
Abstract
Apoptosis, a major form of programmed cell death, is an important mechanism to remove extra or unwanted cells during development. In tissue homeostasis apoptosis also acts as a monitoring machinery to eliminate damaged cells in response to environmental stresses. During these processes, caspases, a group of proteases, have been well defined as key drivers of cell death. However, a wealth of evidence is emerging which supports the existence of many other non-apoptotic functions of these caspases, which are essential not only in proper organism development but also in tissue homeostasis and post-injury recovery. In particular, apoptotic caspases in stress-induced dying cells can activate mitogenic signals leading to proliferation of neighbouring cells, a phenomenon termed apoptosis-induced proliferation. Apparently, such non-apoptotic functions of caspases need to be controlled and restrained in a context-dependent manner during development to prevent their detrimental effects. Intriguingly, accumulating studies suggest that cancer cells are able to utilise these functions of caspases to their advantage to enable their survival, proliferation and metastasis in order to grow and progress. This book chapter will review non-apoptotic functions of the caspases in development and tissue homeostasis with focus on how these cellular processes can be hijacked by cancer cells and contribute to tumourigenesis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Apoptosis
- Caspase
- Non-apoptotic function
- Apoptosis-induced proliferation
- Development
- Tissue homeostasis
- Cancer
4.1 Introduction
Apoptosis was first identified as a form of cell death by its distinct morphological characteristics including cellular shrinkage, chromosome condensation, nuclear fragmentation and formation of apoptotic bodies [1, 2]. Studies in C. elegans then uncovered that apoptosis is genetically controlled and plays critical roles during development to remove unwanted or unnecessary cells [3, 4]. Such function of apoptosis further extends to maintenance of tissue homeostasis by eliminating damaged or unfit cells [5, 6]. Apoptosis has therefore been viewed as a monitoring programme to identify and kill potentially harmful cells that may develop into cancer. Consistent with this idea, evading apoptosis has been considered as a hallmark of cancer [7, 8].
The key components of the apoptotic machinery are caspases, a family of cysteine proteases which cleave their substrates leading to cell death [9, 10]. Recently however, in addition to their functions in apoptosis, caspases are becoming better understood in their multifunctional nature with an increasing number of non-apoptotic functions discovered. We acknowledge the abundance of high quality reviews which have described with clarity the non-apoptotic functions of caspases in the context of development and tissue regeneration [11–14]. This chapter therefore focuses on the roles of caspases in sustaining cancers and promoting their spread which seems to contradict what we know about their roles in apoptosis. There is now certainly a great wealth of evidence to show that the apoptotic caspases actually have multiple functions other than executing cell death, and cancer cells can hijack these activities to directly promote their growth, metastasis and recurrence after therapy. Here we have synthesised the evidence present in the current literature supporting this claim, to highlight that the caspases do indeed have a role in progressing cancers. Issues that may exist in current cancer therapies for particular patient subsets are also discussed.
4.2 The Apoptotic Machinery: Functions of the Apoptotic Caspases
Apoptosis is an evolutionarily conserved mechanism in multicellular organisms, allowing correct pattern formation during development and the removal of cells which are detrimental to the health and survival of the organism [15–18]. The pathways leading to apoptosis have been elucidated in many organisms, including C. elegans, Drosophila and mammals, which are summarised in Fig. 4.1. A noticeable family of key components in these apoptosis pathways are the caspases. By definition, caspases are cysteine-aspartic acid proteases. They cleave their substrates after the aspartic acid residue which features at the end of short tetrapeptide motifs [9, 19]. In addition to their functions in apoptosis, caspases are also well known for their roles in inflammatory responses [20–22]. For example, there are 18 known mammalian caspases among which caspases-2, -3, -6, -7, -8, -9 and -10 function in apoptosis and thus have been classified as apoptotic caspases [14, 23]. This review focuses on these caspases, especially caspases-3, -7, -8 and -9 due to their reported multiple non-apoptotic functions.

Evolutionarily conserved apoptosis pathways. Schematic comparison of major components in the apoptosis pathways in C. elegans (a), Drosophila (b) and mammals (c). Protein homology is indicated in brackets. See text for more details
Under normal cellular conditions all apoptotic caspases are present as inactive pro-caspases, called zymogens, which consist of a prodomain, a small subunit and a large subunit [19, 24]. They require cleavage in apoptotic cellular conditions to become activated. Based on the structure of N-terminal prodomains, apoptotic caspases can be subdivided into the initiator (or apical) and effector (or executioner) caspases. The initiator caspases have elongated prodomains which contain either the death effector domains (DED, e.g. for caspase 8) or the caspase-recruitment domains (CARD, e.g. for caspase 9). In contrast, the effector caspases have small prodomains. These caspases also have distinct functions and substrates during the process of apoptosis [25–27]. The initiator caspases cleave inactive pro-effector caspases and activate them. They are therefore also called apical caspases. In contrast, effector caspases, once activated by the initiator caspases, further cleave their broad range of cellular proteins leading to execution of cell death. They therefore have another name as executioner caspases. For simplicity, terms of initiator and effector caspases are used in this review.
4.2.1 Apoptosis in C. elegans
The caspases were first identified in C. elegans in which 131 cells undergo apoptosis during development by the action of a simple and linear pathway (Fig. 4.1a) [3, 4, 16]. Before an apoptotic stimulus is detected by a cell, CED-4, a homologue of the mammalian adaptor protein apoptosis activating factor 1 (Apaf-1), exists as a dimer, which is sequestered on the outer leaflet of the outer membrane of the mitochondria by contact with a Bcl-2 family member called CED-9 [28]. Upon apoptotic stimulus, Egl-1, a pro-apoptotic BH3-only protein (Bcl-2 homology 3), is expressed, binding CED-9, thus releasing CED-4. CED-4 is then free to form a tetramer. Once the CED-4 tetramer is assembled, it can cleave and activate the caspase CED-3, which in turn activates other downstream apoptotic effector proteins leading to cell death (Fig. 4.1a) [28, 29].
4.2.2 The Intrinsic Apoptosis Pathway in Drosophila and Mammals
Unlike the linear pathway in C. elegans, apoptotic pathways of extrinsic and intrinsic origin have been identified in both Drosophila and mammals (Fig. 4.1). The intrinsic pathway has been extensively studied in Drosophila (Fig. 4.1b). Initially, apoptotic stimuli cause the expression of the pro-apoptotic genes of the RHG family: mainly reaper, hid (head involution defective) and grim [30, 31]. These gene products act to relieve the repression exerted by the inhibitors of apoptosis (IAPs) [32–34], which, under normal cellular conditions, inhibit activities of the Drosophila initiator caspases such as Dronc [35, 36] and effector caspases such as DrICE and Dcp-1 [37, 38]. The major IAP in Drosophila is Diap1 which functions as an E3-ubiquitin ligase. Under no apoptotic stimuli, it binds to Dronc via its own BIR2 domain and causes ubiquitin to be tagged to Dronc [36]. Such ubiquitylation was believed to stimulate degradation of Dronc via the proteasome. However, a recent genetic analysis suggests that Diap1-mediated ubiquitylation blocks processing and activation of Dronc but does not lead to its protein degradation [39]. When RHG proteins antagonise Diap1 by competitively binding to its BIR domains, Diap1 can no longer perform its function on inhibiting Dronc [36, 40, 41]. From here released Dronc, although inactive, can induce formation of the apoptosome by the adapter protein Ark [42, 43]. Upon such interaction Dronc can autocleave and become activated. Activated Dronc further cleaves and activates its downstream effector caspases, mainly DrICE and Dcp-1, leading to apoptosis (Fig. 4.1b) [44–46]. Notably, pro-apoptotic proteins need to localise to the mitochondria and execute their apoptotic functions in Drosophila [47–54]. Two Bcl-2 family members, Debcl and Buffy, have been identified in Drosophila [55–59]. Debcl is localised to the mitochondria and has pro-apoptotic functions, while Buffy may localise to endoplasmic reticula to carry out its own anti-apoptotic roles [60].
In the mammalian intrinsic pathway, the mitochondrion plays a central and more decidedly important role (Fig. 4.1c). The Bcl-2 family protein s can be subdivided into three groups: the BH3-only proteins (such as Bid, Bad, Bik, Bim, Noxa and Puma), the pro-apoptotic Bax subfamily members (such as Bax, Bak and Bok) and the anti-apoptotic Bcl-2 family members (such as Bcl-2 and Bcl-XL) [61–64]. In response to apoptosis, BH3-only proteins either activate the Bax subfamily members or antagonise the anti-apoptotic Bcl-2 members to regulate mitochondrial outer membrane permeabilisation (MOMP) which then leads to release of cytochrome c (cyt c). Released cyt c binds to the adaptor protein Apaf-1, via the WD repeat domain at the carboxy terminus of Apaf-1, forming the apoptosome. Pro-caspase-9 can in turn interact with Apaf-1 in the apoptosome, via their mutual CARD domains [65]. Pro-caspase-9 then autocleaves and becomes active [66]. The activated caspase-9 further cleaves its downstream effector caspases, caspase-3 and -7, to trigger apoptotic cell death [63]. In addition to cyt c, pro-apoptotic proteins such as Smac (or Diablo) and HtrA2 (or Omi) are also released from mitochondria during the process of MOMP [67–69]. Similar to what happens in Drosophila, these pro-apoptotic proteins antagonise IAPs such as XIAP leading to activation of caspase-9, -3 and -7 and apoptosis. In addition to Smac and HtrA2, another mammalian IAP antagonist is ARTS which is not released from mitochondria [70, 71]. Similar to the RHG proteins in Drosophila, it is localised to the mitochondrial outer membrane and inhibits XIAP [72].
4.2.3 The Extrinsic Apoptosis Pathway in Drosophila and Mammals
In contrast to the intrinsic pathway, the extrinsic pathway is initiated by the binding of a death ligand to a death receptor in the cell (Fig. 4.1c). In mammals, examples of the death ligands are tumour necrosis factor (TNF) family members including Fas ligand (FasL) and TNF-related apoptosis-inducing ligand (TRAIL) [73–75]. These ligands bind to their specific receptors Fas and DR4/5, forming complexes. Once such a ligand–receptor complex is formed, the adaptor protein Fas-associated Death Domain (FADD) can bind the cytosolic region of Fas and DR4/5. There, FADD acts as a platform on to which pro-caspase-8 can bind, by interaction of the death effector domain (DED) of FADD with the DED at the extended N-terminus of pro-caspase-8, forming the death-inducing signalling complex (DISC) [76–78]. Due to receptor clustering in the plasma membrane, the pro-caspase-8 monomers are brought within close proximity of each other in DISC complexes, and once in this newly established close proximity they can autocleave and become activated [79, 80]. Upon activation, caspase-8 can then cause the cleavage and activation of effector caspases caspase-3 and -7 leading to cell death [9, 25]. Homologues of death ligands, receptors and their functions in apoptosis induction have also been found in Drosophila (Fig. 4.1b). There is only one TNF homologue, Eiger (Egr), identified in Drosophila so far [81, 82]. Two TNF receptors including Wengen (Wgn) and, more recently, Grindelwald (Grnd) have been reported [83, 84]. Activation of Egr triggers both apoptosis and a type of non-apoptotic cell death through the Jun N-terminal Kinase (JNK) pathway, a stress-response signalling pathway [81, 82, 85, 86]. For the aspect of apoptosis, JNK induces expression of pro-apoptotic genes and activation of the apoptotic machinery [82, 86]. Interestingly, in stress-induced apoptosis, the initiator caspase Dronc can activate not only effector caspases DrICE and Dcp-1 but also JNK which then feedback to the apoptosis pathway to further amplify it [87]. Notably, although different in their nature of inducing apoptosis, connections between the extrinsic and intrinsic pathways also exist in mammals. Caspase-8 can act on the pro-apoptotic BH3-only proteins such as BID leading to activation of the intrinsic pathway which further ensures a robust apoptotic response [88–90].
4.3 Apoptosis, Development and Non-apoptotic Functions of Caspases
Apoptosis and development are interconnected. On the one hand, apoptotic caspases were originally identified as key players in the developmental programme [3]. Their apoptotic functions are critical for removal of extra cells produced at the early stage of development and elimination of unwanted cells in tissue patterning and morphogenesis [16]. A recent study on Apaf-1 knock-out mice suggests that apoptosis is required to remove Fgf8 morphogen-producing cells and terminate Fgf8 production at the correct developmental time, thus ensuring proper development of the forebrain [91]. Apoptotic cells can even actively drive epithelial folding during morphogenesis [92] and cell extrusion during tissue repair [93]. These examples have certainly underlined the developmental role of apoptosis. On the other hand, it is also becoming clear that the developmental programme can modulate cellular apoptotic responses. Many key components in the apoptosis pathway can be targeted by the developmental programme to define distinct cellular susceptibilities to apoptosis. For example, in Drosophila third instar larvae, a pulse of hormone ecdysone increases the whole organismal sensitivity to apoptosis by upregulating the basal level of Ark, Dronc and DrICE [94]. Furthermore, in the developing Drosophila eye tissue, multiple mechanisms were employed to control cellular levels of IAPs as well as pro-apoptotic proteins [95, 96]. Similarly, in mouse embryos, primed stem cells are very sensitive to apoptosis due to their low levels of BIM regulated by microRNAs [97]. Therefore, cellular apoptosis susceptibility can be modulated by developmental programmes. However, the links between apoptotic caspases and development go far beyond death. Increasing evidence is now demonstrating the actual, true multifunctional nature of the caspases with somewhat surprising and fascinating roles in diverse cellular processes. These functions include regulating immune responses, promoting cell proliferation, and regulating cell differentiation and fate specification which have been extensively reviewed elsewhere [1, 11, 13, 14, 21, 25, 98, 99]. Here, we highlight some of these non-apoptotic functions, in particular roles of caspases in tissue homeostasis, in the context of cancer development.
4.4 Caspases in Tissue Homeostasis: Apoptosis-Induced Proliferation (AiP)
Organisms are constantly exposed to environmental stresses. Damaged cells are frequently removed by apoptosis. Meanwhile, new cells are generated by proliferation to compensate for the cell loss thus to maintain tissue homeostasis. For example, up to 60 % of cells in the developing Drosophila wing epithelial tissue can be lost in response to radiation without affecting final adult wing size and morphology [100]. A similar phenomenon has also been found in the processes of wound healing and liver regeneration in mammals [101]. Apparently, tissue homeostasis is important for tissue function to remain optimal and critical to organism survival. Evidence in multiple organisms including Hydra [102], Drosophila [103–105] and mouse [101] is now demonstrating that apoptotic caspases have non-apoptotic functions to trigger compensatory proliferation, a process therefore termed apoptosis-induced proliferation (AiP) or apoptosis-induced compensatory cell proliferation [106–109]. For simplicity we use the term apoptosis-induced proliferation (AiP) in this review. Recent studies in Drosophila have provided mechanistic insights into how AiP occurs (Fig. 4.2a, b) [103–105, 110–113]. Intriguingly, depending on the developmental state of the affected tissue, i.e. proliferating versus differentiating tissues, either initiator or effector caspases drive distinct mechanisms of AiP in Drosophila [103].

Apoptosis-induced proliferation in Drosophila and mammals. Molecular mechanisms of apoptosis-induced proliferation (AiP) in proliferating (a) versus differentiating (cell cycle exited, b) tissues in Drosophila and in mammals (c). A stressed but “undead” cell (a, left, in red) or an apoptotic cell (b and c, left, in red) and a cell that is induced to undergo AiP (right, in green) are shown. See text for more details. iPLA2 calcium-independent phospholipase A2, COX cyclooxygenases, PGE2 prostaglandin E2
4.4.1 The Initiator Caspase-Driven AiP in Drosophila
The molecular mechanism of AiP was first addressed in Drosophila by taking advantage of caspase inhibitors [104, 111, 112]. P35, a baculovirus inhibitor of apoptosis, acts as a peudosubstrate of Drosophila effector caspases, e.g. DrICE and Dcp-1 [114]. Expression of P35 thus blocks activity of DrICE and Dcp-1 and execution of cell death. To determine how stress-induced apoptotic cells may contribute to compensatory proliferation, such cells were kept “undead” by P35 (i.e. the apoptotic machinery is activated but execution of cell death is blocked). Surprisingly, “undead” cells stimulate overgrowth of surrounding tissues despite the presence of P35 [104, 111, 112]. This suggests that dying cells release mitogenic signals to induce AiP independent of effector caspases. Further loss-of-function analyses revealed that the initiator caspase Dronc, which is not inhibited by P35, actually coordinates apoptosis and AiP (Fig. 4.2a). It appears that, at least in the “undead” model of AiP, Dronc activates JNK in dying cells leading to activation of several mitogenic signalling pathways including the Wingless (Wg, a homologue of the mammalian Wnt) and Decapentaplegic (Dpp, a TGF-β-like homologue of the mammalian BMP) signalling pathways which are required for AiP [112, 115]. Drosophila homologue of p53 is also required for AiP, probably through its role in a feedback regulatory loop including JNK, p53 and pro-apoptotic genes [87, 113]. However, one concern of the “undead” model of AiP is that it may not represent what happens in the physiological process of AiP [115–117]. For example, it has been suggested that Wg and Dpp are not required for AiP when there are no “undead” cells [115]. Nevertheless, a Drosophila model of regenerative growth without using P35 has identified Wg as an important factor which is induced in response to tissue damage and is required for tissue regeneration [118]. In addition to these, a recent genetic screen using both an “undead” model and a P35-independent regenerative model has discovered a role of EGFR signalling in AiP and tissue regenerative growth [110]. In this process, JNK transcriptionally induces Spi, one of EGF ligands in Drosophila, in dying cells which then activates proliferation of neighbouring cells via EGFR signalling. JNK can also activate the transcription factor Yorkie (Yki) in the Hippo signalling pathway to regulate AiP in developing Drosophila wing tissues [119, 120]. Interestingly, such a role of Yki in AiP seems to be tissue specific as it is not required for AiP in proliferating eye tissues [110].
4.4.2 The Effector Caspase-Driven AiP in Drosophila
A second form of AiP was identified in the differentiating Drosophila eye tissue which is a monolayer epithelium with differentiated photoreceptor cells at the apical side and cell cycle exited but unspecified cells at the basal side [103]. At the late third instar larval stage, both types of cells have relatively low susceptibility to apoptosis presumably due to their post-mitotic status and protection of survival signals such as high Diap1 and the EGFR signalling [95]. Therefore, under apoptotic stresses, e.g. expression of the pro-apoptotic gene hid, these cells do not die immediately. Instead, the stressed photoreceptor neurons release Hedgehog (Hh), another evolutionarily conserved growth signalling ligand, to trigger cell cycle re-entry of unspecified cells (Fig. 4.2b). Such an AiP event can be blocked by P35 or double mutants of DrICE and Dcp-1 suggesting an effector caspase-driven form of AiP is employed in the differentiating eye tissue [103]. Interestingly, mechanisms of AiP seem to be operated in a context-dependent manner. This is best shown in the developing Drosophila eye tissue. The late third larval eye tissue consists of an anterior proliferating portion where all cells are actively dividing and a posterior differentiating portion where most of the cells present have exited the cell cycle. The initiator caspase-driven AiP appears to be employed in proliferating tissues, while the effector caspase-driven AiP is employed in differentiating tissues [103]. However, what controls such distinction is not yet known.
4.4.3 AiP in Other Organisms Including Mammals
In addition to Drosophila, roles of AiP in regeneration have also been implicated in other multicellular organisms particularly in Hydra, Xenopus and mouse [101, 102, 106, 121]. In the freshwater Hydra, both head and foot can regenerate completely after bisection at the midgastric area. Massive localised apoptosis was observed for the head regenerating tip, but not the foot regenerating counterpart, preceding increase of cell proliferation [102]. Interestingly, ectopic activation of apoptosis at the foot regenerating tip resulted in regeneration of head instead of foot. In this process, caspases activate Wnt3, a homologue of Drosophila Wg, in dying cells leading to regenerative proliferation [102]. This study suggests that apoptosis can direct certain regenerative programmes. Similar requirements of caspases in regeneration were also reported for Xenopus tadpole tail regeneration which is abolished by inhibiting caspase-3 [121]. Notably, in other regeneration models such as planaria and newt, massive apoptosis at the amputation site has been observed [122–124]. However, it is not yet clear whether apoptotic caspases actually drive release of mitogenic signals such as Wnt, TGF-β and Hh in these processes. More recently, roles of AiP in mammals were reported in mouse models of wound healing and liver regeneration [101, 125]. The rate of skin wound healing and liver regrowth after partial hepatectomy was significantly reduced in caspase-3 or -7 deficient mice due to impaired post-injury cell proliferation. It was further revealed that activated caspase-3 and -7 cleave calcium-independent phospholipase A2 (iPLA2) to increase its catalytic ability and promote synthesis of prostaglandin E2 (PGE2). Release of PGE2 from the dying cell then induces compensatory proliferation (Fig. 4.2c) [101]. Although detailed mechanisms on how PGE2 triggers compensatory proliferation are not yet revealed, the link between PGE2 and the Wnt signalling cascade has been established in both zebrafish and mice [126]. PGE2 binds to EP2, a G-protein coupled receptor, leading to activation of β-catenin, a key intracellular transducer of Wnt signalling [127–129].
4.5 Caspases in Cancer Development : Non-apoptotic Functions
Current cancer therapies such as chemo- and radiotherapies frequently aim to activate apoptosis of cancer cells. Therefore, activating apoptosis has long been viewed as an “anti-cancer” process. However, increasing evidence is now suggesting that apoptotic caspases can play oncogenic roles through their non-apoptotic functions (Fig. 4.3). As discussed earlier the roles of the apoptotic caspases are essential in proper organism development and tissue homeostasis. Apparently different functional aspects of caspases needs to be tightly controlled and restrained by cellular contexts in order to prevent their detrimental effects. In the context of cancer, these non-apoptotic functions of caspases can be hijacked to ensure survival of cancer cells and promote their spread. Thus, the multifunctional nature of the apoptotic caspases is becoming clinically important.

Schematic diagram of non-apoptotic functions of caspases that may contribute to various aspects of cancer progression
4.5.1 Caspases Promote Cell Survival and Cell Proliferation
The crucial function of caspases in cell survival and proliferation has been reported during development. Targeted disruption of caspase-8 in mice causes embryonic lethality, a feature not shared by the other caspases [78, 130]. Caspase-8−/− mouse embryos exhibited abnormal phenotypes prior to death, namely hyperaemia, with the number of haematopoietic precursors significantly reduced [78]. This suggests that caspase-8 is required for either maintenance or proliferation of haematopoietic precursors. As further support for this view, depletion of caspase-8 in lymphoid tissues inhibits antigen-induced T and B lymphocyte proliferation [131–133]. Although it was originally thought that caspase-8 regulates cell proliferation in these cases, it is more likely that caspase-8 has pro-survival functions due to its inhibitory role on necroptosis, another form of programmed cell death [130, 134]. The key factors involved in such regulation are caspase-8, the long isoform of cellular FLICE-like inhibitory protein (FLIPL), and two kinases, RIPK1 and RIPK3, which are required for activation of necroptosis. FLIPL is structurally similar to caspase-8 but without its catalytic activity [135]. It can bind to pro-caspase-8, forming a heterodimer which prevents caspase-8 from completing its apoptotic functions by occupying all binding sites of caspase-8 in the DISC. This consequently prevents caspase-8 homodimer formation. Therefore, when the FLIPL levels are low, homodimerisation of pro-caspase-8 occurs which activates caspase-8 for its apoptotic function. In contrast when FLIPL levels are high, e.g. triggered by survival signals mediated by a transcription factor NFκB, formation of the pro-caspase-8-FLIPL heterodimer does not trigger apoptosis. Instead, it can bind to the RIPK1-containing complex to suppress its activation of RIPK3 and necrotic cell death, although the underlying mechanism remains unclear [134]. Hence, the level of FLIPL is crucial for caspase-8-regulated cell survival. Interestingly, an increase in FLIPL expression has been detected in a variety of tumour types, including B-cell chronic lymphocytic leukaemia, pancreatic cancer and ovarian cancer, amongst many others [136, 137]. Down-regulating FLIPL levels in tumours sensitises the cells to apoptosis [136, 138]. This is most likely due to decreased ability for caspase-8-FLIPL heterodimers to form and increased ability of caspase-8 homodimerisation, which can then activate caspase-3 and apoptosis. In a study of cervical cancers, high-grade tumours were found to have higher expression of FLIPL [137]. Moreover, increasing grade of lesions was directly associated with increased c-FLIP expression, where 12.5 % of normal cervical epithelia stained positive for relevant expression of FLIPL compared to 82.1 % of squamous cervical carcinomas stained positive for FLIPL [137]. This shows the significance in correlation of uncontrolled caspase-8-FLIPL dimer formation and cancer progression. Interestingly, infection by high-risk human papillomavirus (HPV), particularly HPV-16, was highly significantly correlated with high expression of FLIPL [139]. Although the viral infection does not explain the cause of high FLIPL expression in other cancer types, high expression of FLIPL was determined to be a marker of early cervical carcinogenesis and therefore has the potential to be utilised for early diagnosis [137, 139]. This evidence highlights that the caspase-8-FLIPL heterodimer can be hijacked by cancer cells to promote tumour survival, by avoiding the apoptotic functions of caspase-8.
In addition to the initiator caspases, the effector caspases have also been implicated in promoting cell survival and cell cycle progression. In cultured cancer cell lines with their origin in leukaemia or hepatocellular or cervical carcinoma, caspase-3 and -7 are found to be required in cell cycle progression through the G1 and G2/M checkpoints [140, 141]. Overexpression of the BIR2 domain of XIAP inhibits caspase-3 and -7, and when added to cells also induced cell cycle arrest. In contrast, inhibition of caspase-9 by expression of the BIR3 domain of XIAP did not cause the same effect, which indicates that caspase-3 and -7 have functions independent of caspase-9 activity. Although it is not yet clear how caspase-3 and -7 may promote cell cycle progression without being cleaved by caspase-9, the anaphase-promoting complex/cyclosome (APC/C) , which regulates degradation of various cell cycle regulators through ubiquitylation, failed to form when caspase-3 and -7 were inhibited [140]. This suggests that pro-caspase-3 and -7 may contribute to cell proliferation. Interestingly, direct substrates of caspases including cell cycle regulators can also promote cell survival or cell cycle progression at least in some circumstances. For example, the cyclin-dependent kinase inhibitor P27Kip1 can be cleaved by caspase-3 which then becomes activated and anti-apoptotic to protect human leukemic cells from death [142]. In addition to this, a more recent study suggested that caspase-3 can act as a sensor to extracellular stresses, therefore determining whether the cells live or not [143]. In this study, caspase-3-knockout mice become more sensitive to UV radiation with increased number of cells undergoing necrosis compared to the control animals. In response to doxorubicin, an anticancer drug inducing apoptosis of cardiomyocytes, the caspase-3-deficient mice also show significantly increased number of apoptotic cardiomyocytes which die through caspase-7 instead [143]. Caspase-3, but not caspase-6 and -7, cleaves the p120 RasGAP protein in vitro to activate a kinase, Akt, leading to survival functions of PI3K signalling [144, 145]. Consistently, Akt activity, indicated by the level of phosphorylated Akt, increases in response to stresses such as UV radiation and doxorubicin injection. But such increase is strongly reduced in caspase-3-knockout mice. Knock-in mice with a RasGAP mutant resistant to caspase-3 cleavage can restore their apoptotic sensitivity [143]. Given these findings of caspases in cell survival and proliferation, could they contribute to tumourigenesis? As discussed later (see Sect. 5.3), the answer becomes clear by discovering roles of AiP in tumour reoccurrence following cytotoxic cancer therapies.
4.5.2 Caspases and Metastasis
Metastasis is a crucial process to understand in cancer progression as it is the cause of approximately 90 % of cancer-related deaths [8]. It is an incredibly complex process consisting of multiple key steps for a cancer cell, or a group of cancer cells, to progress through [146]. These steps include breaking away from a bulk tumour, disseminating in the blood or lymph, exiting the circulation, then establishing and repopulating at a new site, where a secondary tumour forms. Interestingly, caspases have been implicated in aiding some of these steps through their non-apoptotic functions in cell migration, angiogenesis and possibly cell dedifferentiation.
Caspases have been reported for their functions in controlling cell motility during development. In Drosophila, Dronc, the caspase-9 homologue, is required for migration of border cells in the ovary [147], a process critical for oocyte development. In mammals, caspase-8−/− mouse embryos die with a circulatory failure suggesting roles of caspase-8 in migration of endothelial cells [78, 148]. Similarly, in cancer-specific studies, caspase-3 and its downstream targets have been implicated in causing tumour cell migration, thus contributing to achieving metastasis. In ovarian cancer cells, caspase-3 has been shown to be involved in the process of initiating cell migration via activation of arachidonic acid, the precursor of PGE2, similarly as in the context of AiP described earlier (Fig. 4.2c) [128, 149, 150]. Ovarian cancer cells have strong migratory responses towards laminin-10/11, a protein component of the extracellular matrix. This is probably due to the high levels of β1 integrin in ovarian cancer cells, because binding of laminin-10/11 to β1 integrin leads to a moderate increase of caspase-3 activity [150]. Although the intermediate molecules determining caspase-3 activation from integrin–laminin binding are unknown, Zhao et al. [150] determined that moderate increase of caspase-3 activity does not lead to apoptosis, instead, it cleaves iPLA2 and activates its enzymatic activity to produce arachidonic acid and then PGE2. Consistently, pan caspase inhibitors, caspase-3-specific inhibitors or knockdown of iPLA2 inhibits migration of these cells. Interestingly, cleaved iPLA2 also activates Akt survival signalling to protect these cells from apoptosis [150]. This further enhances cancer cell migration. Not surprisingly arachidonic acid has also been implicated to be the driving factor of cell migration in other cancers including prostate cancers [151]. Further support for roles of caspase-3 in cell migration comes from a study on lung cancer metastasis [152]. In this study, however, a protease-independent function of caspase-3 was suggested to promote metastasis. The authors used A549 cells, derived from high malignancy lung adenocarcinoma cells with high levels of caspase-3, for their study. Knockdown of caspase-3 in A549 cells diminishes their metastatic activities in the lungs when these cells were injected into nude mice via the tail vein suggesting roles of caspase-3 in promoting metastasis. Consistently, ectopic expression of caspase-3 in MCF-7 cells, derived from caspase-3-deficient and low malignancy breast cancer cells, enhances metastatic ability of these cells. Following these findings, the authors then found that high levels of caspase-3 actually lead to high activity of the extracellular signal-regulated kinases (ERK) which are required for the observed lung metastasis. However intriguingly, such increased ERK activity and cell migration are not affected by the caspase inhibitor Z-DEVD-FMK. Furthermore, expression of protease-dead mutants of caspase-3 in MCF-7 cells still enhances their migration through increased ERK activities. Although it is not yet clear, the acid sphingomyelinase and its downstream signal molecule ceramide were suggested to be the molecules linking caspase-3 and ERK [152]. Interestingly, another mechanism of caspase-3-dependent cell migration has been reported for the “undead” cells in Drosophila models [153]. In this case, DrICE, a caspase-3 homologue in Drosophila, activates JNK leading to cell migration and tissue invasion. Therefore, cellular contexts may determine how caspase-3 promotes cell motility.
Following migration and invasion of cancer cells, angiogenesis is essential to further cancer progression, enabling tumour growth above a diameter of 1 mm and metastasis [146]. Knockdown of caspase-8 suppressed vascular endothelial growth factor (VEGF)-mediated angiogenic signalling [154]. Interestingly, such requirement of caspase-8 in promoting angiogenesis is not affected by Ac-IETD-cho, a caspase-8 inhibitor that maintains high levels of pro-caspase-8. In contrast, the same study also showed that caspase-8 is required in TRAIL signalling to antagonise angiogenesis which can be inhibited by Ac-IETD-cho [154]. Therefore, pro-caspase-8 and caspase-8 appear to have distinct functions during angiogenesis mediated by VEGF.
Another cellular process that can potentially impact on cancer metastasis is cell dedifferentiation. Although it is still a subject of debate, existence of “cancer stem cells”, a small fraction of stem cell-like cancer progenitor cells, may facilitate or even establish the metastatic colonies for cancer progression [146]. If this is true, maintenance and reprogramming, thus dedifferentiation, of cancer cells may be crucial in the process of metastasis which, again, may involve caspases. Notably, both caspase-8 and caspase-3 are required for the dedifferentiation of murine fibroblasts to form induced pluripotent stem cells (iPSCs) in vitro [155]. Activation of caspase-8 and -3 is induced by expression of Oct-4, one of the four transcription factors used to programme iPSCs . By inhibiting caspase-8 the cells were completely unable to develop into iPSCs , whereas some could if only caspase-3 was inhibited suggesting potential roles of other effector caspases such as caspase-7 in induction of iPSCs . The authors further showed that the caspases act upon retinoblastoma susceptibility protein (Rb), but how from here the phenotype of a pluripotent stem cell is produced is unknown although p53 and its downstream cell cycle regulator p21 have been implicated in the process [155]. Interestingly, studies of human tumours in relation to their Oct-4 expression showed that tumours expressing high levels of Oct-4 resulted in increased metastases, shorter survival and furthered disease progression in comparison to tumours low in Oct-4 expression [156]. A recent study further sorted murine breast cancer 4T1 cells with either high or low Oct-4 expression and tested their tumourigenic potential in vivo by injecting sorted cells into the mouse mammary glands [157]. The results support that Oct-4 can enhance cancer stem cell properties. This fits in vitro data and hypotheses theorising on the capacity of cancer stem cells in disease progression, though more studies are required in a greater range of tumour types.
4.5.3 Caspases in Tumour Repopulation Following Cytotoxic Cancer Therapies
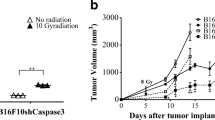
Cytotoxic therapies exert their anti-tumour properties by inducing apoptosis as a result of DNA damage [158]. As discussed earlier, AiP is a process utilised in non-cancerous tissue in order to maintain tissue homeostasis that allows tissue regeneration and recovery from damage. Consequently, this means that cytotoxic therapies can potentially induce not only cell death but also the AiP pathway which may in fact counteract cancer treatment. Tumours, to some extent, are comparable to standard developmental tissues [159], and, conceivably, when damaged they can respond in the same way to regenerate and to compensate for the inflicted damage, thus to repopulate and reoccur. Recent studies on AiP in cancer models have suggested this is the case. In one study, experiments were conducted to find out how caspase-3 is responsible for promoting accelerated tumour repopulation following cytotoxic therapy in 4T1 murine breast cancer cells [160]. It was found that the AiP pathway, activated in either cancer cells or stromal cells, could become hijacked by cancer cells following radiotherapy, causing accelerated tumour repopulation in vitro and in vivo, in nude mice. These were also confirmed with human breast cancer cell lines in nude mice [160]. The results gained in this study were further developed in studies on metastatic melanoma, showing that chemotherapy too can result in AiP and tumour repopulation [161]. As in the tissue regeneration mouse model, it is PGE2 which is secreted from apoptotic cells and stimulates recipient living cells to proliferate in the tumour repopulation model (Fig. 4.2c) [160, 161]. The authors also found that tumours with elevated caspase-3 were more resistant to radiotherapy than those with reduced caspase-3 [160]. This at first seems paradoxical; however, with regards to the AiP model, this observation is logical. Higher caspase-3 expression allows for greater production of PGE2, which in turn stimulates the increased growth rate of surviving cells, thus ensuring the maintenance of a larger tumour mass. Huang et al. found that the therapy sensitive cells were induced to undergo apoptosis, and the release of prostaglandins from the therapy sensitive cells caused the therapy-resistant cells to proliferate at an accelerated rate and repopulate the treated tumour [160].
Statistical studies have been conducted to give a measure of how higher expression of particular caspases in tumours can affect outcome and survival likelihood. In a study of breast cancer, 103 out of 137 tumours were deemed to have high levels of caspase-3, although some activity was noticed in all of the tumours [162]. Increased caspase-3 level significantly correlated with worsened survival of the patients sampled and, in the tumours sampled, caspase-3 was only found in the cytoplasm, not the nucleus where the apoptotic target of caspase-3 resides. This suggests a possible mechanistic block preventing the effector caspase-3 from reaching its target molecule, the inhibitor of caspase-activated DNase (iCAD) , to free caspase-activated DNase (CAD) which can cause DNA fragmentation and subsequent cell death [162]. Another study assessing implications of caspase-3 in gastric, ovarian, cervical and colorectal cancers concluded that patients possessing tumours which expressed higher caspase-3 had shortened survival time and also found that caspase-3 expression was significantly associated with tumour stage [163]. Both studies concluded that higher caspase-3 expression resulted in worsened prognosis. Notably, participants of these studies had not undergone any form of therapy. However, these findings of statistical significance were further confirmed by Huang et al. [160], on patients who had undergone radiotherapy or chemotherapy.
Given these new insights of mechanisms causing tumour repopulation following cytotoxic therapy, if repopulation is to be prohibited in tumours, then the AiP pathway needs to be blocked while still allowing caspase-3 to carry out its apoptotic functions. As described for AiP (Fig. 4.2c), PGE2 is synthesised from arachidonic acid by cyclooxygenases (COX) . Thus, COX inhibitors in theory should prevent the AiP pathway from progressing. This has been shown in practice, where administering a COX inhibitor in conjunction with cytotoxic therapy significantly decreases rate of tumour repopulation [160, 161]. Therefore, use of a COX inhibitor in conjunction with the cytotoxic therapy may benefit patients possessing tumours with high levels of caspase-3. Notably, caspase-3 may not be the only component in the apoptosis pathway that can promote cancer tumourigenesis as suggested by studies on lymphoma [164, 165]. Further mechanistic understanding of AiP in various cellular contexts will be the key to explore its clinical significance. Interestingly, in addition to AiP, engulfment of apoptotic cells by macrophages can create a tumour-promotive microenvironment by releasing signalling molecules [166–168] and regulating various aspects of tumour progression [169]. Again, caspases play key roles here. Activation of iPLA2 by caspase-3 leads to production of lysophosphatidylcholine (LPC) , as well as PGE2, from dying cells [170]. LPCs, together with several other molecules such as sphingosine-1-phosphates (S1Ps) and the nucleotides ATP and UTP, recruit macrophages to engulf apoptotic cells [171]. Therefore, apoptotic caspases can promote tumourigenesis directly through AiP or indirectly through recruiting macrophages [128]. This is further discussed in Chap. 3.
4.6 Concluding Remarks
For many years, the apoptotic function of caspases has been considered, both in developmental settings and in a cancer setting, where activation of apoptotic proteins is considered to be essential in causing cell death and reducing tumour burden [8]. While these considerations of caspase function remain valid, increasing evidence suggests that non-apoptotic functions of the apoptotic caspases exist in a context-dependent manner. These functions are crucial in development and tissue homeostasis, where caspases have been implicated in stem cell pool maintenance by enhancing survival pathways and in AiP for tissue recovery upon cell loss, about which we have learned a lot from Drosophila models. Intriguingly, a wide range of non-apoptotic functions of caspases have been implicated in promoting tumour growth, metastasis and recurrence post-cytotoxic therapy (Fig. 4.3). It is therefore worthwhile to consider not only how to kill the tumour cells, but also how to prevent tumour spread and repopulation in cancer treatments. Further understanding of molecular mechanisms and cellular contexts leading to various non-apoptotic functions of caspases would certainly be beneficial.
References
Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–54.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–29.
Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–98.
Miura M. Active participation of cell death in development and organismal homeostasis. Dev Growth Differ. 2011;53:125–36.
Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–62.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6.
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–6.
Connolly PF, Jager R, Fearnhead HO. New roles for old enzymes: killer caspases as the engine of cell behavior changes. Front Physiol. 2014;5:149.
Kuranaga E. Caspase signaling in animal development. Dev Growth Differ. 2011;53:137–48.
Miura M. Apoptotic and nonapoptotic caspase functions in animal development. Cold Spring Harb Perspect Biol. 2012;4:a008664.
Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ. 2015;22:526–39.
Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol. 2008;9:378–90.
Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–58.
Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345:1250256.
Yamaguchi Y, Miura M. Programmed cell death in neurodevelopment. Dev Cell. 2015;32:478–90.
Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424.
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–22.
Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P. Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 2007;14:44–55.
Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015;25:308–15.
Eckhart L, Ballaun C, Hermann M, Vandeberg JL, Sipos W, Uthman A, Fischer H, Tschachler E. Identification of novel mammalian caspases reveals an important role of gene loss in shaping the human caspase repertoire. Mol Biol Evol. 2008;25:831–41.
Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–67.
Chowdhury I, Tharakan B, Bhat GK. Caspases—an update. Comp Biochem Physiol B Biochem Mol Biol. 2008;151:10–27.
Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43.
Yan N, Shi Y. Mechanisms of apoptosis through structural biology. Annu Rev Cell Dev Biol. 2005;21:35–56.
Lettre G, Hengartner MO. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol. 2006;7:97–108.
Conradt B, Xue D. Programmed cell death. WormBook; 2005. p. 1–13.
Kornbluth S, White K. Apoptosis in Drosophila: neither fish nor fowl (nor man, nor worm). J Cell Sci. 2005;118:1779–87.
Steller H. Regulation of apoptosis in Drosophila. Cell Death Differ. 2008;15:1132–8.
Goyal L, McCall K, Agapite J, Hartwieg E, Steller H. Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J. 2000;19:589–97.
Lisi S, Mazzon I, White K. Diverse domains of THREAD/DIAP1 are required to inhibit apoptosis induced by REAPER and HID in Drosophila. Genetics. 2000;154:669–78.
Wang SL, Hawkins CJ, Yoo SJ, Muller HA, Hay BA. The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell. 1999;98:453–63.
Meier P, Silke J, Leevers SJ, Evan GI. The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 2000;19:598–611.
Wilson R, Goyal L, Ditzel M, Zachariou A, Baker DA, Agapite J, Steller H, Meier P. The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat Cell Biol. 2002;4:445–50.
Hawkins CJ, Wang SL, Hay BA. A cloning method to identify caspases and their regulators in yeast: identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc Natl Acad Sci U S A. 1999;96:2885–90.
Yan N, Wu JW, Chai J, Li W, Shi Y. Molecular mechanisms of DrICE inhibition by DIAP1 and removal of inhibition by Reaper, Hid and Grim. Nat Struct Mol Biol. 2004;11:420–8.
Lee TV, Fan Y, Wang S, Srivastava M, Broemer M, Meier P, Bergmann A. Drosophila IAP1-mediated ubiquitylation controls activation of the initiator caspase DRONC independent of protein degradation. PLoS Genet. 2011;7, e1002261.
Ryoo HD, Bergmann A, Gonen H, Ciechanover A, Steller H. Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nat Cell Biol. 2002;4:432–8.
Yoo SJ, Huh JR, Muro I, Yu H, Wang L, Wang SL, Feldman RM, Clem RJ, Muller HA, Hay BA. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat Cell Biol. 2002;4:416–24.
Chai J, Shi Y. Apoptosome and inflammasome: conserved machineries for caspase activation. Natl Sci Rev. 2014;1:101–18.
Pang Y, Bai XC, Yan C, Hao Q, Chen Z, Wang JW, Scheres SH, Shi Y. Structure of the apoptosome: mechanistic insights into activation of an initiator caspase from Drosophila. Genes Dev. 2015;29:277–87.
Dorstyn L, Kumar S. A biochemical analysis of the activation of the Drosophila caspase DRONC. Cell Death Differ. 2008;15:461–70.
Hawkins CJ, Yoo SJ, Peterson EP, Wang SL, Vernooy SY, Hay BA. The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM. J Biol Chem. 2000;275:27084–93.
Snipas SJ, Drag M, Stennicke HR, Salvesen GS. Activation mechanism and substrate specificity of the Drosophila initiator caspase DRONC. Cell Death Differ. 2008;15:938–45.
Abdelwahid E, Yokokura T, Krieser RJ, Balasundaram S, Fowle WH, White K. Mitochondrial disruption in Drosophila apoptosis. Dev Cell. 2007;12:793–806.
Claveria C, Albar JP, Serrano A, Buesa JM, Barbero JL, Martinez AC, Torres M. Drosophila grim induces apoptosis in mammalian cells. EMBO J. 1998;17:7199–208.
Claveria C, Caminero E, Martinez AC, Campuzano S, Torres M. GH3, a novel proapoptotic domain in Drosophila Grim, promotes a mitochondrial death pathway. EMBO J. 2002;21:3327–36.
Freel CD, Richardson DA, Thomenius MJ, Gan EC, Horn SR, Olson MR, Kornbluth S. Mitochondrial localization of Reaper to promote inhibitors of apoptosis protein degradation conferred by GH3 domain-lipid interactions. J Biol Chem. 2008;283:367–79.
Haining WN, Carboy-Newcomb C, Wei CL, Steller H. The proapoptotic function of Drosophila Hid is conserved in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:4936–41.
Morishita J, Kang MJ, Fidelin K, Ryoo HD. CDK7 regulates the mitochondrial localization of a tail-anchored proapoptotic protein, Hid. Cell Rep. 2013;5:1481–8.
Olson MR, Holley CL, Gan EC, Colon-Ramos DA, Kaplan B, Kornbluth S. A GH3-like domain in reaper is required for mitochondrial localization and induction of IAP degradation. J Biol Chem. 2003;278:44758–68.
Thomenius M, Freel CD, Horn S, Krieser R, Abdelwahid E, Cannon R, Balasundaram S, White K, Kornbluth S. Mitochondrial fusion is regulated by Reaper to modulate Drosophila programmed cell death. Cell Death Differ. 2011;18:1640–50.
Brachmann CB, Jassim OW, Wachsmuth BD, Cagan RL. The Drosophila bcl-2 family member dBorg-1 functions in the apoptotic response to UV-irradiation. Curr Biol. 2000;10:547–50.
Colussi PA, Quinn LM, Huang DC, Coombe M, Read SH, Richardson H, Kumar S. Debcl, a proapoptotic Bcl-2 homologue, is a component of the Drosophila melanogaster cell death machinery. J Cell Biol. 2000;148:703–14.
Igaki T, Kanuka H, Inohara N, Sawamoto K, Nunez G, Okano H, Miura M. Drob-1, a Drosophila member of the Bcl-2/CED-9 family that promotes cell death. Proc Natl Acad Sci U S A. 2000;97:662–7.
Quinn L, Coombe M, Mills K, Daish T, Colussi P, Kumar S, Richardson H. Buffy, a Drosophila Bcl-2 protein, has anti-apoptotic and cell cycle inhibitory functions. EMBO J. 2003;22:3568–79.
Zhang H, Huang Q, Ke N, Matsuyama S, Hammock B, Godzik A, Reed JC. Drosophila pro-apoptotic Bcl-2/Bax homologue reveals evolutionary conservation of cell death mechanisms. J Biol Chem. 2000;275:27303–6.
Doumanis J, Dorstyn L, Kumar S. Molecular determinants of the subcellular localization of the Drosophila Bcl-2 homologues DEBCL and BUFFY. Cell Death Differ. 2007;14:907–15.
Gabriel B, Sureau F, Casselyn M, Teissie J, Petit PX. Retroactive pathway involving mitochondria in electroloaded cytochrome c-induced apoptosis. Protective properties of Bcl-2 and Bcl-XL. Exp Cell Res. 2003;289:195–210.
Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–37.
Xiong S, Mu T, Wang G, Jiang X. Mitochondria-mediated apoptosis in mammals. Protein Cell. 2014;5:737–49.
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–32.
Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–57.
Kumar S, Colussi PA. Prodomains—adaptors—oligomerization: the pursuit of caspase activation in apoptosis. Trends Biochem Sci. 1999;24:1–4.
Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–9.
Silke J, Verhagen AM, Ekert PG, Vaux DL. Sequence as well as functional similarity for DIABLO/Smac and Grim, Reaper and Hid? Cell Death Differ. 2000;7:1275.
Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–21.
Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. 2004;23:1627–35.
Larisch S, Yi Y, Lotan R, Kerner H, Eimerl S, Tony Parks W, Gottfried Y, Birkey Reffey S, De Caestecker MP, Danielpour D, Book-Melamed N, Timberg R, Duckett CS, Lechleider RJ, Steller H, Orly J, Kim SJ, Roberts AB. A novel mitochondrial septin-like protein, ARTS, mediates apoptosis dependent on its P-loop motif. Nat Cell Biol. 2000;2:915–21.
Edison N, Zuri D, Maniv I, Bornstein B, Lev T, Gottfried Y, Kemeny S, Garcia-Fernandez M, Kagan J, Larisch S. The IAP-antagonist ARTS initiates caspase activation upstream of cytochrome C and SMAC/Diablo. Cell Death Differ. 2012;19:356–68.
Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8.
Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci. 2005;118:265–7.
Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol. 1998;10:545–51.
Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol. 1998;8:1001–8.
Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 1997;16:2794–804.
Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–76.
Chang DW, Xing Z, Capacio VL, Peter ME, Yang X. Interdimer processing mechanism of procaspase-8 activation. EMBO J. 2003;22:4132–42.
Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23:1625–37.
Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, Miura M. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002;21:3009–18.
Moreno E, Yan M, Basler K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol. 2002;12:1263–8.
Andersen DS, Colombani J, Palmerini V, Chakrabandhu K, Boone E, Rothlisberger M, Toggweiler J, Basler K, Mapelli M, Hueber AO, Leopold P. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature. 2015;522:482–6.
Kanda H, Igaki T, Kanuka H, Yagi T, Miura M. Wengen, a member of the Drosophila tumor necrosis factor receptor superfamily, is required for Eiger signaling. J Biol Chem. 2002;277:28372–5.
Kanda H, Igaki T, Okano H, Miura M. Conserved metabolic energy production pathways govern Eiger/TNF-induced nonapoptotic cell death. Proc Natl Acad Sci U S A. 2011;108:18977–82.
Ma X, Huang J, Yang L, Yang Y, Li W, Xue L. NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res. 2012;22:425–31.
Shlevkov E, Morata G. A dp53/JNK-dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ. 2012;19:451–60.
Anto RJ, Mukhopadhyay A, Denning K, Aggarwal BB. Curcumin (diferuloylmethane) induces apoptosis through activation of caspase-8, BID cleavage and cytochrome c release: its suppression by ectopic expression of Bcl-2 and Bcl-xl. Carcinogenesis. 2002;23:143–50.
Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta. 2011;1813:558–63.
Tang D, Lahti JM, Kidd VJ. Caspase-8 activation and bid cleavage contribute to MCF7 cellular execution in a caspase-3-dependent manner during staurosporine-mediated apoptosis. J Biol Chem. 2000;275:9303–7.
Nonomura K, Yamaguchi Y, Hamachi M, Koike M, Uchiyama Y, Nakazato K, Mochizuki A, Sakaue-Sawano A, Miyawaki A, Yoshida H, Kuida K, Miura M. Local apoptosis modulates early mammalian brain development through the elimination of morphogen-producing cells. Dev Cell. 2013;27:621–34.
Monier B, Gettings M, Gay G, Mangeat T, Schott S, Guarner A, Suzanne M. Apico-basal forces exerted by apoptotic cells drive epithelium folding. Nature. 2015;518:245–8.
Kuipers D, Mehonic A, Kajita M, Peter L, Fujita Y, Duke T, Charras G, Gale JE. Epithelial repair is a two-stage process driven first by dying cells and then by their neighbours. J Cell Sci. 2014;127:1229–41.
Kang Y, Bashirullah A. A steroid-controlled global switch in sensitivity to apoptosis during Drosophila development. Dev Biol. 2014;386:34–41.
Fan Y, Bergmann A. Multiple mechanisms modulate distinct cellular susceptibilities toward apoptosis in the developing Drosophila eye. Dev Cell. 2014;30:48–60.
Hilgers V, Bushati N, Cohen SM. Drosophila microRNAs 263a/b confer robustness during development by protecting nascent sense organs from apoptosis. PLoS Biol. 2010;8, e1000396.
Pernaute B, Spruce T, Smith KM, Sanchez-Nieto JM, Manzanares M, Cobb B, Rodriguez TA. MicroRNAs control the apoptotic threshold in primed pluripotent stem cells through regulation of BIM. Genes Dev. 2014;28:1873–8.
Kuranaga E. Beyond apoptosis: caspase regulatory mechanisms and functions in vivo. Genes Cells. 2012;17:83–97.
Kuranaga E, Miura M. Nonapoptotic functions of caspases: caspases as regulatory molecules for immunity and cell-fate determination. Trends Cell Biol. 2007;17:135–44.
Haynie J, Bryant P. The effects of X-rays on the proliferation dynamics of cells in the imaginal wing disc of Drosophila melanogaster. Wilhelm Rouxs Arch Dev Biol. 1977;183:85–100.
Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, Li CY. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal. 2010;3:ra13.
Chera S, Ghila L, Dobretz K, Wenger Y, Bauer C, Buzgariu W, Martinou JC, Galliot B. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell. 2009;17:279–89.
Fan Y, Bergmann A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell. 2008;14:399–410.
Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol. 2004;14:1262–6.
Kondo S, Senoo-Matsuda N, Hiromi Y, Miura M. DRONC coordinates cell death and compensatory proliferation. Mol Cell Biol. 2006;26:7258–68.
Bergmann A, Steller H. Apoptosis, stem cells, and tissue regeneration. Sci Signal. 2010;3:re8.
Fan Y, Bergmann A. Apoptosis-induced compensatory proliferation. The cell is dead. Long live the cell! Trends Cell Biol. 2008;18:467–73.
Mollereau B, Perez-Garijo A, Bergmann A, Miura M, Gerlitz O, Ryoo HD, Steller H, Morata G. Compensatory proliferation and apoptosis-induced proliferation: a need for clarification. Cell Death Differ. 2013;20:181.
Ryoo HD, Bergmann A. The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb Perspect Biol. 2012;4:a008797.
Fan Y, Wang S, Hernandez J, Yenigun VB, Hertlein G, Fogarty CE, Lindblad JL, Bergmann A. Genetic models of apoptosis-induced proliferation decipher activation of JNK and identify a requirement of EGFR signaling for tissue regenerative responses in Drosophila. PLoS Genet. 2014;10, e1004131.
Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131:5591–8.
Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501.
Wells BS, Yoshida E, Johnston LA. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol. 2006;16:1606–15.
Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–9.
Perez-Garijo A, Shlevkov E, Morata G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development. 2009;136:1169–77.
Bergantinos C, Corominas M, Serras F. Cell death-induced regeneration in wing imaginal discs requires JNK signalling. Development. 2010;137:1169–79.
Martin FA, Perez-Garijo A, Morata G. Apoptosis in Drosophila: compensatory proliferation and undead cells. Int J Dev Biol. 2009;53:1341–7.
Smith-Bolton RK, Worley MI, Kanda H, Hariharan IK. Regenerative growth in Drosophila imaginal discs is regulated by Wingless and Myc. Dev Cell. 2009;16:797–809.
Sun G, Irvine KD. Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev Biol. 2011;350:139–51.
Sun G, Irvine KD. Ajuba family proteins link JNK to Hippo signaling. Sci Signal. 2013;6:81.
Tseng AS, Adams DS, Qiu D, Koustubhan P, Levin M. Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev Biol. 2007;301:62–9.
Pellettieri J, Fitzgerald P, Watanabe S, Mancuso J, Green DR, Sanchez Alvarado A. Cell death and tissue remodeling in planarian regeneration. Dev Biol. 2010;338:76–85.
Pellettieri J, Sanchez Alvarado A. Cell turnover and adult tissue homeostasis: from humans to planarians. Annu Rev Genet. 2007;41:83–105.
Vlaskalin T, Wong CJ, Tsilfidis C. Growth and apoptosis during larval forelimb development and adult forelimb regeneration in the newt (Notophthalmus viridescens). Dev Genes Evol. 2004;214:423–31.
Jung Y, Witek RP, Syn WK, Choi SS, Omenetti A, Premont R, Guy CD, Diehl AM. Signals from dying hepatocytes trigger growth of liver progenitors. Gut. 2010;59:655–65.
Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, Zon LI. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136–47.
Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–10.
Jager R, Fearnhead HO. “Dead cells talking”: the silent form of cell death is not so quiet. Biochem Res Int. 2012;2012:453838.
Moon RT, Kohn AD, de Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701.
van Raam BJ, Salvesen GS. Proliferative versus apoptotic functions of caspase-8 Hetero or homo: the caspase-8 dimer controls cell fate. Biochim Biophys Acta. 2012;1824:113–22.
Beisner DR, Chen IL, Kolla RV, Hoffmann A, Hedrick SM. Cutting edge: innate immunity conferred by B cells is regulated by caspase-8. J Immunol. 2005;175:3469–73.
Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY, Berry DM, Tamblyn L, Shehabeldin A, Migon E, Wakeham A, Bouchard D, Yeh WC, Mcglade JC, Ohashi PS, Hakem R. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17:883–95.
Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J, Salmena L, Hakem R, Straus S, Lenardo M. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–8.
Oberst A, Green DR. It cuts both ways: reconciling the dual roles of caspase 8 in cell death and survival. Nat Rev Mol Cell Biol. 2011;12:757–63.
Zhou XD, Yu JP, Liu J, Luo HS, Chen HX, Yu HG. Overexpression of cellular FLICE-inhibitory protein (FLIP) in gastric adenocarcinoma. Clin Sci (Lond). 2004;106:397–405.
Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, Los M. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46:497–510.
Ili CG, Brebi P, Tapia O, Sandoval A, Lopez J, Garcia P, Leal P, Sidransky D, Guerrero-Preston R, Roa JC. Cellular FLICE-like inhibitory protein long form (c-FLIPL) overexpression is related to cervical cancer progression. Int J Gynecol Pathol. 2013;32:316–22.
Sharp DA, Lawrence DA, Ashkenazi A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J Biol Chem. 2005;280:19401–9.
Wang W, Wang S, Song X, Sima N, Xu X, Luo A, Chen G, Deng D, Xu Q, Meng L, Lu Y, Ma D. The relationship between c-FLIP expression and human papillomavirus E2 gene disruption in cervical carcinogenesis. Gynecol Oncol. 2007;105:571–7.
Hashimoto T, Kikkawa U, Kamada S. Contribution of caspase(s) to the cell cycle regulation at mitotic phase. PLoS One. 2011;6, e18449.
Hashimoto T, Yamauchi L, Hunter T, Kikkawa U, Kamada S. Possible involvement of caspase-7 in cell cycle progression at mitosis. Genes Cells. 2008;13:609–21.
Eymin B, Sordet O, Droin N, Munsch B, Haugg M, van de Craen M, Vandenabeele P, Solary E. Caspase-induced proteolysis of the cyclin-dependent kinase inhibitor p27Kip1 mediates its anti-apoptotic activity. Oncogene. 1999;18:4839–47.
Khalil H, Peltzer N, Walicki J, Yang JY, Dubuis G, Gardiol N, Held W, Bigliardi P, Marsland B, Liaudet L, Widmann C. Caspase-3 protects stressed organs against cell death. Mol Cell Biol. 2012;32:4523–33.
Yang JY, Michod D, Walicki J, Murphy BM, Kasibhatla S, Martin SJ, Widmann C. Partial cleavage of RasGAP by caspases is required for cell survival in mild stress conditions. Mol Cell Biol. 2004;24:10425–36.
Yang JY, Widmann C. The RasGAP N-terminal fragment generated by caspase cleavage protects cells in a Ras/PI3K/Akt-dependent manner that does not rely on NFkappa B activation. J Biol Chem. 2002;277:14641–6.
Geiger TR, Peeper DS. Metastasis mechanisms. Biochim Biophys Acta. 2009;1796:293–308.
Geisbrecht ER, Montell DJ. A role for Drosophila IAP1-mediated caspase inhibition in Rac-dependent cell migration. Cell. 2004;118:111–25.
Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, Ramakrishnan P, Lapidot T, Wallach D. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol. 2004;173:2976–84.
Portela M, Richardson HE. Death takes a holiday—non-apoptotic role for caspases in cell migration and invasion. EMBO Rep. 2013;14:107–8.
Zhao X, Wang D, Zhao Z, Xiao Y, Sengupta S, Xiao Y, Zhang R, Lauber K, Wesselborg S, Feng L, Rose TM, Shen Y, Zhang J, Prestwich G, Xu Y. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J Biol Chem. 2006;281:29357–68.
Brown M, Roulson JA, Hart CA, Tawadros T, Clarke NW. Arachidonic acid induction of Rho-mediated transendothelial migration in prostate cancer. Br J Cancer. 2014;110:2099–108.
Cheng YJ, Lee CH, Lin YP, Huang JY, Su CC, Chang WT, Yang BC. Caspase-3 enhances lung metastasis and cell migration in a protease-independent mechanism through the ERK pathway. Int J Cancer. 2008;123:1278–85.
Rudrapatna VA, Bangi E, Cagan RL. Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Rep. 2013;14:172–7.
Na HJ, Hwang JY, Lee KS, Choi YK, Choe J, Kim JY, Moon HE, Kim KW, Koh GY, Lee H, Jeoung D, Won MH, Ha KS, Kwon YG, Kim YM. TRAIL negatively regulates VEGF-induced angiogenesis via caspase-8-mediated enzymatic and non-enzymatic functions. Angiogenesis. 2014;17:179–94.
Li F, He Z, Shen J, Huang Q, Li W, Liu X, He Y, Wolf F, Li CY. Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell. 2010;7:508–20.
Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–14.
Kim RJ, Nam JS. OCT4 expression enhances features of cancer stem cells in a mouse model of breast cancer. Lab Anim Res. 2011;27:147–52.
Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–811.
Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–807.
Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O’Sullivan B, He Z, Peng Y, Tan AC, Zhou L, Shen J, Han G, Wang XJ, Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS, Li CY. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17:860–6.
Donato AL, Huang Q, Liu X, Li F, Zimmerman MA, Li CY. Caspase 3 promotes surviving melanoma tumor cell growth after cytotoxic therapy. J Invest Dermatol. 2014;134:1686–92.
Nakopoulou L, Alexandrou P, Stefanaki K, Panayotopoulou E, Lazaris AC, Davaris PS. Immunohistochemical expression of caspase-3 as an adverse indicator of the clinical outcome in human breast cancer. Pathobiology. 2001;69:266–73.
Hu Q, Peng J, Liu W, He X, Cui L, Chen X, Yang M, Liu H, Liu S, Wang H. Elevated cleaved caspase-3 is associated with shortened overall survival in several cancer types. Int J Clin Exp Pathol. 2014;7:5057–70.
Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J, Egle A, Villunger A. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev. 2010;24:1602–7.
Michalak EM, Vandenberg CJ, Delbridge AR, Wu L, Scott CL, Adams JM, Strasser A. Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev. 2010;24:1608–13.
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–5.
Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, Bast MA, Rosenwald A, Muller-Hermelink HK, Rimsza LM, Campo E, Delabie J, Braziel RM, Cook JR, Tubbs RR, Jaffe ES, Lenz G, Connors JM, Staudt LM, Chan WC, Gascoyne RD. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362:875–85.
Willems JJ, Arnold BP, Gregory CD. Sinister self-sacrifice: the contribution of apoptosis to malignancy. Front Immunol. 2014;5:299.
Ford CA, Petrova S, Pound JD, Voss JJ, Melville L, Paterson M, Farnworth SL, Gallimore AM, Cuff S, Wheadon H, Dobbin E, Ogden CA, Dumitriu IE, Dunbar DR, Murray PG, Ruckerl D, Allen JE, Hume DA, van Rooijen N, Goodlad JR, Freeman TC, Gregory CD. Oncogenic properties of apoptotic tumor cells in aggressive B cell lymphoma. Curr Biol. 2015;25:577–88.
Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–30.
Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–55.
Acknowledgments
We apologise to our colleagues for omitting many relevant publications owing to space limitations. This work was supported by Marie Curie Career Integration Grant (CIG) 630846 from the European Union’s Seventh Framework Programme (FP7) and the Birmingham Fellowship, University of Birmingham, UK.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Dabrowska, C., Li, M., Fan, Y. (2016). Apoptotic Caspases in Promoting Cancer: Implications from Their Roles in Development and Tissue Homeostasis. In: Gregory, C. (eds) Apoptosis in Cancer Pathogenesis and Anti-cancer Therapy. Advances in Experimental Medicine and Biology, vol 930. Springer, Cham. https://doi.org/10.1007/978-3-319-39406-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-39406-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39404-6
Online ISBN: 978-3-319-39406-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)