
Abstract
The stimulation of cancer immunosurveillance requires malignant cells to differ from their normal counterparts at two distinct levels. First, cancer cells need to express tumor-associated antigens including neoantigens resulting from somatic mutations, meaning that they are antigenically distinct from parental cells. Second, in response to cell-intrinsic or external stress signals, malignant cells must exhibit or release danger-associated molecular patterns (DAMPs) that function as adjuvant signals. The combination of antigenicity and adjuvanticity results in immunogenicity. Some widely used chemotherapeutics, as well as radiotherapy, can stimulate immunogenic cell death (ICD) to set off an anticancer immune response accounting for long-term therapeutic responses. Here we discuss DAMPs, such as annexin A1, ATP, calreticulin, HMGB1, and type-1 interferons, which are exposed or released from stressed and dying cancer cells in response to ICD inducers and the signal transduction pathways leading to their activation. ICD-associated DAMPs act on receptors that are mostly expressed by immune cells, allowing for the orchestration of an intratumoral immune response that involves the recruitment of dendritic cell precursors, the cross-presentation of tumor antigens, and the induction of a cognate anticancer immune response mediated by interferon-γ producing CD8+ T cells. Failure to emit DAMPs due to cancer cell-intrinsic defects or failure to perceive DAMPs due to mutations or inactivation of the receptors required for their perception (such as the annexin A1 receptor FPR1, the HMGB1 receptor TLR4, or the type-1 interferon receptor IFNAR) weakens anticancer immunosurveillance and has a negative prognostic impact in cancer patients.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Before the renaissance of the immunosurveillance theory that accompanied the approval of immune checkpoint blockers [1], cancer was generally viewed as a cell-autonomous disease that is solely caused by genetic and epigenetic alterations of the malignant cells [2]. Therapeutic interventions hence were conceived to take advantage of the cancer cell-intrinsic vulnerabilities (making them particularly susceptible to antiproliferative and cytotoxic insults) or to target pathways that would be specifically activated in malignant cells yet absent in their normal counterparts (much like antibiotics that affect bacterial enzymes but not those of their host). Based on this paradigm, cytotoxic and targeted therapies have been developed following a workflow in which anticancer agents were first identified on cultured human cell lines, then tested on immunodeficient mice carrying human cancers, and, finally, introduced into the clinics [2, 3]. Seemingly supporting this strategy, several successful chemotherapeutics have been developed. In particular, combination therapies involving several distinct cytotoxic agents have been highly successful in reducing the risk of relapse after adjuvant chemotherapy in breast and colorectal cancers [4,5,6]. Moreover, the success of the first targeted anticancer agent, imatinib mesylate, which targets several oncogenic tyrosine kinases (such as the BCR-ABL kinase activated in chronic myeloid leukemia and KIT activated in gastrointestinal stromal tumors) [7,8,9,10,11,12] apparently comforted the idea that cancer can be treated with specific agents (although it turned out later that the therapeutic efficacy of imatinib relies on NK and T lymphocytes) [13].
Our laboratory has been adhering to this cell-autonomous vision of cancer therapy until 2004 when we performed a stunningly simple experiment. We subcutaneously implanted a mouse colorectal cancer cell line, CT26, either in immunocompetent BALB/c mice (the strain from which CT26 was originally derived from) or in immunodeficient nu/nu mice (which are athymic and hence lack thymus-derived T lymphocytes) and treated the emerging tumors with chemotherapy based on the anthracycline doxorubicin. To our dismay, the growth of CT26 cancers was only reduced if they evolved in an immunocompetent setting (in BALB/c mice), not if they grew on nu/nu mice [14]. Hence, the efficacy of chemotherapy turned out to depend on a cellular immune response.
The next surprise came when we analyzed the cell death modality induced by doxorubicin in CT26 cells. At that time, only two major cell death pathways were known, namely, apoptosis and necrosis [15]. Apoptosis was conceived to constitute a physiological pathway accounting for cellular demise in developmental cell death and adult tissue homeostasis [16]. Necrosis was conceived as a purely pathological pathway resulting in pro-inflammatory tissue reaction due to the uncontrolled spilling of the cellular content through the permeabilized plasma membrane [17]. In CT26 cells, doxorubicin induced two hallmarks of the apoptotic pathway, namely, an early loss of the mitochondrial inner transmembrane potential as well as the activation of caspases [14]. Addition of a pharmacological caspase inhibitor prevented the cells to adopt an apoptotic morphology with nuclear condensation and fragmentation and led to a more necrotic phenotype. When doxorubicin-treated apoptotic CT26 cells were injected subcutaneously into BALB/c mice, they induced an immune response that protected the mice against a subsequent challenge with live CT26 cells that were injected 1 week later into the opposite flank. In contrast, doxorubicin-treated necrotic CT26 cells (that were killed in the presence of Z-VAD-fmk) failed to stimulate such an immune response [14]. These results pleaded in favor of a novel caspase-dependent modality of apoptosis that could stimulate anticancer immunosurveillance and that we dubbed “immunogenic cell death” (ICD) [14]. Later, it turned out that CT26 cells lack the expression of receptor-interacting serine/threonine-protein kinase 3 (RIPK3), a protein required for necroptosis (which is a regulated version of necrosis) and that other mouse cancer cell lines that possess the entire molecular machinery required for necroptosis can undergo ICD in response to necroptotic stimuli [18,19,20] including anthracyclines. Hence, different forms of regulated cell death (apoptosis and necroptosis) can contribute to ICD.
Based on the aforementioned results that were replicated in multiple different cancer cell types and mouse strains [21,22,23,24,25,26], we have been postulating that ICD would constitute an important mechanism to convert the cell-autonomous chemotherapeutic response, leading to focal apoptosis and necroptosis within the tumor, into a systemic immune-mediated response that can amplify and prolong the anticancer effects of chemotherapy [19, 27,28,29]. In other words, ICD would convert cancer into its own vaccine. We also found that not all chemotherapeutic agents are equally potent in causing ICD (and hence in provoking an antitumor immune response) observing that anthracyclines and oxaliplatin are particularly efficient in doing so while many other cytotoxicants are unable to do so [27,28,29]. In subsequent studies, we observed that ICD inducers are able to trigger premortem stress responses such as autophagy and endoplasmic reticulum (ER) stress that lead to the release and exposure of DAMPs required for ICD [28, 30]. Hence, it is not only cell death as such but a constellation of stress pathways and lethal events that yields ICD. These pathways and their connection to the exposure or release of DAMPs, as well as their clinical implications, will be discussed in this chapter.
2 Annexin A1
Annexin A1 (ANXA1) is a relatively abundant and ubiquitously expressed cytoplasmic protein [31] that is released from dying cancer cells responding to chemotherapy with anthracyclines or oxaliplatin in vitro [32]. The exact mode of release is not known, although a relative of ANXA1, annexin A2 (ANXA2), has been shown to be secreted by an unconventional pathway [33]. Alternatively, ANXA1 may be released passively, via the permeabilized plasma membrane as cells die. Mouse cancer cell lines from which ANXA1 was removed by CRISPR/Cas9 technology failed to undergo ICD in vitro (meaning that, if they were cultured with anthracyclines and then injected in vivo, they would fail to induce a protective anticancer immune response). Cancers arising from such ANXA1-deficient cancer cell also failed to reduce their growth in vivo, in response to systemic injections of anthracyclines or oxaliplatin [32].
ANXA1 can bind to formyl peptide receptor-1 (FPR1), a seven transmembrane G protein-coupled receptor mostly expressed by myeloid cells [34]. Knockout of FPR1 in the host immune system (as well as transfer of FPR1-deficient hematopoietic stem cells into FPR1-sufficient irradiated hosts) led to the incapacity of the host to mount an anticancer immune response against dying cancer cells. Moreover, the absence of FPR1 from the immune system led to a failure to control the growth of cancers treated with anthracyclines or oxaliplatin in vivo [32]. These results underscore the importance of the interaction between ANXA1 and FPR1 for the chemotherapy-triggered dialogue between cancer cells and the immune system. Mechanistically, it turned out that FPR1 guides differentiating dendritic cells within the tumor into the proximity of dying cancer cells, allowing the dendritic cell-mediated uptake of tumor-associated antigens and their subsequent presentation to T cells (Fig. 12.1) [32]. As a result, FPR1-deficient hosts are unable to mount an immune response mediated by CD8+ T cells against tumor antigens.

Annexin A1-mediated homing of dendritic cells. Annexin A1 (ANXA1) is released from cancer cells in response to certain therapeutic approaches including the anthracycline- and oxaliplatin-based chemotherapeutic induction of immunogenic cell death. Driven by chemotaxis, immature dendritic cells (iDCs) are homed in on their target in a FPR1-dependent fashion, finally leading to a close proximity of dying cancer cells and antigen-presenting cells. FPR1, formyl peptide receptor 1
The aforementioned findings, which have been obtained in mice, are supported by epidemiological studies in cancer patients. A loss-of-function mutation in FPR1 (A299G), which affects the intracellular domain of the protein within its N-terminus abolishing the dimerization of the receptor required for its activation [35], had negative prognostic features in two types of cancer. Breast cancer patients bearing one loss-of-function allele of FPR1 exhibited a shorter progression-free and overall survival upon adjuvant anthracycline-based chemotherapy than patients bearing two normal alleles of FPR1. This finding was obtained for two independent cohorts of breast cancer patients [32]. Moreover, colorectal cancer patients bearing two loss-of-function alleles of FPR1 had a statistically shorter survival upon adjuvant oxaliplatin-based chemotherapy than patients bearing one or two normal alleles of FPR1 [32]. The mechanistic bases for these differences are not understood yet. In addition, it appears that mammary carcinoma cells express lower ANXA1 levels than their normal epithelial counterparts [32], perhaps reflecting immunoselection in favor of cancers that lack the DAMP ANXA1.
3 ATP
In response to treatment with chemotherapeutics in vitro, cancer cells release adenosine triphosphate (ATP) into the culture supernatant, an event that can be visualized by a reduction in quinacrine-labeled, ATP-containing lysosomal compartments [36]. The accompanying increase in extracellular levels of ATP can be measured by means of a firefly luciferase construct that is tethered to the cancer cell surface and that detects pericellular ATP upon addition of d-luciferin [37, 38]. This latter system is suitable for measuring extracellular ATP in vivo in tumor-bearing mice, in which the luminescence signal strongly increases 2 days post-chemotherapy [25]. The mechanism of ATP release has not been entirely elucidated yet appears to involve a lysosomal secretion mechanism that depends on at least two processes, namely, a premortem autophagy response and caspase activation. Autophagy must occur to allow ATP to redistribute from lysosomes to autolysosomes and to be secreted by a mechanism that requires the lysosomal-associated membrane protein 1 (LAMP1), which translocates to the plasma membrane in a caspase-dependent manner. The release of ATP additionally involves the caspase-mediated activation of the Rho-associated coiled-coil-containing protein kinase (ROCK1) resulting in myosin II-dependent membrane blebbing as well as the opening of pannexin 1 (PANX1) channels, subsequent to their cleavage by caspases. While autophagy and LAMP1 do not affect PANX1 channel opening, PANX1 is required for the ICD-associated translocation of LAMP1 to the plasma membrane [39]. Hence, apoptosis-associated ATP release is a complex process that is abolished in autophagy-deficient tumors, knowing that inactivation of autophagy occurs rather frequently, especially during early oncogenesis [25, 40,41,42,43]. Necroptotic signaling via RIPK3 and the mixed lineage kinase domain-like (MLKL) pseudokinase may also contribute to ATP release [18], although it is not known whether this process also requires premortem autophagy to be induced. In any case, it appears that cancer cells manipulated to suppress the autophagic process fail to undergo ICD and do not reduce their growth upon treatment with anthracyclines or oxaliplatin in vivo [25]. A similar abolition of ICD and chemotherapeutic responses can be obtained by overexpressing the ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, also known as the ectoATPase CD39) on the cancer cells [25, 44].
Extracellular ATP acts on two classes of purinergic receptors, namely, the metabotropic P2Y2 and the ionotropic P2X7 receptors. P2Y2 receptors facilitate the ATP-mediated chemotaxis of myeloid cells (dendritic cell precursors, neutrophils and macrophages) into the tumor bed post-chemotherapy (Fig. 12.2). Both autophagy-deficient and CD39-overexpressing cancers fail to accumulate myeloid cells post-chemotherapy in the tumor bed [25], and a similar effect can be obtained upon pharmacological inhibition of P2Y2 [44]. P2X7 receptors facilitate the ATP-stimulated activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome in dendritic cells, which then triggers the secretion of interleukin-1β (IL1β) and the IL1β-dependent priming of tumor antigen-specific CD8+ T cells [45]. Indeed, neutralization of P2Y2 or IL1 receptors and knockout of P2X7, NLRP3, or caspase-1 abolish the capacity of the immune system to mount a protective immune response against cancer cells that succumb to ICD [45].

ATP-dependent recruitment and activation of dendritic cells. The autophagy-dependent lysosomal secretion of ATP from cancer cells that undergo immunogenic cell death leads to the recruitment and activation of immature dendritic cells (iDCs). Extracellular ATP acts on purinergic receptors of the metabotropic P2Y2 and the ionotropic P2X7 type. Most prominently P2Y2 receptors drive the ATP-mediated chemotaxis of myeloid cells including immature dendritic cells (iDCs) into the tumor bed post-chemotherapy. In summary, ATP release from the dying cancer cells leads to an enrichment of the tumor bed with immune cells. P2RX7, purinergic receptor P2X7; P2RY2, purinergic receptor P2Y2
The aforementioned interaction between extracellular ATP and purinergic receptors again appears clinically relevant. Indeed, in breast cancer patients treated with adjuvant chemotherapy, the absence of autophagy has a negative impact on the local immune response with an unfavorable ratio of CD8+ T lymphocytes over forkhead box P3+ (FOXP3+) regulatory T cells. Such observation correlates with poor patient survival [46]. Similarly, high expression of ATP-degrading ectoenzymes such as CD39 and the ecto-5′ nucleotidase NT5E (best known as CD73) indicates poor prognosis in multiple distinct cancers including breast and ovarian cancers [47, 48]. Finally, a loss-of-function mutation in P2X7 has been linked to poor prognosis in a segment of breast cancer patients that are treated with anthracycline-based adjuvant chemotherapy [45].
Experimentally, it is possible to stimulate autophagy, ATP release, and consequent myeloid cell recruitment and anticancer immune responses by fasting or by non-immunosuppressive autophagy inducers that fall into the class of “caloric restriction mimetics” (CRMs) [49,50,51,52]. Several CRMs including hydroxycitrate can be used in mouse models to improve anticancer immunosurveillance and to boost the anticancer immune responses elicited by ICD-inducing chemotherapeutics [49]. Whether this strategy is applicable to cancer patients awaits urgent clarification.
4 Calreticulin
Calreticulin (CALR) is the most abundant protein in the lumen of the endoplasmic reticulum (ER). In the context of ICD, a fraction of CALR translocates to the surface of the plasma membrane (and it is possible that another fraction of CALR is secreted as well) [23, 26, 53, 54]. The complex mechanisms that underlie CALR exposure are linked to the apical phosphorylation of eukaryotic initiation factor-2α (eIF2α) in the context of an ER stress response that culminates in the activation of an eIF2α kinase (EIF2K) such as EIF2K2 (best known as PKR) and EIF2K3 (best known as PERK) and/or in the inhibition of the corresponding phosphatase (composed by the catalytic subunit PP1 and the regulatory subunit GADD34) [26, 55, 56]. Downstream of eIF2α phosphorylation, caspases (and in particular caspase-8, CASP8) are activated, and calreticulin is transported to the cell surface following anterograde ER-Golgi traffic and soluble N-ethylmaleimide-sensitive factor attachment receptors (SNARE)-dependent exocytosis that involves the vesicle-associated membrane protein 1 (VAMP1) and the synaptosomal-associated protein 23 (SNAP23) [55].
Once on the cell surface, CALR acts as an “eat-me” signal to facilitate the transfer of tumor-associated antigens to dendritic cells [57], which express the CALR receptor low-density lipoprotein receptor-related protein 1 (LRP1, best known as CD91; Fig. 12.3) [58]. CALR can be locally antagonized by CD47, which is constitutively expressed on cancer cells and can function as a “don’t eat me” signal [59]. Knockdown of PERK, CASP8, CALR, or SNAP23 as well as pharmacological inhibition of caspases and anterograde ER-Golgi transport is sufficient to abolish ICD in vitro and in vivo [55]. Conversely, stimulation of eIF2α phosphorylation by thapsigargin (which activates PERK) or inhibitors of PP1 can stimulate CALR exposure and enhance anticancer immune responses in vivo, in the context of chemotherapy [55, 60]. Coating of cancer cells that are deficient in the CALR exposure pathway with recombinant CALR protein (which binds to the plasma membrane surface, presumably via interaction between its lectin domain and the glycocalyx) can restore deficient ICD [26, 61]. Similarly, intratumoral injection of CALR can enhance the chemotherapy-elicited immune response and improve tumor growth inhibition in vivo, in mouse models [62].

Calreticulin as de novo uptake signal for dendritic cells. The endoplasmic reticulum (ER) stress-mediated exposure (or release) of the ER chaperone calreticulin (CALR) to the surface of the plasma membrane in the course of immunogenic cell death (ICD) serves as de novo uptake signal for dendritic cells. The binding of CALR to the low-density lipoprotein receptor-related protein 1 (LRP1) receptor expressed on dendritic cells (DC) serves the transfer of tumor-associated antigens to DC
There is widespread evidence that CALR expression and exposure contribute to anticancer immunosurveillance in vivo, in cancer patients. Low intracellular CALR expression levels have been correlated with a low presence of CALR on the cell surface, both in acute myeloid leukemia (AML) and in non-small cell lung cancer (NSCLC), as well as reduced phosphorylation of eIF2α [59, 63, 64]. In AML, reduced expression of CALR protein has a negative impact on progression-free and overall survival post-chemotherapy, correlating with poor T cell-mediated immune responses against AML-associated tumor antigens [59, 64]. In NSCLC, approximately 15% of the patients have barely detectable CALR protein in cancer cells, correlating with dismal prognosis, reduced infiltration by DC-LAMP+ dendritic cells and CD8+ T lymphocytes [63]. Of note, low CALR expression supersedes in importance the TNM classification of NSCLC with respect to prognosis, meaning that patients with CALRlow stage 1 NSCLC exhibit a poorer survival than stage 3 and stage 4 patients bearing CALRhigh cancers [63]. These results have been confirmed for two distinct NSCLC cohorts by detecting CALR protein with immunohistochemistry [64], as well as for an additional NSCLC cohort by measuring CALR mRNA levels and its correlations with metagenes reflecting the presence of CTL and dendritic cells [65]. Similarly, in ovarian cancer, high levels of CALR mRNA expression have a favorable impact on patient survival, if combined with the analysis of activated dendritic cells [65]. Conversely, high CD47 expression has a negative impact on the prognosis of multiple distinct cancers [66,67,68]. Mutations in the CALR gene have been described in myeloproliferative neoplasms [69,70,71], causing mislocalization of the corresponding gene product [72], although the exact impact of these mutations on tumor immunosurveillance remains elusive. Regardless, the clinical data validate the importance of the CALR exposure pathway for tumor biology.
5 HMGB1
High molecular group B1 protein (HMGB1) is the most abundant nonhistone chromatin-binding protein [73,74,75]. HMGB1 is usually found in an exclusively nuclear location yet can translocate to the cytoplasm, for instance, after inhibition of histone deacetylases [76]. Moreover, HMGB1 is usually released from cells that undergo necroptosis or secondary necrosis [77, 78]. Mouse cancer cells in which either RIPK3 or MLKL have been knocked out release lower amounts of HMGB1 in response to anthracyclines than their necroptosis-competent controls [18].
Experiments on tumors implanted in mice revealed that HMGB1 is released from cancer cells upon chemotherapy in vivo [79,80,81,82,83]. Cancer cells from which HMGB1 has been depleted by RNA interference are unable to undergo ICD and become resistant to chemotherapy in vivo. Similarly, injection of neutralizing anti-HMGB1 antibodies abolished the anticancer immune response elicited by ICD-inducing chemotherapy in vivo and hence compromised tumor growth reduction [81]. These results support the importance of extracellular HMGB1 as a DAMP in tumor immunology.
Once present in the extracellular space, HMGB1 can interact with multiple additional factors including nucleic acids and bacterial polysaccharides [84,85,86,87,88]. HMGB1 also binds to several receptors including toll-like receptor 4 (TLR4), which is expressed on multiple immune cell types including dendritic cells, in which it stimulates maturation and antigen presentation (Fig. 12.4) [81, 89, 90]. Knockout of TLR4, or that of its adaptor MYD88, from the host immune system abolishes the perception of ICD as well as tumor growth reduction by anthracyclines or oxaliplatin [22, 80, 81, 91]. This defect has been linked to a reduced antigen presentation by dendritic cells and can be partially rescued by treatment with the lysosomal inhibitor chloroquine [81].

HMGB1 facilitates antigen presentation by dendritic cells. High-mobility group box 1 (HMGB1), normally secluded in the nucleus, is released at later stages of immunogenic cell death in response to treatments such as anthracycline-based chemotherapy or ionizing irradiation. Extracellular HMGB1 serves as a ligand for TLR4 on dendritic cells (DCs) and triggers a MYD88-dependent signaling that stimulates DC maturation and antigen presentation to cytotoxic T cells (CTLs). TLR4, toll-like receptor 4
In human breast cancer, reduced HMGB1 expression has been linked to the advancement of the disease and increased tumor size [46]. Reduced HMGB1 expression is a negative prognostic feature in breast cancer and correlates with an intratumoral infiltration by fewer CD8+ cytotoxic T lymphocytes and more immunosuppressive populations of FOXP3+ regulatory T cells and CD68+ tumor-associated macrophages [46]. Moreover, there are at least two cancer types in which a loss-of-function allele of TLR4 compromises patient prognosis, namely, (1) breast cancer and (2) colorectal cancer treated with adjuvant chemotherapy based on anthracyclines and oxaliplatin, respectively [92]. These findings underscore the likely importance of the HMGB1/TLR4 interaction for the fate of cancer patients.
In the case of HMGB1-negative cancers, artificial supply of a synthetic TLR4 ligand, dendrophilin, can compensate for the HMGB1 defect and restore anticancer immune responses elicited by chemotherapy in mouse models. Whether such a strategy might also work in cancer patients bearing HMGB1-negative neoplasia remains to be investigated.
6 Type-1 Interferons and Chemokines
In response to chemotherapeutics, tumor cells liberate nucleic acids including DNA and double-stranded RNA that may activate intracellular or extracellular sensors for ectopic molecules of this kind. One example for such nucleic acid sensor is the toll-like receptor-3 (TLR3) [93], although other sensors including the GAS/STING pathway might be involved as well [94]. In response to these stimuli that resemble those induced by a viral infection and hence can be referred to as “viral mimicry,” cancer cells transcriptionally activate one or several type-1 interferon genes, secrete the corresponding gene products, and then stimulate their type-1 interferon receptor (IFNAR) to induce a multipronged type-1 interferon response consisting in the activation of multiple antiviral and immunostimulatory gene products (Fig. 12.5) [95]. One quintessential antiviral gene product is myxovirus resistance 1 (MX1), and one well-known immunostimulatory gene product is the C-X-C motif chemokine ligand 10 (CXCL10), which acts on the C-X-C motif chemokine receptor 3 (CXCR3) to attract T lymphocytes into the tumor bed [95]. Cancer cells that lack TLR3, IFNAR, or CXCL10 are unable to elicit anticancer immune responses upon chemotherapy and hence become refractory to the treatment [95]. Local injection of recombinant type-1 interferons and CXCL10 can overcome this defect [95], underscoring the importance of the type-1 interferon response for therapeutic outcome in mouse tumor models.

Type I interferon-dependent chemokine release triggers T cell priming. In response to chemotherapeutics that mimic a viral infection, with regard to the liberation of nucleic acids from the dying cells, sensors for such molecules like the toll-like receptor-3 (TLR3), transcriptionally activate one or several type-1 interferon genes. Once secreted, the interferon stimulates the type-1 interferon receptor (IFNAR) to induce a multipronged type-1 interferon response including the production of C-X-C motif chemokine ligand 10 (CXCL10), which acts on the C-X-C motif chemokine receptor 3 (CXCR3) to recruit T lymphocytes into the tumor bed finally leading to a γδ T cell-mediated priming of αβ T cells. CXCR3, CXC-chemokine receptor 3; IFNAR1; interferon α/β-receptor subunit 1; IL, interleukin
At least in breast cancer patients, the aforementioned pathway seems to be therapeutically relevant. Thus, MX1 expression is induced by chemotherapy in vivo. The absence of signaling through IFNAR, indicated by the lack of signal transducer and activator of transcription 1 (STAT1) phosphorylation [96] or low MX1 expression, constitutes a poor prognostic feature, in particular in the context of anthracycline-based adjuvant chemotherapy [95]. Moreover, a polymorphism that affects the function of TLR3 reportedly influences the fate of breast cancer patients [97]. These results have to be interpreted in the context of mounting clinical evidences that type-1 interferons can be injected into patients to stimulate anticancer immune responses in the context of renal cancer and chronic myeloid leukemia (CML) [98].
7 Concluding Remarks and Perspective
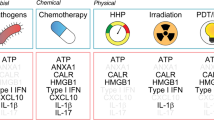
As mentioned above, there are multiple DAMPs (such as ANXA1, ATP, CALR, HMGB1, and type-1 interferons) that function as adjuvant signals in the context of immunogenic chemotherapies (Fig. 12.6). It is important to note that these DAMPs do not act in a redundant fashion (in which case they would be able to replace each other) but in a non-redundant way, meaning that removal of one single DAMP (or its receptor) from the system is sufficient to undermine anticancer immunosurveillance elicited by immunogenic chemotherapies. One possibility to look at this problem is to postulate that each of the DAMPs must come into action following a defined spatiotemporary sequence, perhaps within a narrow range of intensity, following the “key-lock principle” [99]. Only if the DAMPs are expressed in the correct order, at the correct intensity, they are able to form the “key” that opens the vault that normally precludes an immune response [99]. Speculatively, this particular design of the system may reduce the probability of unwarranted autoinflammatory and autoimmune reactions in normal tissues [100]. On the other hand, this means that suppression of one single DAMP due to mutation (perhaps driven by immunoselection) or inhibition of one single DAMP receptor is sufficient to subvert anticancer immunosurveillance and to reduce the chance of cancer patients to control their disease upon chemotherapy.

Mechanisms of immunogenic cell death in therapy-induced immunosurveillance. Cancer cells undergoing immunogenic cell death (ICD) in response to chemotherapeutic treatments, such as doxorubicin or oxaliplatin, exhibit or release certain danger-associated molecular patterns (DAMPs) such as calreticulin (CALR) ATP, type I interferon (IFN), high-mobility group box 1 (HMGB1), and annexin A1 (ANXA1). Ligation of cognate receptors on the surface of myeloid or lymphoid cells facilitates the recruitment and activation of dendritic cells, their homing to the dying cancer cells, subsequent tumor antigen uptake, and final presentation (upon maturation of the dendritic cells). The production of immunostimulatory cytokines eventually leads to the onset of an adaptive immune response involving αβ and γδ T cells that reestablishes cancer immunosurveillance
Irrespective of these speculations, it is possible to measure all known DAMPs in cultured cells exposed to libraries of anticancer agents to identify ICD inducers. In practical terms, this is achieved by generating biosensor cell lines that express fluorescent versions of ANXA1, CALR, or HMGB1 that have been fused to green fluorescent protein (GFP) or its derivatives. ATP release can be measured upon staining with chloroquine. The activation of the type-1 interferon response can be determined by placing GFP under the control of the MX1 promoter. Using this battery of biosensors, it is hence possible to select anticancer agents that stimulate all aspects of ICD. We have successfully used this approach to identify ICD inducers that are effective in stimulating anticancer immune responses in vivo, in mouse models [101,102,103].
It is tempting to speculate that such an approach may become even more useful in selecting successful anticancer drugs based on their ICD-stimulatory potential. Obviously, this approach would require additional in vivo experimentations in preclinical models while carefully avoiding the use of immunodeficient mice carrying xenotransplants like it was done in the past (see Sect. 12.1). Rather, anticancer drug candidates should always be evaluated in immunocompetent rodent models, including humanized mouse models. It is tempting to predict that this kind of approach will greatly reduce the attrition rate that has been characterizing the traditional drug development pipeline.
References
Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73. doi:10.1186/s12916-016-0623-5.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
Kelland LR. Of mice and men: values and liabilities of the athymic nude mouse model in anticancer drug development. Eur J Cancer. 2004;40(6):827–36. doi:10.1016/j.ejca.2003.11.028.
Schmoll HJ, Tabernero J, Maroun J, de Braud F, Price T, Van Cutsem E, Hill M, Hoersch S, Rittweger K, Haller DG. Capecitabine plus Oxaliplatin compared with fluorouracil/Folinic acid as adjuvant therapy for stage III Colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol. 2015;33(32):3733–40. doi:10.1200/JCO.2015.60.9107.
Yothers G, O'Connell MJ, Allegra CJ, Kuebler JP, Colangelo LH, Petrelli NJ, Wolmark N. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses. J Clin Oncol. 2011;29(28):3768–74. doi:10.1200/JCO.2011.36.4539.
Andre T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, Bonetti A, Clingan P, Bridgewater J, Rivera F, de Gramont A. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol. 2009;27(19):3109–16. doi:10.1200/JCO.2008.20.6771.
Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, Fletcher CD, Crowley JJ, Borden EC. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–32. doi:10.1200/JCO.2007.13.4452.
Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD, Roberts PJ, Heinz D, Wehre E, Nikolova Z, Joensuu H. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26(4):620–5. doi:10.1200/JCO.2007.13.4403.
Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, Letvak L, Larson RA, Investigators I. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–17. doi:10.1056/NEJMoa062867.
Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, Bertulli R, Judson I. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127–34. doi:10.1016/S0140-6736(04)17098-0.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80. doi:10.1056/NEJMoa020461.
Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–7. doi:10.1056/NEJM200104053441401.
Zitvogel L, Rusakiewicz S, Routy B, Ayyoub M, Kroemer G. Immunological off-target effects of imatinib. Nat Rev Clin Oncol. 2016;13(7):431–46. doi:10.1038/nrclinonc.2016.41.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Metivier D, Pichard E, Aucouturier P, Pierron G, Garrido C, Zitvogel L, Kroemer G. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202(12):1691–701. doi:10.1084/jem.20050915.
Kanduc D, Mittelman A, Serpico R, Sinigaglia E, Sinha AA, Natale C, Santacroce R, Di Corcia MG, Lucchese A, Dini L, Pani P, Santacroce S, Simone S, Bucci R, Farber E. Cell death: apoptosis versus necrosis (review). Int J Oncol. 2002;21(1):165–70.
Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147(4):742–58. doi:10.1016/j.cell.2011.10.033.
Rello S, Stockert JC, Moreno V, Gamez A, Pacheco M, Juarranz A, Canete M, Villanueva A. Morphological criteria to distinguish cell death induced by apoptotic and necrotic treatments. Apoptosis. 2005;10(1):201–8. doi:10.1007/s10495-005-6075-6.
Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, Pietrocola F, Vacchelli E, Souquere S, Sauvat A, Zitvogel L, Kepp O, Kroemer G. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology. 2016;5(6):e1149673. doi:10.1080/2162402X.2016.1149673.
Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2016; doi:10.1038/nri.2016.107.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–14. doi:10.1038/nrm2970.
Michaud M, Sukkurwala AQ, Di Sano F, Zitvogel L, Kepp O, Kroemer G. Synthetic induction of immunogenic cell death by genetic stimulation of endoplasmic reticulum stress. Oncoimmunology. 2014;3:e28276. doi:10.4161/onci.28276.
Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, Sistigu A, Prada N, Adjemian S, Catani JP, Freudenberg M, Galanos C, Andre F, Kroemer G, Zitvogel L. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ. 2014;21(1):69–78. doi:10.1038/cdd.2013.72.
Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, Annaert W, Golab J, de Witte P, Vandenabeele P, Agostinis P. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31(5):1062–79. doi:10.1038/emboj.2011.497.
Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, Boucontet L, Apetoh L, Ghiringhelli F, Casares N, Lasarte JJ, Matsuzaki G, Ikuta K, Ryffel B, Benlagha K, Tesniere A, Ibrahim N, Dechanet-Merville J, Chaput N, Smyth MJ, Kroemer G, Zitvogel L. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med. 2011;208(3):491–503. doi:10.1084/jem.20100269.
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, di Virgilio F, Zitvogel L, Kroemer G. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573–7. doi:10.1126/science.1208347.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi:10.1038/nm1523.
Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, Sautes-Fridman C, Fucikova J, Galon J, Spisek R, Tartour E, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology. 2015;4(4):e1008866. doi:10.1080/2162402X.2015.1008866.
Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187. doi:10.3389/fimmu.2015.00187.
Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res. 2015;3(5):436–43. doi:10.1158/2326-6066.CIR-15-0064.
Gebremeskel S, Johnston B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: impact on clinical studies and considerations for combined therapies. Oncotarget. 2015;6(39):41600–19. doi:10.18632/oncotarget.6113.
D’Acunto CW, Gbelcova H, Festa M, Ruml T. The complex understanding of Annexin A1 phosphorylation. Cell Signal. 2014;26(1):173–8. doi:10.1016/j.cellsig.2013.09.020.
Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, Yang H, Adjemian S, Chaba K, Semeraro M, Signore M, De Ninno A, Lucarini V, Peschiaroli F, Businaro L, Gerardino A, Manic G, Ulas T, Gunther P, Schultze JL, Kepp O, Stoll G, Lefebvre C, Mulot C, Castoldi F, Rusakiewicz S, Ladoire S, Apetoh L, Bravo-San Pedro JM, Lucattelli M, Delarasse C, Boige V, Ducreux M, Delaloge S, Borg C, Andre F, Schiavoni G, Vitale I, Laurent-Puig P, Mattei F, Zitvogel L, Kroemer G. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science. 2015;350(6263):972–8. doi:10.1126/science.aad0779.
Danielsen EM, van Deurs B, Hansen GH. "Nonclassical" secretion of annexin A2 to the lumenal side of the enterocyte brush border membrane. Biochemistry. 2003;42(49):14670–6. doi:10.1021/bi0355239.
Gavins FN, Hickey MJ. Annexin A1 and the regulation of innate and adaptive immunity. Front Immunol. 2012;3:354. doi:10.3389/fimmu.2012.00354.
Wenzel-Seifert K, Seifert R. Functional differences between human formyl peptide receptor isoforms 26, 98, and G6. Naunyn Schmiedeberg's Arch Pharmacol. 2003;367(5):509–15. doi:10.1007/s00210-003-0714-7.
Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Seror C, Metivier D, Perfettini JL, Zitvogel L, Kroemer G. Chemotherapy induces ATP release from tumor cells. Cell Cycle. 2009;8(22):3723–8. doi:10.4161/cc.8.22.10026.
Di Virgilio F, Pinton P, Falzoni S. Assessing extracellular ATP as danger signal in vivo: the pmeLuc system. Methods Mol Biol. 2016;1417:115–29. doi:10.1007/978-1-4939-3566-6_7.
Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One. 2008;3(7):e2599. doi:10.1371/journal.pone.0002599.
Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Metivier D, Galluzzi L, Perfettini JL, Zitvogel L, Kroemer G. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014;21(1):79–91. doi:10.1038/cdd.2013.75.
Ko A, Kanehisa A, Martins I, Senovilla L, Chargari C, Dugue D, Marino G, Kepp O, Michaud M, Perfettini JL, Kroemer G, Deutsch E. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014;21(1):92–9. doi:10.1038/cdd.2013.124.
Martins I, Michaud M, Sukkurwala AQ, Adjemian S, Ma Y, Shen S, Kepp O, Menger L, Vacchelli E, Galluzzi L, Zitvogel L, Kroemer G. Premortem autophagy determines the immunogenicity of chemotherapy-induced cancer cell death. Autophagy. 2012;8(3):413–5. doi:10.4161/auto.19009.
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi:10.1101/gad.2016211.
Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282(25):18573–83. doi:10.1074/jbc.M701194200.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–6. doi:10.1038/nature08296.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–8. doi:10.1038/nm.2028.
Ladoire S, Enot D, Senovilla L, Ghiringhelli F, Poirier-Colame V, Chaba K, Semeraro M, Chaix M, Penault-Llorca F, Arnould L, Poillot ML, Arveux P, Delaloge S, Andre F, Zitvogel L, Kroemer G. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy. 2016;12(5):864–75. doi:10.1080/15548627.2016.1154244.
Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J, Chen GM, Gendoo DMA, Haibe-Kains B, Karn T, Rahimi K, Le Page C, Provencher D, Mes-Masson AM, Stagg J. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015;75(21):4494–503. doi:10.1158/0008-5472.Can-14-3569.
Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, Stagg J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci U S A. 2013;110(27):11091–6. doi:10.1073/pnas.1222251110.
Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, Levesque S, Castoldi F, Jacquelot N, Yamazaki T, Senovilla L, Marino G, Aranda F, Durand S, Sica V, Chery A, Lachkar S, Sigl V, Bloy N, Buque A, Falzoni S, Ryffel B, Apetoh L, Di Virgilio F, Madeo F, Maiuri MC, Zitvogel L, Levine B, Penninger JM, Kroemer G. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30(1):147–60. doi:10.1016/j.ccell.2016.05.016.
Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng CW, Cacciottolo M, Martin-Montalvo A, de Cabo R, Wei M, Morgan TE, Longo VD. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell. 2016;30(1):136–46. doi:10.1016/j.ccell.2016.06.005.
Marino G, Pietrocola F, Madeo F, Kroemer G. Caloric restriction mimetics: natural/physiological pharmacological autophagy inducers. Autophagy. 2014;10(11):1879–82. doi:10.4161/auto.36413.
Madeo F, Pietrocola F, Eisenberg T, Kroemer G. Caloric restriction mimetics: towards a molecular definition. Nat Rev Drug Discov. 2014;13(10):727–40. doi:10.1038/nrd4391.
Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. 2015;25(1):11–7. doi:10.1016/j.semradonc.2014.07.005.
Sukkurwala AQ, Martins I, Wang Y, Schlemmer F, Ruckenstuhl C, Durchschlag M, Michaud M, Senovilla L, Sistigu A, Ma Y, Vacchelli E, Sulpice E, Gidrol X, Zitvogel L, Madeo F, Galluzzi L, Kepp O, Kroemer G. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2014;21(1):59–68. doi:10.1038/cdd.2013.73.
Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P, Yuan J, Zitvogel L, Madeo F, Williams DB, Kroemer G. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28(5):578–90. doi:10.1038/emboj.2009.1.
Kepp O, Galluzzi L, Giordanetto F, Tesniere A, Vitale I, Martins I, Schlemmer F, Adjemian S, Zitvogel L, Kroemer G. Disruption of the PP1/GADD34 complex induces calreticulin exposure. Cell Cycle. 2009;8(23):3971–7. doi:10.4161/cc.8.23.10191.
Lu YC, Weng WC, Lee H. Functional roles of calreticulin in cancer biology. Biomed Res Int. 2015;2015:526524. doi:10.1155/2015/526524.
Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123(2):321–34. doi:10.1016/j.cell.2005.08.032.
Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, Weiskopf K, Willingham SB, Raveh T, Park CY, Majeti R, Weissman IL. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2(63):63ra94. doi:10.1126/scitranslmed.3001375.
Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N, Tesniere A, Zitvogel L, Kroemer G. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30(10):1147–58. doi:10.1038/onc.2010.500.
Garg AD, Elsen S, Krysko DV, Vandenabeele P, de Witte P, Agostinis P. Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget. 2015;6(29):26841–60. doi:10.18632/oncotarget.4754.
Obeid M, Tesniere A, Panaretakis T, Tufi R, Joza N, van Endert P, Ghiringhelli F, Apetoh L, Chaput N, Flament C, Ullrich E, de Botton S, Zitvogel L, Kroemer G. Ecto-calreticulin in immunogenic chemotherapy. Immunol Rev. 2007;220:22–34. doi:10.1111/j.1600-065X.2007.00567.x.
Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D, Alifano M, Devi P, Biton J, Germain C, Lupo A, Fridman WH, Dieu-Nosjean MC, Kroemer G, Sautes-Fridman C, Cremer I. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and Favorable prognosis. Cancer Res. 2016;76(7):1746–56. doi:10.1158/0008-5472.Can-15-1142.
Fucikova J, Truxova I, Hensler M, Becht E, Kasikova L, Moserova I, Vosahlikova S, Klouckova J, Church SE, Cremer I, Kepp O, Kroemer G, Galluzzi L, Salek C, Spisek R. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood. 2016;128(26):3113–24. doi:10.1182/blood-2016-08-731737.
Stoll G, Iribarren K, Michels J, Leary A, Zitvogel L, Cremer I, Kroemer G. Calreticulin expression: interaction with the immune infiltrate and impact on survival in patients with ovarian and non-small cell lung cancer. Oncoimmunology. 2016;5(7):e1177692. doi:10.1080/2162402X.2016.1177692.
Zhao H, Wang J, Kong X, Li E, Liu Y, Du X, Kang Z, Tang Y, Kuang Y, Yang Z, Zhou Y, Wang Q. CD47 promotes tumor invasion and metastasis in non-small cell lung cancer. Sci Rep. 2016;6:29719. doi:10.1038/srep29719.
Wang H, Tan M, Zhang S, Li X, Gao J, Zhang D, Hao Y, Gao S, Liu J, Lin B. Expression and significance of CD44, CD47 and c-met in ovarian clear cell carcinoma. Int J Mol Sci. 2015;16(2):3391–404. doi:10.3390/ijms16023391.
Yoshida K, Tsujimoto H, Matsumura K, Kinoshita M, Takahata R, Matsumoto Y, Hiraki S, Ono S, Seki S, Yamamoto J, Hase K. CD47 is an adverse prognostic factor and a therapeutic target in gastric cancer. Cancer Med. 2015;4(9):1322–33. doi:10.1002/cam4.478.
Malcovati L, Rumi E, Cazzola M. Somatic mutations of calreticulin in myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms. Haematologica. 2014;99(11):1650–2. doi:10.3324/haematol.2014.113944.
Lundberg P, Nienhold R, Ambrosetti A, Cervantes F, Perez-Encinas MM, Skoda RC. Somatic mutations in calreticulin can be found in pedigrees with familial predisposition to myeloproliferative neoplasms. Blood. 2014;123(17):2744–5. doi:10.1182/blood-2014-01-550863.
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Casetti IC, Sant'Antonio E, Ferretti V, Elena C, Schischlik F, Cleary C, Six M, Schalling M, Schonegger A, Bock C, Malcovati L, Pascutto C, Superti-Furga G, Cazzola M, Kralovics R. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–90. doi:10.1056/NEJMoa1311347.
Araki M, Yang Y, Masubuchi N, Hironaka Y, Takei H, Morishita S, Mizukami Y, Kan S, Shirane S, Edahiro Y, Sunami Y, Ohsaka A, Komatsu N. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood. 2016;127(10):1307–16. doi:10.1182/blood-2015-09-671172.
Watson M, Stott K, Fischl H, Cato L, Thomas JO. Characterization of the interaction between HMGB1 and H3-a possible means of positioning HMGB1 in chromatin. Nucleic Acids Res. 2014;42(2):848–59. doi:10.1093/nar/gkt950.
Thomas JO, Stott K. H1 and HMGB1: modulators of chromatin structure. Biochem Soc Trans. 2012;40(2):341–6. doi:10.1042/BST20120014.
Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5(4):331–42. doi:10.1038/nri1594.
Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H, Robinson M, Van Zoelen MA, Yang H, Li J, Erlandsson-Harris H, Chavan SS, Wang H, Andersson U, Tracey KJ. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A. 2014;111(8):3068–73. doi:10.1073/pnas.1316925111.
Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie W, Feng Y, Socorburam T, Wu G, Xia Z, Wu Q. Receptor interacting protein 3-mediated necroptosis promotes lipopolysaccharide-induced inflammation and acute respiratory distress syndrome in mice. PLoS One. 2016;11(5):e0155723. doi:10.1371/journal.pone.0155723.
Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–23. doi:10.1016/j.immuni.2013.02.003.
Guerriero JL, Ditsworth D, Fan Y, Zhao F, Crawford HC, Zong WX. Chemotherapy induces tumor clearance independent of apoptosis. Cancer Res. 2008;68(23):9595–600. doi:10.1158/0008-5472.CAN-08-2452.
Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, Andre F, Tursz T, Kroemer G, Zitvogel L. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi:10.1111/j.1600-065X.2007.00573.x.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–9. doi:10.1038/nm1622.
Dong Xda E, Ito N, Lotze MT, Demarco RA, Popovic P, Shand SH, Watkins S, Winikoff S, Brown CK, Bartlett DL, Zeh HJ 3rd. High mobility group box I (HMGB1) release from tumor cells after treatment: implications for development of targeted chemoimmunotherapy. J Immunother. 2007;30(6):596–606. doi:10.1097/CJI.0b013e31804efc76.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–5. doi:10.1038/nature00858.
Hreggvidsdottir HS, Ostberg T, Wahamaa H, Schierbeck H, Aveberger AC, Klevenvall L, Palmblad K, Ottosson L, Andersson U, Harris HE. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol. 2009;86(3):655–62. doi:10.1189/jlb.0908548.
Qin YH, Dai SM, Tang GS, Zhang J, Ren D, Wang ZW, Shen Q. HMGB1 enhances the proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J Immunol. 2009;183(10):6244–50. doi:10.4049/jimmunol.0900390.
Youn JH, Oh YJ, Kim ES, Choi JE, Shin JS. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J Immunol. 2008;180(7):5067–74.
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–96. doi:10.1038/ni1457.
Ivanov S, Dragoi AM, Wang X, Dallacosta C, Louten J, Musco G, Sitia G, Yap GS, Wan Y, Biron CA, Bianchi ME, Wang H, Chu WM. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110(6):1970–81. doi:10.1182/blood-2006-09-044776.
Saenz R, Futalan D, Leutenez L, Eekhout F, Fecteau JF, Sundelius S, Sundqvist S, Larsson M, Hayashi T, Minev B, Carson D, Esener S, Messmer B, Messmer D. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med. 2014;12:211. doi:10.1186/1479-5876-12-211.
Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174(12):7506–15.
Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ. A critical cysteine is required for HMGB1 binding to toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107(26):11942–7. doi:10.1073/pnas.1003893107.
Vacchelli E, Galluzzi L, Rousseau V, Rigoni A, Tesniere A, Delahaye N, Schlemmer FD, Menger L, Sukkurwala AQ, Adjemian S, Martins I, Michaud M, Dunant A, Kepp O, Brambilla E, Soria JC, Zitvogel L, Kroemer G. Loss-of-function alleles of P2RX7 and TLR4 fail to affect the response to chemotherapy in non-small cell lung cancer. Oncoimmunology. 2012;1(3):271–8. doi:10.4161/onci.18684.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11(5):373–84. doi:10.1038/ni.1863.
Liang Y, Peng H. STING-cytosolic DNA sensing: the backbone for an effective tumor radiation therapy. Ann Transl Med. 2016;4(3):60. doi:10.3978/j.issn.2305-5839.2015.12.48.
Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, Fend L, Hannani D, Aymeric L, Ma Y, Niso-Santano M, Kepp O, Schultze JL, Tuting T, Belardelli F, Bracci L, La Sorsa V, Ziccheddu G, Sestili P, Urbani F, Delorenzi M, Lacroix-Triki M, Quidville V, Conforti R, Spano JP, Pusztai L, Poirier-Colame V, Delaloge S, Penault-Llorca F, Ladoire S, Arnould L, Cyrta J, Dessoliers MC, Eggermont A, Bianchi ME, Pittet M, Engblom C, Pfirschke C, Preville X, Uze G, Schreiber RD, Chow MT, Smyth MJ, Proietti E, Andre F, Kroemer G, Zitvogel L. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–9. doi:10.1038/nm.3708.
Legrier ME, Bieche I, Gaston J, Beurdeley A, Yvonnet V, Deas O, Thuleau A, Chateau-Joubert S, Servely JL, Vacher S, Lassalle M, Depil S, Tucker GC, Fontaine JJ, Poupon MF, Roman-Roman S, Judde JG, Decaudin D, Cairo S, Marangoni E. Activation of IFN/STAT1 signalling predicts response to chemotherapy in oestrogen receptor-negative breast cancer. Br J Cancer. 2016;114(2):177–87. doi:10.1038/bjc.2015.398.
Wang BG, Yi DH, Liu YF. TLR3 gene polymorphisms in cancer: a systematic review and meta-analysis. Chin J Cancer. 2015;34(6):272–84. doi:10.1186/s40880-015-0020-z.
Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16(3):131–44. doi:10.1038/nrc.2016.14.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi:10.1146/annurev-immunol-032712-100008.
Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–37. doi:10.1038/nri2873.
Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016;16(12):759–73. doi:10.1038/nrc.2016.91.
Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, Schlemmer F, Sulpice E, Locher C, Gidrol X, Ghiringhelli F, Modjtahedi N, Galluzzi L, Andre F, Zitvogel L, Kepp O, Kroemer G. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med. 2012;4(143):143ra199. doi:10.1126/scitranslmed.3003807.
Sukkurwala AQ, Adjemian S, Senovilla L, Michaud M, Spaggiari S, Vacchelli E, Baracco EE, Galluzzi L, Zitvogel L, Kepp O, Kroemer G. Screening of novel immunogenic cell death inducers within the NCI mechanistic diversity set. Oncoimmunology. 2014;3:e28473. doi:10.4161/onci.28473.
Acknowledgments
GK is supported by the French Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Institut Universitaire de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LeDucq Foundation; the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Kepp, O., Pol, J., Zitvogel, L., Kroemer, G. (2018). Immunogenic Stress and Death of Cancer Cells in Natural and Therapy-Induced Immunosurveillance. In: Zitvogel, L., Kroemer, G. (eds) Oncoimmunology. Springer, Cham. https://doi.org/10.1007/978-3-319-62431-0_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-62431-0_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62430-3
Online ISBN: 978-3-319-62431-0
eBook Packages: MedicineMedicine (R0)