
Abstract
For many years it has been thought that apoptotic cells rapidly cleared by phagocytic cells do not trigger an immune response but rather have anti-inflammatory properties. However, accumulating experimental data indicate that certain anticancer therapies can induce an immunogenic form of apoptosis associated with the emission of damage-associated molecular patterns (DAMPs), which function as adjuvants to activate host antitumor immune responses. In this review, we will first discuss recent advances and the significance of danger signaling pathways involved in the emission of DAMPs, including calreticulin, ATP, and HMGB1. We will also emphasize that switching on a particular signaling pathway depends on the immunogenic cell death stimulus. Further, we address the role of ER stress in danger signaling and the classification of immunogenic cell death inducers in relation to how ER stress is triggered. In the final part, we discuss the role of radiotherapy-induced immunogenic apoptosis and the relationship of its immunogenicity to the fraction dose and concomitant chemotherapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
6.1 Introduction
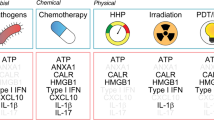
In the human body close to 500 billion cells die each day by apoptosis, and they are continuously recognized and removed by the phagocytic system without causing inflammation or scars. The process of clearing—dying cells play a critical role in development, maintenance of tissue homeostasis, control of immune responses, and resolution of inflammation. Immunological responses elicited by apoptotic cells have been studied extensively in the last two decades. Back in the nineties it was shown that uptake of apoptotic neutrophils or eosinophils by human monocyte-derived macrophages does not induce secretion of granulocyte macrophage colony stimulating factor (GM-CSF) or thromboxane B2 [1, 2]. In later studies it was shown that apoptotic cells actually inhibit the production of many proinflammatory cytokines by antigen-presenting cells (Fig. 6.1) [1, 3–10]. Cells undergoing apoptosis are known to modulate their tissue microenvironments either by acting on phagocytes and thereby inhibiting immunological and inflammatory responses and promoting “healing” signaling pathways and/or by releasing immunomodulatory signals. Indeed, in the context of anticancer therapy it is generally accepted that most chemotherapeutic drugs elicit apoptotic cell death. Phagocytosis of apoptotic cells maintains an anti-inflammatory state in the extracellular environment and thereby contributes to an immunosuppressive network in a primary tumor site to promote further tumorigenesis [11]. Several studies have confirmed this notion. It has been shown that apoptotic tumor cells promote coordinated tumor growth, angiogenesis, and accumulation of tumor-associated macrophages (TAMs) in aggressive B cell lymphomas [12]. It has also been demonstrated that radiotherapy induces caspase-3-dependent release from apoptotic cells of arachidonic acid and prostaglandin E2, which then promote the growth of the tumor cells that survive radiation activation [13]. This correlates with observations in cancer patients that tumors with elevated levels of activated caspase-3 are associated with a poor disease outcome [13]. All these studies indeed demonstrate that cancer cells undergoing apoptosis can promote tumor progression. However, in the late nineties it was reported that dendritic cells (DCs) internalize apoptotic cells and process them for presentation to both MHC class I- and class II-restricted T cells with an efficiency that is dependent on the number of apoptotic cells [14]. Later, it was discovered that certain types of anticancer treatments, such as chemotherapeutics (e.g., anthracyclines) [15], γ-irradiation [16, 17], and photodynamic therapy [18–21] (Table 6.1) can induce a specific form of apoptosis, which was named immunogenic apoptosis (IA) due to its immunostimulatory or adjuvant-like properties (Fig. 6.1). When cancer cell lines exposed to lethal doses of inducers of immunogenic apoptosis in vitro are used to vaccinate syngenic mice, they protect them against a subsequent challenge with live cancer cells of the same type. The immunogenicity of apoptotic cancerous cells relies on the spatiotemporal emission of specific signals called danger-associated molecular patterns (DAMPs) , such as calreticulin (CRT), ATP, and HMGB-1. Most of these molecules have predominantly nonimmunological functions inside the cell but they become immunogenic after they are emitted extracellularly. DAMPs are derived from different subcellular compartments, including the plasma membrane, nucleus, ER, and cytosol, and they can often be modified by the proteolysis and/or oxidation associated with cell death mechanisms [22, 23]. DAMPs exert their immunostimulatory effects upon their recognition by membrane-bound or cytoplasmic pattern-recognition receptors (PRRs, e.g., Toll-like Receptor-4, TLR4), phagocytic receptors or scavenger receptors (e.g., LDL-receptor-related protein, LRP1/CD91), and purinergic receptors (e.g., P2RX7/P2RY2). These danger signals, in combination with cancer antigens, induce maturation of dendritic cells (DCs) and can lead to an adaptive immune response against tumor cells, thereby mediating anticancer immunity. This review covers recent advances in our understanding of the molecular mechanisms involved in danger signaling, DAMPs emission, the role of ER stress, and classification of immunogenic cell death inducers in relation to the way ER stress is triggered. In the final part, we discuss the role of radiotherapy-induced immunogenic apoptosis and the relationship of its immunogenicity to the fraction dose and concomitant chemotherapy.

Timeline of the key milestones in the development of the immunogenic cell death concept. Of note that immunotherapy in the treatment of cancer was first successfully used in 1891 by William B. Coley, who injected streptococcal products into patients with inoperable cancer. These products became known as Coley’s Toxins. The following references are used to make this figure: [14–16, 20, 25, 32, 33, 42, 43, 46, 92–104]
6.2 ER Stress and ROS: Crucial Players in Danger Signaling
Immunogenic anticancer drugs and treatments can trigger IA in dying cancer cells via the combined action of ER stress and ROS production, which activate danger signaling pathways and mediate the trafficking of DAMPs to the extracellular space [20, 24, 25]. ER stress was proposed to be a crucial component because the emission of DAMPs (e.g., calreticulin and ATP) and subsequent immunogenicity of cell death in vivo was found to be diminished when molecular effectors of the ER stress pathway were silenced [20, 25]. Anticancer drugs that do not induce ER stress (e.g., cisplatin) are poor inducers of IA [26]. Notably, the immunogenicity of drugs such as cisplatin could be restored by combining it with thapsigargin or tunicamycin [26] or by expression of the ER resident protein reticulon-1 [27]. ROS was also proposed to be required for immunogenicity of cell death because antioxidants (N-Acetyl cysteine, glutathione ethyl ester, and l-histidine) decrease its immunogenicity [20, 25]. As many immunogenic cell death inducers are diverse both biologically and chemically (reviewed in detail in [24, 28]), there seems to be no simple structure–function relationship that could explain the ability of these agents to induce IA. Therefore, we proposed that immunogenic cell death inducers can be classified into two categories (Type I and Type II) based on their distinct mode of action in the induction of ER stress and apoptosis [24]. Most of immunogenic cell death inducers (Table 6.1) are categorized as type I immunogenic cell death inducers that primarily trigger cell death via targeting cytosolic proteins, plasma membranes, or nucleic proteins rather than primary targeting ER mechanisms [24, 29, 30]. The type II immunogenic cell death inducers preferentially target the ER and include hypericin-based PDT and oncolytic coxsackievirus B3 (CVB3, Table 6.1). Although ER stress and ROS are essential in the immunogenicity of cell death, it is still not clear how these two signaling modules cooperate to efficiently induce immunogenic cell death. Therefore, further studies to elucidate the precise interplay between the ER stress and ROS is required to modulate antitumor immune responses.
6.3 Main Effectors of Immunogenic Cell Death: CRT, ATP, and HMGB1
Calreticulin (CRT) is an ER chaperone and its function is usually linked with Ca2+ homeostasis [31]. The role of CRT in the clearance of apoptotic cells was first described by Gardai et al. [32], who showed that CRT acts as a recognition ligand (“eat me” signal) on the surface of apoptotic cells by binding and activating LRP1/CD91 on the engulfing cell (Fig. 6.1). However, a new life was given to CRT by studies showing that CRT exposure is a key determinant of immunogenicity of dying cells and anticancer immune responses [33]. In that study, the authors found that anthracyclines induce rapid preapoptotic translocation of CRT to the cell surface and that blockade or knockdown of CRT suppresses the immunogenicity of apoptotic cancerous cells in mice. Several signaling pathways triggered by immunogenic cell death inducers have been described (Fig. 6.2). One pathway is induced by anthracyclines and relies on the phosphorylation of eukaryotic initiation factor 2a (eIF2a) by the ER stress-sensing kinase, PKR-related ER kinase (PERK), the activation of caspase-8, BAX and BAK, the transport of ER-derived vesicles through the Golgi apparatus, and the SNAP receptor (SNARE)-dependent exocytosis of these vesicles [25]. It has also been shown that paracrine signals that involve the chemokine CXCL8 contribute to CRT exposure on the cell surface [34]. The second pathway for CRT exposure is more rapid and relies on PERK-mediated trafficking of ecto-CRT by regulation of the proximal secretory pathway [20]. In this signaling pathway, eIF2a phosphorylation and caspase-8 signaling were not required for CRT exposure. Vaccination of mice with cells deficient in any of the proteins required for CRT exposure or with cells in which CRT was knocked down reduced the immunogenicity of the cancer cells [20, 33]. All these results underline the key role of CRT exposure on the cell surface to the efficacy of anticancer therapy.

An overview of the danger signaling pathways involved in surface CRT exposure and ATP secretion and their relation to different apoptotic stages. Signaling pathways responsible for surface exposure of CRT and secretion of ATP depend on immunogenic cell death stimuli [24]
ATP is involved in various cellular metabolic processes and intracellular responses. However, it has become clear that APT is also actively secreted or passively released from dying cancerous cells, and that it is modulating the immunogenicity of dying cancerous cells (Fig. 6.1) [22, 23, 35, 36] via activation of purinergic P2X7 and P2X2 receptors [37]. The mechanisms of ATP secretion are strongly dependent on the type of immunogenic cell death inducer. Anthracyclines induce ATP secretion by a mechanism involving the caspase-dependent activation of pannexin 1 channels, lysosomal exocytosis, and plasma membrane blebbing [36, 38, 39]. Moreover, cancer cells undergoing IA in response to anthracycline secrete ATP in an autophagy-dependent manner [40–42]. Autophagy-deficient tumors exposed to chemotherapy cannot attract tumor-infiltrating leukocytes and therefore do not induce therapeutic anticancer immune responses [42]. However, in contrast to anthracyclines, hypericin-based PDT-induced ATP secretion is independent of autophagy [43] and involves the classical and PERK-regulated proximal secretory pathway, as well as PI3K-dependent exocytosis [20]. All these studies suggest that the mechanisms of ATP secretion might vary from one immunogenic cancer cell death inducer to another (Fig. 6.2).
HMGB1 is a broadly expressed and highly abundant nonhistone chromatin-binding protein expressed constitutively by all eukaryotic cells, and it has various cytosolic and extracellular functions [44, 45]. It was found that the immunogenicity of IA also depends on the passive release of HMGB1 from cells undergoing immunogenic death and on its binding to TLR-4 [46]. Nevertheless, the role of HMGB1 in anticancer immunity is complex, and the diversity of HMGB1 extracellular functions can also be partially explained by the posttranslation modifications, including different redox states and cell death types [23, 47, 48].
6.4 Immunostimulatory Effects of Chemotherapeutics Not Related to DAMPs
In addition to the induction of danger signaling and modulation of DAMPs emission in cancer cells (discussed earlier), many chemotherapeutics can induce immunostimulation by targeting other elements of anticancer immunity [36]. Chemotherapeutic drugs can increase the expression or presentation of tumor-associated antigens (TAA) on the surface of cancer cells and increase their so-called antigenicity by inducing antigen presentation of both dominant and subdominant epitopes. It has been shown that the variety of TAA eliciting cytotoxic T lymphocytes (CTL) can be increased by cisplatin and gemcitabine [49]. The authors showed that chemotherapy reveals weaker tumor antigens to the immune system, resulting in the induction of specific CTLs. The antigenicity of cancer cells can be enhanced by increasing the expression of MHC class I molecules (e.g., cyclophosphamide, gemcitabine, oxaliplatin, paclitaxel, and γ-irradiation) [36, 50, 51]. In addition, some anticancer drugs can increase the expression of TAA, including carcinoembryonic antigen (induced by 5-fluorouracil), multiple cancer testis antigens (increased by 5-aza-20deoxycytidine and γ-irradiation), and melanoma-associated antigens (increased by vemurafenib) [36, 50, 52, 53]. It is of interest that subtoxic doses of paclitaxel and doxorubicin increased the expression of components of the MHC class I antigen processing machinery (calmodulin, LMP2, LMP7, TAP1, and tapasin) in cancer cells [54]. Chemotherapeutic agents also cause immunopotentiation by directly stimulating immune cells. It has been shown that low doses of paclitaxel, doxorubicin, mitomycin C, and methotrexate that do not cause cell death up-regulate the ability of DCs to present antigens to antigen-specific T cells [55]. Recently, we demonstrated that intraperitoneal injection of doxorubicin in mice triggers the signs of acute inflammatory response (accumulation of neutrophils and increased levels of IL6, TNF, and MCP-1) [56–58]. Of interest is that the inflammatory response was significantly reduced in mice deficient in myeloid differentiation primary response gene 88 (MyD88), TLR-2 or TLR-9 [58], or tumor necrosis factor receptor-1 (TNFR1) [57]. These studies provide important new insights into how the innate immune system is modulated by immunogenic drugs such as doxorubicin (Table 6.1). It was also shown that the percentage of regulatory T cells among the CD4+ lymphocytes was decreased by cyclophosphamide , which allowed a whole tumor cell vaccine or costimulatory receptor OX40 (OX86) immunotherapy to eradicate established tumors in colon carcinoma or melanoma models [59, 60]. The number of myeloid-derived suppressor cells (MDSCs) was reduced by gemcitabine in the spleen of mice bearing large tumors but did not affect CD4 and CD8 T cells, NK cells, macrophages, and B cells [61–63]. The bisphosphonate zoledronate , a drug that has been approved by the FDA for the treatment of bone metastases, was shown to induce caspase-1 activation in DC-like cells, which then provide mature IL-18 and IL-1β for the activation of IL-2-primed NK cells [64]. All these data suggest that some chemotherapeutics can directly stimulate immune cell functions and that their therapeutic efficacy could be at least partly explained by their ability to modulate the host immune system.
6.5 Radiotherapy-Induced Immunogenic Cell Death: Fraction Dose and Concomitant Chemotherapy
Together with surgery and chemotherapy, gamma-irradiation (RT) is important in the treatment of cancer. For decades, its main antitumor activity was believed to result from a direct and local cytotoxic effect on malignant cells within the irradiated area [65]. Nowadays, there is growing evidence for the occurrence of immune-mediated systemic effects resulting from local RT. Clinical proof of principle for such abscopal effects is provided by regression of distant metastases after local RT. Abscopal effects have been observed with various dose and fractionation regimens in melanoma (3 × 8 Gy to 3 × 18 Gy) [66–68] and lung adenocarcinoma (5 × 6 Gy) [69]. The necessity of combining RT with immunotherapy (in these cases CTLA4 blockade) to achieve these abscopal effects indicates that proimmunogenic effects are often dampened by the immune-suppressive microenvironment that characterizes cancer [70–73].
As for other immunogenic agents [74], radiation-induced immunogenic cell death is characterized, in cell cultures, by preapoptotic exposure on the extracellular surface of the “eat-me” signal CRT [25, 75, 76] and emission of ATP [75, 77, 78], and by late-apoptotic release of the “find-me” signal HMGB-1 [46, 75, 77, 79]. Animal and clinical experimental evidence supporting the ability of RT to induce immunogenic cell death remains scarce [77], and the clinical relevance of these pathways to the therapeutic efficacy of RT has yet to be validated.
Induction of immunogenic cell death is most likely highly dependent on total dose and fractionation. Golden et al. showed, in cell cultures, that the clinically used single doses between 2 and 20 Gy (1 × 2–20 Gy) effectively induce the signals for each individual component of immunogenic cell death in a dose-dependent manner [75]. Gameiro et al. showed the same, albeit with a clinically irrelevant single dose of 100 Gy [77]. Demaria et al. overviewed the literature and found immunogenic cell death to be often detected in tumor cell cultures exposed to mid-to-high doses of RT (1 × >5–10 Gy) [80]. They initiated animal experiments using three RT regimens (1 × 20 Gy, 3 × 8 Gy and 5 × 6 Gy) combined with CTLA-4 antibody treatment in syngeneic mice with breast and colorectal carcinoma. While anti-CTLA-4 treatment on its own and its combination with a single-dose RT were not able to induce an abscopal effect, the fractioned regimens did [81]. This could explain why a single 8-Gy fraction treatment of bone metastases in prostate cancer patients failed to induce an abscopal effect when combined with anti-CTLA-4 treatment [82], whereas the above described clinical trials succeeded [66–69].
In addition to the induction of immunogenic cell death, other components up- or downregulated in response to RT are involved in antitumor immunity [71]. Tumor cell surface expression of MHC Class I molecules increases and CD47 (a “don’t eat-me” signal for DCs) decreases in a dose-dependent manner in cell cultures [83–85]. Additionally, it was shown in a murine model that RT (2 × 12 Gy) increases the expression on tumor cell surface of RAE-1, a ligand for natural killer cell group 2D [86]. Distinct radiation fraction doses also have a direct effect on the irradiated tumor microenvironment. Clinical observations showed that immune-suppressing Treg cells are more radioresistant than CD8+ T cells [87, 88]. In a xenotransplant mouse model, a lower RT dose (1 × 2 Gy) reprograms macrophages toward an iNOS+/M1 phenotype, allowing them to recruit tumor-specific T cells [89].
The above-mentioned data support the growing consensus that hypofractionated regimens (a limited number but >1 fraction high doses per fraction) are more effective at inducing the proimmunogenic effects of RT than single high doses or normofractionation (2 Gy per fraction or “×” times × 2 Gy) [90]. The hypofractionated regimens are mostly used to treat small (often oligo-) metastatic lesions, whereas for treatment of the primary tumor, normofractionation combined with chemotherapy is often the standard treatment. Concomitant use of both treatments has been shown to be superior to sequential chemo-RT in numerous clinical trials. It should be considered that concomitant chemo-RT causes a tumor cell death that is both qualitatively and quantitatively different from that achieved by each therapy alone [83]. Frey et al. showed that combining 5-FU, oxaliplatin, and irinotecan with RT could induce immunogenic cell death in colorectal cancer cells [91]. Golden et al. designed a cell culture assay to examine the effect on immunogenic cell death when combining RT (1 × 2 Gy) with paclitaxel and found that all three components of immunogenic cell death (i.e., CRT, ATP, and HMGB1; discussed earlier) to be increased significantly when chemotherapy and RT were used together as compared to separate treatments [75, 83]. Animal and clinical experiments are awaited to validate these interesting findings.
6.6 Conclusions
Only one decade ago, apoptotic cell death was presented as anti-inflammatory and tolerogenic, or even as a silent mode of cell death. However, insights over the last decade increasingly support the view that under specific conditions certain types and regimens of anticancer therapy can induce an immunogenic form of apoptosis that can be beneficial for the induction of anticancer immunity and long-lasting remission in cancer patients. Many questions remain regarding what determines the difference between immunogenic aspects of apoptosis and the danger signaling subroutines in the various types of cancers. Deeper insight into the molecular mechanisms of immunogenicity of apoptotic cells will lead to novel experimental immunotherapies for cancer, and is therefore a challenging research area. This work highlights the need for careful preclinical testing of the immunological effects of chemotherapies, alone and in combination with partner cytotoxic agents and immunotherapies, before proceeding to clinical investigations.
References
Meagher LC, Savill JS, Baker A, Fuller RW, Haslett C. Phagocytosis of apoptotic neutrophils does not induce macrophage release of thromboxane B2. J Leukoc Biol. 1992;52:269–73.
Stern M, Savill J, Haslett C. Human monocyte-derived macrophage phagocytosis of senescent eosinophils undergoing apoptosis. Mediation by alpha v beta 3/CD36/thrombospondin recognition mechanism and lack of phlogistic response. Am J Pathol. 1996;149:911–21.
Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu M, Haegeman G, D’Herde K, Vandenabeele P. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell. 2004;15:1089–100.
Cocco RE, Ucker DS. Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol Biol Cell. 2001;12:919–30.
Cvetanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J Immunol. 2004;172:880–9.
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8.
Krysko DV, D’Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis. 2006;11:1709–26.
Krysko DV, Vandenabeele P. From regulation of dying cell engulfment to development of anti-cancer therapy. Cell Death Differ. 2008;15:29–38.
Krysko DV, Vandenabeele P. Clearance of dead cells: mechanisms, immune responses and implication in the development of diseases. Apoptosis. 2010;15:995–7.
Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1.
Gregory CD, Pound JD. Cell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol. 2011;223:177–94.
Ford CA, Petrova S, Pound JD, Voss JJ, Melville L, Paterson M, Farnworth SL, Gallimore AM, Cuff S, Wheadon H, et al. Oncogenic properties of apoptotic tumor cells in aggressive B cell lymphoma. Curr Biol. 2015;25:577–88.
Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O’Sullivan B, He Z, Peng Y, Tan AC, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17:860–6.
Rovere P, Vallinoto C, Bondanza A, Crosti MC, Rescigno M, Ricciardi-Castagnoli P, Rugarli C, Manfredi AA. Bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J Immunol. 1998;161:4467–71.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–701.
Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P, Zitvogel L, Kroemer G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14:1848–50.
Scheithauer H, Belka C, Lauber K, Gaipl US. Immunological aspects of radiotherapy. Radiat Oncol. 2014;9:185.
Garg AD, Krysko DV, Vandenabeele P, Agostinis P. The emergence of phox-ER stress induced immunogenic apoptosis. Oncoimmunology. 2012;1:786–8.
Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother. 2012;61:215–21.
Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31:1062–79.
Panzarini E, Inguscio V, Fimia GM, Dini L. Rose Bengal acetate photodynamic therapy (RBAc-PDT) induces exposure and release of Damage-Associated Molecular Patterns (DAMPs) in human HeLa cells. PLoS One. 2014;9, e105778.
Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32:157–64.
Krysko O, Love Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013;4, e631.
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75.
Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28:578–90.
Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30:1147–58.
Michaud M, Sukkurwala AQ, Di Sano F, Zitvogel L, Kepp O, Kroemer G. Synthetic induction of immunogenic cell death by genetic stimulation of endoplasmic reticulum stress. Oncoimmunology. 2014;3, e28276.
Dudek AM, Garg AD, Krysko DV, De Ruysscher D, Agostinis P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013;24:319–33.
Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. 2014;21:26–38.
Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014;21:39–49.
Gelebart P, Opas M, Michalak M. Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int J Biochem Cell Biol. 2005;37:260–6.
Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Sukkurwala AQ, Martins I, Wang Y, Schlemmer F, Ruckenstuhl C, Durchschlag M, Michaud M, Senovilla L, Sistigu A, Ma Y, et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2014;21:59–68.
Wang Y, Martins I, Ma Y, Kepp O, Galluzzi L, Kroemer G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy. 2013;9:1624–5.
Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–8.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–6.
Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Metivier D, Galluzzi L, Perfettini JL, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014;21:79–91.
Martins I, Michaud M, Sukkurwala AQ, Adjemian S, Ma Y, Shen S, Kepp O, Menger L, Vacchelli E, Galluzzi L, et al. Premortem autophagy determines the immunogenicity of chemotherapy-induced cancer cell death. Autophagy. 2012;8:413–5.
Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Seror C, Metivier D, Perfettini JL, Zitvogel L, Kroemer G. Chemotherapy induces ATP release from tumor cells. Cell Cycle. 2009;8:3723–8.
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–7.
Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV, Mathieu C, Agostinis P. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy. 2013;9:1292–307.
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116.
Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol. 2015;6:93.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9.
Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32.
Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–28.
Jackaman C, Majewski D, Fox SA, Nowak AK, Nelson DJ. Chemotherapy broadens the range of tumor antigens seen by cytotoxic CD8(+) T cells in vivo. Cancer Immunol Immunother. 2012;61:2343–56.
Chen G, Emens LA. Chemoimmunotherapy: reengineering tumor immunity. Cancer Immunol Immunother. 2013;62:203–16.
Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. 2010;102:115–23.
Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–31.
Sharma A, Bode B, Wenger RH, Lehmann K, Sartori AA, Moch H, Knuth A, Boehmer L, Broek M. gamma-Radiation promotes immunological recognition of cancer cells through increased expression of cancer-testis antigens in vitro and in vivo. PLoS One. 2011;6, e28217.
Kaneno R, Shurin GV, Kaneno FM, Naiditch H, Luo J, Shurin MR. Chemotherapeutic agents in low noncytotoxic concentrations increase immunogenicity of human colon cancer cells. Cell Oncol. 2011;34:97–106.
Shurin GV, Tourkova IL, Kaneno R, Shurin MR. Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J Immunol. 2009;183:137–44.
Kaczmarek A, Brinkman BM, Heyndrickx L, Vandenabeele P, Krysko DV. Severity of doxorubicin-induced small intestinal mucositis is regulated by the TLR-2 and TLR-9 pathways. J Pathol. 2012;226:598–608.
Kaczmarek A, Krysko O, Heyndrickx L, Love Aaes T, Delvaeye T, Bachert C, Leybaert L, Vandenabeele P, Krysko DV. TNF/TNF-R1 pathway is involved in doxorubicin-induced acute sterile inflammation. Cell Death Dis. 2013;4, e961.
Krysko DV, Kaczmarek A, Krysko O, Heyndrickx L, Woznicki J, Bogaert P, Cauwels A, Takahashi N, Magez S, Bachert C, Vandenabeele P. TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death Differ. 2011;18:1316–25.
Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336–44.
Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, Cohen AD, Avogadri F, Lesokhin AM, Weinberg AD, Wolchok JD, Houghton AN. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. J Exp Med. 2009;206:1103–16.
Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–26.
Ko HJ, Kim YJ, Kim YS, Chang WS, Ko SY, Chang SY, Sakaguchi S, Kang CY. A combination of chemoimmunotherapies can efficiently break self-tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer Res. 2007;67:7477–86.
Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–21.
Nussbaumer O, Gruenbacher G, Gander H, Thurnher M. DC-like cell-dependent activation of human natural killer cells by the bisphosphonate zoledronic acid is regulated by gammadelta T lymphocytes. Blood. 2011;118:2743–51.
Bernier J, Hall EJ, Giaccia A. Radiation oncology: a century of achievements. Nat Rev Cancer. 2004;4:737–47.
Hiniker SM, Chen DS, Reddy S, Chang DT, Jones JC, Mollick JA, Swetter SM, Knox SJ. A systemic complete response of metastatic melanoma to local radiation and immunotherapy. Transl Oncol. 2012;5:404–7.
Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366:925–31.
Stamell EF, Wolchok JD, Gnjatic S, Lee NY, Brownell I. The abscopal effect associated with a systemic anti-melanoma immune response. Int J Radiat Oncol Biol Phys. 2013;85:293–5.
Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res. 2013;1:365–72.
De Meerleer G, Khoo V, Escudier B, Joniau S, Bossi A, Ost P, Briganti A, Fonteyne V, Van Vulpen M, Lumen N, et al. Radiotherapy for renal-cell carcinoma. Lancet Oncol. 2014;15:e170–7.
De Wolf K, Vermaelen K, De Meerleer G, Lambrecht BN, Ost P. The potential of radiotherapy to enhance the efficacy of renal cell carcinoma therapy. Oncoimmunology. 2015;4, e1042198.
Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105:256–65.
Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. 2015;25:11–7.
Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3, e955691.
Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3, e28518.
Obeid M, Tesniere A, Panaretakis T, Tufi R, Joza N, van Endert P, Ghiringhelli F, Apetoh L, Chaput N, Flament C, et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol Rev. 2007;220:22–34.
Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget. 2014;5:403–16.
Ohshima Y, Tsukimoto M, Takenouchi T, Harada H, Suzuki A, Sato M, Kitani H, Kojima S. gamma-Irradiation induces P2X(7) receptor-dependent ATP release from B16 melanoma cells. Biochim Biophys Acta. 2010;1800:40–6.
Suzuki Y, Mimura K, Yoshimoto Y, Watanabe M, Ohkubo Y, Izawa S, Murata K, Fujii H, Nakano T, Kono KA. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012;72:3967–76.
Demaria S, Formenti SC. Radiation as an immunological adjuvant: current evidence on dose and fractionation. Front Oncol. 2012;2:153.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379–88.
Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–12.
Formenti SC, Demaria S. Effects of chemoradiation on tumor-host interactions: the immunologic side. J Clin Oncol. 2008;26:1562–3; author reply 1563.
Hauser SH, Calorini L, Wazer DE, Gattoni-Celli S. Radiation-enhanced expression of major histocompatibility complex class I antigen H-2Db in B16 melanoma cells. Cancer Res. 1993;53:1952–5.
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–71.
Ruocco MG, Pilones KA, Kawashima N, Cammer M, Huang J, Babb JS, Liu M, Formenti SC, Dustin ML, Demaria S. Suppressing T cell motility induced by anti–CTLA-4 monotherapy improves antitumor effects. J Clin Invest. 2012;122:3718–30.
Kachikwu EL, Iwamoto KS, Liao YP, DeMarco JJ, Agazaryan N, Economou JS, McBride WH, Schaue D. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys. 2011;81:1128–35.
Qinfeng S, Depu W, Xiaofeng Y, Shah W, Hongwei C, Yili W. In situ observation of the effects of local irradiation on cytotoxic and regulatory T lymphocytes in cervical cancer tissue. Radiat Res. 2013;179:584–9.
Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24:589–602.
Formenti SC. Is classical stereotactic radiotherapy the optimal partner for immunotherapy? Oncology 2015;29:340, 347, 387.
Frey B, Stache C, Rubner Y, Werthmoller N, Schulz K, Sieber R, Semrau S, Rodel F, Fietkau R, Gaipl US. Combined treatment of human colorectal tumor cell lines with chemotherapeutic agents and ionizing irradiation can in vitro induce tumor cell death forms with immunogenic potential. J Immunotoxicol. 2012;9:301–13.
Burnstock G. Purinergic nerves. Pharmacol Rev. 1972;24:509–81.
Kaufmann SH. Immunology’s foundation: the 100-year anniversary of the Nobel Prize to Paul Ehrlich and Elie Metchnikoff. Nat Immunol. 2008;9:705–12.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Lohmann K. Über die Pyrophosphatfraktion im Muskel. Naturwissenschaften. 1929;17:624–5.
Maccubbin DL, Wing KR, Mace KF, Ho RL, Ehrke MJ, Mihich E. Adriamycin-induced modulation of host defenses in tumor-bearing mice. Cancer Res. 1992;52:3572–6.
MacLennan DH, Yip CC, Iles GH, Seeman P. Isolation of sarcoplasmic reticulum proteins. Cold Spring Harb Symp Quant Biol. 1972;37:469–77.
Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045.
McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–8.
Medzhitov R, Preston-Hurlburt P, Janeway Jr CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7.
Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–61.
Rapaport E, Fishman RF, Gercel C. Growth inhibition of human tumor cells in soft-agar cultures by treatment with low levels of adenosine 5′-triphosphate. Cancer Res. 1983;43:4402–6.
Rapaport E, Fontaine J. Anticancer activities of adenine nucleotides in mice are mediated through expansion of erythrocyte ATP pools. Proc Natl Acad Sci U S A. 1989;86:1662–6.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5.
Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, Sanchez M, Lorenzi S, D’Urso MT, Belardelli F, et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011;71:768–78.
Davies AM, Lara Jr PN, Mack PC, Gandara DR. Incorporating bortezomib into the treatment of lung cancer. Clin Cancer Res. 2007;13:s4647–51.
Tseng LM, Liu CY, Chang KC, Chu PY, Shiau CW, Chen KF. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012;14:R68.
Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem. 2003;278:33714–23.
Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–45.
Cirone M, Di Renzo L, Lotti LV, Conte V, Trivedi P, Santarelli R, Gonnella R, Frati L, Faggioni A. Primary effusion lymphoma cell death induced by bortezomib and AG 490 activates dendritic cells through CD91. PLoS One. 2012;7, e31732.
Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4:143ra199.
Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011;30:4297–306.
Chen HM, Wang PH, Chen SS, Wen CC, Chen YH, Yang WC, Yang NS. Shikonin induces immunogenic cell death in tumor cells and enhances dendritic cell-based cancer vaccine. Cancer Immunol Immunother. 2012;61:1989–2002.
Garrido G, Rabasa A, Sanchez B, Lopez MV, Blanco R, Lopez A, Hernandez DR, Perez R, Fernandez LE. Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a specific antibody. J Immunol. 2011;187:4954–66.
Yang Y, Li XJ, Chen Z, Zhu XX, Wang J, Zhang LB, Qiang L, Ma YJ, Li ZY, Guo QL, You QD. Wogonin induced calreticulin/annexin A1 exposure dictates the immunogenicity of cancer cells in a PERK/AKT dependent manner. PLoS One. 2012;7, e50811.
Fucikova J, Moserova I, Truxova I, Hermanova I, Vancurova I, Partlova S, Fialova A, Sojka L, Cartron PF, Houska M, et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int J Cancer. 2014;135:1165–77.
Sonnemann J, Gressmann S, Becker S, Wittig S, Schmudde M, Beck JF. The histone deacetylase inhibitor vorinostat induces calreticulin exposure in childhood brain tumour cells in vitro. Cancer Chemother Pharmacol. 2010;66:611–6.
West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, Johnstone RW. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res. 2013;73:7265–76.
Guillot F, Boutin B, Blanquart C, Fonteneau JF, Robard M, Gregoire M, Pouliquen D. Vaccination with epigenetically treated mesothelioma cells induces immunisation and blocks tumour growth. Vaccine. 2011;29:5534–43.
Bugaut H, Bruchard M, Berger H, Derangere V, Odoul L, Euvrard R, Ladoire S, Chalmin F, Vegran F, Rebe C, et al. Bleomycin exerts ambivalent antitumor immune effect by triggering both immunogenic cell death and proliferation of regulatory T cells. PLoS One. 2013;8, e65181.
Calvet CY, Famin D, Andre FM, Mir LM. Electrochemotherapy with bleomycin induces hallmarks of immunogenic cell death in murine colon cancer cells. Oncoimmunology. 2014;3, e28131.
Sukkurwala AQ, Adjemian S, Senovilla L, Michaud M, Spaggiari S, Vacchelli E, Baracco EE, Galluzzi L, Zitvogel L, Kepp O, Kroemer G. Screening of novel immunogenic cell death inducers within the NCI Mechanistic Diversity Set. Oncoimmunology. 2014;3, e28473.
Liu SH, Lee WJ, Lai DW, Wu SM, Liu CY, Tien HR, Chiu CS, Peng YC, Jan YJ, Chao TH, et al. Honokiol confers immunogenicity by dictating calreticulin exposure, activating ER stress and inhibiting epithelial-to-mesenchymal transition. Mol Oncol. 2015;9:834–49.
Sanovic R, Verwanger T, Hartl A, Krammer B. Low dose hypericin-PDT induces complete tumor regression in BALB/c mice bearing CT26 colon carcinoma. Photodiagnosis Photodyn Ther. 2011;8:291–6.
Liu Z, Zhang HM, Yuan J, Ye X, Taylor GA, Yang D. The immunity-related GTPase Irgm3 relieves endoplasmic reticulum stress response during coxsackievirus B3 infection via a PI3K/Akt dependent pathway. Cell Microbiol. 2012;14:133–46.
Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, Okazaki T, Marumoto T, Takahashi A, Takayama K, et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012;72:2609–21.
Acknowledgments
We thank A. Bredan for editing the manuscript. This work was supported by project grants from the Fund for Scientific Research Flanders (FWO-Vlaanderen: G060713N, 3G067512, G0A5413N to DVK) and UGent Special Research Fund (BOF14/GOA/019 to D.V.K. and C.B.). Vandenabeele’s group is supported by VIB, Ghent University (GROUP-ID Consortium of the UGent MRP initiative), FWO-Vlaanderen (G.0875.11, G.0973.11, G.0A45.12N, FWO G.0787.13N, FWO G.0C31.14N, FWO G0E04.16N), Federal Research Program (IAP 7/32). PV holds a Methusalem grant BOF09/01M00709 and BOF16/MET_V/007 from the Flemish Government, by Belgian grants (Interuniversity Attraction Poles, IAP 7/32) and grant from the ‘Foundation against Cancer, 2012-188’. DVK is paid by the Methusalem grant.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Vandenabeele, P., Vandecasteele, K., Bachert, C., Krysko, O., Krysko, D.V. (2016). Immunogenic Apoptotic Cell Death and Anticancer Immunity. In: Gregory, C. (eds) Apoptosis in Cancer Pathogenesis and Anti-cancer Therapy. Advances in Experimental Medicine and Biology, vol 930. Springer, Cham. https://doi.org/10.1007/978-3-319-39406-0_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-39406-0_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39404-6
Online ISBN: 978-3-319-39406-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)