers. b Purified LysAB2 and its derivative mutants were electrophoresed on a 12% SDS gel, followed by staining with coomassie brilliant blue R-250. Lane M, markers; lane 1, purified LysAB2; lane 2, purified mutant LysAB2-D1; lane 3, purified mutant LysAB2-D2
Enzyme activity of purified LysAB2
To determine whether the cloned sequence obtained from the phage ϕAB2 did indeed encode a protein with lytic activity toward bacterial cell walls, an in situ assay was performed. Partially purified LysAB2, LysAB2-D1 and LysAB2-D2 were separated on an SDS-polyacrylamide gel containing either crude A. baumannii or S. aureus cell walls. After enzyme renaturation in the gel, the lytic activity corresponding to a protein with a molar mass of about 26 kDa was observed. However, there were no clear translucent bands shown on the molar mass of about 19 or 10 kDa. Interestingly, LysAB2 showed lysozyme activity toward A. baumannii and S. aureus. These results indicate that only the intact LysAB2 could lyse the peptidoglycan of A. baumannii and the S. aureus cell wall. We also calculated the activity of LysAB2 and CEWL on chloroform-treated A. baumannii cells at the experiment conditions described in the lysozyme activity assay in the materials and methods. These values are approximately 518 ± 143 U/mg and 1,685 ± 603 U/mg for LysAB2 and CEWL, respectively. The lysozyme activity of the native LysAB2 was only about 30% of that of CEWL. In addition, the thermostability analysis showed that LysAB2 was stable between 20°C and 40°C. The pH optimum analysis showed the effective pH for LysAB2 ranged from 4 to 8, with the optimum value at 6.0.
Antimicrobial activity of LysAB2
To study the antibacterial activity of LysAB2, multidrug-resistant A. baumannii (BCRC 80276), E. coli (Top 10) and methicillin-resistant S. aureus (BCRC 80277), were examined for their susceptibility to LysAB2. The concentrations of LysAB2 and of the control enzyme CEWL were 500 μg ml−1. The results of the antimicrobial activities of LysAB2 towards these three bacteria are shown in Fig. 3 and Table 2. The viabilities of the two Gram-negative bacteria, A. baumannii (BCRC 80276) and E. coli (Top 10), dropped to less than 1% following the incubation with LysAB2 for 1 h. On the other hand, the viability of methicillin-resistant S. aureus (BCRC 80277) retained 18% following incubation with LysAB2 for 1 h. Interestingly, in contrast to the weaker lysozyme activity of LysAB2 relative to CWEL shown earlier, LysAB2 exhibited stronger antibacterial activity towards A. baumannii, E. coli and S. aureus than CWEL. To further analyze the antibacterial spectrum of LysAB2, another four Gram-negative and two Gram-positive bacteria were included for the antimicrobial assay. The antimicrobial activities of LysAB2 towards these nine bacteria are shown in Table 2. We noticed that LysAB2 had a wide spectrum of antimicrobial activities against seven out of the nine tested bacteria (Table 2).

Antimicrobial activities. The antibacterial rates of LysAB2 (filled bar) and lysozyme (open bar) with different kinds of microorganisms are indicated
To elucidate the antimicrobial action of LysAB2 against A. baumannii further, partially purified LysAB2, and two deletion derivatives LysAB2-D1 and LysAB2-D2 were tested. The results showed that the antibacterial rate of the intact LysAB2 was greater than 99%. However, when the C-terminal region of LysAB2 was deleted, the antibacterial activity was significantly reduced. For the mutant LysAB2-D1, without the C-terminal putative amphipathic helix domains but keeping the putative catalysis and sugar binding regions, it retained only 40% of the antibacterial activity. As for the mutant LysAB2-D2, which contains only the first 41 amino acids with all known putative functional domains being removed, it remained less than 20% antibacterial activity. These results suggest that the existence of the C terminus of LysAB2 is essential for the antibacterial action against A. baumannii.
Lysis of viable cells by purified endolysin LysAB2
To determine the exogenous lytic activity of LysAB2, a suspension of viable A. baumannii was mixed with purified LysAB2 in phosphate buffer, and the lytic activity was determined by measuring the turbidity at OD600 at various times. The lytic activity of LysAB2 is shown in Fig. 4. The results clearly show that LysAB2 has an exogenous lytic effect on the viable cells because the relative absorbance decreased compared with that of cells incubated with phosphate buffer as a control.

Lysis of viable cells by purified LysAB2 protein. Filled square (black square) indicates Acinetobacter baumannii incubated with phosphate buffer, and an open square (white square) indicates A. baumannii incubated with 500 μg ml−1 of purified LysAB2 protein
Electron microscopy experiments
The effects of LysAB2 on the morphology of A. baumannii and S. aureus were visualized using scanning electron microscopy (SEM). The exposure of bacteria to 500 μg ml−1 LysAB2 for 60 min caused a remarkable modification of their cell shape, as shown by SEM. Untreated bacteria displayed a rough bright surface with no apparent cellular debris (Fig. 5a, c). In contrast, LysAB2-exposed cells exhibited a wide range of significant abnormalities, including deep roughening of the cell surface, the collapse of the cell structure and resulting cell debris (Fig. 5b, d).

Scanning electron microscopy of LysAB2-treated Acinetobacter baumannii and Staphylcoccus aureus. Cells were grown in LB medium and incubated with 500 μg ml−1 of purified LysAB2 for 60 min. Controls were run in deionized water alone. a Untreated A. baumannii cells. b A. baumannii treated with 500 μg ml−1 LysAB2 for 60 min. c Untreated S. aureus cells. d S. aureus treated with 500 μg ml−1 LysAB2 for 60 min
Permeation of the bacterial cytoplasmic membrane by LysAB2
To visualize the membrane-perturbing activity of LysAB2 on bacteria, we used FITC as a probe. FITC is a low-molecular-mass (389.4 Da) green fluorochrome which is unable to traverse the cytoplasmic membrane of intact cells. Indeed, when A. baumannii cells were incubated with the FITC probe without pretreatment with LysAB2, no appreciable fluorescent signal was discerned, as visualized by comparing the brightfield and fluorescence microscopy images (Figs. 6a, b). In contrast, FITC was readily accumulated in the bacteria after their exposure to 500 μg ml−1 LysAB2 (Figs. 6c, d), suggesting that LysAB2 increases the permeability of the bacterial membrane.

Bacterial membrane permeation induced by LysAB2 and visualized by fluorescein isothiocyanate (FITC) fluorescence. Acinetobacter baumannii were grown, treated with or without LysAB2 and stained with FITC, as indicated in the “Materials and methods.” Brightfield images of the untreated bacteria (a); fluorescent images of the untreated bacteria (b); brightfield images of A. baumannii treated with LysAB2 (c); fluorescent images of A. baumannii treated with LysAB2 (d)
Discussion
MDRAB are increasingly being reported worldwide. Bacteriophage therapy is a potential alternative treatment for MDR bacterial infections. Although A. baumannii infections have been experimentally treated with phages, no A. baumannii-specific phage endolysin has been characterized. In our previous work, ten phages with differing host ranges and lysis efficacy for MDRAB were isolated; one of these, ϕAB2, was further studied. From the ϕAB2 genome, we have successfully amplified and cloned an open reading frame encoding putative endolysin and named it lysAB2. Instead of being identified as a phage-type lysozyme (endolysin), BLAST results for the encoding product LysAB2 showed a strong sequence similarity to GH 19 chitinases. These enzymes, together with bacterial chitosanases and lysozymes from geese, phages and hens, were regarded as a superfamily of hydrolases based on their common catalytic core structure (Monzingo et al. 1996). Multiple sequence alignment of LysAB2 and its homologues showed the matching of some essential residues and the predicted secondary structures to the GH 19 chitinases. Therefore, according to the multiple sequence alignment features, we suspected that the ORF in ϕAB2 was expressing a novel type of endolysin that possesses a chitinase-like sequence and is capable of hydrolyzing polysaccharide in bacterial peptidoglycan. The subsequent experiments in this work supported this presumption. In contrast to chitin, which is a homopolymer of N-acetylglucosamine, the sugar component in bacterial peptidoglycan consists of alternating N-acetylglucosamine and N-acetylmuramic acid. This may possibly explain why among the conserved residues that were suggested to involve substrate interaction in chitinases (Hart et al. 1995), some residues (such as Gln118 and Asn124 in 2BAA numbering) were found in LysAB2, whereas other residues (such as Tyr123, Gln162, and Lys165 in 2BAA numbering) were absent.
In general, exogenous endolysin is highly active against many Gram-positive species but ineffective against Gram-negative bacteria. The different susceptibilities are probably due to the barrier function of the outer membrane existing in Gram-negative bacteria, which is impermeable to macromolecules and allows only limited diffusion of hydrophobic substances through bacterial lipopolysaccharide-covered surfaces (Vaara 1992). The antibacterial activity of endolysins is commonly ascribed to their enzymatic function, that is, cleavage of the covalent bonds in peptidoglycan. However, some endolysins, especially those from phages of Gram-negative bacteria, are capable of affecting bacterial cells by means of a mechanism completely independent of their enzymatic activity (During et al. 1999; Orito et al. 2004). In these cases, it was found that helix-forming amphipathic peptides containing basic amino acid residues seem to interact with negatively charged membrane elements, such as lipopolysaccharide in Gram-negative bacteria (During et al. 1999). Enhancement of lysozyme penetration through the outer membrane of Gram-negative bacteria has been attempted using permeabilizers, such as chelating agents, and the attachment of hydrophobic peptides to lysozymes (Morita et al. 2001). In this study, LysAB2 showed lysozyme activity toward both Gram-negative and Gram-positive bacteria, including A. baumannii and S. aureus. Moreover, it showed wide spectrum of antimicrobial activities against almost all the tested bacterial species (Table 2) and displayed stronger antimicrobial activities than CEWL (Fig. 3). Although the enzymatic activity of LysAB2 was less than that of CEWL, LysAB2 showed strong antibacterial activity against A. baumannii. This observation suggests that in addition to the enzymatic activity, extra functions are required for the antibacterial activity of LysAB2 against A. baumannii. To further elucidate the function of the antimicrobial action of LysAB2 against A. baumannii, we also compared the antimicrobial activities between partially purified intact LysAB2 and its two deletion derivatives LysAB2-D1 and LysAB-D2.
The antibacterial rate of intact LysAB2 was more than 99%. In contrast, the deletion of the C-terminal region of LysAB2 significantly reduced its antibacterial activity. Mutant LysAB2-D1, which contains the first 118 aa of LysAB2, keeping the putative catalysis and sugar binding domain but losing the putative amphipathic helix domains, retained only 40% of the antibacterial activity.
Considering A. baumannii being a Gram-negative bacterium with inner and outer membrane in the cell envelop, LysAB2 has to permeate through the bacterial outer membrane to reach the peptidoglycan layer in the envelope. Therefore, we suggest that in LysAB2, the C-terminal region possibly plays the roles to enhance the permeability of bacterial outer membrane and assist its N-terminal enzymatic domain to reach the periplasm and gain access to the peptidoglycan layer. Our preliminary experiments have shown that the synthesized potential cationic antimicrobial peptide (aa 113–145) of LysAB2 exhibited membrane permeability and antibacterial activities (data not shown). Based on our results shown in this study and the above presumption, we speculate that the mechanism of antibacterial function of LysAB2 is as follows: the C-terminal region interacts with or penetrates the cell envelope; subsequently, the N terminus of endolysin, which may harbor the catalytic domain, approaches the peptidoglycan layer, causing the lysis of the bacterial cell. Further characterization on LysAB2 is now under investigation. We hope that this study will provide further insight on controlling multidrug-resistant strains of pathogens by phage endolysins.
References
Barrow PA, Soothill JS (1997) Bacteriophage therapy and prophylaxis: rediscovery and renewed assessment of potential. Trends Microbiol 5:268–271
Bergogne-Berezin E, Towner KJ (1996) Acinetobacter spp. as nosocomial pathogens: microbiological, clinical, and epidemiological features. Clin Microbiol Rev 9:148–165
Borysowski J, Weber-Dabrowska B, Gorski A (2006) Bacteriophage endolysins as a novel class of antibacterial agents. Exp Biol Med (Maywood) 231:366–377
Brunner F, Stintzi A, Fritig B, Legrand M (1998) Substrate specificities of tobacco chitinases. Plant J 14:225–234
Ceyssens PJ, Lavigne R, Mattheus W, Chibeu A, Hertveldt K, Mast J, Robben J, Volckaert G (2006) Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: establishment of the phiKMV subgroup within the T7 supergroup. J Bacteriol 188:6924–6931
Dijkshoorn L, Nemec A, Seifert H (2007) An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol 5:939–951
During K, Porsch P, Mahn A, Brinkmann O, Gieffers W (1999) The non-enzymatic microbicidal activity of lysozymes. FEBS Lett 449:93–100
Entenza JM, Loeffler JM, Grandgirard D, Fischetti VA, Moreillon P (2005) Therapeutic effects of bacteriophage Cpl-1 lysin against Streptococcus pneumoniae endocarditis in rats. Antimicrob Agents Chemother 49:4789–4792
Gille C, Frommel C (2001) STRAP: editor for STRuctural alignments of proteins. Bioinformatics 17:377–378
Hahn M, Hennig M, Schlesier B, Hohne W (2000) Structure of jack bean chitinase. Acta Crystallogr D Biol Crystallogr 56:1096–1099
Hart PJ, Pfluger HD, Monzingo AF, Hollis T, Robertus JD (1995) The refined crystal structure of an endochitinase from Hordeum vulgare L. seeds at 1.8 a resolution. J Mol Biol 248:402–413
Hermoso JA, Garcia JL, Garcia P (2007) Taking aim on bacterial pathogens: from phage therapy to enzybiotics. Curr Opin Microbiol 10:461–472
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36 (Web Server issue):W5–9
Kakikawa M, Yokoi KJ, Kimoto H, Nakano M, Kawasaki K, Taketo A, Kodaira K (2002) Molecular analysis of the lysis protein Lys encoded by Lactobacillus plantarum phage phig1e. Gene 299:227–234
Lavigne R, Burkal'tseva MV, Robben J, Sykilinda NN, Kurochkina LP, Grymonprez B, Jonckx B, Krylov VN, Mesyanzhinov VV, Volckaert G (2003) The genome of bacteriophage phiKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology 312:49–59
Lin NT, Chiou PY, Chang KC, Chen LK, Lai MJ (2010) Isolation and characterization of phi AB2: a novel bacteriophage of Acinetobacter baumannii. Res Microbiol 161:308–314
Mangoni ML, Papo N, Barra D, Simmaco M, Bozzi A, Di Giulio A, Rinaldi AC (2004) Effects of the antimicrobial peptide temporin L on cell morphology, membrane permeability and viability of Escherichia coli. Biochem J 380:859–865
Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Tasneem A, Thanki N, Yamashita RA, Zhang D, Zhang N, Bryant SH (2009) CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res 37 (Database issue):D205–D210
McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16:404–405
Monzingo AF, Marcotte EM, Hart PJ, Robertus JD (1996) Chitinases, chitosanases, and lysozymes can be divided into procaryotic and eucaryotic families sharing a conserved core. Nat Struct Biol 3:133–140
Morita M, Tanji Y, Orito Y, Mizoguchi K, Soejima A, Unno H (2001) Functional analysis of antibacterial activity of Bacillus amyloliquefaciens phage endolysin against Gram-negative bacteria. FEBS Lett 500:56–59
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217
Orito Y, Morita M, Hori K, Unno H, Tanji Y (2004) Bacillus amyloliquefaciens phage endolysin can enhance permeability of Pseudomonas aeruginosa outer membrane and induce cell lysis. Appl Microbiol Biotechnol 65:105–109
Patil RS, Ghormade VV, Deshpande MV (2000) Chitinolytic enzymes: an exploration. Enzyme Microb Technol 26:473–483
Peleg AY, Seifert H, Paterson DL (2008) Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582
Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA (2007) Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 51:3471–3484
Stone R (2002) Bacteriophage therapy. Stalin’s forgotten cure. Science 298:728–731
Sulakvelidze A, Alavidze Z, Morris JG Jr (2001) Bacteriophage therapy. Antimicrob Agents Chemother 45:649–659
Ubhayasekera W, Rawat R, Ho SW, Wiweger M, Von Arnold S, Chye ML, Mowbray SL (2009) The first crystal structures of a family 19 class IV chitinase: the enzyme from Norway spruce. Plant Mol Biol 71:277–289
Vaara M (1992) Agents that increase the permeability of the outer membrane. Microbiol Rev 56:395–411
van Hengel AJ, Tadesse Z, Immerzeel P, Schols H, van Kammen A, de Vries SC (2001) N-acetylglucosamine and glucosamine-containing arabinogalactan proteins control somatic embryogenesis. Plant Physiol 125:1880–1890
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191
Wroblewska M (2006) Novel therapies of multidrug-resistant Pseudomonas aeruginosa and Acinetobacter spp. infections: the state of the art. Arch Immunol Ther Exp (Warsz) 54:113–120
Acknowledgments
We thank the Electron Microscopy Laboratory of the Department of Anatomy of Tzu Chi University for technical assistance. This work was supported by grants TCSP-0302 from the Buddhist Tzu Chi General Hospital and TCIRP99002-03 from Tzu Chi University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Meng-Jiun Lai and Nien-Tsung Lin contributed equally to this work.
Rights and permissions
About this article
Cite this article
Lai, MJ., Lin, NT., Hu, A. et al. Antibacterial activity of Acinetobacter baumannii phage ϕAB2 endolysin (LysAB2) against both Gram-positive and Gram-negative bacteria. Appl Microbiol Biotechnol 90, 529–539 (2011). https://doi.org/10.1007/s00253-011-3104-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3104-y