
Abstract
ABCG2 is an ATP-binding cassette (ABC) half-transporter localized to the cell membrane and important in normal physiology, in normal tissue protection including in the maternal-fetal barrier and the blood brain barrier, and potentially in multidrug resistance. Dimerization is required for function. Polymorphic variants have been described that impair trafficking to the cell surface, and one, Q141K, has been associated with a higher incidence of gout in individuals who are carriers. Working in concert with ABCB1 (P-glycoprotein), ABCG2 is expressed at high levels in the vascular endothelium of the brain and pumps xenobiotics and chemotherapeutics back into the bloodstream. ABCG2 expression is also found at high levels in some cancer types including pancreatic and liver cancers. Whether its expression in these tumor types contributes to drug resistance via drug efflux and reduction in drug accumulation is a question that remains unanswered. Studies of drug accumulation, such as through imaging solid tumors in patients, are needed to answer this question.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Breast Cancer Resistance Protein
- Brain Endothelial Cell
- Brain Capillary Endothelial Cell
- ABCG2 Expression
- ABCG2 Protein
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction to ABCG2
The human ATP-binding cassette (ABC) transporters are transmembrane efflux transporters that belong to a large superfamily of 48 members including ABCG2, or human breast cancer resistance protein (BCRP), encoded by the ABCG2 gene located on chromosome 4q22.
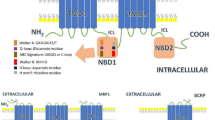
ABCG2 is a 655 amino acid, 72 kDa protein consisting of an N-terminal nucleotide-binding domain (NBD) containing an ABC signature motif (ALSGGQ) and a C-terminal transmembrane domain (TMD) with six hydrophobic segments and an extracellular loop between TM5 and TM6. Its structure is in reverse configuration to most other ABC transporters containing one or two NBDs that are C-terminal to the TMDs. ABCG2 is a half-transporter (one NBD and one TMD), requiring at least two NBDs to function as an efflux pump; thus, functional ABCG2 exists as either homodimers or homomultimers (Bhatia et al. 2005; Doyle and Ross 2003; Kage et al. 2002; Nakanishi et al. 2003; Xu et al. 2004). Homodimer formation involves a disulfide bridge linkage at cysteine 603 located in the extracellular loop (Henriksen et al. 2005; Wakabayashi et al. 2007). Crystallization studies reveal a tetrameric complex comprised of four homodimers (McDevitt et al. 2006) and other studies have shown that it can also form higher oligomeric states (Xie et al. 2008; Xu et al. 2007). It is speculated that ABCG2 dimerizes in the endoplasmic reticulum (ER), exits to the Golgi apparatus for posttranslational processing, and is finally then trafficked to the apical membrane of the cell (Graf et al. 2003).
This chapter is about ABCG2—its expression in normal tissues, substrate/inhibitor specificity, role in cancer multidrug resistance, and particularly its role in the blood–brain barrier (BBB). Understanding how genetic variations affect transporter function or substrate and inhibitor specificity and drug–transporter interactions at the BBB will shed light on improving drug delivery to the brain. The contribution of ABCG2 to multidrug resistance in the tumor vasculature versus the BBB will be addressed followed by a brief discussion on prospects for transporter inhibition at the BBB.
Tissue Expression
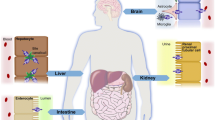
Since ABCG2 was first discovered in drug-resistant human cancer cells, subsequent studies focused on its pattern of expression in various normal tissues, where ABCG2 was found to be involved in the absorption, distribution, metabolism, and elimination of xenobiotics and endogenous chemicals. The location and level of expression of ABCG2 highlighted major roles for ABCG2 in forming the maternal–fetal barrier, the blood–testis cell barrier, and the blood–brain barrier; controlling the absorption and efflux of xenobiotics and endogenous metabolic products within the gastrointestinal tract; and more recently its role in urate transport in the kidney.
Placenta and Mammary Gland
The highest levels of ABCG2 expression are found in placental tissue, specifically in the placental syncytiotrophoblast at the apical surface of the chorionic villi. Due to this specific localization, it is thought that ABCG2 helps form the barrier between the maternal and fetal circulation systems, and thus protects the fetus from endogenous and exogenous toxins (Fetsch et al. 2006). It is also possible that ABCG2 transports steroid hormones produced in the placenta (Doyle and Ross 2003), since the major estrogens produced and secreted by the placenta are ABCG2 substrates (Suzuki et al. 2003).
While ABCG2 is expressed at low levels in the non-lactating adult breast, its expression is elevated in lactating mammary tissue, localized in the lobules and lactiferous ducts of the mammary gland, as well as in venous/capillary endothelium (Maliepaard et al. 2001; Faneyte et al. 2002). As opposed to the protective role ABCG2 may play in the placenta, the functional role of ABCG2 here would be to concentrate substrates such as vitamins—as has been demonstrated for riboflavin—or other essential factors into breast milk; however, enrichment of toxins in breast milk has also been demonstrated (Jonker et al. 2005; van Herwaarden et al. 2006, 2007).
Testis
High expression of ABCG2 has been reported in normal testis tissue, where it is expressed in the Sertoli–Leydig cells, in the myoid cell layer, as well as in endothelial cells (Fetsch et al. 2006; Bart et al. 2004). Due to its localization on the luminal side of the endothelium as well as the apical side of myoid cells, it probably transports substrates out of the seminiferous tubules, implying a potential role in protecting the germ cells.
Blood–Brain Barrier
ABCG2 was found to be expressed on the luminal surface of brain microvessel endothelium, suggesting a role in the blood–brain barrier (Cooray et al. 2002). Since then, numerous researchers have explored the role of ABCG2 in forming the blood–brain barrier (BBB), predominantly in murine models. ABCG2 was found to be expressed along with P-gp (ABCB1) in the microvasculature of both normal and malignant tissues from the central nervous system, but their relative functional contribution still needs to be determined. Not only do studies indicate that ABCG2 plays a role in the efflux of toxic xenobiotics in the BBB, but it also appears that it could play a role in efflux of endogenous substrates, since its activity in the transport of efflux of amyloid-β peptide was recently reported (Do et al. 2012; Tai et al. 2009).
Liver and the Gastrointestinal Tract
When Taipalensuu et al. (2001) examined jejunal mucosa from 13 normal volunteers, and compared expression levels of 10 drug efflux transporters from the ABC family, the highest level detected was for the mRNA encoding ABCG2. Other studies indicated that ABCG2 mRNA was detected at high levels in the duodenum, decreasing along the gastrointestinal tract, and reaching lowest levels in the rectum (Gutmann et al. 2005; Hilgendorf et al. 2007). ABCG2 was localized by immunohistochemistry at the apical membrane of the small intestine, the colon, and the liver bile canaliculi. A recent quantification of intestinal ABCG2 from 14 human donors indicates that the average BCRP density in the human duodenum is 305 ± 248 fmol/cm2 and it was found at 2.6 pmol per gram in the liver (Tucker et al. 2012).
The presence of ABCG2 in the apical membrane of the small intestine and bile canniculi implies a role for the protein in xenobiotic and endogenous substrate efflux from the liver and substrate absorption in the GI tract (Adachi et al. 2005). Another role for ABCG2 in the intestine is in the secretion of endogenous uric acid, even though renal excretion is the most important factor in uric acid elimination (Hosomi et al. 2012).
Kidneys
BCRP is expressed at the apical membrane of proximal tubules, suggesting a role for BCRP in renal excretion of xenobiotics (Fetsch et al. 2006; Huls et al. 2008). In 2008, a genome-wide association study (GWAS) reported that a common single nucleotide polymorphism of ABCG2 was linked to elevated blood urate level and gout (Dehghan et al. 2008). Further studies revealed that ABCG2 was indeed involved in excretion of urate in urine, again highlighting the importance of ABCG2 in the transport of endogenous compounds (Woodward et al. 2009).
Other Sites
BCRP is expressed in a wide variety of stem cells including those of hematopoietic origin and is the molecular determinant of the side population (SP) phenotype (Zhou et al. 2001; Scharenberg et al. 2002). SP cells have been identified by their ability to effectively exclude the fluorescent vital dye Hoechst 33342 and form a distinct population, definable by flow cytometry (Goodell et al. 1996).
ABCG2 mRNA expression was reported in multiple other tissues (see Fig. 1), and various studies confirmed ABCG2 protein expression in alveolar pneumocytes, sebaceous glands, transitional epithelium of the bladder, prostate epithelium, uterine endocervical cells, cervical squamous epithelia, small and large intestinal mucosa/epithelial cells, pancreatic islet and acinar cells, zona reticularis layer of the adrenal gland, gall bladder, conjunctival epithelium, retinal pigment epithelium, spinal cord, and hepatocytes (Fetsch et al. 2006; Maliepaard et al. 2001; Aust et al. 2004; Chen et al. 2013).

Log2 median-centered ABCG2 expression data obtained from analysis of 502 normal tissues, source GEO accession number GSE7307 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE7307). Selected samples were grouped by organ and median expression per organ was calculated
Polymorphisms and ABCG2 Null Humans
Structural and functional studies reveal that ABCG2 transporter activity is affected by both posttranslational modifications and genetic polymorphisms. During its transit through the endoplasmic reticulum–Golgi pathway to the plasma membrane, ABCG2 undergoes N-linked glycosylation at asparagine 596 (Diop and Hrycyna 2005) and formation of an intramolecular disulfide bond between C592 and C608, where mutating these cysteine residues results in impaired localization and function (Henriksen et al. 2005; Wakabayashi et al. 2007). The oligomeric glycosylated ABCG2 undergoes lysosomal degradation whereas the underglycosylated, misfolded protein lacking the intramolecular disulfide bond is degraded via the proteasome (Wakabayashi et al. 2007; Nakagawa et al. 2009; Wakabayashi-Nakao et al. 2009). Certain non-synonymous single nucleotide polymorphisms (SNPs), such as Q141 K, F208S, and S441 N, affect ABCG2 protein stability resulting in enhanced ubiquitin-mediated proteasomal degradation (Furukawa et al. 2009; Nakagawa et al. 2008). Moreover, Q141 K and F208S variants are found sequestered in the aggresome followed by subsequent degradation via autophagy (Basseville et al. 2012; Wakabayashi-Nakao et al. 2010). In addition, mutational studies of ABCG2 identified a GXXXG motif (residues 406–410), responsible for dimerization, where mutation of the glycines to leucines affected the protein’s function (Polgar et al. 2004). Mutational analysis of threonine 402, three residues from the GXXXG motif, reveals a role for this residue in modulating protein folding and processing as well as dimerization (Polgar et al. 2010). Furthermore, functional variations in ABCG2 have been shown to affect substrate binding and specificity. Mutating Arg482 and Pro485, located in the drug-binding pocket within TM3, can affect the efflux of some substrates (Ni et al. 2011).
More than 80 single nucleotide polymorphisms in the ABCG2 gene have been identified across ethnically diverse subpopulations. Among them, Q141K (421C > A, rs2231142) and V12M (34G > A, rs2231137) occur most frequently in Asians (~25–35 %) and with lower frequencies in Caucasians (~10 %), and African-American populations (~<5 %), while the remaining SNPs have allelic frequencies of less than 2 % (Backstrom et al. 2003; de Jong et al. 2004; Iida et al. 2002; Imai et al. 2002; Kobayashi et al. 2005; Kondo et al. 2004; Zamber et al. 2003). Functional studies revealed that the Q141K and D620N mutations decrease ABCG2 activity, while V12M, I206L, and N590Y do not appear to alter protein function (Morisaki et al. 2005; Vethanayagam et al. 2005). Other non-synonymous SNPs, F208S (623T > C, rs1061018) and S441 N (1321G > A), lead to reduced protein stability by enhancing proteasomal proteolysis (Furukawa et al. 2009). In fact, S441N displayed lower protein expression and impaired membrane localization (Kondo et al. 2004). Moreover, an in vitro study found 6 of the 18 ABCG2 variants to exhibit defective or impaired transport of the substrates hematoporphyrin or methotrexate: Q126stop (376C > T, rs72552713); F208S; S248P (742T > C, rs3116448); E334stop (1000G > T, rs3201997); S441 N; and F489L (1465T > C) (Tamura et al. 2006). Additional SNPs reported in the ABCG2 promoter region, −15622C > T and −1379A > G, may be involved in transcriptional control of the gene (Noguchi et al. 2014). The nonsense mutation Q126X (376C > T, rs72552713) introduces a stop codon resulting in a non-functional transporter (Matsuo et al. 2009).
Q141K and Drug Disposition/Clinical Outcome
Pharmacogenetics studies of ABCG2 have mainly focused on the Q141K variant. Q141K is associated with reduced protein expression (Imai et al. 2002; Morisaki et al. 2005) possibly due to increased ubiquitin-mediated proteasomal degradation (Furukawa et al. 2009), as well as reduced protein function. The SNP is located in the ATP-binding region between the Walker A and B motifs of ABCG2 and has also been associated with reduced ATPase activity, impaired membrane localization, and instability in the nucleotide-binding domain (Imai et al. 2002; Kondo et al. 2004; Mizuarai et al. 2004; Woodward et al. 2013). These findings indicate that this SNP may result in altered transporter function and have important implications for regulating drug disposition. However, studies evaluating the clinical relevance of this SNP have been inconsistent largely due to the small sample size of the study populations. Patients with the variant genotype (AA or CA) had significantly higher plasma AUC or Cmax levels for statins (Keskitalo et al. 2009a, b; Lee et al. 2013; Zhang et al. 2006; Zhou et al. 2013), TKIs (sunitinib Mizuno et al. 2012 and imatinib Takahashi et al. 2010), sulfasalazine (Urquhart et al. 2008; Yamasaki et al. 2008) as well as the camptothecin derivatives diflomotecan (Sparreboom et al. 2004) and 9-aminocamptothecin (Zamboni et al. 2006). However, no significant effects of the Q141 K variant on pharmacokinetic parameters were observed for irinotecan (de Jong et al. 2004; Han et al. 2007; Jada et al. 2007), docetaxel (Chew et al. 2011), or nitrofurantoin (Adkison et al. 2008). Furthermore, this SNP has also been linked with increased drug-induced diarrhea for Korean patients with diffuse large B-cell lymphoma receiving rituximab plus cyclophosphamide/doxorubicin/vincristine/prednisone (Kim et al. 2008) or patients on gefitinib therapy (Cusatis et al. 2006). The 421AA genotype correlated with risk of grade 3 or grade 4 thrombocytopenia and neutropenia in Korean patients with metastatic renal cell carcinoma on sunitinib therapy (Kim et al. 2013).
The association of Q141 K with clinical outcome has also been mixed with a study demonstrating longer progression free survival for the variant allele in advanced ovarian cancer patients (Tian et al. 2012) and other studies showing poor outcome for patients with acute myeloid leukemia being treated with idarubicin-based therapy (Tiribelli et al. 2013) or lung cancer patients on a platinum-based regimen (Muller et al. 2009).
The other common ABCG2 SNP, 34G > A, has not been reported to significantly affect drug pharmacokinetics. The ABCG2 (−15622C/T) polymorphism and the ABCG2 (1143C/T, −15622C/T) haplotype were associated with gefitinib-induced moderate-to-severe diarrhea, but their functional impact is still unknown (Lemos et al. 2011). Other genetic polymorphisms have been identified in the coding region of ABCG2 but their clinical relevance remains to be determined (Noguchi et al. 2014; Zhao et al. 2009).
ABCG2 Null Alleles
ABCG2 was identified as the genetic basis of a blood group system named Junior, Jr or Jr(a), such that null alleles of ABCG2 cause the Jr(a−) blood type. Individuals who do not express Jr(a) antigen are rare and are mainly found in Japanese and other Asian populations. SNP analysis on genomic DNA from 6 Jr(a−) subjects led to the identification of three nonsense mutations (Q126X, Q246X, and R236X) where all three SNPs resulted in a premature stop codon in the ATP-binding domain of ABCG2 (Zelinski et al. 2012). A separate study also sequenced ABCG2 in 18 Jr(a−) individuals and found eight mutations: three nonsense mutations described above (Q126X, R236X, and Q246X) and five frameshift mutations (I63Yfs, F182Vfs, L264Hfs, F293Lfs, and T371Lfs) (Saison et al. 2012). Individuals were either homozygous for a single ABCG2 mutation or heterozygous for two mutations. Both studies concluded that the ABCG2 mutations identified in the Jr(a−) individuals correspond to null alleles of ABCG2 and are responsible for the Jr(a−) blood type. Ongoing studies have continued to identify additional new null alleles in Jr(a−) patients (Hue-Roye et al. 2013a, b; Ogasawara et al. 2014; Tanaka et al. 2014) and the functional significance is still unknown.
Substrates and Inhibitors of ABCG2
Substrates
The ABCG2 substrate spectrum has increased exponentially over the years to include physiological compounds, cancer and non-cancer therapeutics, molecularly targeted drugs, and common dietary xenobiotics. ABCG2 is involved in the physiologic efflux of multiple endogenous substrates that include conjugated organic anions (Mao and Unadkat 2015; Polgar et al. 2008); porphyrin/heme (Jonker et al. 2002; Krishnamurthy et al. 2004); folates (Chen et al. 2003); and urate (Woodward et al. 2009) as well as amyloid-β peptides across the BBB (Xiong et al. 2009).
Since it was originally discovered in drug-resistant cells, the first compounds reported as (exogenous) substrates of ABCG2 were chemotherapeutics that include mitoxantrone (Doyle et al. 1998), anthracyclines, methotrexate, camptothecin derivatives, and flavopiridol as well as tyrosine kinase inhibitors (TKIs) such as imatinib and gefitinib (Mao and Unadkat 2015; Polgar et al. 2008). Drug-selected cell lines that express mutant forms of ABCG2 (R482G and R482T) were found to modify substrate selectivity resulting in enhanced efflux of rhodamine 123 and increased resistance to anthracyclines (Chen et al. 2003; Allen et al. 2002a; Honjo et al. 2001). Subsequent site-directed mutagenesis studies at amino acid 482 further demonstrated its importance in determining substrate recognition and transport (Miwa et al. 2003; Ozvegy-Laczka et al. 2005; Robey et al. 2003). However, it should be noted that mutations of Arg482 have never been identified in clinical samples to date.
Other substrate classes have been identified and include certain antibiotics (fluoroquinolones and erythromycin), antiretrovirals (nucleoside analogs, zidovudine and abacavir), carcinogens, flavonoids (genistein and quercetin), and HMG-CoA reductase inhibitors (Mao and Unadkat 2015; Polgar et al. 2008). Fluorescent probes have been reported to be ABCG2 substrates and include Hoechst 33342, BODIPY-prazosin, LysoTracker Green, and pheophorbide A (Litman et al. 2000; Robey et al. 2004). The transport of photosensitizers by ABCG2 further implicates this transporter as a possible cause of cellular resistance to photodynamic therapy (Robey et al. 2005). More recently, D-luciferin was identified as a specific ABCG2 substrate that can be used as a probe for (bioluminescent) imaging of ABCG2 function at the BBB in vivo (Bakhsheshian et al. 2013a).
ABCG2 has a broad substrate specificity that is distinct, but substantially overlaps, with that of P-gp and multidrug resistance protein (MRP1). Although the number of ABCG2 substrates exceeds 200, few studies have been conducted to determine structure–activity relationship (SAR) for prediction of ABCG2 substrates. One SAR study conducted with camptothecin analogs showed that ABCG2 transports compounds with high polarity at carbon positions 10 and 11 (Yoshikawa et al. 2004). A recent study by Hazai et al. (2013) developed a predictive model for wild-type ABCG2 substrates, using a support vector machine (SVM) method based on 263 known BCRP substrates and non-substrates, with an overall prediction accuracy of ~76 % for identifying potential novel substrates.
Inhibitors
The first ABCG2 specific inhibitor identified was the diketopiperazine fumitremorgin C (FTC) isolated from the fungi Aspergillus fumigatus (Rabindran et al. 1998). FTC was reported to inhibit mitoxantrone resistance in drug-selected cell lines even before ABCG2 was cloned, but had no effect on resistance mediated by P-gp or MRP1 (Rabindran et al. 1998). Because of its neurotoxic effects, FTC analogs (Kol43) were soon developed with more potent inhibitory activity and high selectivity for ABCG2 and low neurotoxicity (Allen et al. 2002b; van Loevezijn et al. 2001).
Many ABCG2 inhibitors are not selective and can inhibit other ABC transporters, such as the potent dual P-gp/ABCG2 inhibitors elacridar (de Bruin et al. 1999) and tariquidar (Robey et al. 2004) as well as the growing list of TKIs (Durmus et al. 2015). Additional classes of inhibitors include flavonoids, steroids, nucleoside analogs, and immunosuppressants (tacrolimus, cyclosporine A). Chromone derivatives and tariquidar-like structures are considered the most potent and selective inhibitors of ABCG2, inducing high inhibition of ABCG2 with low effect on ABCC1 and ABCB1 (Lecerf-Schmidt et al. 2013). Given the promiscuous nature of ABCG2, the list of inhibitors (and substrates) will continue to expand in the coming years.
The mechanism of inhibition divides ABCG2 inhibitors into two groups: “general” inhibitors that inhibit ATPase activity of the transporter such as FTC and Ko143; and “substrate-dependent” inhibitors that are ABCG2 substrates and thus act as competitive inhibitors. The latter group results in substrate-dependent inhibition where the inhibitor may interact with ABCG2 on (i) binding sites of one class of substrates but not others, or (ii) on allosteric sites to induce conformational changes in the large binding pocket that subsequently affects transport of certain substrates. For example, nelfinavir has been shown to inhibit the efflux of zidovudine and abacavir with no effect on efflux of prazosin and imatinib, suggesting that the nucleoside analogs possibly bind to ABCG2 at sites that do not overlap with those for prazosin or imatinib (Giri et al. 2009). These findings support the notion that substrates interact with ABCG2 at multiple binding regions in the protein. For this reason, predicting and screening for ABCG2 inhibitors remains a challenge in drug discovery.
Role of ABCG2 in Cancer Multidrug Resistance
The story of ABCG2 in multidrug resistance is inextricably linked with that of P-gp/ABCB1. Discovered in 1976 in drug-resistant Chinese hamster ovary cells, the therapeutic potential was quickly recognized when verapamil was found to overcome resistance by blocking drug efflux (Tsuruo et al. 1981; Juliano and Ling 1976). Other inhibitors were rapidly discovered and the idea was translated to the clinic with the first trial result reported in 1987, a trial combining verapamil with adriamycin in ovarian cancer (Ozols et al. 1987) relying on data showing that high levels of resistance due to P-gp-mediated efflux could be overcome by addition of P-gp inhibitors. For over a decade, many publications began with some variation of ‘drug resistance is a major problem in the treatment of cancer’, and then went on to report the detection of P-gp expression in a new tumor type or the identification of a new inhibitor to overcome resistance. Eventually it became apparent that there were drug-resistant cell lines with high levels of drug efflux but in which P-gp expression was not detectable. MRP was discovered as a mediator of drug efflux in some of those cell lines (Schneider et al. 1994), and then ABCG2 (Ross et al. 1999). However, because the discovery of P-gp preceded those of MRP and ABCG2 by two decades, it was translated to the clinic on its own.
From the first clinical trial in 1987, a series of studies testing a variety of P-gp substrates and inhibitors in patients with a variety of tumor types was carried out. Although many trials were conducted with off-the-shelf agents developed for another purpose, a number of later P-gp inhibitors were developed specifically for that purpose. In sum, a handful of trials had a statistically significant positive outcome, but most of the studies were unconvincing and did not support a role for overcoming resistance by adding a P-gp inhibitor. In retrospect, there were mistakes in the development that may have obscured a positive signal. These included a delay in the conduct of randomized trials, failure to selectively enroll only patients whose tumors had detectable P-gp expression prior to enrollment, and utilization of inhibitors that also altered the pharmacokinetics of the anticancer drug in the combination such that lower drug doses had to be used (Amiri-Kordestani et al. 2012; Shaffer et al. 2012). A large number of trials were conducted with inconclusive results. Two randomized trials were launched in 2002—with tariquidar, a very potent P-gp inhibitor, combined with vinorelbine or paclitaxel plus carboplatin in non-small cell lung cancer. The trials were conducted at about 100 centers around the U.S., and were closed within a year for toxicity in the experimental arm, with only 304 of an expected 1000 patients enrolled (Fox and Bates 2007). The most positive signal had come from an acute leukemia study with cyclosporine A in 2001, with multiple intervening studies inconclusive (Shaffer et al. 2012). A third generation P-gp inhibitor, zosuquidar, was conclusively negative (Cripe et al. 2010). In none of the trials were patients selected because their tumors demonstrated expression of P-gp—and in the ensuing years we have learned just how important it is that the target be demonstrated in the tumor before expecting any activity from a targeted agent (Bates et al. 2012). In these studies, P-gp was the obvious target.
In this climate of disappointment, ABCG2 and ABCC1 were discovered and came of age as potential targets for cancer therapy. It was not long before inhibitors of both were discovered and with that the need to identify which tumors were ABCG2-expressing and which were P-gp-expressing. Given that the P-gp clinical trials were turning up negative, and that a clinically validated assay for P-gp had never been developed, the road to an ABCG2 inhibitor in the clinic seemed very long indeed. VX-710, biricodar, was an P-gp and ABCC1 modulator that was thought to also inhibit ABCG2 (Minderman et al. 2004). But a single arm Phase II trial of VX-710, doxorubicin, and vincristine did not meet a level of interest to be pursued (Gandhi et al. 2007). Nor were studies in ovarian, breast, or prostate cancer (Seiden et al. 2002; Toppmeyer et al. 2002; Bramwell et al. 2002; Rago et al. 2003). Notably all these studies enrolled patients with refractory or relapsed cancers and none selected patients based on transporter expression. VX-710 also required dose reduction for the anticancer agent (Toppmeyer et al. 2002; Rago et al. 2003). This is as close to an ABCG2 inhibitor trial as the field came. The disappointment with P-gp inhibitors led to a halt of almost all such clinical development, although the FDA and industry continued to be interested in the role of ABCG2 and its polymorphic variants in the disposition of substrate drugs (Mao and Unadkat 2015). Furthermore, this impasse diverted interest in drug transporters to their role in normal tissue protection, including a focus on their role in the blood–brain barrier.
Perhaps time and improving technology will allow a reassessment of the role of drug transporters in cancer drug resistance. The sequencing methodologies employed by The Cancer Genome Atlas (TCGA) also yield gene expression quantitation. The greater sensitivity of this method has allowed detection of ABCG2 and ABCB1 in a large number of clinical cancer samples. As shown in Fig. 2a, the range of ABCG2 expression is over 1000-fold among eight different tumor types shown in the dot plot: breast, pancreas, kidney, liver cancers, and glioblastomas and sarcomas. The range of expression is striking and suggests that for some tumor types the transporter expression may be higher and potentially more important than recently considered. Further, the co-expression with ABCB1 is very striking in these tumor types, with r = 0.659. In Fig. 2b, colorectal cancer samples have been added. Here the correlation has fallen to 0.479 because the colon cancer samples express ABCB1 more than ABCG2. In Fig. 2c, data for glioblastoma and low-grade gliomas are shown. Here a tight correlation between ABCG2 and ABCB1 is shown. Interestingly, relative to other tumor types, the level of expression in the glioblastomas is high, with low-grade glioma higher still, perhaps linked to normal tissue expression.

Expression data for ABCB1 and ABCG2 in tumor samples in the TCGA dataset. a Tumor sample expression of ABCB2 and ABCB1 in the TCGA dataset is shown, with tissue of origin color-coded. Number of samples from TCGA depicted: breast cancer = 994; pancreatic cancer = 56; kidney cancer = 480; hepatocellular cancer = 134; glioblastoma = 169; sarcoma = 77. b Colorectal samples (n = 233) shown in addition to the samples depicted in (a). c Expression data for glioblastoma (n = 169, squares) and low-grade glioma (n = 275, circles) are shown; darker color represents higher transport expression with highest levels found in low-grade glioma. Data for all three graphs were acquired from CellMiner, courtesy of Rajapakse (2015)
This striking analysis raises several questions. One is whether inhibition of one transporter could ever be enough in tumors that express both pumps (and see the data for murine BBB knockout models below). A second is whether the correlative studies reporting poor outcome with expression of only one transporter may have been reflecting the totality of transporter presence. Note that the question of whether drug transporters impact drug accumulation in tumor tissue has never been satisfactorily answered; studies measuring drug in tumor tissue are generally old and generally show wide interpatient variability. Clinical imaging tools are critically needed to address the question. Asking whether an P-gp/ABCG2 inhibitor could improve outcome was not the right question. Rather, the question that needs to be addressed is whether the presence of drug transporters reduces drug accumulation in tumor tissue. Certainly data shown below, gathered in murine models for normal brain penetration, imply that the P-gp/ABCG2 combination could have a tremendous impact.
The Blood–Brain Barrier
Introduction to the Blood–Brain Barrier
The blood–brain barrier (BBB) is a dynamic network of cells and proteins that regulates the molecular communication between the blood and most of the central nervous system (CNS) (Pardridge 2012). The brain and spinal cord actively adjust to daily variations of nutritional and oxygen requirements while maintaining the capacity to respond to pathologic alterations in the microenvironment such as increased intracranial pressure and toxic materials in the systemic circulation (Saunders et al. 2014). The BBB presents clinical challenges in the context of vasogenic edema, neuro-imaging, and drug delivery for various pathologic conditions including tumors, epilepsy, and infection (Löscher and Potschka 2005).
A small molecule can traverse a biological barrier in several ways: by passing through the capillary cells by either passive diffusion or facilitated transport (transcellular), by passing between the endothelial cells (paracellular), or by being taken up by the cell (pinocytosis) and across the cell (transcytosis) (Pardridge 2012; Abbott 2013). The BBB protects the brain by regulating and restricting each of the described pathways. Firstly, brain capillary cells form a physical barrier preventing paracellular transport. This is achieved by capillary endothelial cells forming tight junctions via proteins such as the connexins and claudins, and are characterized by the visualization of ‘kissing points’ via electron microscopy (Bauer et al. 2014). An underlying basement membrane and astrocytic end-feet have also been postulated to provide an additional barrier to permeability, and the maintenance of the BBB architecture has been shown to be regulated by astrocytes and pericytes that are immediately basolateral to the basement membrane (Cabezas et al. 2014; Muoio et al. 2014). Furthermore, BBB sites have fewer pores between the endothelial cells and decreased pinocytic activity compared to peripheral endothelial cells (Georgieva et al. 2014). Therefore, in a normal physiological setting, drug delivery to the brain parenchyma relies primarily on transcytosis, and this is consistent with the observation that for many organic small molecules, brain penetrance positively correlates with the partition coefficient (logP) for those molecules (Levin 1980). However, there are outliers to this relationship. The classic example is d-glucose, whose uptake to the brain is much greater than anticipated based on logP, due to the expression of high-affinity glucose uptake transporters (such as GLUT1) at the apical surface of brain endothelial cells, and carrier-mediated transport is responsible for the uptake of amino acids and other molecules essential for metabolic function (Cornford and Hyman 2005).
The other critical role for the BBB is that of active defense, precluding entry into the brain of unwanted small molecules that are capable of passive diffusion or ‘hijacking’ passive transporters. This is achieved by the expression of ATP-Binding Cassette (ABC) transporters at the apical surface, primarily the drug transporters ABCG2 and ABCB1 (see next section) (Kannan et al. 2009). Here, transporters can intercept small molecules at the apical cell membrane (often referred to as the ‘lipid-membrane vacuum cleaner’ model), and efflux them back into the blood plasma against the concentration gradient (Gottesman et al. 2002). Drug metabolizing cytochromes are also expressed in brain endothelial cells, and ABCC1 and ABCC4 are also expressed at the apical surface—these have been associated with the transport of secondary metabolites, such as glutathione conjugates (Cole 2014). These protective processes are highly energy (ATP) dependent, and this is one of the reasons that brain endothelial cells are mitochondria-rich (Hicks et al. 1983). We present a summary of transporters expressed at the BBB in Fig. 3.

Schematic representation of a the blood-brain barrier and b the blood-CSF barrier. The blood-brain barrier (a) is formed primarily by brain endothelial cells in capillaries, and is regulated by surrounding pericytes and astrocytes at the basolateral side of the endothelial cells. The endothelial cells form tight junctions, mediated by connexin, occludin and claudin family proteins. At the apical cell surface, ABC transporters such as P-gp (P-glycoprotein, ABCB1), ABCG2 (also breast cancer resistance protein) and MRP4 (multidrug resistance protein 4, ABCC4) transport small molecules back into the lumen. Ingress of nutrients from the blood supply is mediated by facilitative solute carrier SLC transporters, such as glutamate (excitatory amino acid transporter 1, Eaat1, SLC1A3) and d-glucose (glucose uptake transporter 1, Glut1, SLC2A1). LRP1 (low-density lipoprotein receptor-related protein 1 receptor, APOER) mediates receptor-mediated transcytosis across brain endothelial cells, and is one of several receptor targets present on the apical side of the BBB being evaluated for brain-targeted therapeutics. Lining the apical surface and projecting into the lumen is the glycocalyx (not shown), composed of glycoprotein and polysaccharide. The blood-CSF barrier (b) is distinct from the BBB and exists as part of the choroid plexus, responsible for producing CSF. It is comprised of epithelial cells forming tight junctions, while the endothelial capillary cells that supply nutrients for CSF production are fenestrated, allowing protein access to the choroid plexus epithelium. ABC transporter expression is a matter of some debate, but there appears to be general agreement that MRP (ABCC) transporters are oriented in the epithelium towards the blood, and that P-gp and ABCG2 are oriented towards the CSF. This has led to the hypothesis that the choroid plexus supplies drug to the CSF, and that this is a ‘back door’ for drug access to the brain parenchyma, and the follow-on conclusion that drug CSF levels are an acceptable surrogate indicator of drug brain concentrations. However, there is now clear evidence that there is a large divergence between brain and CSF levels for most drugs. Figure was prepared with the assistance of Alan Hoofring of NIH Medical Arts and Printing
ABC Transporter Expression at the BBB
Numerous studies have examined the expression of ABC transporters in brain capillary cells. It was recognized very early on that P-glycoprotein was expressed in brain endothelial cells, and ABCG2 expression was reported shortly after the gene itself was identified. The functional contribution of ABCG2 in protecting the BBB has been examined for a range of therapeutics. The most direct insights have been derived from gene knockout mice (see elsewhere in this chapter). For many agents the cross-recognition of substrates by P-gp and ABCG2 indicates a cooperativity in their protective role at the BBB. This is significant given the recognition that a population of humans of the Jr(a) blood group carry two missense copies of ABCG2 and do not express functional ABCG2 protein. Given the lack of reported drug-related adverse neurologic events in these patients, it is likely that redundancy with P-gp minimizes the impact of the loss of ABCG2.
The absolute levels of ABC transporter expression have been assessed, and the work of Terasaki and co-workers will be highlighted here. They have measured the expression of transporters in isolated brain capillary endothelial cells and in cell culture using quantitative liquid chromatography-tandem mass spectrometry (Kamiie et al. 2008). This method allows for measurement of protein levels, usually given as fmol/μg of cellular protein in endothelial cells isolated from brain capillaries of species including mice, monkeys, and humans. Primary findings regarding expression will be conveyed here, along with specific observations related to ABCG2. Assessment is carried out by first isolating brain capillaries from brains of the species of interest, exclusion of large vessels (that are unlikely to be capillaries), and subsequently isolating protein from the resulting capillaries (Ohtsuki et al. 2014).
A summary of expression of ABC transporters is shown in Table 1. It should be noted that ABCG2 is comprised of two functional protein units and as such two protein molecules form one functional transporter, so, for example, 14 fmol/µg ABCG2 protein results in 7 fmol/µg functional ABCG2 protein. Given that the functional capacity of ABC transporters is so high, the functional consequences could be concluded to be irrelevant (the so-called ‘spare transporter’ effect) (Kalvass and Pollack 2007). In mouse brain capillaries, P-gp (mdr1a, 12–15 fmol/µg protein) expression was higher than Abcg2 (4–5 fmol/µg protein), and MRP4 (ABCC4) was also detectable (Kamiie et al. 2008). The second mouse P-gp, Mdr1b, was not detectable. As a means of comparison, the highest transporter protein expression was for Glut1 (90 fmol/µg protein). Expression in adult and younger cynomolgus monkeys revealed differences from mouse, the primary difference being that ABCG2 expression (14 fmol/µg protein) was higher than P-gp (5–6 fmol/µg protein) (Ito et al. 2011). There was no difference in ABCG2 expression between adult and infant monkeys, though P-gp expression was slightly higher in infants. In Sprague-Dawley and Wistar rats, levels of P-gp (Abcb1, 19 fmol/µg protein) and Abcg2 (2–3 fmol/µg protein) were approximately equivalent between the two breeds, and Abcc4 (MRP4, 1–2 fmol/µg protein) was also detectable (Hoshi et al. 2013). In porcine brain capillaries, BCRP was more highly expressed than P-gp, but comparison with other species is complicated by an alternative analysis technique (Kubo et al. 2015). The expression of fractionated lysate was assessed to determine the localization of transporters toward the luminal (apical) or abluminal (basolateral) fraction (Kubo et al. 2015). P-gp and ABCG2 were almost exclusively located in the luminal fraction, but this does not indicate all transporters are at the cell surface, and a significant portion may be located in recycling endosomal compartments below the apical cell surface. Curiously, the cholesterol transporter ABCA1 was also detectable, but its expression was toward the abluminal surface of capillary cells. It is worth noting that in the above studies multiple other ABC transporters were assessed for expression and were not detectable, such as ABCB5, MRP1-3, ABCG5, and ABCG8 (Ito et al. 2011).
Two studies examining absolute transport protein levels in human brain microvessels have been reported; the first utilizing capillaries from brains of male patients who died of peripheral disease (Uchida et al. 2011), the second from patients with brain pathologies such as epilepsy or glioma (Shawahna et al. 2011). Major challenges in working with human brain samples are the time from death till autopsy, the tissue harvesting protocols employed, and freezer storage time, which are known to generally impact protein expression levels (Hynd et al. 2003; Harrison et al. 1995). In brains without a brain pathology, ABCG2 was more highly expressed (3–11 fmol/µg protein but with less functional protein than P-gp as explained above) than P-gp (4–9 fmol/µg protein) and MRP4 (0.1–0.3 fmol/µg protein). Of note, the lysosomal transporter ABCA2 and the proposed lipid transporter ABCA8 were also detectable (Uchida et al. 2011). Capillaries derived from brain pathologies did not deviate significantly from those of a healthy brain (Shawahna et al. 2011). An informative comparison was made between human brain microvessels and the human brain capillary endothelial cell line hCMEC/D3 (Ohtsuki et al. 2013). Strikingly, P-gp was undetectable in cells, and ABCG2 expression (1–2 fmol/µg protein) was reduced compared with fresh brain microvessels. Furthermore, ABCA2 expression (11 fmol/µg protein) was greater than brain microvessels (2–3 fmol/µg protein), and overall the correlation between primary tissue and cell culture was poor.
A controversial area of study has been that of the expression of ABC transporters in astrocytes, and whether expression occurs in the end-feet processes and is part of the BBB transport capacity. Early immunohistochemical studies of human brain reported that P-gp localized to cells also expressing glial fibrillary acidic protein (GFAP), a marker for astrocytes, but not with the brain endothelial cell marker GLUT1. This led to the hypothesis that transporters are found predominantly at the astrocyte foot processes and not in the endothelial luminal membrane (Golden and Pardridge 2000). Multiple issues arose from this model, that required drug to cross endothelial cells into the brain extracellular fluid before being pushed back across endothelial cells into the vasculature. The observations were critically discussed in the literature (Schinkel 1999), and multiple follow-up studies have demonstrated that high expression of P-gp occurs on the apical surface of capillary cells, with little to no P-gp expression in astrocytes (Loscher and Potschka 2005). Several pathologies associated with neuroinflammation, such as drug-resistant epilepsy, have led to increases in expression of P-gp in astrocytes, but this is not generally proposed to contribute to the barrier function of the neurovascular unit (Loscher and Potschka 2005).
Drug–Transporter Interactions at the BBB
Understanding the role of ABCB1 and ABCG2 at the blood–brain barrier has been aided by the study of brain penetration of various substrate drugs in murine knockout experiments. First, studies were performed in mice in which Abcb1a and Abcb1b had been deleted—the human ABCB1 equivalent. Further exploration demonstrated that when Abcg2 was deleted the effects on drug accumulation in the CNS were modest, but that when both were deleted there could be markedly greater brain penetration of substrates. Recently a study went one further step, deleting Abcc4 as well and evaluating topotecan brain accumulation (Lin et al. 2013). This was important because ABCC4 is a multidrug transporter also expressed at the blood–brain barrier. The studies demonstrated the additive effect of ABCC4 together with P-gp and ABCG2 in restricting brain penetration. The heat map in Fig. 4 depicts the results with the double knockout, Abcb1a/b and Abcg2, and shows how neither knockout alone has a major impact on the accumulation of most substrates, but that together the impact is clinically important and statistically significant for most substrates.

Role of ABCG2 and ABCB1 in plasma or brain concentrations of chemotherapy drugs. Values shown are fold increase compared to wild-type mice, regardless of the parameter examined. Inspection of the heat map indicates that transporter knockout (in the murine model) seldom affects pharmacokinetics, and single transporter knockout seldom affects brain penetration. But double knockout has a marked effect on the brain accumulation of substrates. Data abstracted and calculated from references—using absolute concentrations where possible, to calculate fold change over wild-type (Lin et al. 2013; Schinkel et al. 1994; van Asperen et al. 1999; Choo et al. 2014; Mittapalli et al. 2012; Vaidhyanathan et al. 2014; Kort et al. 2015; Lagas et al. 2010; Oberoi et al. 2013; Durmus et al. 2013; Chen et al. 2009; Poller et al. 2011; Zhou et al. 2009; Sane et al. 2013; Lin et al. 2014; Chuan Tang et al. 2014; Durmus et al. 2015; Wang et al. 2012; Polli et al. 2009; de Vries et al. 2007; Salphati et al. 2010)
Brain Metastases and Tumor Vasculature
One area still in need of investigation is that of expression of ABCG2 and P-gp in brain tumors and brain metastases. The blood–brain barrier becomes partially disrupted in patients with both glioblastomas and brain metastases from different solid tumors. This is readily demonstrated by the MRI visualization of gadolinium contrast agent, which represents the leakage of gadolinium into the tumor. This leakage has caused some investigators to conclude there is no blood–brain barrier left in individuals with these malignancies. However, a careful observation shows that certain areas of tumors are enhanced with contrast while others are not; just as certain drugs work better than others in treating malignancies involving the CNS. The best evidence suggests that some components of the BBB remain (endothelial cells, astrocytes, basal lamina, pericytes, etc.) and contribute to the multidrug resistance seen in malignant disease in the CNS. This evidence has been gathered by studying drug concentrations in metastatic lesions in the brain in preclinical models, and reinforced by clinical studies. In brain metastases derived from MDA-MB-231 cells in nude mice, a range of paclitaxel concentrations was detected, from 22 ng/g—approximating normal brain at 14 ng/g—to 1400 ng/g, and everything in between (Lockman et al. 2010). Only 15 % of tumors had levels at the low end approximating normal brain, while 10 % of tumors had levels at the highest end, still far below the 10,000 range measured in systemic metastases. These data convincingly show that the vestiges of the BBB remain in intracranial metastases, and explain why the occasional patient shows a marked and unexpected response with an agent not thought to cross the blood–brain barrier. A recent clinical study suggested a similar range of drug penetration in metastases from patients (Morikawa et al. 2015). Levels of lapatinib varied from 1.0 to 6.5 µM and capecitabine from 0.12 to 1.97 µM. Together, these data suggest that the blood–brain barrier is leaky and aberrant in individuals with brain tumors, and the regulation of the ABC transporters in that setting not really known.
Prospects for Transporter Inhibition at the BBB
The question then becomes whether we could improve cancer therapy in the CNS by combining therapy with an efflux inhibitor. Certainly an efflux inhibitor would be most valuable when the blood–brain barrier is still intact—at a time when preventing the early development of CNS metastases would be most likely to succeed. Such a prevention strategy has been successfully explored in animal models (Palmieri et al. 2014).
Drug delivery to the CNS represents a major challenge for chemotherapeutics due to the existence of efflux transporters such as P-gp and ABCG2 at the BBB that affect drug distribution. Studies have shown that these two transporters function in concert, or even synergistically (Zhou et al. 2009). A rational approach is the deliberate modulation of these transporters to mediate the permeability of substrates across the BBB (the “modulation of efflux” approach). However, it has also been suggested that clinical inhibition of P-gp and ABCG2 function at the BBB may not be pharmacologically achievable (Kalvass et al. 2013).
Development of approaches to successfully modulate ABCG2 activity at the BBB depends largely on improving and validating reliable BBB models for extrapolation of rodent data to humans. First, the overlapping substrate and inhibitor specificity of human and mouse ABCG2 supports the use of mouse models to evaluate the clinical, physiological, and pharmacological roles of ABCG2, especially at the BBB (Bakhsheshian et al. 2013b). Second, ABCG2 levels are reported to be similar (less than twofold difference) between murine and human BBB (Uchida et al. 2011). Third, any model must take into account the overlapping substrate specificity of ABCG2 and ABCB1 and ABCC4, all at the BBB. A recent study identified d-luciferin as a specific probe for bioluminescence imaging of ABCG2 function in vivo at the BBB (Bakhsheshian et al. 2013a). This novel method will allow for better understanding of the kinetics of transporter activity at the BBB. The study further demonstrated that ABCG2 plays a role in limiting the biodistribution of a specific substrate, and the possibility for saturable transporter inhibition at the BBB using pharmacologic levels of inhibitor, which was not possible before for ABCG2. Such tools will aid drug discovery studies with novel modulators of the transporter and/or new methods to increase drug delivery to the brain.
References
Abbott NJ (2013) Blood-brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis 36(3):437–449. doi:10.1007/s10545-013-9608-0
Adachi Y, Suzuki H, Schinkel AH, Sugiyama Y (2005) Role of breast cancer resistance protein (Bcrp1/Abcg2) in the extrusion of glucuronide and sulfate conjugates from enterocytes to intestinal lumen. Mol Pharmacol 67(3):923–928
Adkison KK, Vaidya SS, Lee DY, Koo SH, Li L, Mehta AA et al (2008) The ABCG2 C421A polymorphism does not affect oral nitrofurantoin pharmacokinetics in healthy Chinese male subjects. Br J Clin Pharmacol 66(2):233–239 doi:BCP3184 [pii]. 10.1111/j.1365-2125.2008.03184.x
Allen JD, Jackson SC, Schinkel AH (2002a) A mutation hot spot in the Bcrp1 (Abcg2) multidrug transporter in mouse cell lines selected for Doxorubicin resistance. Cancer Res 62(8):2294–2299
Allen JD, van Loevezijn A, Lakhai JM, van der Valk M, van Tellingen O, Reid G et al (2002b) Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther 1(6):417–425
Amiri-Kordestani L, Basseville A, Kurdzeil K, Fojo A, Bates S (2012) Targeting MDR in breast and lung cancer: discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist Updat
Aust S, Obrist P, Jaeger W, Klimpfinger M, Tucek G, Wrba F et al (2004) Subcellular localization of the ABCG2 transporter in normal and malignant human gallbladder epithelium. Lab Invest 84(8):1024–1036
Backstrom G, Taipalensuu J, Melhus H, Brandstrom H, Svensson AC, Artursson P et al (2003) Genetic variation in the ATP-binding cassette transporter gene ABCG2 (BCRP) in a Swedish population. Eur J Pharm Sci 18(5):359–364 doi:S0928098703000381 [pii]
Bakhsheshian J, Wei BR, Chang KE, Shukla S, Ambudkar SV, Simpson RM et al (2013a) Bioluminescent imaging of drug efflux at the blood-brain barrier mediated by the transporter ABCG2. Proc Natl Acad Sci USA 110(51):20801–20806 doi:1312159110 [pii]. 10.1073/pnas.1312159110
Bakhsheshian J, Hall MD, Robey RW, Herrmann MA, Chen JQ, Bates SE et al (2013b) Overlapping substrate and inhibitor specificity of human and murine ABCG2. Drug Metab Dispos 41(10):1805–1812 doi:dmd.113.053140 [pii]. 10.1124/dmd.113.053140
Bart J, Hollema H, Groen HJ, de Vries EG, Hendrikse NH, Sleijfer DT et al (2004) The distribution of drug-efflux pumps, P-gp, BCRP, MRP1 and MRP2, in the normal blood-testis barrier and in primary testicular tumours. Eur J Cancer 40(14):2064–2070
Basseville A, Tamaki A, Ierano C, Trostel S, Ward Y, Robey RW et al (2012) Histone deacetylase inhibitors influence chemotherapy transport by modulating expression and trafficking of a common polymorphic variant of the ABCG2 efflux transporter. Cancer Res 72(14):3642–3651 doi:0008-5472.CAN-11-2008 [pii]. 10.1158/0008-5472.CAN-11-2008
Bates SE, Amiri-Kordestani L, Giaccone G (2012) Drug development: portals of discovery. Clin Cancer Res 18(1):23–32. doi:10.1158/1078-0432.CCR-11-1001
Bauer HC, Krizbai IA, Bauer H, Traweger A (2014) “You Shall Not Pass”-tight junctions of the blood brain barrier. Front Neurosci 8:392. doi:10.3389/fnins.2014.00392
Bhatia A, Schafer HJ, Hrycyna CA (2005) Oligomerization of the human ABC transporter ABCG2: evaluation of the native protein and chimeric dimers. Biochemistry 44(32):10893–10904. doi:10.1021/bi0503807
Bramwell VH, Morris D, Ernst DS, Hings I, Blackstein M, Venner PM et al (2002) Safety and efficacy of the multidrug-resistance inhibitor biricodar (VX-710) with concurrent doxorubicin in patients with anthracycline-resistant advanced soft tissue sarcoma. Clin Cancer Res 8(2):383–393
Cabezas R, Avila M, Gonzalez J, El-Bachá RS, Báez E, García-Segura LM et al (2014) Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci 8:211. doi:10.3389/fncel.2014.00211
Chen ZS, Robey RW, Belinsky MG, Shchaveleva I, Ren XQ, Sugimoto Y et al (2003) Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res 63(14):4048–4054
Chen Y, Agarwal S, Shaik N, Chen C, Yang Z, Elmquist W (2009) P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther 330(3):956–963 doi:jpet.109.154781 [pii]. 10.1124/jpet.109.154781
Chen P, Chen H, Zang X, Chen M, Jiang H, Han S et al (2013) Expression of efflux transporters in human ocular tissues. Drug Metab Dispos 41(11):1934–1948. doi:10.1124/dmd.113.052704
Chew SC, Singh O, Chen X, Ramasamy RD, Kulkarni T, Lee EJ et al (2011) The effects of CYP3A4, CYP3A5, ABCB1, ABCC2, ABCG2 and SLCO1B3 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of docetaxel in nasopharyngeal carcinoma patients. Cancer Chemother Pharmacol 67(6):1471–1478. doi:10.1007/s00280-011-1625-9
Choo EF, Ly J, Chan J, Shahidi-Latham SK, Messick K, Plise E et al (2014) Role of P-glycoprotein on the brain penetration and brain pharmacodynamic activity of the MEK inhibitor cobimetinib. Mol Pharm 11(11):4199–4207. doi:10.1021/mp500435s
Chuan Tang S, Nguyen LN, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH (2014) Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Int J Cancer 134(6):1484–94. doi:10.1002/ijc.28475
Cole SP (2014) Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J Biol Chem 289(45):30880–30888. doi:10.1074/jbc.R114.609248
Cooray HC, Blackmore CG, Maskell L, Barrand MA (2002) Localisation of breast cancer resistance protein in microvessel endothelium of human brain. NeuroReport 13(16):2059–2063
Cornford EM, Hyman S (2005) Localization of brain endothelial luminal and abluminal transporters with immunogold electron microscopy. NeuroRx 2(1):27–43. doi:10.1602/neurorx.2.1.27
Cripe LD, Uno H, Paietta EM, Litzow MR, Ketterling RP, Bennett JM et al (2010) Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood 116(20):4077–85. doi:blood-2010-04-277269 [pii]. 10.1182/blood-2010-04-277269
Cusatis G, Gregorc V, Li J, Spreafico A, Ingersoll RG, Verweij J et al (2006) Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst 98(23):1739–1742 doi:98/23/1739 [pii]. 10.1093/jnci/djj469
de Bruin M, Miyake K, Litman T, Robey R, Bates SE (1999) Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter. MXR. Cancer Lett 146(2):117–126 doi:S0304383599001822 [pii]
de Jong FA, Marsh S, Mathijssen RH, King C, Verweij J, Sparreboom A et al (2004) ABCG2 pharmacogenetics: ethnic differences in allele frequency and assessment of influence on irinotecan disposition. Clin Cancer Res 10(17):5889–5894. doi:10.1158/1078-0432.CCR-04-0144 10/17/5889 [pii]
de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O (2007) P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res 13(21):6440–6449
Dehghan A, Köttgen A, Yang Q, Hwang S, Kao W, Rivadeneira F et al (2008) Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet 372(9654):1953–1961
Diop NK, Hrycyna CA (2005) N-Linked glycosylation of the human ABC transporter ABCG2 on asparagine 596 is not essential for expression, transport activity, or trafficking to the plasma membrane. Biochemistry 44(14):5420–5429. doi:10.1021/bi0479858
Do TM, Noel-Hudson MS, Ribes S, Besengez C, Smirnova M, Cisternino S et al (2012) ABCG2- and ABCG4-mediated efflux of amyloid-β peptide 1-40 at the mouse blood-brain barrier. J Alzheimers Dis 30(1):155–166. doi:10.3233/JAD-2012-112189
Doyle L, Ross DD (2003) Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22(47):7340–7358. doi:10.1038/sj.onc.1206938 1206938 [pii]
Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK et al (1998) A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA 95(26):15665–15670
Durmus S, Xu N, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH (2013) P-glycoprotein (MDR1/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) restrict brain accumulation of the JAK1/2 inhibitor, CYT387. Pharmacol Res 76:9–16. doi:10.1016/j.phrs.2013.06.009
Durmus S, Hendrikx JJ, Schinkel AH (2015a) Apical ABC transporters and cancer chemotherapeutic drug disposition. Adv Cancer Res 125:1–41 doi:S0065-230X(14)00002-5 [pii]. 10.1016/bs.acr.2014.10.001
Durmus S, Sparidans RW, van Esch A, Wagenaar E, Beijnen JH, Schinkel AH (2015b) Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699). Pharm Res 32(1):37–46. doi:10.1007/s11095-014-1442-z
Faneyte IF, Kristel PM, Maliepaard M, Scheffer GL, Scheper RJ, Schellens JH et al (2002) Expression of the breast cancer resistance protein in breast cancer. Clin Cancer Res 8(4):1068–1074
Fetsch P, Abati A, Litman T, Morisaki K, Honjo Y, Mittal K et al (2006) Localization of the ABCG2 mitoxantrone resistance-associated protein in normal tissues. Cancer Lett 235(1):84–92 doi:S0304-3835(05)00353-8 [pii]. 10.1016/j.canlet.2005.04.024
Fox E, Bates SE (2007) Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther 7(4):447–459. doi:10.1586/14737140.7.4.447
Furukawa T, Wakabayashi K, Tamura A, Nakagawa H, Morishima Y, Osawa Y et al (2009) Major SNP (Q141 K) variant of human ABC transporter ABCG2 undergoes lysosomal and proteasomal degradations. Pharm Res 26(2):469–479. doi:10.1007/s11095-008-9752-7
Gandhi L, Harding MW, Neubauer M, Langer CJ, Moore M, Ross HJ et al (2007) A phase II study of the safety and efficacy of the multidrug resistance inhibitor VX-710 combined with doxorubicin and vincristine in patients with recurrent small cell lung cancer. Cancer 109(5):924–932. doi:10.1002/cncr.22492
Georgieva JV, Hoekstra D, Zuhorn IS (2014) Smuggling Drugs into the Brain: An Overview of Ligands Targeting Transcytosis for Drug Delivery across the Blood-Brain Barrier. Pharmaceutics 6(4):557–583. doi:10.3390/pharmaceutics6040557
Giri N, Agarwal S, Shaik N, Pan G, Chen Y, Elmquist WF (2009) Substrate-dependent breast cancer resistance protein (Bcrp1/Abcg2)-mediated interactions: consideration of multiple binding sites in in vitro assay design. Drug Metab Dispos 37(3):560–570 doi:dmd.108.022046 [pii]. 10.1124/dmd.108.022046
Golden PL, Pardridge WM (2000) Brain microvascular P-glycoprotein and a revised model of multidrug resistance in brain. Cell Mol Neurobiol 20(2):165–181
Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC (1996) Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 183(4):1797–1806
Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nature Rev Cancer 2(1):48–58
Graf GA, Yu L, Li WP, Gerard R, Tuma PL, Cohen JC et al (2003) ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem 278(48):48275–48282. doi:10.1074/jbc.M310223200 M310223200 [pii]
Gutmann H, Hruz P, Zimmermann C, Beglinger C, Drewe J (2005) Distribution of breast cancer resistance protein (BCRP/ABCG2) mRNA expression along the human GI tract. Biochem Pharmacol 70(5):695–699
Han JY, Lim HS, Yoo YK, Shin ES, Park YH, Lee SY et al (2007) Associations of ABCB1, ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and clinical outcome in patients with advanced non-small cell lung cancer. Cancer 110(1):138–147. doi:10.1002/cncr.22760
Harrison PJ, Heath PR, Eastwood SL, Burnet PW, McDonald B, Pearson RC (1995) The relative importance of premortem acidosis and postmortem interval for human brain gene expression studies: selective mRNA vulnerability and comparison with their encoded proteins. Neurosci Lett 200(3):151–154
Hazai E, Hazai I, Ragueneau-Majlessi I, Chung SP, Bikadi Z, Mao Q (2013) Predicting substrates of the human breast cancer resistance protein using a support vector machine method. BMC Bioinformatics 14:130 doi:1471-2105-14-130 [pii]. 10.1186/1471-2105-14-130
Henriksen U, Fog JU, Litman T, Gether U (2005) Identification of intra- and intermolecular disulfide bridges in the multidrug resistance transporter ABCG2. J Biol Chem 280(44):36926–36934 doi:M502937200 [pii]. 10.1074/jbc.M502937200
Hicks P, Rolsten C, Brizzee D, Samorajski T (1983) Age-related changes in rat brain capillaries. Neurobiol Aging 4(1):69–75
Hilgendorf C, Ahlin G, Seithel A, Artursson P, Ungell A, Karlsson J (2007) Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab Dispos 35(8):1333–1340 doi:dmd.107.014902 [pii]. 10.1124/dmd.107.014902
Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A et al (2001) Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res 61(18):6635–6639
Hoshi Y, Uchida Y, Tachikawa M, Inoue T, Ohtsuki S, Terasaki T (2013) Quantitative atlas of blood-brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J Pharm Sci 102(9):3343–3355. doi:10.1002/jps.23575
Hosomi A, Nakanishi T, Fujita T, Tamai I (2012) Extra-renal elimination of uric acid via intestinal efflux transporter BCRP/ABCG2. PLoS ONE 7(2):e30456. doi:10.1371/journal.pone.0030456
Hue-Roye K, Lomas-Francis C, Coghlan G, Zelinski T, Reid ME (2013a) The JR blood group system (ISBT 032): molecular characterization of three new null alleles. Transfusion 53(7):1575–1579. doi:10.1111/j.1537-2995.2012.03930.x
Hue-Roye K, Zelinski T, Cobaugh A, Lomas-Francis C, Miyazaki T, Tani Y et al (2013b) The JR blood group system: identification of alleles that alter expression. Transfusion. 53(11):2710–2714. doi:10.1111/trf.12118
Huls M, Brown CD, Windass AS, Sayer R, van den Heuvel JJ, Heemskerk S et al (2008) The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int 73(2):220–225
Hynd MR, Lewohl JM, Scott HL, Dodd PR (2003) Biochemical and molecular studies using human autopsy brain tissue. J Neurochem 85(3):543–562
Iida A, Saito S, Sekine A, Mishima C, Kitamura Y, Kondo K et al (2002) Catalog of 605 single-nucleotide polymorphisms (SNPs) among 13 genes encoding human ATP-binding cassette transporters: ABCA4, ABCA7, ABCA8, ABCD1, ABCD3, ABCD4, ABCE1, ABCF1, ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8. J Hum Genet 47(6):285–310. doi:10.1007/s100380200041
Imai Y, Nakane M, Kage K, Tsukahara S, Ishikawa E, Tsuruo T et al (2002) C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141 K protein and low-level drug resistance. Mol Cancer Ther 1(8):611–616
Ito K, Uchida Y, Ohtsuki S, Aizawa S, Kawakami H, Katsukura Y et al (2011) Quantitative membrane protein expression at the blood-brain barrier of adult and younger cynomolgus monkeys. J Pharm Sci 100(9):3939–3950. doi:10.1002/jps.22487
Jada SR, Lim R, Wong CI, Shu X, Lee SC, Zhou Q et al (2007) Role of UGT1A1*6, UGT1A1*28 and ABCG2 c.421C > A polymorphisms in irinotecan-induced neutropenia in Asian cancer patients. Cancer Sci 98(9):1461–1467 doi:CAS541 [pii]. 10.1111/j.1349-7006.2007.00541.x
Jonker JW, Buitelaar M, Wagenaar E, Van Der Valk MA, Scheffer GL, Scheper RJ et al (2002) The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci USA 99(24):15649–15654. doi:10.1073/pnas.202607599 202607599 [pii]
Jonker JW, Merino G, Musters S, van Herwaarden AE, Bolscher E, Wagenaar E et al (2005) The breast cancer resistance protein BCRP (ABCG2) concentrates drugs and carcinogenic xenotoxins into milk. Nat Med 11(2):127–129
Juliano RL, Ling V (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 455:152–162
Kage K, Tsukahara S, Sugiyama T, Asada S, Ishikawa E, Tsuruo T et al (2002) Dominant-negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S-S dependent homodimerization. Int J Cancer 97(5):626–630. doi:10.1002/ijc.10100 [pii]
Kalvass JC, Pollack GM (2007) Kinetic considerations for the quantitative assessment of efflux activity and inhibition: implications for understanding and predicting the effects of efflux inhibition. Pharm Res 24(2):265–276. doi:10.1007/s11095-006-9135-x
Kalvass JC, Polli JW, Bourdet DL, Feng B, Huang SM, Liu X et al (2013) Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: the ITC evidence-based position. Clin Pharmacol Ther 94(1):80–94 doi:clpt201334 [pii]. 10.1038/clpt.2013.34
Kamiie J, Ohtsuki S, Iwase R, Ohmine K, Katsukura Y, Yanai K et al (2008) Quantitative atlas of membrane transporter proteins: development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm Res 25(6):1469–1483. doi:10.1007/s11095-008-9532-4
Kannan P, John C, Zoghbi S, Halldin C, Gottesman M, Innis R et al (2009) Imaging the function of P-glycoprotein with radiotracers: pharmacokinetics and in vivo applications. Clin Pharmacol Ther 86(4):368–377 doi:clpt2009138 [pii]. 10.1038/clpt.2009.138
Keskitalo JE, Pasanen MK, Neuvonen PJ, Niemi M (2009a) Different effects of the ABCG2 c.421C > A SNP on the pharmacokinetics of fluvastatin, pravastatin and simvastatin. Pharmacogenomics 10(10):1617–1624. doi:10.2217/pgs.09.85
Keskitalo JE, Zolk O, Fromm MF, Kurkinen KJ, Neuvonen PJ, Niemi M (2009b) ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther 86(2):197–203 doi:clpt200979 [pii]. 10.1038/clpt.2009.79
Kim IS, Kim HG, Kim DC, Eom HS, Kong SY, Shin HJ et al (2008) ABCG2 Q141 K polymorphism is associated with chemotherapy-induced diarrhea in patients with diffuse large B-cell lymphoma who received frontline rituximab plus cyclophosphamide/doxorubicin/vincristine/prednisone chemotherapy. Cancer Sci 99(12):2496–2501 doi:CAS985 [pii]. 10.1111/j.1349-7006.2008.00985.x
Kim HR, Park HS, Kwon WS, Lee JH, Tanigawara Y, Lim SM et al (2013) Pharmacogenetic determinants associated with sunitinib-induced toxicity and ethnic difference in Korean metastatic renal cell carcinoma patients. Cancer Chemother Pharmacol 72(4):825–835. doi:10.1007/s00280-013-2258-y
Kobayashi D, Ieiri I, Hirota T, Takane H, Maegawa S, Kigawa J et al (2005) Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab Dispos 33(1):94–101 doi:dmd.104.001628 [pii]. 10.1124/dmd.104.001628
Kondo C, Suzuki H, Itoda M, Ozawa S, Sawada J, Kobayashi D et al (2004) Functional analysis of SNPs variants of BCRP/ABCG2. Pharm Res 21(10):1895–1903
Kort A, Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH (2015) Brain and Testis Accumulation of Regorafenib is Restricted by Breast Cancer Resistance Protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1). Pharm Res. doi:10.1007/s11095-014-1609-7
Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE et al (2004) The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem 279(23):24218–24225. doi:10.1074/jbc.M313599200 M313599200 [pii]
Kubo Y, Ohtsuki S, Uchida Y, Terasaki T (2015) Quantitative Determination of Luminal and Abluminal Membrane Distributions of Transporters in Porcine Brain Capillaries by Plasma Membrane Fractionation and Quantitative Targeted Proteomics. J Pharm Sci. doi:10.1002/jps.24398
Lagas J, van Waterschoot R, Sparidans R, Wagenaar E, Beijnen J, Schinkel A (2010) Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther 9(2):319–326 doi:1535-7163.MCT-09-0663 [pii]. 10.1158/1535-7163.MCT-09-0663
Lecerf-Schmidt F, Peres B, Valdameri G, Gauthier C, Winter E, Payen L et al (2013) ABCG2: recent discovery of potent and highly selective inhibitors. Future Med Chem 5(9):1037–1045. doi:10.4155/fmc.13.71
Lee HK, Hu M, Lui S, Ho CS, Wong CK, Tomlinson B (2013) Effects of polymorphisms in ABCG2, SLCO1B1, SLC10A1 and CYP2C9/19 on plasma concentrations of rosuvastatin and lipid response in Chinese patients. Pharmacogenomics 14(11):1283–1294. doi:10.2217/pgs.13.115
Lemos C, Giovannetti E, Zucali PA, Assaraf YG, Scheffer GL, van der Straaten T et al (2011) Impact of ABCG2 polymorphisms on the clinical outcome and toxicity of gefitinib in non-small-cell lung cancer patients. Pharmacogenomics 12(2):159–170. doi:10.2217/pgs.10.172
Levin VA (1980) Relationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeability. J Med Chem 23(6):682–684
Lin F, Marchetti S, Pluim D, Iusuf D, Mazzanti R, Schellens JH et al (2013) Abcc4 together with abcb1 and abcg2 form a robust cooperative drug efflux system that restricts the brain entry of camptothecin analogues. Clin Cancer Res 19(8):2084–2095. doi:10.1158/1078-0432.CCR-12-3105
Lin F, de Gooijer MC, Roig EM, Buil LC, Christner SM, Beumer JH et al (2014) ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin Cancer Res 20(10):2703–2713. doi:10.1158/1078-0432.CCR-14-0084
Litman T, Brangi M, Hudson E, Fetsch P, Abati A, Ross DD et al (2000) The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2). J Cell Sci 113(Pt 11):2011–2021
Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA et al (2010) Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res 16(23):5664–5678. doi:10.1158/1078-0432.CCR-10-1564
Loscher W, Potschka H (2005) Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2(1):86–98
Löscher W, Potschka H (2005) Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 6(8):591–602. doi:10.1038/nrn1728
Maliepaard M, Scheffer GL, Faneyte IF, van Gastelen MA, Pijnenborg AC, Schinkel AH et al (2001) Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res 61(8):3458–3464
Mao Q, Unadkat JD (2015) Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport–an update. AAPS J 17(1):65–82. doi:10.1208/s12248-014-9668-6
Matsuo H, Takada T, Ichida K, Nakamura T, Nakayama A, Ikebuchi Y et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci Transl Med 2009;1(5):5ra11. doi:1/5/5ra11 [pii]. 10.1126/scitranslmed.3000237
McDevitt CA, Collins RF, Conway M, Modok S, Storm J, Kerr ID et al (2006) Purification and 3D structural analysis of oligomeric human multidrug transporter ABCG2. Structure 14(11):1623–1632 doi:S0969-2126(06)00387-X [pii]. 10.1016/j.str.2006.08.014
Minderman H, O’Loughlin KL, Pendyala L, Baer MR (2004) VX-710 (biricodar) increases drug retention and enhances chemosensitivity in resistant cells overexpressing P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Clin Cancer Res 10(5):1826–1834
Mittapalli RK, Vaidhyanathan S, Sane R, Elmquist WF (2012) Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel BRAF inhibitor: vemurafenib (PLX4032). J Pharmacol Exp Ther 342(1):33–40. doi:10.1124/jpet.112.192195
Miwa M, Tsukahara S, Ishikawa E, Asada S, Imai Y, Sugimoto Y (2003) Single amino acid substitutions in the transmembrane domains of breast cancer resistance protein (BCRP) alter cross resistance patterns in transfectants. Int J Cancer 107(5):757–763. doi:10.1002/ijc.11484
Mizuarai S, Aozasa N, Kotani H (2004) Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer 109(2):238–246. doi:10.1002/ijc.11669
Mizuno T, Fukudo M, Terada T, Kamba T, Nakamura E, Ogawa O et al (2012) Impact of genetic variation in breast cancer resistance protein (BCRP/ABCG2) on sunitinib pharmacokinetics. Drug Metab Pharmacokinet 27(6):631–639 doi:DN/JST.JSTAGE/dmpk/DMPK-12-RG-026 [pii]
Morikawa A, Peereboom DM, Thorsheim HR, Samala R, Balyan R, Murphy CG et al (2015) Capecitabine and lapatinib uptake in surgically resected brain metastases from metastatic breast cancer patients: a prospective study. Neuro Oncol 17(2):289–295. doi:10.1093/neuonc/nou141
Morisaki K, Robey RW, Ozvegy-Laczka C, Honjo Y, Polgar O, Steadman K et al (2005) Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol 56(2):161–172. doi:10.1007/s00280-004-0931-x
Muller PJ, Dally H, Klappenecker CN, Edler L, Jager B, Gerst M et al (2009) Polymorphisms in ABCG2, ABCC3 and CNT1 genes and their possible impact on chemotherapy outcome of lung cancer patients. Int J Cancer 124(7):1669–1674. doi:10.1002/ijc.23956
Muoio V, Persson PB, Sendeski MM (2014) The neurovascular unit- concept review. Acta Physiol (Oxf) 210(4):790–798. doi:10.1111/apha.12250
Nakagawa H, Tamura A, Wakabayashi K, Hoshijima K, Komada M, Yoshida T et al (2008) Ubiquitin-mediated proteasomal degradation of non-synonymous SNP variants of human ABC transporter ABCG2. Biochem J 411(3):623–631 doi:BJ20071229 [pii]. 10.1042/BJ20071229
Nakagawa H, Wakabayashi-Nakao K, Tamura A, Toyoda Y, Koshiba S, Ishikawa T (2009) Disruption of N-linked glycosylation enhances ubiquitin-mediated proteasomal degradation of the human ATP-binding cassette transporter ABCG2. FEBS J 276(24):7237–7252 doi:EJB7423 [pii]. 10.1111/j.1742-4658.2009.07423.x
Nakanishi T, Doyle LA, Hassel B, Wei Y, Bauer KS, Wu S et al (2003) Functional characterization of human breast cancer resistance protein (BCRP, ABCG2) expressed in the oocytes of Xenopus laevis. Mol Pharmacol 64(6):1452–1462. doi:10.1124/mol.64.6.1452 64/6/1452 [pii]
Ni Z, Bikadi Z, Shuster DL, Zhao C, Rosenberg MF, Mao Q (2011) Identification of proline residues in or near the transmembrane helices of the human breast cancer resistance protein (BCRP/ABCG2) that are important for transport activity and substrate specificity. Biochemistry 50(37):8057–8066. doi:10.1021/bi200573t
Noguchi K, Katayama K, Sugimoto Y (2014) Human ABC transporter ABCG2/BCRP expression in chemoresistance: basic and clinical perspectives for molecular cancer therapeutics. Pharmgenomics Pers Med 7:53–64. doi:10.2147/PGPM.S38295 pgpm-7-053 [pii]
Oberoi RK, Mittapalli RK, Elmquist WF (2013) Pharmacokinetic assessment of efflux transport in sunitinib distribution to the brain. J Pharmacol Exp Ther 347(3):755–764. doi:10.1124/jpet.113.208959
Ogasawara K, Osabe T, Suzuki Y, Tsuneyama H, Isa K, Kawai M et al (2014) A new ABCG2 null allele with a 27-kb deletion including the promoter region causing the Jr(a-) phenotype. Transfusion. doi:10.1111/trf.12969
Ohtsuki S, Ikeda C, Uchida Y, Sakamoto Y, Miller F, Glacial F et al (2013) Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood-brain barrier model. Mol Pharm 10(1):289–296. doi:10.1021/mp3004308
Ohtsuki S, Hirayama M, Ito S, Uchida Y, Tachikawa M, Terasaki T (2014) Quantitative targeted proteomics for understanding the blood-brain barrier: towards pharmacoproteomics. Expert Rev Proteomics 11(3):303–313. doi:10.1586/14789450.2014.893830
Ozols RF, Cunnion RE, Klecker RW, Hamilton TC, Ostchega Y, Parrillo JE et al (1987) Verapamil and adriamycin in the treatment of drug-resistant ovarian cancer patients. J Clin Oncol 5:641–647
Ozvegy-Laczka C, Koblos G, Sarkadi B, Varadi A (2005) Single amino acid (482) variants of the ABCG2 multidrug transporter: major differences in transport capacity and substrate recognition. Biochim Biophys Acta 1668(1):53–63 doi:S0005-2736(04)00291-3 [pii]. 10.1016/j.bbamem.2004.11.005
Palmieri D, Duchnowska R, Woditschka S, Hua E, Qian Y, Biernat W et al (2014) Profound prevention of experimental brain metastases of breast cancer by temozolomide in an MGMT-dependent manner. Clin Cancer Res 20(10):2727–2739. doi:10.1158/1078-0432.CCR-13-2588
Pardridge WM (2012) Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 32(11):1959–1972. doi:10.1038/jcbfm.2012.126
Polgar O, Robey RW, Morisaki K, Dean M, Michejda C, Sauna ZE et al (2004) Mutational analysis of ABCG2: role of the GXXXG motif. Biochemistry 43(29):9448–9456. doi:10.1021/bi0497953
Polgar O, Robey RW, Bates SE (2008) ABCG2: structure, function and role in drug response. Expert Opin Drug Metab Toxicol 4(1):1–15. doi:10.1517/17425255.4.1.1
Polgar O, Ierano C, Tamaki A, Stanley B, Ward Y, Xia D et al (2010) Mutational analysis of threonine 402 adjacent to the GXXXG dimerization motif in transmembrane segment 1 of ABCG2. Biochemistry 49(10):2235–2245. doi:10.1021/bi902085q
Poller B, Iusuf D, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH (2011) Differential impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on axitinib brain accumulation and oral plasma pharmacokinetics. Drug Metab Dispos 39(5):729–735. doi:10.1124/dmd.110.037317
Polli J, Olson K, Chism J, John-Williams L, Yeager R, Woodard S et al (2009) An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos 37(2):439–442
Rabindran SK, He H, Singh M, Brown E, Collins KI, Annable T et al (1998) Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res 58(24):5850–5858
Rago RP, Einstein A, Lush R, Beer TM, Ko YJ, Henner WD et al (2003) Safety and efficacy of the MDR inhibitor Incel (biricodar, VX-710) in combination with mitoxantrone and prednisone in hormone-refractory prostate cancer. Cancer Chemother Pharmacol 51(4):297–305. doi:10.1007/s00280-003-0573-4
Rajapakse VN, Luna A, Ohashi H, Onishi N, Sasa K, Sousa FG, Varma S, Kohn KW, Reinhold WC, Pommier Y (2015) tcgaMiner (web application in development), NCI-Developmental Therapeutics Branch, Genomics and Bioinformatics Group
Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M et al (2003) Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer 89(10):1971–1978. doi:10.1038/sj.bjc.6601370 6601370 [pii]
Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P et al (2004) Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res 64(4):1242–1246
Robey RW, Steadman K, Polgar O, Bates SE (2005) ABCG2-mediated transport of photosensitizers: potential impact on photodynamic therapy. Cancer Biol Ther 4(2):187–194 doi:1440 [pii]
Ross DD, Yang W, Abruzzo LV, Dalton WS, Schneider E, Lage H et al. (1999) A typical multidrug resistance: breast cancer resistance protein messenger RNA expression in mitoxantrone-selected cell lines. J Natl Cancer Inst 91(5):429–433
Saison C, Helias V, Ballif BA, Peyrard T, Puy H, Miyazaki T et al (2012) Null alleles of ABCG2 encoding the breast cancer resistance protein define the new blood group system Junior. Nat Genet 44(2):174–177 doi:ng.1070 [pii]. 10.1038/ng.1070
Salphati L, Lee LB, Pang J, Plise EG, Zhang X (2010) Role of P-glycoprotein and breast cancer resistance protein-1 in the brain penetration and brain pharmacodynamic activity of the novel phosphatidylinositol 3-kinase inhibitor GDC-0941. Drug Metab Dispos 38(9):1422–1426. doi:10.1124/dmd.110.034256
Sane R, Agarwal S, Mittapalli RK, Elmquist WF (2013) Saturable active efflux by p-glycoprotein and breast cancer resistance protein at the blood-brain barrier leads to nonlinear distribution of elacridar to the central nervous system. J Pharmacol Exp Ther 345(1):111–124. doi:10.1124/jpet.112.199786
Saunders NR, Dreifuss JJ, Dziegielewska KM, Johansson PA, Habgood MD, Møllgård K et al (2014) The rights and wrongs of blood-brain barrier permeability studies: a walk through 100 years of history. Front Neurosci 8:404. doi:10.3389/fnins.2014.00404
Scharenberg CW, Harkey MA, Torok-Storb B (2002) The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 99(2):507–512
Schinkel AH (1999) P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv Drug Deliv Rev 36(2–3):179–194
Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L et al (1994) Disruption of mouse mdr-1a p-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77:491–502
Schneider E, Horton JK, Yang CH, Nakagawa M, Cowan KH (1994) Multidrug resistance-associated protein gene overexpression and reduced drug sensitivity of topoisomerase II in a human breast carcinoma MCF7 cell line selected for etoposide resistance. Cancer Res 54:152–158
Seiden MV, Swenerton KD, Matulonis U, Campos S, Rose P, Batist G et al (2002) A phase II study of the MDR inhibitor biricodar (INCEL, VX-710) and paclitaxel in women with advanced ovarian cancer refractory to paclitaxel therapy. Gynecol Oncol 86(3):302–310
Shaffer B, Gillet J, Patel C, Baer M, Bates S, Gottesman M (2012) Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist Updat.
Shawahna R, Uchida Y, Declèves X, Ohtsuki S, Yousif S, Dauchy S et al (2011) Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm 8(4):1332–1341. doi:10.1021/mp200129p
Sparreboom A, Gelderblom H, Marsh S, Ahluwalia R, Obach R, Principe P et al (2004) Diflomotecan pharmacokinetics in relation to ABCG2 421C > A genotype. Clin Pharmacol Ther 76(1):38–44. doi:10.1016/j.clpt.2004.03.003 S0009923604000931 [pii]
Suzuki M, Suzuki H, Sugimoto Y, Sugiyama Y (2003) ABCG2 transports sulfated conjugates of steroids and xenobiotics. J Biol Chem 278(25):22644–22649
Tai LM, Loughlin AJ, Male DK, Romero IA (2009) P-glycoprotein and breast cancer resistance protein restrict apical-to-basolateral permeability of human brain endothelium to amyloid-beta. J Cereb Blood Flow Metab 29(6):1079–1083. doi:10.1038/jcbfm.2009.42
Taipalensuu J, Tornblom H, Lindberg G, Einarsson C, Sjoqvist F, Melhus H et al (2001) Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial Caco-2 cell monolayers. J Pharmacol Exp Ther 299(1):164–170
Takahashi N, Miura M, Scott SA, Kagaya H, Kameoka Y, Tagawa H et al (2010) Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J Hum Genet 55(11):731–737 doi:jhg201098 [pii]. 10.1038/jhg.2010.98
Tamura A, Watanabe M, Saito H, Nakagawa H, Kamachi T, Okura I et al (2006) Functional validation of the genetic polymorphisms of human ATP-binding cassette (ABC) transporter ABCG2: identification of alleles that are defective in porphyrin transport. Mol Pharmacol 70(1):287–296 doi:mol.106.023556 [pii]. 10.1124/mol.106.023556
Tanaka M, Kamada I, Takahashi J, Kimura K, Matsukura H, Tani Y (2014) Defining the Jr(a-) phenotype in the Japanese population. Transfusion 54(2):412–417. doi:10.1111/trf.12277
Tian C, Ambrosone CB, Darcy KM, Krivak TC, Armstrong DK, Bookman MA et al (2012) Common variants in ABCB1, ABCC2 and ABCG2 genes and clinical outcomes among women with advanced stage ovarian cancer treated with platinum and taxane-based chemotherapy: a Gynecologic Oncology Group study. Gynecol Oncol 124(3):575–581 doi:S0090-8258(11)00940-1 [pii]. 10.1016/j.ygyno.2011.11.022
Tiribelli M, Fabbro D, Franzoni A, Fanin R, Damante G, Damiani D (2013) Q141 K polymorphism of ABCG2 protein is associated with poor prognosis in adult acute myeloid leukemia treated with idarubicin-based chemotherapy. Haematologica 98(3):e28–e29 doi:haematol.2012.075895 [pii]. 10.3324/haematol.2012.075895
Toppmeyer D, Seidman AD, Pollak M, Russell C, Tkaczuk K, Verma S et al (2002) Safety and efficacy of the multidrug resistance inhibitor Incel (biricodar; VX-710) in combination with paclitaxel for advanced breast cancer refractory to paclitaxel. Clin Cancer Res 8(3):670–678
Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y (1981) Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res 41(5):1967–1972
Tucker TG, Milne AM, Fournel-Gigleux S, Fenner KS, Coughtrie MW (2012) Absolute immunoquantification of the expression of ABC transporters P-glycoprotein, breast cancer resistance protein and multidrug resistance-associated protein 2 in human liver and duodenum. Biochem Pharmacol 83(2):279–285. doi:10.1016/j.bcp.2011.10.017
Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J et al (2011) Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem 117(2):333–345. doi:10.1111/j.1471-4159.2011.07208.x
Urquhart BL, Ware JA, Tirona RG, Ho RH, Leake BF, Schwarz UI et al (2008) Breast cancer resistance protein (ABCG2) and drug disposition: intestinal expression, polymorphisms and sulfasalazine as an in vivo probe. Pharmacogenet Genomics 18(5):439–448. doi:10.1097/FPC.0b013e3282f974dc 01213011-200805000-00008 [pii]
Vaidhyanathan S, Mittapalli RK, Sarkaria JN, Elmquist WF (2014) Factors influencing the CNS distribution of a novel MEK-1/2 inhibitor: implications for combination therapy for melanoma brain metastases. Drug Metab Dispos 42(8):1292–1300. doi:10.1124/dmd.114.058339
van Asperen J, van Tellingen O, Tijssen F, Schinkel AH, Beijnen JH (1999) Increased accumulation of doxorubicin and doxorubicinol in cardiac tissue of mice lacking mdr1a P-glycoprotein. Br J Cancer 79(1):108–113
van Herwaarden AE, Wagenaar E, Karnekamp B, Merino G, Jonker JW, Schinkel AH (2006) Breast cancer resistance protein (Bcrp1/Abcg2) reduces systemic exposure of the dietary carcinogens aflatoxin B1, IQ and Trp-P-1 but also mediates their secretion into breast milk. Carcinogenesis 27(1):123–130
van Herwaarden AE, Wagenaar E, Merino G, Jonker JW, Rosing H, Beijnen JH et al (2007) Multidrug transporter ABCG2/breast cancer resistance protein secretes riboflavin (vitamin B2) into milk. Mol Cell Biol 27(4):1247–1253
van Loevezijn A, Allen JD, Schinkel AH, Koomen GJ (2001) Inhibition of BCRP-mediated drug efflux by fumitremorgin-type indolyl diketopiperazines. Bioorg Med Chem Lett 11(1):29–32 doi:S0960-894X(00)00588-6 [pii]
Vethanayagam RR, Wang H, Gupta A, Zhang Y, Lewis F, Unadkat JD et al (2005) Functional analysis of the human variants of breast cancer resistance protein: I206L, N590Y, and D620N. Drug Metab Dispos 33(6):697–705 doi:dmd.105.003657 [pii]. 10.1124/dmd.105.003657
Wakabayashi K, Nakagawa H, Tamura A, Koshiba S, Hoshijima K, Komada M et al (2007) Intramolecular disulfide bond is a critical check point determining degradative fates of ATP-binding cassette (ABC) transporter ABCG2 protein. J Biol Chem 282(38):27841–27846 doi:C700133200 [pii]. 10.1074/jbc.C700133200
Wakabayashi-Nakao K, Tamura A, Furukawa T, Nakagawa H, Ishikawa T (2009) Quality control of human ABCG2 protein in the endoplasmic reticulum: ubiquitination and proteasomal degradation. Adv Drug Deliv Rev 61(1):66–72 doi:S0169-409X(08)00258-5 [pii]. 10.1016/j.addr.2008.08.008
Wakabayashi-Nakao K, Tamura A, Koshiba S, Toyoda Y, Nakagawa H, Ishikawa T (2010) Production of cells with targeted integration of gene variants of human ABC transporter for stable and regulated expression using the Flp recombinase system. Methods Mol Biol 648:139–159. doi:10.1007/978-1-60761-756-3_9
Wang T, Agarwal S, Elmquist WF (2012) Brain distribution of cediranib is limited by active efflux at the blood-brain barrier. J Pharmacol Exp Ther 341(2):386–395. doi:10.1124/jpet.111.190488
Woodward OM, Kottgen A, Coresh J, Boerwinkle E, Guggino WB, Kottgen M (2009) Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci USA 106(25):10338–10342 doi:0901249106 [pii]. 10.1073/pnas.0901249106
Woodward OM, Tukaye DN, Cui J, Greenwell P, Constantoulakis LM, Parker BS et al (2013) Gout-causing Q141 K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc Natl Acad Sci USA 110(13):5223–5228 doi:1214530110 [pii]. 10.1073/pnas.1214530110
Xie Y, Xu K, Linn DE, Yang X, Guo Z, Shimelis H et al (2008) The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J Biol Chem 283(6):3349–3356 doi:M707773200 [pii]. 10.1074/jbc.M707773200
Xiong H, Callaghan D, Jones A, Bai J, Rasquinha I, Smith C et al (2009) ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J Neurosci 29(17):5463–5475 doi:29/17/5463 [pii]. 10.1523/JNEUROSCI.5103-08.2009
Xu J, Liu Y, Yang Y, Bates S, Zhang JT (2004) Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J Biol Chem 279(19):19781–19789. doi:10.1074/jbc.M310785200 M310785200 [pii]
Xu J, Peng H, Chen Q, Liu Y, Dong Z, Zhang JT (2007) Oligomerization domain of the multidrug resistance-associated transporter ABCG2 and its dominant inhibitory activity. Cancer Res 67(9):4373–4381 doi:67/9/4373 [pii]. 10.1158/0008-5472.CAN-06-3169
Yamasaki Y, Ieiri I, Kusuhara H, Sasaki T, Kimura M, Tabuchi H et al (2008) Pharmacogenetic characterization of sulfasalazine disposition based on NAT2 and ABCG2 (BCRP) gene polymorphisms in humans. Clin Pharmacol Ther 84(1):95–103 doi:6100459 [pii]. 10.1038/sj.clpt.6100459
Yoshikawa M, Ikegami Y, Hayasaka S, Ishii K, Ito A, Sano K et al (2004) Novel camptothecin analogues that circumvent ABCG2-associated drug resistance in human tumor cells. Int J Cancer 110(6):921–927. doi:10.1002/ijc.20216
Zamber CP, Lamba JK, Yasuda K, Farnum J, Thummel K, Schuetz JD et al (2003) Natural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestine. Pharmacogenetics. 13(1):19–28
Zamboni WC, Ramanathan RK, McLeod HL, Mani S, Potter DM, Strychor S et al (2006) Disposition of 9-nitrocamptothecin and its 9-aminocamptothecin metabolite in relation to ABC transporter genotypes. Invest New Drugs 24(5):393–401. doi:10.1007/s10637-006-6335-5
Zelinski T, Coghlan G, Liu XQ, Reid ME (2012) ABCG2 null alleles define the Jr(a-) blood group phenotype. Nat Genet 44(2):131–132 doi:ng.1075 [pii]. 10.1038/ng.1075
Zhang W, Yu BN, He YJ, Fan L, Li Q, Liu ZQ et al (2006) Role of BCRP 421C > A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin Chim Acta 373(1–2):99–103 doi:S0009-8981(06)00266-X [pii]. 10.1016/j.cca.2006.05.010
Zhao R, Raub TJ, Sawada GA, Kasper SC, Bacon JA, Bridges AS et al (2009) Breast cancer resistance protein interacts with various compounds in vitro, but plays a minor role in substrate efflux at the blood-brain barrier. Drug Metab Dispos 37(6):1251–1258 doi:dmd.108.025064 [pii]. 10.1124/dmd.108.025064
Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ et al (2001) The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7(9):1028–1034
Zhou L, Schmidt K, Nelson FR, Zelesky V, Troutman MD, Feng B (2009) The effect of breast cancer resistance protein and P-glycoprotein on the brain penetration of flavopiridol, imatinib mesylate (Gleevec), prazosin, and 2-methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)phenyl)propanoic acid (PF-407288) in mice. Drug Metab Dispos 37(5):946–955 doi:dmd.108.024489 [pii]. 10.1124/dmd.108.024489
Zhou Q, Ruan ZR, Yuan H, Xu DH, Zeng S (2013) ABCB1 gene polymorphisms, ABCB1 haplotypes and ABCG2 c.421c > A are determinants of inter-subject variability in rosuvastatin pharmacokinetics. Pharmazie 68(2):129–134
Acknowledgements
This research was funded by the Intramural Research Program of the National Institutes of Health (Center for Cancer Research).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Basseville, A. et al. (2016). The ABCG2 Multidrug Transporter. In: George, A. (eds) ABC Transporters - 40 Years on. Springer, Cham. https://doi.org/10.1007/978-3-319-23476-2_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-23476-2_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23475-5
Online ISBN: 978-3-319-23476-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)