
Abstract
Ewing's sarcoma of bone is a primary bone sarcoma found predominantly in patients during their second decade of life. It is a high-grade aggressive small round blue cell tumor that is part of the Ewing's family of tumors. Its exact eitiology is unknown but it commonly demonstrates reproducible staining of CD99 and translocations of the EWS gene. Historically, this diagnosis was associated with near certain metastasis and subsequent mortality. However, current management consists of extensive chemotherapy in addition to local control with surgical resection and/or radiation. As a result, survival has improved to the 55–75% range in those patients who present without known metastases. Current research aims to continue this improvement by looking further into the assocated gene abnormalities and possibly targeted therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Ewing’s sarcoma of bone is a part of the Ewing’s sarcoma family of tumors, which includes primitive neuroectodermal tumors, Ewing’s soft tissue sarcomas, and Askins’ tumors. Each shares similar molecular and histologic findings. It is found predominantly in the diaphysis of long bones and pelvis of patients in their second decade of life. It is a high-grade aggressive lesion that most commonly originates in bone and is associated with large soft tissue masses and frequent metastases. Histologically, it presents as sheets of small round blue cells which almost universally stain for CD99 and demonstrate common translocations of the EWS gene on chromosome 22. Treatment consists of multiagent systemic chemotherapy and local control with surgery and/or radiation. Current management has improved survival to the 55–75 % range in those who present without metastases.
2 History
The sarcoma that we currently call Ewing’s sarcoma was first described in detail by famed pathologist James Ewing in 1921 [1]. He noted seeing a form of bony neoplasm that did not fit with the appearance or behavior of other known lesions such as osteosarcoma or myeloma. He described his first case of a teenage girl who presented with a pathologic fracture of her forearm in which the tumor had an impressive response to radium, which was unlike osteosarcoma. Similarly, the Bence-Jones protein was never found in her urine to suggest myeloma. He goes on to report six additional cases of teenagers with permeative lesions in the shaft of long bones. Histology showed small polyhedral cells with hyperchromic nuclei, pale cytoplasm, and a lack of intercellular stromal material. All of the tumors seemed to at least temporarily resolve after radiation was administered. Given their appearance, he surmised the tumors may have originated from the endothelium, and he named them diffuse endotheliomas of bone [1]. It is impressive that with the exception of their relationship to endothelium, nearly all of the characteristics he described remain pathognomonic for Ewing’s sarcoma to this day.
The application of the name Ewing’s sarcoma would come 4 years later in 1925 by Ernest Codman [2]. Codman was one of the first surgeons to promote the use of registries to further the understanding of rare diseases and to promote the use of outcomes in guiding surgical practice [3]. As a result, he created the first sarcoma registry and within its description he refers to the sarcoma as described by Ewing as a Ewing’s sarcoma. Of note, Codman would be probably best known for his description of the way aggressive bone tumors elevate periosteum leading to the radiographic finding of a Codman’s Triangle [4].
While the understanding of Ewing’s sarcoma evolved, there were other neoplasms that were felt to be clinically unique and different based on their behavior and histology that are now known to be related and part of the Ewing’s sarcoma family of tumors.
The first was described in 1918 by Arthur Stout as a tumor of the ulnar nerve composed of undifferentiated round cells which formed rosettes. This later became known as a primitive neuroectodermal soft tissue tumor or PNET. Similarly, Askin et al. in 1979 described a soft tissue tumor found in the thoracopulmonary region of adolescents, which was composed of small round cells and was associated with high rates of recurrence and mortality, which would come to be known as Askin’s tumor. Nonosseous forms of Ewing’s sarcoma have also been documented, but are rare compared to osseous forms.
When molecular studies showed similar genetic profiles and translocations for these three tumors, they were subsequently felt to be related, as opposed to distinct entities. As a result, currently they are all considered to be a part of the Ewing’s Sarcoma Family of tumors. For the purpose of this review, the focus will be on Ewing’s sarcoma of bone.
3 Epidemiology and Etiology
As with other bone cancers, much of the current understanding of the epidemiology of Ewing’s sarcoma in the United States comes from the Surveillance, Epidemiology, and End Results (SEER) Program from the National Institute of Health. This is a database of cancer statistics drawn from a variety of cancer centers throughout the United States in an attempt to provide an accurate cross-section of the population. Esiashvili et al. have performed the most recent review of the SEER data from 1973 to 2004 looking specifically at Ewing’s sarcoma. They found an average annual incidence of about 3 per 1 million, which has been stable over the past 40 years. The incidence peaks within the second decade of life, with more than 50 % of cases being diagnosed between the ages of 10–20. Less than 23 % are found in those younger than 10 and the incidence declines rapidly as age increases beyond 20 years. Ewing’s sarcoma has a slight predominance in males at 61 % of the cases diagnosed, and is found almost exclusively in Caucasians who represented 92 % of the cases [5, 6].
In terms of location within the body, Ewing’s sarcoma has a predilection for the diaphysis of tubular bones and the pelvis. The most common location is the extremities at 46 % of cases, with the lower extremity being more common than the upper. This is followed by the pelvis at 25 %, trunk including ribs or spine at 22 %, and other sites including soft tissue Ewing’s at 6 % [5, 6]. These epidemiological findings in the United States are similar to those of Europe based on a study by Stiller et al. who used the Automated Childhood Cancer Information System European database to demonstrate concurrent findings regarding the epidemiology of Ewing’s sarcoma [7].
The etiology of Ewing’s sarcoma remains unknown. Despite most cases being associated with reproducible genetic abnormalities such as translocations, most seem to be sporadic in nature as no hereditary link has been found. Similarly, an association with environmental factors has yet to be demonstrated.
4 Patient Presentation
As with other primary bone sarcomas, pain is the most common initial symptom of patients with Ewing’s sarcoma of bone. As the tumor destroys bone, patients may notice a deep, dull, aching pain in the involved region or extremity [8]. While antiinflammatories and pain medicines may initially offer some relief, often their effect diminishes as the tumor grows. Although some may notice the pain to be more severe at night, this is certainly not a universal feature with only about 20 % noting it in one study [9]. If the bone is sufficiently weakened to alter its mechanical properties, it is common for pain to worsen with activities which put increased stress on the remaining bone.
Unfortunately, many patients who initially present with pain are initially misdiagnosed as having more common benign conditions such as strains or tendinitis. More than 25 % of Ewing’s patients may have a delay in diagnosis of over 6 months from the time of their first appointment with a physician. Those whose tumors are sufficiently large may have a palpable mass, leading to a quicker workup and diagnosis [9].
Systemic symptoms tend to be more commonplace in Ewing’s sarcoma compared to osteosarcoma. It is not uncommon for patients to present with fevers or weight loss, which in the presence of bone pain may mislead the physician into misdiagnosing the cause as osteomyelitis. Laboratory findings can promote this as many Ewing’s patients will have mildly elevated inflammatory markers such as erythrocyte sedimentation rate, creactive protein, and other cytokines [9–13]. It is important to note that in general these lab values, while elevated, are lower than those in patients with true osteomyelitis.
Other abnormal laboratory findings include the presence of anemia as well as elevated markers of bone turnover such as lactate dehydrogenase (LDH) and alkaline phosphatase (AP) [10]. The trend in the LDH and AP levels may offer some indication as to treatment response, but their current utility and role in standard care is debatable.
5 Histologic and Molecular Pathology
Similar to other bone sarcomas, a definitive diagnosis of Ewing’s sarcoma is based on tissue biopsy. If possible, it is preferred that the definitive treatment team participates in the biopsy or its planning in order to assure that sound oncologic principles are used in obtaining the biopsy, facilitating surgical resection, reconstruction, and ideally limb-salvage [14].
Grossly, Ewing’s sarcoma has the classic grayish/fleshy appearance of other sarcomas [1]. It may occasionally be associated with necrosis.
Histologically, Ewing’s sarcoma appears as sheets of homogenous densely packed small round blue cells. They have a high nuclear to cytoplasm ratio and the nucleus is associated with fine granular chromatin and pinpoint nucleoli. The cytoplasm typically has few or small organelles and abundant glycogen [15] (Figs. 1c, 2d and 3c). Unfortunately, they appear very histologically similar to other blue cell tumors so the differential includes other diagnoses such as lymphoma, leukemia, small cell carcinoma, rhabdomyosarcoma, neuroblastoma, and others.



Seventeen-year old male with left hip and groin pain. a Anteroposterior radiographs of the pelvis reveal a subtle, but apparent, radiolucent lesion centered on the left superior pubic ramus. b T1 and STIR Axial images on MRI reveal a large soft tissue mass centered on the left superior pubic ramus. c Representative H&E histology demonstrates sheets of small blue cells typical of Ewing’s sarcoma. d Axial postcontrast T1 Fat Saturation image after radiation and neo-adjuvant chemotherapy reveal significant local response with a significantly reduced soft tissue mass. e Surgically excised specimen also reveals no discernable soft tissue mass remaining after neoadjuvant treatment. f Anteroposterior radiographs of the pelvis 5 years after treatment. He has remained disease free



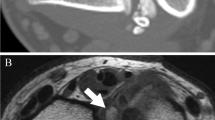
Sixteen-year old female with left thigh pain. a Anteroposterior and lateral radiographs of the left femur reveal an eccentric, poorly marginated radiolucent lesion involving the mid-diaphysis of the femur. b T1 and STIR Axial images on MRI reveal extensive marrow changes around the lesion with a soft tissue mass not appreciated on the plain radiographs. c T1 and STIR Coronal images on MRI. d A biopsy revealed a small blue cell tumor and immunohistochemical staining that was positive for FLI-1. e Axial T1 and sagittal STIR imaging of the lesion after neoadjuvant chemotherapy with visible shrinkage of the soft tissue mass. f Intraoperative images of the surgical removal of the diaphyseal segment of the left femur with the tumor followed by allograft reconstruction. g Anteroposterior radiographs 2 years after surgery reveal healing of the allograft


Thirteen-year old female with right ankle pain and swelling. a Anteroposterior radiographs of the right ankle before (left) and after (right) neoadjuvant chemotherapy. b T2 Axial MRI images of the right ankle before (left) and after (right) neoadjuvant chemotherapy. c A biopsy revealed a small blue cell tumor and immunohistochemical staining and subsequent FISH (not shown) revealed the 11:22 EWS-FLI-1 fusion gene. d Intraoperative images of the surgical removal of the right distal fibula with no subsequent reconstruction. e Anteroposterior radiographs 10 years after surgery reveal no local recurrence or ankle instability
One of the main ways to distinguish Ewing’s sarcoma from these other diagnoses is through the use of immunohistochemistry. Most Ewing’s cells stain strongly for CD99 which is a cell surface glycoprotein encoded by the MIC2 gene [16]. While CD99 staining is very sensitive for Ewing’s sarcoma, it is not specific as nearly all small blue cell tumors will at least partially stain for CD99. This makes it imperative to use it as part of an immunohistochemical panel in order to differentiate from diagnoses such as lymphoma or rhabdomyoscaroma [17]. Also, this is why cytogenetic and molecular findings are typically used to confirm the diagnosis.
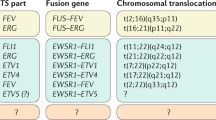
Cytogenetically there are a small number of characteristic and reproducible translocations associated with the Ewing’s sarcoma family of tumors. All of them involve the EWS gene on chromosome 22, which encodes an RNA binding protein, whose exact role in cellular function is unknown. It is subsequently upregulated through translocations with the ETS family of transcription factor genes, the most common being FLI1 on chromosome 11 [18]. This t(11;22)(q24;q12) translocation produces the EWS-FLI1 fusion gene which is found in about 85 % of Ewing’s tumors [19]. The next most common is the t(21;22)(q22;q12) translocation of the EWS-ERG gene found in another 5–10 % [20]. Other translocations that are much more rare have also been reported including EWS-ETV1, EWS-FEV, and EWS-EIAF amongst other translocations and cytogenetic abnormalities [21, 22].
At present, the prognostic value of one translocation over another remains controversial. Some studies have suggested an improved prognosis with the EWS-FLI1 translocation, [23] although others have found there to be no discernable difference in terms of phenotype or prognosis [24, 25].
Currently, these translocations are determined through the use of fluorescent in situ hybridization (FISH) and reverse transcriptase polymerase chain reaction (RT-PCR) methods. Both of these methods can be employed to detect the presence of micrometastases in bone marrow biopsies obtained for staging purposes [26, 27]. The benefit to FISH is that it has been shown to be more sensitive and specific compared to RT-PCR, but the techniques are complementary [28].
While the EWS translocations remain highly sensitive and specific for the Ewing’s family of tumors, it is important to note that they have been reported in other tumors as well, underscoring the importance of the consensus between the microscopic, immunohistochemical, and molecular characteristics in the diagnosis of Ewing’s sarcoma [29].
6 Imaging
Imaging studies are critical in the diagnosis, staging, and surveillance of Ewing’s sarcoma. Even with many advanced imaging techniques available, standard radiographs remain the first-line choice. As mentioned in the epidemiology section, Ewing’s sarcoma of bone is found predominantly in the diaphyseal region of long bones as well as the pelvis and ribs [5]. Its radiographic appearance can vary, but it will typically demonstrate aggressive features. The tumor margin is often poorly defined and permeative in nature [30]. The bone may show areas of radiulucency, or may demonstrate a mixed lytic/sclerotic appearance [31] (Figs. 1a, 2a and 3a). In the majority of patients, a soft tissue mass will be present at diagnosis, but it can be very difficult to distinguish on radiographs since it does not demonstrate ossification as is routinely seen in osteosarcoma [30]. As a result, it is not uncommon for plain radiographs to appear normal, especially when a comparison study is unavailable, leading to a delay in diagnosis [9]. In fact, an unremarkable plain X-ray of a bone with an MRI demonstrating a bony lesion with an associated large soft tissue mass can be highly suggestive of a blue cell tumor such as Ewing’s.
The soft tissue mass may be visualized on X-ray via its interaction with the periosteum. As it expands it will elevate normal periosteum which will subsequently ossify, leading to an “onion-skinning” appearance, or similarly, Codman’s triangle [30–32]. Codman’s triangle occurs when the soft tissue component of the tumor elevates the periosteum of the involved bone, causing new bone to form in the apex where the periosteum contacts the bone and where it has been elevated by tumor [4]. These periosteal reactions are not unique to Ewing’s sarcoma, but rather demonstrate its aggressive nature.
MRI is the most sensitive imaging technique for evaluating Ewing’s sarcoma, and can be especially helpful in cases where the radiographs are indeterminate. Ewing’s is often heterogenous in its appearance, and is dark on T1 sequences, and mostly bright or heterogeneous on T2. It will enhance if the study is performed with gadolinium (Figs. 1b,d, 2b,c and 3b). MRI is also helpful in determining the extent of the soft tissue mass and its relationship to adjacent structures. These factors become critical if a surgical resection and reconstruction is to be considered. It is important to include the entire involved bone in the MRI study to evaluate for skip metastases, which are noncontiguous tumors present within the same bone, and may be present in 10–20 % of patients [31].
While radiographs and an MRI of the involved bone are essential in the evaluation of Ewing’s sarcoma, CT of the tumor is generally less helpful. Its main advantage is the ability to look at the degree of bony destruction, or if combined with angiography, to evaluate vascular structures that may be altered by the tumor.
Radiographic studies are also important to evaluate for distant sites of disease. As with other sarcomas, the most common site of Ewing’s metastases are the lungs, followed by other bones or soft tissues [8].
Unfortunately, the studies used in the staging of Ewing’s sarcoma are somewhat institution dependent. The National Comprehensive Cancer Network guidelines for Ewing’s staging account for some of this variability [14]. They recommend that all patients should have at least an MRI of the known primary tumor, plus X-rays or a CT if indicated. For pulmonary disease, they recommend a CT scan of the chest and to evaluate for osseous metastases, either a PET scan or bone scan. In addition, they also recommend either an MRI of the spine or a bone marrow biopsy, and possibly molecular studies to look for micrometastases.
Much of the staging debate currently revolves around the role and accuracy of PET scans. Position emission tomography (PET) scanning represents a newer modality which has shown promise in the diagnosis and monitoring of Ewing’s sarcoma. They rely on radio-labeled glucose molecules, which are taken up preferentially in tumors with higher metabolic rates. Increased PET uptake at diagnosis has been shown to be associated with a worse prognosis and improvement in PET uptake after treatment can be suggestive of tumor necrosis [33–35].
Many studies have been performed directly comparing the sensitivity of detecting osseous metastases of both PET and bone scans. Most support that PET scans are similar if not superior to bone scans in terms of accuracy [36–38]. However, PET scans are much more expensive, and studies also exist which demonstrate that bone scans continue to be more sensitive [39].
7 Staging and Workup
One of the first steps after a patient has been diagnosed with a Ewing’s sarcoma is to determine if there are other sites of disease as this impacts both their future therapies as well as prognosis. Ewing’s is similar to osteosarcoma in the sense that even though most patients do not initially present with overt metastatic disease, the majority have subclinical micrometastases that will become apparent in the future if the patient does not receive systemic treatment. This is known because prior to systemic treatments, radical surgical excision alone resulted in dismal cure rates of about 10 % [40].
The staging workup of a patient diagnosed with Ewing’s sarcoma starts with a thorough history and physical exam. Baseline laboratory studies are ordered including a CBC, BMP, ESR, and LDH. There are a variety of imaging studies performed to characterize the primary tumor and look for sites of metastatic disease.
One unique staging aspect of Ewing’s sarcoma compared to other primary bone sarcomas is the evaluation of micrometastatic disease. Traditionally, this has been achieved by performing either a unilateral or bilateral bone marrow biopsy or aspiration looking for malignant cells. There is little evidence to suggest the utility of this and its use is somewhat institution dependent. Some authors have recently argued that PET scans and or MRI’s of the entire body may be as accurate as a bone marrow biopsy in detecting metastatic disease with much less morbidity [36, 38]. Also, molecular tests as described in the pathology section are increasingly being used in determining the presence of micrometastases.
In regards to staging systems, there is no system that is unique to Ewing’s sarcoma. Rather the two most commonly used systems for Ewing’s sarcoma of bone are designed for bone tumors in general. The first was created by Enneking et al. in 1980 and is the Surgical Staging System of Musculoskeletal Tumors [41]. The second was later created by the American Joint Committee on Cancer (AJCC) based on an adaptation of its system for carcinomas which relies on a TNM or tumor, node, metastasis methodology [42]. These systems are similar in that they are predominantly concerned with tumor size, grade, and the presence of metastases.
Both consider low-grade tumors to be Stage I and high-grade tumors to be at least Stage II. Ewing’s sarcoma by nature is a high-grade tumor, so all are at least Stage II. These stages are further divided into A or B based on the size of the tumor with IIA/B being intra or extra-compartmental in the Enneking system or less than or greater than 8 cm in the AJCC system. In the Enneking system, Stage III implies metastatic disease; whereas, Stage III disease in the AJCC system is used to describe patients with skip metastases to the same bone, with no other sites of disease. Finally, stage IV in the AJCC implies distant metastases and is further subdivided based on the location. In general, a higher stage is suggestive of a poorer prognosis [5].
8 Treatment
Since the time of Ewing’s description of Ewing’s sarcoma, treatment and subsequent prognosis have improved dramatically. Surgery and radiation continue to play an important role in the control of local disease. However, major advances in survival have occurred with the addition of systemic chemotherapy. Prior to the use of systemic treatment, almost 80–90 % of patients would develop distant metastases despite the use of aggressive local control measures such as amputation [5]. Currently, the standard treatment of Ewing’s sarcoma of bone involves neoadjuvant chemotherapy, followed by local treatment with either surgery and/or radiation depending on tumor characteristics such as size, proximity to critical structures, and resectability. This is then followed by a course of adjuvant chemotherapy.
8.1 Chemotherapy
Since the majority of patients who present with a localized Ewing’s sarcoma will develop distant metastases with the use of local control alone, systemic chemotherapy is crucial to killing subclinical micrometastatic cells within the body in an attempt to cure.
Chemotherapy was first used to treat Ewing’s sarcoma in the early 1960s, when it was discovered that cyclophosphamide therapy provided a survival benefit [43, 44]. Subsequent randomized trials throughout the 1970s and 1980s looked at the benefit of adding additional systemic agents. These studies found that survival was increased with the use of multiagent regimens incorporating vincristine, doxorubicin, cyclophosphamide, and dactinomycin (VACD), with reported 5 year survival rates in the 50–60 % range for localized disease [45–48]. VACD therapy has become the mainstay of systemic treatment to this day. Subsequent studies have examined the benefit of using additional drugs, including ifosfamide and etoposide and have shown a modest improvement in survival. For example, Grier et al. showed an improvement in 5 year event-free survival from 54 to 69 % in patients who underwent alternating cycles of ifosfamide and etoposide with VACD, compared to VACD alone for those with localized disease. Interestingly, no improvement in survival has been shown for those who presented with metastatic disease [49, 50].
Despite significant improvement in survival with the use of multiagent therapies, patients who present with known metastases continue to have poor 5 year survival figures in the 25 % range [45]. One of the methods explored to overcome this was the use of dose-intensive regimens where chemotherapy cycles were given in either higher doses or more rapidly. Included in this was the use of very high-dose treatments with a subsequent bone marrow transplant. In general, these techniques subjected patients to very high toxicities and complications with very little survival benefit [51–53]. One area which has shown some promise is the use of granulocyte colony stimulating factor in between cycles of treatment in order to offset bone marrow toxicity and restore blood counts more rapidly to decrease the time between chemotherapy cycles [54].
Like other malignancies, the latest area of interest has been the application of targeted therapies. Given that Ewing’s sarcoma demonstrates common genetic translocations and abnormalities, it would seem an ideal disease for molecular therapies. However, much remains unknown about the role of the EWS fusion genes or their cellular pathways. As a result, it has been difficult to exploit the unique fusion protein in Ewing’s sarcoma as a target for treatment and at this time no drugs have been approved for clinical use outside of trials [55, 56]. Certain drugs have shown promise in clinical trials, such as molecular targets for the insulin-like growth factor-I receptor. These have demonstrated the ability to decrease or stabilize some tumors, but have had little effect on others [57]. Conversely, other drugs such as the tyrosine kinase inhibitor Imatinib, which has been so efficacious in other malignancies, has demonstrated little effect in the treatment of Ewing’s sarcoma [58]. Despite this, targeted therapy development continues to be an area of intense research.
8.2 Local Control
Ewing’s sarcoma of bone is unique compared to other common primary bone sarcomas such as osteosarcoma or chondrosarcoma in that it is very radiosensitive. This was an observation that initially helped James Ewing distinguish it from osteosarcoma [1]. As a result, prior to the use of routine chemotherapy, it was primarily treated with external beam radiation. Radiation was often successful in halting the progression of the tumor and even causing it to shrink. However, most patients eventually succumbed to metastatic disease. Since chemotherapy has significantly improved overall survival, the complications of radiation have become more apparent, especially since patients are treated at a young age.
Most patients who receive radiation therapy will receive a total dose of about 60 Gy fractionated over 6 weeks [59]. The main advantage of radiation is that it avoids the morbidity associated with surgical intervention. However, it is associated with many complications both short-term and long-term. The short-term side-effects are often transient and include dermatitis, fatigue, and nausea. The long-term effects include fracture, growth arrest, joint stiffness, and secondary malignancies, all of which can have devastating effects on function and are particularly concerning in skeletally immature patients [59–61].
In the past, surgical resection was recommended for “expendable” bones. With more data on the long-term effects of radiation, this opinion has evolved. Controversy has developed over what defined an “expendable” bone as well as which local treatment, radiation or surgery, results in improved survival and local control. Most of the data regarding this comes from the pelvis, since its anatomic location makes it difficult to resect with negative margins and equally difficult to reconstruct.
A multitude of studies have examined outcomes in pelvic Ewing’s sarcomas and they generally demonstrate improved survival and local control rates when surgical resection is performed compared to radiation alone [45, 62–67]. Yang et al. found that the overall survival in pelvic cases was 51 % with surgical resection compared to 18 % with radiation alone [62]. Similarily Frassica et al. showed 5 year overall survival was 75 % with surgery versus 25 % for radiation [63]. Local control also appears improved with surgery at 83 % compared to 67 % [66]. Surgical resection also has been shown to be superior to radiation alone with improved survival in the extremities [67, 68, 69].
However, caution should be used when interpreting these studies as there is definite selection bias in their design. All of them are retrospective, nonrandomized studies where radiation alone was often reserved for those cases where surgical resection with negative margins would be unlikely. Unfortunately, a randomized clinical trial evaluating this would be difficult to justify in light of existing data.
Therefore, the current treatment strategy employed by most orthopedic oncologists is to surgically resect Ewing’s sarcoma of bone when adequate margins are obtainable and the reconstructive result will leave the patient with a satisfactorily functional limb. If there are positive margins after resection, then postoperative radiation should be considered. Radiation alone is typically reserved for tumors where the resection offers no meaningful reconstructive options necessitating amputation, or in certain cases of certain pelvic or spinal tumors.
In terms of postresection reconstruction, modern techniques make limb-salvage feasible in the majority of cases. Common reconstructive options include the use of large endoprostheses, bulk allografts, and allograft-prosthetic composites (APCs).
While endoprostheses required custom manufacturing in the past, most are currently modular and can be assembled at the time of surgery using off-the-shelf components. These endoprostheses are typically reserved for tumors involving the metaphyseal or epiphyseal sections of bones in which the articular surface must be resected with the tumor. Therefore, these devices can be used to reconstruct the joint. Like all implants, they are prone to wear and failure over time and have high rates of infection depending on surgical and anatomic factors such as soft tissue coverage [70–72].
Bulk allografts are commonly employed for tumors in the diaphysis of long bones, where resection can be performed and an intercalary allograft can be put in its place (Figs. 2f,g). The advantage with this technique is that is spares the patient’s articular surfaces and once it is incorporated can allow for full activities. However, they also have high rates of complications including resorption, nonunions, fractures, and infections [73–75]. Another option employed at some centers, primarily in Asia, is to resect the involved bone, submit it to high extracorporeal doses of radiation to kill the tumor cells, and subsequently use this autograft bone to reconstruct the defect. This has been shown to result in good local control, but is associated with many of the same complications associated with cadaveric allografts [76].
Allograft-prosthetic composites are felt to be a compromise between allografts and endoprosthetics, in which articular segments are reconstructed with an allograft junction at the metaphysis and a prosthetic joint. These are most commonly employed in Ewing’s sarcomas of the pelvis whereby the bone is reconstructed with allograft and the hip is replaced with a total hip prosthesis. While they have advantages, they suffer from similar complications unique to both allografts and prostheses [77, 78].
8.3 Metastatic Disease
Patients with who present with metastatic disease or who develop it later have much worse survival outcomes. However, their survival can be improved with aggressive management of metastatic lesions, especially if there is only a single site. The most common sites of metastasis are the lungs, bone, and soft tissues [5, 6]. Lung metastases can be treated with thoracotomy if there is a single lesion, or whole lung radiation if there are multiple lesions [79]. Similarly, a single bony metastasis should be treated as if it is a primary tumor with radiation or surgery depending on its location.
9 Prognosis
Overall the prognosis of a patient diagnosed with Ewing’s sarcoma of bone has improved dramatically since it was first described. Prior to the use of systemic chemotherapy and local control, the overall survival was minimal at best. However, modern methods have increased the 5 year event-free survival statistics for those who present without metastases to the 55–75 % range, with overall survival being slightly less [45, 61, 62, 80–82]. Those who present with metastases or who subsequently develop a recurrence have much worse survival in the 20 % range. Also, not all metastases are equal, as metastases to the lungs have shown a survival advantage compared to those in bone [61].
In regards to negative prognostic factors, advanced age, large tumor volume, axial skeleton involvement, and lack of surgical resection have all been associated with worse outcomes [61, 80–82].
10 Summary
In summary, since first described by James Ewing almost 95 years ago, there has been a dramatic increase in our understanding and treatment of Ewing’s sarcoma. Despite major improvements in survival with systemic chemotherapy and local control with surgery and/or radiation, there continues to be much room for improvement, especially in those patients who present with metastases or develop a recurrence. Current research aims to characterize and target the EWS fusion protein in order to develop new treatments that will improve survival and reduce the toxicity and morbidity associated with current treatment options.
References
Ewing J (1921) Diffuse endothelioma of bone. Proc NY Pathol Soc. 21:17–24
Codman E (1925) Bone sarcoma: an interpretation of the nomenclature used by the committee on the Registry of Bone Sarcomas of the American College of Surgeons. Paul B. Hoeber, Inc., New York
Codman EA (2009) The Classic: The Registry of Bone Sarcomas as an example of the End-Result Idea in Hospital Organization. Clin Orthop Relat Res 467(11):2766–2770. doi:10.1007/s11999-009-1048-7
Codman E (1909) The use of the x-ray and radiation in surgery. In: Keen WB (ed) Surgery. Its principles and practice by various authors, vol 1909. WB Saunders, Philadelphia, p. 1170
Esiashvili N, Goodman M, Marcus RB Jr (2008) Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30(6):425–430. doi:10.1097/MPH.0b013e31816e22f3
Dorfman HD, Czerniak B (1995) Bone cancers. Cancer 75(1 Suppl):203–210
Stiller CA, Desandes E, Danon SE et al (2006) Cancer incidence and survival in European adolescents (1978–1997). Report from the Automated Childhood Cancer Information System project. Eur J Cancer Oxf Engl 1990 2(13):2006–2018. doi:10.1016/j.ejca.2006.06.002
Patricio MB, Vilhena M, Neves M et al (1991) Ewing’s sarcoma in children: twenty-five years of experience at the Instituto Portugês de Oncologia de Francisco Gentil (I.P.O.F.G.). J Surg Oncol 47(1):37–40
Widhe B, Widhe T (2000) Initial symptoms and clinical features in osteosarcoma and Ewing sarcoma. J Bone Joint Surg Am 82(5):667–674
Bacci G, Balladelli A, Forni C et al (2007) Ewing’s sarcoma family tumours. Differences in clinicopathological characteristics at presentation between localised and metastatic tumours. J Bone Joint Surg Br 89(9):1229–1233. doi:10.1302/0301-620X.89B9.19422
Rutkowski P, Kamińska J, Kowalska M, Ruka W, Steffen J (2003) Cytokine and cytokine receptor serum levels in adult bone sarcoma patients: correlations with local tumor extent and prognosis. J Surg Oncol 84(3):151–159. doi:10.1002/jso.10305
Nakamura T, Grimer RJ, Gaston CL, Watanuki M, Sudo A, Jeys L (2013) The prognostic value of the serum level of C-reactive protein for the survival of patients with a primary sarcoma of bone. Bone Jt J 95-B(3):411–418. doi:10.1302/0301-620X.95B3.30344
Ilić I, Manojlović S, Cepulić M, Orlić D, Seiwerth S (2004) Osteosarcoma and Ewing’s sarcoma in children and adolescents: retrospective clinicopathological study. Croat Med J 45(6):740–745
Biermann JS, Adkins DR, Agulnik M et al (2013) Bone cancer. J Natl Compr Cancer Netw JNCCN 11(6):688–723
Suh C-H, Ordóñez NG, Hicks J, Mackay B (2002) Ultrastructure of the Ewing’s sarcoma family of tumors. Ultrastruct Pathol 26(2):67–76. doi:10.1080/01913120252959236
Fellinger EJ, Garin-Chesa P, Triche TJ, Huvos AG, Rettig WJ (1991) Immunohistochemical analysis of Ewing’s sarcoma cell surface antigen p30/32MIC2. Am J Pathol 139(2):317–325
Folpe AL, Goldblum JR, Rubin BP et al (2005) Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol 29(8):1025–1033
Delattre O, Zucman J, Plougastel B et al (1992) Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 359(6391):162–165. doi:10.1038/359162a0
Delattre O, Zucman J, Melot T et al (1994) The Ewing family of tumors—a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331(5):294–299. doi:10.1056/NEJM199408043310503
Peter M, Mugneret F, Aurias A, Thomas G, Magdelenat H, Delattre O (1996) An EWS/ERG fusion with a truncated N-terminal domain of EWS in a Ewing’s tumor. Int J Cancer J Int Cancer 67(3):339–342. doi:10.1002/(SICI)1097-0215(19960729)67:3<339:AID-IJC6>3.0.CO;2-S
Wang L, Bhargava R, Zheng T et al (2007) Undifferentiated small round cell sarcomas with rare EWS gene fusions: identification of a novel EWS-SP3 fusion and of additional cases with the EWS-ETV1 and EWS-FEV fusions. J Mol Diagn JMD 9(4):498–509. doi:10.2353/jmoldx.2007.070053
Shulman SC, Katzenstein H, Bridge J et al (2012) Ewing sarcoma with 7;22 translocation: three new cases and clinicopathological characterization. Fetal Pediatr Pathol 31(6):341–348. doi:10.3109/15513815.2012.659397
De Alava E, Kawai A, Healey JH et al (1998) EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. J Clin Oncol Off J Am Soc Clin Oncol 16(4):1248–1255
Ginsberg JP, Alava E de, Ladanyi M et al (1999) EWS-FLI1 and EWS-ERG gene fusions are associated with similar clinical phenotypes in Ewing’s Sarcoma. J Clin Oncol 17(6):1809
Le Deley M-C, Delattre O, Schaefer K-L et al (2010) Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol Off J Am Soc Clin Oncol 28(12):1982–1988. doi:10.1200/JCO.2009.23.3585
Wagner LM, Smolarek TA, Sumegi J, Marmer D (2012) Assessment of minimal residual disease in Ewing sarcoma. Sarcoma 2012:780129. doi:10.1155/2012/780129
Dubois SG, Epling CL, Teague J, Matthay KK, Sinclair E (2010) Flow cytometric detection of Ewing sarcoma cells in peripheral blood and bone marrow. Pediatr Blood Cancer 54(1):13–18. doi:10.1002/pbc.22245
Bridge RS, Rajaram V, Dehner LP, Pfeifer JD, Perry A (2006) Molecular diagnosis of Ewing sarcoma/primitive neuroectodermal tumor in routinely processed tissue: a comparison of two FISH strategies and RT-PCR in malignant round cell tumors. Mod Pathol Off J U S Can Acad Pathol Inc 19(1):1–8. doi:10.1038/modpathol.3800486
Thorner P, Squire J, Chilton-MacNeil S et al (1996) Is the EWS/FLI-1 fusion transcript specific for Ewing sarcoma and peripheral primitive neuroectodermal tumor? A report of four cases showing this transcript in a wider range of tumor types. Am J Pathol 148(4):1125–1138
Kuleta-Bosak E, Kluczewska E, Machnik-Broncel J et al (2010) Suitability of imaging methods (X-ray, CT, MRI) in the diagnostics of Ewing’s sarcoma in children—analysis of own material. Pol J Radiol Pol Med Soc Radiol 75(1):18–28
Peersman B, Vanhoenacker FM, Heyman S et al (2007) Ewing’s sarcoma: imaging features. JBR-BTR Organe Société R Belge Radiol SRBR Orgaan Van K Belg Ver Voor Radiol KBVR 90(5):368–376
Van der Woude HJ, Bloem JL, Hogendoorn PC (1998) Preoperative evaluation and monitoring chemotherapy in patients with high-grade osteogenic and Ewing’s sarcoma: review of current imaging modalities. Skeletal Radiol 27(2):57–71
Eary JF, Mankoff DA (1998) Tumor metabolic rates in sarcoma using FDG PET. J Nucl Med Off Publ Soc Nucl Med 39(2):250–254
Eary JF, O’Sullivan F, Powitan Y et al (2002) Sarcoma tumor FDG uptake measured by PET and patient outcome: a retrospective analysis. Eur J Nucl Med Mol Imaging 29(9):1149–1154. doi:10.1007/s00259-002-0859-5
Sharma P, Khangembam BC, Suman KCS et al (2013) Diagnostic accuracy of 18F-FDG PET/CT for detecting recurrence in patients with primary skeletal Ewing sarcoma. Eur J Nucl Med Mol Imaging 40(7):1036–1043. doi:10.1007/s00259-013-2388-9
Newman EN, Jones RL, Hawkins DS (2013) An evaluation of [F-18]-fluorodeoxy-D-glucose positron emission tomography, bone scan, and bone marrow aspiration/biopsy as staging investigations in Ewing sarcoma. Pediatr Blood Cancer 60(7):1113–1117. doi:10.1002/pbc.24406
Treglia G, Salsano M, Stefanelli A, Mattoli MV, Giordano A, Bonomo L (2012) Diagnostic accuracy of 18F-FDG-PET and PET/CT in patients with Ewing sarcoma family tumours: a systematic review and a meta-analysis. Skeletal Radiol 41(3):249–256. doi:10.1007/s00256-011-1298-9
Kumar J, Seith A, Kumar A et al (2008) Whole-body MR imaging with the use of parallel imaging for detection of skeletal metastases in pediatric patients with small-cell neoplasms: comparison with skeletal scintigraphy and FDG PET/CT. Pediatr Radiol 38(9):953–962. doi:10.1007/s00247-008-0921-y
Franzius C, Sciuk J, Daldrup-Link HE, Jürgens H, Schober O (2000) FDG-PET for detection of osseous metastases from malignant primary bone tumours: comparison with bone scintigraphy. Eur J Nucl Med 27(9):1305–1311
Chan RC, Sutow WW, Lindberg RD, Samuels ML, Murray JA, Johnston DA (1979) Management and results of localized Ewing’s sarcoma. Cancer 43(3):1001–1006
Enneking WF, Spanier SS, Goodman MA (1980) A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop 153:106–120
American Joint Committee on Cancer (2010) AJCC cancer staging manual, 7th edn. Springer, New York
Sutow WW, Sullivan MP (1962) Cyclophosphamide therapy in children with Ewing’s sarcoma. Cancer Chemother Rep 23:55–60
Pinkel D (1962) Cyclophosphamide in children with cancer. Cancer 15:42–49
Donaldson SS, Torrey M, Link MP et al (1998) A multidisciplinary study investigating radiotherapy in Ewing’s sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 42(1):125–135
Bacci G, Toni A, Avella M et al (1989) Long-term results in 144 localized Ewing’s sarcoma patients treated with combined therapy. Cancer 63(8):1477–1486
Bacci G, Ferrari S, Avella M et al (1991) Non-metastatic Ewing’s sarcoma: results in 98 patients treated with neoadjuvant chemotherapy. Ital J Orthop Traumatol 17(4):449–465
Nesbit ME Jr, Gehan EA, Burgert EO Jr et al (1990) Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup study. J Clin Oncol Off J Am Soc Clin Oncol. 8(10):1664–1674
Grier HE, Krailo MD, Tarbell NJ et al (2003) Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348(8):694–701. doi:10.1056/NEJMoa020890
Miser JS, Krailo MD, Tarbell NJ et al (2004) Treatment of metastatic Ewing’s sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide—a Children’s Cancer Group and Pediatric Oncology Group study. J Clin Oncol Off J Am Soc Clin Oncol. 22(14):2873–2876. doi:10.1200/JCO.2004.01.041
Laurence V, Pierga J-Y, Barthier S et al (2005) Long-term follow up of high-dose chemotherapy with autologous stem cell rescue in adults with Ewing tumor. Am J Clin Oncol 28(3):301–309
Kushner BH, Meyers PA (2001) How effective is dose-intensive/myeloablative therapy against Ewing’s sarcoma/primitive neuroectodermal tumor metastatic to bone or bone marrow? The Memorial Sloan-Kettering experience and a literature review. J Clin Oncol Off J Am Soc Clin Oncol 19(3):870–880
Navid F, Santana VM, Billups CA et al (2006) Concomitant administration of vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide for high-risk sarcomas: the St Jude Children’s Research Hospital experience. Cancer 106(8):1846–1856. doi:10.1002/cncr.21810
Womer RB, Daller RT, Fenton JG, Miser JS (2000) Granulocyte colony stimulating factor permits dose intensification by interval compression in the treatment of Ewing’s sarcomas and soft tissue sarcomas in children. Eur J Cancer Oxf Engl 1990 36(1):87–94
Ludwig JA (2008) Ewing sarcoma: historical perspectives, current state-of-the-art, and opportunities for targeted therapy in the future. Curr Opin Oncol 20(4):412–418. doi:10.1097/CCO.0b013e328303ba1d
Lissat A, Chao MM, Kontny U (2012) Targeted therapy in Ewing sarcoma. ISRN Oncol 2012:609439. doi:10.5402/2012/609439
Olmos D, Postel-Vinay S, Molife LR et al (2010) Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751, 871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol 11(2):129–135. doi:10.1016/S1470-2045(09)70354-7
Bond M, Bernstein ML, Pappo A et al (2008) A phase II study of imatinib mesylate in children with refractory or relapsed solid tumors: a Children’s Oncology Group study. Pediatr Blood Cancer 50(2):254–258. doi:10.1002/pbc.21132
Donaldson SS (2004) Ewing sarcoma: radiation dose and target volume. Pediatr Blood Cancer 42(5):471–476. doi:10.1002/pbc.10472
Dalinka MK, Edeiken J, Finkelstein JB (1974) Complications of radiation therapy: adult bone. Semin Roentgenol 9(1):29–40
Cotterill SJ, Ahrens S, Paulussen M et al (2000) Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol Off J Am Soc Clin Oncol 18(17):3108–3114
Yang RS, Eckardt JJ, Eilber FR et al (1995) Surgical indications for Ewing’s sarcoma of the pelvis. Cancer 76(8):1388–1397
Frassica FJ, Frassica DA, Pritchard DJ, Schomberg PJ, Wold LE, Sim FH (1993) Ewing sarcoma of the pelvis. Clinicopathological features and treatment. J Bone Joint Surg Am 75(10):1457–1465
Thorpe SW, Weiss KR, Goodman MA, Heyl AE, McGough RL (2012) Should aggressive surgical local control be attempted in all patients with metastatic or pelvic Ewing’s sarcoma? Sarcoma 2012:953602. doi:10.1155/2012/953602
Puri A, Gulia A, Jambhekar NA, Laskar S (2012) Results of surgical resection in pelvic Ewing’s sarcoma. J Surg Oncol 106(4):417–422. doi:10.1002/jso.23107
Donati D, Yin J, Di Bella C et al (2007) Local and distant control in non-metastatic pelvic Ewing’s sarcoma patients. J Surg Oncol 96(1):19–25. doi:10.1002/jso.20752
Sucato DJ, Rougraff B, McGrath BE et al (2000) Ewing’s sarcoma of the pelvis. Long-term survival and functional outcome. Clin Orthop 373:193–201
Bacci G, Ferrari S, Longhi A et al (2004) Role of surgery in local treatment of Ewing’s sarcoma of the extremities in patients undergoing adjuvant and neoadjuvant chemotherapy. Oncol Rep 11(1):111–120
Sluga M, Windhager R, Lang S et al (2001) The role of surgery and resection margins in the treatment of Ewing’s sarcoma. Clin Orthop 392:394–399
Henderson ER, Groundland JS, Pala E et al (2011) Failure mode classification for tumor endoprostheses: retrospective review of five institutions and a literature review. J Bone Joint Surg Am 93(5):418–429. doi:10.2106/JBJS.J.00834
Horowitz SM, Glasser DB, Lane JM, Healey JH (1993) Prosthetic and extremity survivorship after limb salvage for sarcoma. How long do the reconstructions last? Clin Orthop 293:280–286
Malawer MM, Chou LB (1995) Prosthetic survival and clinical results with use of large-segment replacements in the treatment of high-grade bone sarcomas. J Bone Joint Surg Am 77(8):1154–1165
Mankin HJ, Gebhardt MC, Jennings LC, Springfield DS, Tomford WW (1996) Long-term results of allograft replacement in the management of bone tumors. Clin Orthop 324:86–97
Berrey BH Jr, Lord CF, Gebhardt MC, Mankin HJ (1990) Fractures of allografts. Frequency, treatment, and end-results. J Bone Joint Surg Am 72(6):825–833
Getty PJ, Peabody TD (1999) Complications and functional outcomes of reconstruction with an osteoarticular allograft after intra-articular resection of the proximal aspect of the humerus. J Bone Joint Surg Am 81(8):1138–1146
Hong AM, Millington S, Ahern V et al (2013) Limb preservation surgery with extracorporeal irradiation in the management of malignant bone tumor: the oncological outcomes of 101 patients. Ann Oncol Off J Eur Soc Med Oncol ESMO 24(10):2676–2680. doi:10.1093/annonc/mdt252
Benedetti MG, Bonatti E, Malfitano C, Donati D (2013) Comparison of allograft-prosthetic composite reconstruction and modular prosthetic replacement in proximal femur bone tumors: functional assessment by gait analysis in 20 patients. Acta Orthop 84(2):218–223. doi:10.3109/17453674.2013.773119
Donati D, Di Bella C, Frisoni T, Cevolani L, DeGroot H (2011) Alloprosthetic composite is a suitable reconstruction after periacetabular tumor resection. Clin Orthop 469(5):1450–1458. doi:10.1007/s11999-011-1799-9
Bölling T, Schuck A, Paulussen M et al (2008) Whole lung irradiation in patients with exclusively pulmonary metastases of Ewing tumors. Toxicity analysis and treatment results of the EICESS-92 trial. Strahlenther Onkol Organ Dtsch Röntgenges Al 4(4):193–197. doi:10.1007/s00066-008-1810-x
Rodríguez-Galindo C, Navid F, Liu T, Billups CA, Rao BN, Krasin MJ (2008) Prognostic factors for local and distant control in Ewing sarcoma family of tumors. Ann Oncol Off J Eur Soc Med Oncol ESMO 19(4):814–820. doi:10.1093/annonc/mdm521
Ahrens S, Hoffmann C, Jabar S et al (1999) Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol 32(3):186–195
Lee J, Hoang BH, Ziogas A, Zell JA (2010) Analysis of prognostic factors in Ewing sarcoma using a population-based cancer registry. Cancer 116(8):1964–1973. doi:10.1002/cncr.24937
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Moore, D.D., Haydon, R.C. (2014). Ewing’s Sarcoma of Bone. In: Peabody, T., Attar, S. (eds) Orthopaedic Oncology. Cancer Treatment and Research, vol 162. Springer, Cham. https://doi.org/10.1007/978-3-319-07323-1_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-07323-1_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-07322-4
Online ISBN: 978-3-319-07323-1
eBook Packages: MedicineMedicine (R0)