
Abstract
Improved understanding of tumor biology through molecular alteration and genetic advances has resulted in a number of major changes in the 2020 World Health Organization’s (WHO) classification of bone tumors. These changes include the reclassification of the existing tumors and the introduction of several new entities. A new chapter on undifferentiated small round cell sarcomas of bone and soft tissue was added to classify Ewing sarcoma and the family of Ewing-like sarcomas, which share similar histologies but different molecular and clinical behaviors. Knowledge of the current classification of bone tumors is essential to ensure the appropriate recognition of the inherent biological potential of individual osseous lesions for optimal treatment, follow-up, and overall outcome. This article reviews the major changes to the 2020 WHO’s classification of primary bone tumors and the pertinent imaging of selected tumors to highlight these changes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reproducible and consistent diagnostic criteria are essential for accurate classification and proper clinical management of bone tumors. Since 1967, the World Health Organization (WHO) classification of tumors has provided practical guidance to pathologists, radiologists, and clinicians involved in oncologic multidisciplinary teams [1]. Improved understanding of tumor genetics led the WHO to reclassify selected bone tumors in 2020. Although pathology sets the gold standard for bone tumor diagnosis, radiologic-pathologic correlation remains an essential component in tumor evaluation and is crucial to minimizing diagnostic error and achieving optimal clinical outcomes. Therefore, up-to-date knowledge is essential to ensure optimal recognition of the biological behavior of bone tumors and consistent oncologic treatment. This article reviews major changes to the 2020 WHO’s classification of primary bone tumors and pertinent imaging of selected tumors to enhance such up-to-date knowledge.
Classification based on histologic families and behavioral categories
The 2020 WHO classification of bone tumors includes eight histologic families of bone tumors (Table 1). Each family is further classified into individual tumor types based on histologic, immunohistochemical, and molecular characteristics. The WHO further classifies bone tumors into four categories based on biological behavior, including the risk for local recurrence and metastasis. These are benign, intermediate (locally aggressive), intermediate (rarely metastasizing), and malignant (Table 2) [1, 2]. Knowing such categories of biological behavior is crucial for effective clinical treatment, surgical planning, and surveillance.
In the current 5th ed, the WHO has deleted a few families of the tumors from the previous classification (Table 3). It added the new family “other mesenchymal tumors,” which includes some of the entities from the deleted family tumor types (Tables 1, 3). Beyond primary lymphoma and solitary plasmacytoma of bone, the family of lesions termed “hematopoietic neoplasm of bone” now also includes Langerhans cell histiocytosis, Erdheim-Chester disease, and Rosai-Dorfman disease.
The “undifferentiated small round cell sarcomas of bone and soft tissue tumors” is a new category that includes small round cell tumors with similar histologic features but distinct molecular and clinical behaviors (Table 4). These include Ewing sarcoma, round cell sarcoma with EWSR1-non-ETS fusion, CIC-rearranged sarcoma, and sarcoma with BCOR genetic alteration. The three new entities added to prototypical Ewing Sarcoma occur either in bone or soft tissue, with prevalence to one or other depending on their molecular characteristics.
Chondrogenic tumor
Enchondroma and periosteal chondroma, previously listed together under “chondromas,” are now separate benign entities. Chondroblastoma, previously categorized intermediate (rarely metastasizing), is reclassified benign due to its favorable outcomes and low surgical recurrence rate (≤ 5%) without distant metastases [4]. Chondromyxoid fibroma, previously labeled intermediate and locally aggressive, is also reclassified benign due to its excellent clinical prognosis despite a wide range (3–22%) of local recurrence rates [5, 6]. Conversely, synovial chondromatosis has shifted from benign to intermediate (locally aggressive) due to its propensity for local recurrence and unfavorable clinical outcomes.
Synovial chondromatosis
Synovial chondromatosis (SC) is a neoplasm that produces hyaline cartilaginous nodules in joints or extra-articular synovium, most often in the third to fifth decades, with a propensity for men [7]. In a study of 20 patients with SC in the knee, the recurrence rate was 10%, and one patient underwent amputation due to malignant transformation [8]. In a study of 26 patients with SC of the hip treated with total hip arthroplasty, the recurrence rate was 12%. Complication rate was high (50%), with the most common complication being aseptic loosening [9]. Malignant transformation of SC to synovial chondrosarcoma has been reported in 1–6.4%, with a median transformation time of 20 years from diagnosis [10]. Similar clinico-radiologic presentation and histologic overlap render the distinction of SC from chondrosarcoma challenging [10].
Imaging appearance of primary SC is characterized by multiple calcifications of similar size and shape with calcified chondroid matrix (70–95%) and extrinsic erosion (20–50%) [11]. Because enchondral ossification is not present in all cartilage nodules, the extent of calcification and fat in SC varies, accounting for distinct MRI appearance (Figs. 1, 2) [11]. The cartilage nodules show intermediate T1 and high T2 signals in the absence of calcification, as well as low signal in all pulse sequences in the presence of enchondral ossification [11]. It is challenging to differentiate recurrent SC and malignant chondrosarcomatous transformation as they appear similar. However, marrow invasion and cortical destruction in the setting of multiple recurrences may help identify chondrosarcomatous transformation (Fig. 3) [11].

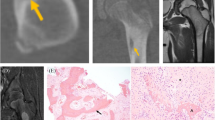
A 25-year-old man with synovial chondromatosis. (A) Axial CT image shows calcified nodules. (B) Axial T1-weighted image shows highT1 signal (arrow) consistent with marrow fat in the nodule

A 13-year-old boy with synovial chondromatosis. (A) AP view of the left hip shows a lytic lesion in the left femoral neck causing a thin sclerotic rim (arrows) and without soft tissue calcifications. (B) Coronal T2-weighted fat-suppressed image of the left hip shows diffusely high T2 signal lesion, which causes considerable erosion (arrows) in the left femoral neck. Due to lack of calcification, synovial chondromatosis mimics periosteal chondroma

A 30-year-old woman with chondrosarcomatous transformation of synovial chondromatosis following three synovectomies four years after the initial diagnosis. The most recent synovectomy reveals chondrosarcoma grade 2 arising from synovial chondromatosis. Sagittal T2-weighted fat-suppressed image shows multiple chondroid nodules causing multifocal bone marrow invasion (arrows) in the intercondylar femur and tibial spine
Enchondroma vs. central atypical cartilaginous tumor/chondrosarcoma grade 1
Enchondromas are usually discovered in the 3rd and 4th decades of life [12]. They most likely result from a separated fragment of the physis, which is incorporated into the metaphysis during growth and development, and a fragment becomes more metadiaphyseal or diaphyseal as growth continues [13, 14]. Enchondromas are commonly detected as incidental lesions on MRI. Reported prevalence of enchondroma at MRI includes 2.8% in knees [15], 0.7% in proximal femora [16], and 2.2% in shoulders [17]. Enchondromas have variable imaging appearance depending on the location of the lesions within the bones as well as within the skeleton and skeletal maturity [14, 18]. In the diaphysis of long bones, enchondroma may contains varying amounts of ring and arc-like mineralized matrix, variable rim sclerosis, and may also show mild endosteal scalloping.
Central atypical cartilaginous tumor (ACT) and chondrosarcoma grade 1 (CS1) are designations used to describe histologically identical chondroid lesions, which the WHO now distinguishes based on their anatomic location and the differences in the clinical outcomes associated with their anatomic location. Both occur in adults of a wide age range (median: 49 years) [19]. The designation intermediate (locally aggressive) ACT is reserved for the long and short tubular bones, while the designation CS1 is reserved for the axial skeleton and flat bones. ACT/CS1 most commonly occurs in the femur, the humerus, and flat bones like the ilium and rarely the short tubular bones [19]. The distinction between enchondroma and ACT/CS1 remains challenging due to the lack of diagnostic criteria and poor interobserver variability amongst pathologists, radiologists, and surgical oncologists [18,19,20,21].
The prevalence of cartilaginous tumors, especially enchondroma and ACT, has grown due to increased medical imaging [22,23,24]. Davies et al. reported a 68% increase in annual referral rate from 1985–2018 primarily due to ACT with no increase in higher-grade CS, osteosarcoma, or Ewing sarcoma [23]. There has been a growing consensus favoring surveillance of enchondroma/ACT due to low risk of higher grade transformation (< 1%) and improved functional outcome through observation alone [22, 23, 25].
Imaging features that distinguish enchondroma from ACT/CS1 include tumor size (> 5 cm), deep endosteal scalloping (> 2/3 of cortical thickness), expansile bone remodeling, cortical destruction, cortical thickening, soft tissue extension, radiotracer uptake in a bone scan, and pain [26,27,28,29]. Douis et al. reported that endosteal scalloping > 2/3 of the cortex by MRI is highly effective in distinguishing enchondroma from ACT/CS1 (Fig. 4) [28]. In their meta-analysis of 14 MRI studies, Deckers et al. added loss of entrapped fat as signs of high-grade chondrosarcoma on MRI [30]. Another study by Deckers et al. demonstrated that 87% of lesions remained stable or showed regression on MRI, and the marrow fat signal was present in 87% of lesions that underwent regression [22]. Therefore, the presence of marrow fat and lack of deep endosteal scalloping potentially differentiate enchondromas from ACT or signal regression of cartilaginous lesions (Fig. 5). Brien et al. proposed that a confluent mass histologically led to CS, whereas enchondromas grew as clustered cartilage deposits without forming a single mass [13]. Studies show variable utility of dynamic contrast-enhanced MRI in distinguishing enchondroma from ACT, while diffusion-weighted imaging shows little value [18, 29].

A 46-year-old woman with atypical cartilaginous tumor and pain in the knee. (A) T2-weighted fat-suppressed images shows a chondroid lesion in the distal femoral metaphysis with marked endosteal scalloping (arrows) in the posterior cortex. (B) On the sagittal CT image, the lesion contains multiple curvilinear calcifications with deep endosteal scalloping (arrows) in the anterior and posterior cortex. Bone scan shows mild uptake in the left distal femur (not shown). Pain resolved after curettage of the lesion

A 57-year-old woman with regressing enchondroma. (A) Coronal T1-weighted and (B) coronal T2-weighted fat-suppressed images of MRI performed 15 years ago, showed a lobulated chondroid lesion occupying the entire medullary canal and without endosteal scalloping in the proximal humerus. Fat (arrow) was interspersed between chondroid lobules. Bone scan showed radiotracer uptake (not shown). In the current MRI, (C) coronal T1-weighted shows increased fat (arrows) between lobules, consistent with what has been described as a regressing enchondroma
Central chondrosarcoma grades 2, 3
The location of high-grade chondrosarcoma is similar to that of ACT/CS1. Half of high-grade chondrosarcoma share IDH1 or IDH2 mutations with enchondroma/ACT, suggesting a clonal relationship with enchondroma or ACT/CS grade 1 [2]. The MRI features distinguishing high-grade chondrosarcoma from low-grade CS include loss of entrapped marrow fat, cortical destruction, and extraosseous extension (Fig. 6). The value of contrast enhancement and diffusion-weighted imaging remains clinically inconclusive [28, 30, 31].

A 27-year-old woman with grade 2 and 3 chondrosarcoma in the right ilium. (A) Axial CT image shows a lytic lesion causing endosteal scalloping, expansile remodeling, and cortical destruction with soft tissue extension (arrowheads) in the right sacral neuroforamen and gluteal muscles. The lesion contains multiple linear and curvilinear calcifications (arrows). (B) Axial T2-weighted fat-suppressed image shows multiple high-T2 signal lobules without internal fat, extraosseous mass (arrowheads) with sacral marrow edema (arrow)
Secondary peripheral ACT/CS1 and secondary peripheral high-grade chondrosarcoma
Secondary peripheral CS arises in the cartilaginous cap of an osteochondroma. The vast majority (> 90%) are low-grade chondrosarcoma [2]. Currently, 2.0 cm is the proposed cut-off measurement for the thickest portion of the cartilaginous cap as perpendicular to the tidemark, to distinguish secondary chondrosarcoma from osteochondroma [32, 33]. The current WHO classification proposes that tumors in the appendicular skeleton can be called secondary peripheral ACT, and tumors in the axial skeleton can be called peripheral CS1 [32].
Osteogenic tumors
Osteoid osteoma and osteoblastoma
Osteoid osteoma (OO) and osteoblastoma are distinct bone forming lesions that, when small, can have similar microscopic features. The lesions have different biological potentials with OO (benign) and osteoblastoma (intermediate, locally aggressive), reflecting several differences in clinical and radiologic presentations. The usual age of onset is in children and adolescents for OO and the 2nd–3rd decades of life for osteoblastoma [34, 35]. Typical locations for OO and osteoblastoma are long bones and posterior spinal elements, but flat bones are more common in osteoblastoma [34, 35]. Pain is usually relieved with NSAIDs in OO but not in osteoblastoma, and growth potential is limited in OO while increased in OB [34, 35]. Currently these tumors are diagnosed according to their size; lesions less than 2 cm are diagnosed as OO, and those 2 cm or larger as osteoblastoma [34, 35].
Jaffee initially described OO as a lesion consisting of two components: the “core or nidus-like focus (the osteoid-osteoma proper)” and the peripheral bone thickening [36]. However, now the term “nidus” is typically used to describe the entire lesion, including the mineralized center and non-mineralized peripheral zone [37]. OO typically appears as a lytic lesion with or without central calcification, cortical thickening, periosteal reaction at CT, and perilesional edema at MRI. However, when OO is intraarticular, cortical thickening may be minimal due to the lack of a periosteal layer to produce bone [36]. In contrast, osteoblastoma presents as a lytic, sclerotic, or mixed lesion with perilesional edema (> 90%) on MRI, and it can present with fractures and soft tissue extension [38].
Osteosarcoma
The WHO now reclassifies osteosarcoma (OS) into six subtypes: OS not otherwise specified (NOS), low-grade central OS, parosteal OS, periosteal OS, high-grade surface OS, and secondary OS. Osteosarcoma NOS includes three subtypes: conventional OS, telangiectatic OS and small cell OS [39]. Conventional OS account for the majority of OS (93% of all OS) and telangiectatic OS (TOS) (4.5%), and small cell OS (< 1%) are rare [40]. Based on dominant matrix, OS NOS can be subdivided into osteoblastic, chondroblastic, and fibroblastic histologic types, resulting in variable appearance of the tumor at imaging. However, there is no relationship between the histologic patterns and prognosis [39]. Metaphyses of long bones are common sites for conventional OS and TOS with male predilection, while diaphyses of long bones are affected by small cell OS [39]. Secondary osteosarcoma is now classified as a separate entity and categorized into six subtypes based on underlying conditions: Paget disease, radiation-associated OS, infarct-related OS, OS due to chronic osteomyelitis, implant-related OS, and OS secondary to early postzygotic disorders such as fibrous dysplasia [41]. Imaging studies are crucial to assess underlying conditions.
Fibrogenic tumors
This category remains unchanged and includes desmoplastic fibroma and fibrosarcoma of bone. Desmoplastic fibroma is an intermediate (locally aggressive) entity typically occurring in the mandible and long bones [42]. The tumor is expansile and lytic, often with marginal sclerosis on radiograph and decreased T1 and heterogeneous T2 signals on MRI [42, 43]. Fibrosarcoma is a diagnosis of exclusion because it shares similar spindle cell histology with other sarcomas [44].
Vascular tumors
Primary vascular tumors of bone include hemangioma, epithelioid hemangioma (EH), epithelioid hemangioendothelioma (EHE), and angiosarcoma. Although these tumors may behave benign (hemangioma), intermediate (EH), or malignant (EHE, angiosarcoma), overlap among their histologic and imaging features result in diagnostic challenges [45].
Previously labeled an intermediate locally aggressive and rarely metastasizing tumor, EH was reclassified as an intermediate locally aggressive tumor [46]. Some regional nodal and soft tissue involvement cases have been reported, but the prognosis remains good without distant metastases or disease-related death [45,46,47]. EH usually occurs in adults with a slight male predilection. Typical locations are the long bones, followed by flat bones and spine. EH is multifocal in 18–25% of cases [2, 47]. The tumor typically presents as a well-defined, expansile lytic lesion causing bone erosion with bony septae (Fig. 7) [45, 47]. MRI appearance is similar to other vascular tumors, with high T2 signal and contrast enhancement (Fig. 7), and multifocality of bone lesions at imaging is a helpful feature [45, 47].

A 66-year-old woman with an epithelioid hemangioma. (A) Coronal T2-weighted fat-suppressed image shows increased signal with mild soft tissue extension (arrow). There is a small amount of fluid in the iliopsoas bursa (IB). (B) Corona CT image shows diffuse contrast enhancement and thickened trabeculae (arrow) in the lesion, as well as mild soft tissue extension (arrowhead). Bone scan shows radiotracer uptake in the lesion (not shown)
Osteoclastic giant cell-rich tumors
This family of lesions contains entities that are osteoclast-rich lesions and includes non-ossifying fibroma (NOF), aneurysmal bone cyst (ABC), giant cell tumor of bone (GCTB), and malignant GCTB (MGCTB). The new changes include the deletion of the “giant cell lesion of small bones,” which is no longer addressed by the WHO, and the addition of ABC and non-ossifying fibroma, which are readily diagnosed at imaging. The previous giant cell lesion of small bones is considered a solid variant of the ABC in the current WHO classification [2, 49], and the terminology “giant cell lesion of small bones” and the related “giant cell reparative granuloma of small bone” are not recommend.
The distinction of primary and secondary ABC has often been problematic; however, the identification of the USP6 gene rearrangement now serves as a diagnostic marker seen in 70% of primary ABC [2, 49]. While the distinction of primary and secondary lesions is often apparent on imaging, identifying the USP6 gene rearrangement imaging findings can be crucial in confirming the diagnosis as the clinical management of primary and secondary lesions may be radically different.
Giant cell tumor of bone
GCTB is an intermediate, locally aggressive, rarely metastasizing tumor typically affecting the ends of long bones, such as the distal femur and proximal tibia. The WHO added MGCTB as a separate malignant entity which may be primary or secondary, and MGCTB accounts for < 10% of all GCTB [2, 50]. Secondary MGCTB is more common than primary, accounting for 62% of MGCTB, while primary MGCTB has a more favorable prognosis [2, 51]. On imaging, GCTB is typically an eccentrically located well-circumscribed lytic lesion that arises in the metaphysis and extends toward the articular cartilage. Still, there is no specific radiographic or cross-sectional imaging appearance to differentiate GCTB from MGCTB [51]. In secondary MGCTB, mixed lytic and sclerotic appearance, dense ossification, or calcification can be seen likely due to osteosarcoma arising from treated GCTB [52]. Malignancy arising from GCTB treated with denosumab has been also reported [2, 50].
Notochordal tumors
The family contains benign notochordal cell tumor and chordoma. The WHO now classifies chordoma into conventional chordoma, dedifferentiated chordoma (DC), and poorly differentiated chordoma (PDC) [53,54,55]. PDC is a new subtype, occurring in children and young adults, with a worse prognosis than conventional chordoma [55,56,57]. Conventional chordomas predominantly occur in the skull base, spine, and sacrococcygeal bones [53]. The typical location of DC is sacrococcygeal and PDC in the skull base [54, 55].
Chordoma is a midline lytic lesion with cortical destruction, soft tissue extension, and occasional calcification. Multilevel involvement is typical and pathological fractures are more frequent in the spine than in other sites [58]. Chordoma tumor exhibits isointense T1 signal and hyperintense T2 signal on MRI with heterogeneous contrast enhancement. Olson et al. reported that foci of hyperintense T1 signal are common (72% of cases), probably secondary to hemorrhage or proteinaceous material and multilevel involvement (86%) in the spine [58]. Chordomas show moderate metabolic activities (SUVmax ≥ 5) on PET, but there was no relationship between tumor size and metabolic activity to local recurrence or metastatic disease. Nor was there a statistically significant association between the degree of contrast enhancement or enhancement pattern on MRI and SUVmax on PET [58].
The imaging appearance of DC and PDC is similar to conventional chordoma in terms of bone destruction and soft tissue mass. However, DC typically shows bimorphic appearance depicting conventional and dedifferentiated components on MRI (Fig. 8). PDC generally demonstrates avid contrast enhancement and intermediate T2 signal than conventional chordoma (Fig. 9) [57].

A 73-year-old male with a dedifferentiated chordoma. (A) T2-weighted fat-suppressed, and (B) T1-weighted post-contrast images show a large destructive lesion in the sacrum with presacral and epidural extraosseous extension. The lesion has a bimorphic appearance; the caudal component (s) is solid with diffuse contrast enhancement and the cranial component (f) near the sacrum shows fluid-like signal with hemorrhage and less-avid contrast enhancement. The enhancing dedifferentiated component (s) has a malignant fibrous histiocytoma-like growth pattern in the histology

A 25-year-old woman with a poorly differentiated chordoma of the sacrum. (A) Sagittal T1-weighted image and (B) T2-weighted fat-suppressed image of the sacrum shows a destructive multilevel lesion at S3 and S4 vertebrae with epidural extension, hemorrhage (arrows), and low T2 signal (arrowhead). The low T2 signal area showed little enhancement (not shown). Despite the therapy, the tumor increased, and the patient died of metastatic disease three years after initial diagnosis
Other mesenchymal tumors
The WHO's new family of “other mesenchymal tumors” includes a number of the entities from deleted previous tumor families (Table 3). Fibrocartilaginous mesenchymoma and hibernoma of bone were newly added in the WHO's 2020 classification. Adamantinoma is further subtyped into classic, osteofibrous dysplasia-like, and dedifferentiated adamantinoma.
Fibrocartilaginous mesenchymoma
Fibrocartilaginous mesenchymoma, initially described by Dahlin et al. in 1984, is a rare and locally aggressive neoplasm characterized by spindle cells with mild cytological atypia, bone formation, and hyaline cartilage nodules and can demonstrate a growth plate-like appearance [59]. Fibrocartilaginous mesenchymoma affects patients under 30 years [59, 60]. Lack of GNAS, IDH mutations, and MDM2 amplification support the distinction of fibrocartilaginous mesenchymoma from other tumors such as fibrous dysplasia, chondrosarcoma, and low-grade osteosarcoma [60]. Fibrocartilaginous mesenchymoma is typically located in metaphysis of long bones and pelvis. Although complete regression and spontaneous regression have been reported, surgical resection is the primary treatment [60]. Fibrocartilaginous mesenchymoma is a lytic lesion frequently associated with a sclerotic rim, cortical destruction, and extraosseous extension (Fig. 10) [60]. On MRI, the tumor shows low T1 signal and heterogenous high T2 signal with contrast enhancement and increased radiotracer uptake on bone scan [60].

An 11-year-old patient with fibrocartilaginous mesenchymoma in the left humerus. (A) Frontal view radiograph of left humerus shows a lytic lesion with a sclerotic rim (arrows) in the proximal humeral metaphysis and diaphysis. Follow-up radiographs (B) shows the lesion's increased size, internal calcification (arrowheads), cortical destruction, and marked endosteal scalloping (arrows). Courtesy of Dr. Michael Klein
Hibernoma
Newly classified benign, hibernoma of bone is a tumor of brown adipocytes. It is more common in older women than its soft tissue counterpart, which is common in young men [61,62,63]. The typical location is the spine and pelvis [62,63,64]. Hibernoma is frequently sclerotic (64%) and occasionally lytic (18%) on CT [63]. It shows hypointense T1 signal and hyperintense T2 signal compared to muscle in MRI, variable presence of intralesional fat, and heterogeneous contrast enhancement (Fig. 11) [63]. Hibernoma shows minimal uptake in lytic lesions or elevated radiotracer uptake in sclerotic lesions on bone scan and mild metabolic activity (SUVmax 3.0–4.1) on PET [63, 63].

A 52-year-old woman with a hibernoma. (A) Coronal CT image shows a ground glass dense lesion with a thin sclerotic rim (arrowheads) in the right upper sacrum. (B) Fused axial PET-CT image shows an elevated FDG uptake with SUVmax 5.0. (C) Coronal T1-weighted image shows high T1 signal focus (arrow) consistent with macroscopic fat. The lesion showed heterogenous T2 signal and contrast enhancement (not shown)
Adamantinoma of long bones
Adamantinoma of long bones is classified into three diagnostic entities: (1) the intermediate (locally aggressive) osteofibrous dysplasia-like adamantinoma (OFD-LA), (2) the malignant adamantinoma of long bone, considered the classic form, and (3) dedifferentiated adamantinoma, a newly recognized and rare type with a poor prognosis [65].
Previously malignant, OFD-LA is now reclassified by the WHO as an intermediate and locally aggressive tumor because of its potential for local recurrence (20%) [65]. Dedifferentiated adamantinoma is characterized by classic adamantinoma and gradual sarcomatoid transition in histology [2]. Metastases were reported in two-thirds of patients, one-third of whom died of metastases within two years of initial metastasis [66]. In contrast, classic adamantinoma displays a lower rate of metastasis (30%) and longer survival (> 4 years after first metastasis) [67].
There is considerable overlap in clinical presentation between benign osteofibrous dysplasia (OFD) and OFD-LA, and between adamantinoma and dedifferentiated adamantinoma. Both OFD and OFD-LA present in young children with slight female predilection, and classic adamantinoma and dedifferentiated adamantinoma occur in adults with male predilection [65]. Tibial diaphysis is the typical site of OFD and all types of adamantinoma. Synchronous fibula lesions are reported in 12% of OFD and OFD-LA and 10–50% of classic adamantinoma [68]. The radiologic similarity of these entities may also lead to sampling error in biopsy and misdiagnosis, prompting a change of diagnosis to higher grade tumor after surgical resection [69]. There have been reports that 60% of OFD-LA was initially diagnosed as benign OFD, and nearly 90% of dedifferentiated adamantinoma were initially diagnosed as classic adamantinoma or other sarcomatous lesions at biopsy [69, 70].
At imaging, all types are typically intracortical, well-circumscribed, multilobulated, and lytic with sclerosis [66,67,68] (Fig. 12). Complete marrow involvement, cortical destruction, and extraosseous extension are more common in classic adamantinoma and dedifferentiated adamantinoma than OFD-LA [66,67,68,69]. Anterior bowing is more common in OFD and OFD-LA than in adamantinoma [68], but it may not be specific to these entities [69].

A 15-year-old male with osteofibrous dysplasia-like adamantinoma. The patient was diagnosed with osteofibrous dysplasia (OFD) at the age of six. (A) Lateral view of the left tibia and fibula at the time of OFD diagnosis shows multifocal mixed lytic and sclerotic lesions in the anterior tibial cortex causing mild anterior bowing. (B) Sagittal T1-weighted shows marked narrowing of the medullary canal caused by the tumor. The lesion gradually increased in size and lytic components. (C) Pasted lateral radiograph nine years later showed increased mixed lytic and sclerotic lesion with persistent anterior bowing and a focal lytic component (arrow) causing marked endosteal scalloping
Undifferentiated pleomorphic sarcoma
The former “undifferentiated high-grade pleomorphic sarcoma” is now termed undifferentiated pleomorphic sarcoma [71]. Undifferentiated pleomorphic sarcoma may be primary or secondary, associated with other conditions, such as infarct, Paget disease, radiation, orthopedic prosthesis, and diaphyseal medullary stenosis [71]. UPS is usually lytic at imaging with cortical destruction and soft tissue extension mimicking primary bone sarcomas or metastases [43].
Hematopoietic neoplasm of bone
This category has undergone significant revision and expansion (Table 1). Multiple myeloma has been removed from the WHO classification of Soft Tissue and Bone and included in the 4th edition of WHO Classification of Haematopoietic and Lymphoid tissues published in 2017 [2]. The current category includes solitary plasmacytoma of bone, primary lymphoma with its types, Langerhans cell histiocytosis (LCH), Erdheim-Chester disease, and Rosai-Dorfman disease. LCH, Erdheim-Chester disease, and Rosai-Dorfman disease are histiocytosis that share somatic mutations in the common MAPK pathway, which may be treated with the same targeted therapies using BRAF or MEK inhibitors [72].
Langerhans cell histiocytosis
The WHO classifies LCH into two types depending on a single organ or multiorgan involvement. Single organ LCH typically involves bone and most often presents as a single lesion. Single organ LCH with focal or multifocal lesions remains categorized as intermediate (locally aggressive) [2, 73, 74]. The disseminated multiorgan type is recognized for its higher mortality rates and poor prognosis [2, 74].
Skeletal LCH is lytic on radiograph with a predilection for the skull and spine. LCH may appear aggressive with periosteal reaction in the acute phase, mimicking Ewing sarcoma or osteomyelitis [75]. Typical MRI appearance is a low T1, and high T2 signal lesion associated with extensive bone and soft tissue edema [74, 75]. A skeletal survey is the primary imaging study for staging of LCH, although PET and whole-body MRI detect more bone lesions [74].
Erdheim-Chester disease
Erdheim-Chester disease is a chronic progressive disease that leads to multiorgan failure and affects adults between 50–60 years of age with a male prediction (3:1) [2, 76, 77]. Cardiac involvement and retroperitoneal fibrosis are identified in more than half of patients, potentially causing cardiac arrhythmias and hydronephrosis [78]. Erdheim-Chester disease is no longer considered an intermediate locally aggressive tumor due to its unfavorable clinical outcomes by multiorgan involvement and is now classified as a hematopoietic neoplasm of bone [2, 76].
Erdheim-Chester disease is characterized by bilateral medullary osteosclerosis of long bones and increased radiotracer uptake in bone scans. Osteosclerosis is seen mainly in diaphysis or metaphysis in long bones, sparing the axial skeleton, hands, and feet (Fig. 13) [77]. They are nodular or diffuse enhancing marrow lesions on MRI, reflecting progressive stages of the disease [77]. FDG uptake values are variable in PET (Fig. 13) [77].

A 46-year-old man with Erdheim-Chester disease. (A) Coronal CT image shows infiltrative pericardial soft tissue (arrows) encasing the aorta, pulmonary artery, and vein, as well as bilateral retroperitoneal soft tissue encasing the kidneys (arrowheads). (B) Bone scan shows intense radiotracer uptake in the clavicles, diaphyses and distal metaphyses of right humerus, and bilateral femora. (C) PET MIP image shows diffuse mild FDG uptake in mediastinum and bone and soft tissue of lower extremities (arrows). The patient was treated with dabrafenib, BRAF inhibitor. Subsequent PET shows decrease in FDG uptake and sclerosis (not shown)
Rosai-Dorfman disease
Rosai-Dorfman disease is formally termed “sinus histiocytosis with massive lymphadenopathy,” and extranodal Rosai-Dorfman disease accounts for 43% of cases [2, 79, 80]. Although previously considered benign with a good prognosis, Rosai-Dorfman disease is no longer classified as benign in terms of its biological behavior in the current WHO classification. Bone involvement is seen in 5–10% of Rosai-Dorfman disease with a slight predilection for women (mean age: 31 years) and correlated with worse prognosis with fatal cases [80]. The typical locations are metaphyses or epiphyses of long and craniofacial bones [2, 77, 80]. The lesions are typically intramedullary lytic with cortical destruction and extraosseous extension and can mimic malignant tumors or metastases [77, 80]. MRI appearance consists of focal lesions with low T1 and high T2 signal and contrast enhancement [77].
Undifferentiated small round cell sarcomas of bone and soft tissue (Table 4)
Despite their histologic similarity, small round cell sarcomas are diverse entities which arise either from bone or soft tissue and exhibit unique genetic mutations and clinical behaviors. The 2020 WHO classification recognizes these distinct tumors under a new category in a separate chapter titled “undifferentiated small round cell sarcomas of bone and soft tissue” [2, 81,82,83,84]. Diagnostic molecular profiles and clinical features separate this category into four types: Ewing sarcoma, CIC-rearranged sarcoma, sarcoma with BCOR genetic alterations, and EWSR1-non-ETS fusions. Ewing sarcoma is the second most common primary malignancy of bone in children and young adults after osteosarcoma. Ewing sarcoma is characterized by gene fusion involving the FET (FUS, EWSR1 and TAF15 genes) family of genes with a member of the ETS (Erythroblast Transformation Specific) transcription factors. The other three Ewing-like sarcoma entities lack EWSR1-ETS gene fusion [2, 82,83,84,85].
CIC-rearranged sarcoma
Discovered in 2016, the CIC::DUX4 fusion is the most common genetic alteration in CIC-rearranged sarcoma and comprises the majority lacking EWSR1 fusions [2, 82, 85]. CIC-rearranged sarcomas peak in the third decade of life and rarely involve bones [85]. Their clinical behavior is aggressive, with a five-year survival rate ranging from 17–44% [85]. CIC-rearranged sarcoma demographics and anatomic sites are similar to extraskeletal ES [86]. Imaging features of CIC::DUX4 sarcomas also overlap with those of extraskeletal ES, including isodense to hypodense attenuation to skeletal muscles in non-contrast CT and heterogenous contrast enhancement with necrosis on CT and MRI (Fig. 14) [86]. Brady et al. reported that the average CIC-rearranged sarcoma FDG uptake was higher than those of extraskeletal Ewing sarcoma, which likely accounts for their aggressive clinical course [86]. Flow voids and hemorrhage are common with occasional fluid levels, likely due to hemorrhage (Fig. 14).

A 21-year-old woman with undifferentiated small round cell sarcoma with CIC-DUX4 gene fusion. (A) In the axial T2 image, a few fluid levels (arrowheads) are evident in the mass. (B) Axial-fused PET MRI image shows high metabolic activity (arrow) in the tumor. The tumor showed contrast enhancement in the area with high metabolic activity on MRI (not shown)
Sarcoma with BCOR genetic alterations
Sarcomas with BCOR genetic alterations account for about 5% of Ewing-like sarcoma [85]. They are due to gene fusion (most commonly BCOR::CCNB3) or BCOR-internal tandem duplication (BCOR-ITD). BCOR::CCNB3 sarcoma affects children with male predilections and usually arises from the pelvis and lower extremities, affecting more bone than soft tissue [2, 83]. The prognosis of BCOR::CCNB3 is similar to that of Ewing sarcoma and better than other Ewing-like sarcomas [56, 57]. BCOR-ITD usually occurs in infancy and mainly arises in the soft tissue of the trunk, retroperitoneum, and head and neck [1, 56]. Prognosis is not known [56].
Imaging features of BCOR::CCNB3 include either lytic or sclerotic bone lesions. Soft tissue calcifications on CT are seen in 40% of cases (Fig. 15). Flow voids and necrosis are also frequently seen [86, 87]. These tumors are hypermetabolic on PET [86]. BCOR-sarcomas in soft tissue are often large masses involving deep soft tissue with or without well-demarcated borders and heterogeneous T2 signal at MRI. These tumors may invade bone [87]. Imaging features of BCOR-ITD are little known. The tumor can be aggressive invading spinal canals and show low T1 signal and high T2 signal with heterogeneous enhancement and non-enhancing areas, probably due to variable degrees of cellularity and myxoid matrix (Fig. 16).

Undifferentiated small round cell sarcoma with BCOR::CNNB3 genetic alteration in two different patients. (A) Sagittal CT of a 2-year-old boy shows a lytic L3 vertebral lesion (arrow) causing severe pathologic compression fracture and a large low-attenuation extraosseous paraspinal mass (arrowheads). (B) Sagittal CT image of an 18-year-old man shows a sclerosing T8 vertebral lesion with a partially calcified epidural soft-tissue mass (arrow). Cord compression was present on MRI (not shown)

An 8-month-old male infant with undifferentiated small round cell sarcoma with BCOR-ITD genetic alteration. (A) Sagittal T2-weighted image demonstrates a large retroperitoneal and pelvic tumor (T) with diffuse high T2 signal. The tumor extends into the lumbar and sacral spinal canal (t) and bilateral neural foramens (arrows). (B) Sagittal T1-weighted fat-suppressed image shows diffuse contrast enhancement in the tumor (T), as well as its posterior extension (arrow). However, the epidural component (t) shows no contrast enhancement. The tumor compresses on the urinary bladder (ub) and causes hydronephrosis (arrowheads). The tumor subsequently increased in size despite chemotherapy and the patient died of disease within a year following initial diagnosis
EWSR1-non-ETS fusions
Sarcomas with EWSR1-non-ETS fusions include EWSR1::NFATC2, FUS::NFATC2, and EWSR::PATZ1 sarcomas. EWSR1::NFATC2 sarcomas occur more frequently in long bones than soft tissue, while FUS::NFATC2 have been reported only in long bones with a male predilection and affecting a wide age range [84]. EWSR::PATZ1 sarcomas tend to involve deep soft tissue of the chest and abdominal wall with a broad age range and equal gender distribution [85]. The imaging appearance of these tumors is little known. The tumor can appear similar to other primary bone malignancies, causing bone destruction and extraosseous extension with necrosis (Fig. 17).

A 28-year-old woman with undifferentiated small round cell sarcoma with FUS::NFATC2 fusion. (A) Sagittal T1-weighted fat-suppressed image demonstrates an expansile lesion in the proximal humerus. Within the lesion, there is thick peripheral contrast enhancement with central non-enhancement (N), due to necrosis. (B) Axial fused CT-PET image demonstrates intense FDG uptake in the lesion
Conclusion
This article reviewed major changes in the WHO’s 2020 classification of bone tumors and pertinent imaging findings. These changes include reclassification of existing bone tumors, including benign (chondroblastoma, chondromyxoid fibroma, ABC), intermediate (OFD-LA, synovial chondromatosis), and malignant entities (disseminated LCH, Erdheim-Chester disease). The current WHO classification also introduced new entities according to tumor genetics and biological behavior. A new chapter on the category of undifferentiated small round cell sarcomas of bone and soft tissue classifies Ewing sarcoma and Ewing-like sarcoma, according to their different molecular and clinical behavior. Up-to-date knowledge of both terminology and classification is essential for recognizing the biological behavior of each disease entity and providing consistent oncologic treatment options for better clinical outcomes.
References
The WHO Classification of Tumours Editorial Board. WHO classification of tumours soft tissue and bone tumours. 5th ed. Lyon: IARC Press; 2020.
Choi JH, Ro JY. The 2020 WHO classification of tumors of bone: an updated review. Adv Anat Pathol. 2021;28(3):119–38. https://doi.org/10.1097/PAP.0000000000000293.
Flanagan AM, Blay JY, Bovée JVMG, Bredella A, Cool P, Nielsen GP, Yoshida A. 2020 Bone tumours: introduction. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:340–344.
Xu H, Nugent D, Monforte HL, Binitie OT, Ding Y, Letson GD, et al. Chondroblastoma of bone in the extremities: a multicenter retrospective study. J Bone Joint Surg Am. 2015;97(11):925–31. https://doi.org/10.2106/JBJS.N.00992.
Hogendoorn PCW, Bloem JL, Bridge JA. Chondromyxoid fibroma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:362–364.
Bhamra JS, Al-Khateeb H, Dhinsa BS, Gikas PD, Tirabosco R, Pollock RC, et al. Chondromyxoid fibroma management: a single institution experience of 22 cases. World J Surg Oncol. 2014;12(12):283. https://doi.org/10.1186/1477-7819-12-283.
Flanagan AM, Bloem JL, Cates JMM, O’Donnell PG. Synovial Chondromatosis. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:368–369.
Houdek MT, Wyles CC, Rose PS, Stuart MJ, Sim FH, Taunton MJ. High rate of local recurrence and complications following total knee arthroplasty in the setting of synovial chondromatosis. J Arthroplasty. 2017;32(7):2147–50. https://doi.org/10.1016/j.arth.2017.02.040.
Tibbo ME, Wyles CC, Rose PS, Sim FH, Houdek MT, Taunton MJ. Long-term outcome of hip arthroplasty in the setting of synovial chondromatosis. J Arthroplasty. 2018 Jul;33(7):2173–6. https://doi.org/10.1016/j.arth.2018.02.027.
Evans S, Boffano M, Chaudhry S, Jeys L, Grimer R. Synovial chondrosarcoma arising in synovial chondromatosis. Sarcoma. 2014;2014:647939. https://doi.org/10.1155/2014/647939.
Murphey MD, Vidal JA, Fanburg-Smith JC, Gajewski DA. Imaging of synovial chondromatosis with radiologic-pathologic correlation. Radiographics. 2007;27(5):1465–88. https://doi.org/10.1148/rg.275075116.
Bovée JVMG, Bloem JL, Flanagan AM, Nielsen GP, Yoshida A. Enchondroma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:353–355.
Brien EW, Mirra JM, Kerr R. Benign and malignant cartilage tumors of bone and joint: their anatomic and theoretical basis with an emphasis on radiology, pathology, and clinical biology I The intramedullary cartilage tumors. Skeletal Radiol. 1997;26(6):325–53. https://doi.org/10.1007/s002560050246.
Brien EW, Mirra JM, Luck JV Jr. Benign and malignant cartilage tumors of bone and joint: their anatomic and theoretical basis with an emphasis on radiology, pathology, and clinical biology. II Juxtacortical cartilage tumors Skeletal Radiol. 1999;28(1):1–20. https://doi.org/10.1007/s002560050466.
Stomp W, Reijnierse M, Kloppenburg M, de Mutsert R, Bovée JV, den Heijer M, NEO study group, et al. Prevalence of cartilaginous tumours as an incidental finding on MRI of the knee. Eur Radiol. 2015;25(12):3480–7. https://doi.org/10.1007/s00330-015-3764-6.
Davies AM, Patel A, Azzopardi C, James SL, Botchu R. Prevalence of Enchondromas of the Proximal Femur in Adults as an Incidental Finding on MRI of the Pelvis. Indian J Radiol Imaging. 2021;31(3):582–5. https://doi.org/10.1055/s-0041-1735915.
Hong ED, Carrino JA, Weber KL, Fayad LM. Prevalence of shoulder enchondromas on routine MR imaging. Clin Imaging. 2011;35(5):378–84. https://doi.org/10.1016/j.clinimag.2010.10.012.
Douis H, Saifuddin A. The imaging of cartilaginous bone tumours. I Benign lesions Skeletal Radiol. 2012;41(10):1195–212. https://doi.org/10.1007/s00256-012-1427-0.
Bovée JVMG, Bloem JL, Flanagan AM, Nielsen GP, Yoshida A. Central atypical cartilaginous tumour/chondrosarcoma grade 1. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:370–372.
Eefting D, Schrage YM, Geirnaerdt MJ, Le Cessie S, Taminiau AH, Bovée JV, et al. EuroBoNeT consortium. Assessment of interobserver variability and histologic parameters to improve reliability in classification and grading of central cartilaginous tumors. Am J Surg Pathol. 2009;33(1):50–7. https://doi.org/10.1097/PAS.0b013e31817eec2b.
Zamora T, Urrutia J, Schweitzer D, Amenabar PP, Botello E. Do orthopaedic oncologists agree on the diagnosis and treatment of cartilage tumors of the appendicular skeleton? Clin Orthop Relat Res. 2017;475(9):2176–86. https://doi.org/10.1007/s11999-017-5276-y.
Deckers C, Rooy JWJ, Flucke U, Schreuder HWB, Dierselhuis EF, Geest ICMV. Midterm MRI follow-up of untreated enchondroma and atypical cartilaginous tumors in the long bones. Cancers (Basel). 2021;13(16):4093. https://doi.org/10.3390/cancers13164093.
Davies AM, Patel A, Botchu R, Azzopardi C, James S, Jeys L. The changing face of central chondrosarcoma of bone. One UK-based orthopaedic oncology unit’s experience of 33 years referrals. J Clin Orthop Trauma. 2021;17:106–11. https://doi.org/10.1016/j.jcot.2021.02.017.
van PraagVeroniek VM, Rueten-Budde AJ, Ho V, Dijkstra PDS, Fiocco M, van de Sande MAJ, Study group bone and soft tissue tumours (WeBot). Incidence, outcomes and prognostic factors during 25 years of treatment of chondrosarcomas. Surg Oncol. 2018;27(3):402–8. https://doi.org/10.1016/j.suronc.2018.05.009.
Omlor GW, Lohnherr V, Lange J, Gantz S, Mechtersheimer G, Merle C, et al. Outcome of conservative and surgical treatment of enchondromas and atypical cartilaginous tumors of the long bones: retrospective analysis of 228 patients. BMC Musculoskelet Disord. 2019;20(1):134. https://doi.org/10.1186/s12891-019-2502-7.
Geirnaerdt MJ, Hermans J, Bloem JL, Kroon HM, Pope TL, Taminiau AH, et al. Usefulness of radiography in differentiating enchondroma from central grade 1 chondrosarcoma. AJR Am J Roentgenol. 1997;169(4):1097–104. https://doi.org/10.2214/ajr.169.4.9308471.
Murphey MD, Walker EA, Wilson AJ, Kransdorf MJ, Temple HT, Gannon FH. From the archives of the AFIP: imaging of primary chondrosarcoma: radiologic-pathologic correlation. Radiographics. 2003;23(5):1245–78. https://doi.org/10.1148/rg.235035134.
Douis H, Parry M, Vaiyapuri S, Davies AM. What are the differentiating clinical and MRI-features of enchondromas from low-grade chondrosarcomas? Eur Radiol. 2018;28(1):398–409. https://doi.org/10.1007/s00330-017-4947-0.
Sharif B, Lindsay D, Saifuddin A. The role of imaging in differentiating low-grade and high-grade central chondral tumours. Eur J Radiol. 2021;137:109579. https://doi.org/10.1016/j.ejrad.2021.109579.
Deckers C, Steyvers MJ, Hannink G, Schreuder HWB, de Rooy JWJ, Van Der Geest ICM. Can MRI differentiate between atypical cartilaginous tumors and high-grade chondrosarcoma? A systematic review Acta Orthop. 2020;91(4):471–8. https://doi.org/10.1080/17453674.2020.1763717.
Alhumaid SM, Alharbi A 4th, Aljubair H. Magnetic resonance imaging role in the differentiation between atypical cartilaginous tumors and high-grade chondrosarcoma: an updated systematic review. Cureus. 2020;12(10):e11237. https://doi.org/10.7759/cureus.11237.
Bovée JVMG, Bloem JL, Flanagan AM, Nielsen GP, Yoshida A. Secondary peripheral atypical cartilaginous tumour/chondrosarcoma grade 1. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:373–374.
Bernard SA, Murphey MD, Flemming DJ, Kransdorf MJ. Improved differentiation of benign osteochondromas from secondary chondrosarcomas with standardized measurement of cartilage cap at CT and MR imaging. Radiology. 2010;255(3):857–65. https://doi.org/10.1148/radiol.10082120.
Amary F, Bredella MA, Horvai AE, Mahar AM. Osteoid osteoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:394–396.
Amary F, Bredella MA, Horvai AE, Mahar AM. Osteoblastoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:397–399.
Chai JW, Hong SH, Choi JY, Koh YH, Lee JW, Choi JA, Kang HS. Radiologic diagnosis of osteoid osteoma: from simple to challenging findings. Radiographics. 2010 May;30(3):737-49. https://doi.org/10.1148/rg.303095120. Erratum in: Radiographics. 2010 Jul-Aug;30(4):1156 https://doi.org/10.1148/rg.303095120
Jaffe HL. Osteoid-osteoma. Proc R Soc Med. 1953;46(12):1007–12.
Liu J, Han S, Li J, Yuan Y, Guo W, Yuan H. Spinal osteoblastoma: a retrospective study of 35 patients’ imaging findings with an emphasis on MRI. Insights Imaging. 2020;11(1):122. https://doi.org/10.1186/s13244-020-00934-y.
Baumhoer D, Böhling TO, Cates JMM, Cleton-Jansen AM, Hogendoorn PCW, O’Donnell PG, Rosenberg AE. Osteosarcoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:403–409.
Smeland S, Bielack SS, Whelan J, Bernstein M, Hogendoorn P, Krailo MD, et al. Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer. 2019;109:36–50. https://doi.org/10.1016/j.ejca.2018.11.027.
Flanagan AM, Bridge JA, O’Donnell PG. Secondary osteosarcoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:419–421.
Suurmeijer AJH, Cleton-Jansen AM. Desmoplastic fibroma of bone. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:422–423.
Berkeley R, Andrei V, Saifuddin A. The rare primary bone sarcomas: imaging pathological correlation. Skeletal Radiol. 2021;50(8):1491–511. https://doi.org/10.1007/s00256-020-03692-6.
Dei Tos AP, Czerniak B, Inwards CY. Fibrosarcoma of bone. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:424–425.
Errani C, Vanel D, Gambarotti M, Alberghini M, Picci P, Faldini C. Vascular bone tumors: a proposal of a classification based on clinicopathological, radiographic and genetic features. Skeletal Radiol. 2012;41(12):1495–507. https://doi.org/10.1007/s00256-012-1510-6.
Bovée JVMG, Rosenberg AE. Epitheliod haemangioma of bone. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:428–430.
Zhou Q, Lu L, Fu Y, Xiang K, Xu L. Epithelioid hemangioma of bone: a report of two special cases and a literature review. Skeletal Radiol. 2016;45(12):1723–7. https://doi.org/10.1007/s00256-016-2482-8.
Tsuda Y, Suurmeijer AJH, Sung YS, Zhang L, Healey JH, Antonescu CR. Epithelioid hemangioma of bone harboring FOS and FOSB gene rearrangements: A clinicopathologic and molecular study. Genes Chromosomes Cancer. 2021;60(1):17–25. https://doi.org/10.1002/gcc.22898.
Agaram NP, Bredella MA. Aneurysmal bone cyst. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:437–439.
Flanagan AM, Larousserie F,O’Donnell PG, Yoshida A. Giant cell tumour of bone. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:440–446.
Tahir I, Andrei V, Pollock R, Saifuddin A. Malignant giant cell tumour of bone: a review of clinical, pathological, and imaging features. Skeletal Radiol. 2021. https://doi.org/10.1007/s00256-021-03913-6.
Liu W, Chan CM, Gong L, Bui MM, Han G, Letson GD, et al. Malignancy in giant cell tumor of bone in the extremities. J Bone Oncol. 2020 Nov;5(26):100334. https://doi.org/10.1016/j.jbo.2020.100334.
Tirabosco R, O’Donnell PG, Yamaguchi T. Conventional chordoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:451–453.
Tirabosco R, Hameed M. Dedifferentiated chordoma. In: Lokuhetty D, White V, Cree I, editors. WHO classification of tumours, soft tissue and bone tumors. 5th ed. International Agency for Research on Cancer (IARC): Lyon Cedex, France; 2020. p. 454–5.
Nielsen GP, Dickson BC, Tirabosco R. Poorly differentiated chordoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:456–457.
Shih AR, Cote GM, Chebib I, Choy E, DeLaney T, Deshpande V, et al. Clinicopathologic characteristics of poorly differentiated chordoma. Mod Pathol. 2018;31(8):1237–45. https://doi.org/10.1038/s41379-018-0002-1.
Rekhi B, Michal M, Ergen FB, Roy P, Puls F, Haugland HK, et al. Poorly differentiated chordoma showing loss of SMARCB1/INI1: Clinicopathological and radiological spectrum of nine cases, including uncommon features of a relatively under-recognized entity. Ann Diagn Pathol. 2021;55:151809. https://doi.org/10.1016/j.anndiagpath.2021.151809.
Olson JT, Wenger DE, Rose PS, Petersen IA, Broski SM. Chordoma: 18F-FDG PET/CT and MRI imaging features. Skeletal Radiol. 2021;50(8):1657–66. https://doi.org/10.1007/s00256-021-03723-w.
Gambarotti M, Inwards CY. Fibrocartilaginous mesenchymoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:470–471.
Gambarotti M, Righi A, Vanel D, Cocchi S, Benini S, Elli FM, et al. Fibrocartilaginous mesenchymoma of bone: a single-institution experience with molecular investigations and a review of the literature. Histopathology. 2017;71(1):134–42. https://doi.org/10.1111/his.13201.
Rosenberg AE, Bloem JL, Sumathi VP. Lipoma and hibernoma of bone. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:475–477.
Bonar SF, Watson G, Gragnaniello C, Seex K, Magnussen J, Earwaker J. Intraosseous hibernoma: characterization of five cases and literature review. Skeletal Radiol. 2014;43(7):939–46. https://doi.org/10.1007/s00256-014-1868-8.
Myslicki FA, Rosenberg AE, Chaitowitz I, Subhawong TK. Intraosseous Hibernoma: Five Cases and a Review of the Literature. J Comput Assist Tomogr. 2019;43(5):793–8. https://doi.org/10.1097/RCT.0000000000000912.
Gitto S, Doeleman T, van de Sande MAJ, van Langevelde K. Intraosseous hibernoma of the appendicular skeleton. Skeletal Radiol. 2021. https://doi.org/10.1007/s00256-021-03956-9.
Nielsen GP, Hogendoorn PCW. Adamantinoma of long bones. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:463–465.
Rekhi B, Sahay A, Puri A. Clinicopathologic Features of Two Rare Cases of Dedifferentiated Adamantinomas, Including Diagnostic Implications. Int J Surg Pathol. 2019;27(2):193–202. https://doi.org/10.1177/1066896918790388.
Sharifai N, Runyon R, Friedman M, Cipriano C, Chrisinger. Adamantinoma of the Femur with High-Grade Epithelial and Sarcomatoid Components: Case Report and Review of the Literature. AJSP Reviews & Reports. 2020;25(1):19–25. https://doi.org/10.1097/PCR.0000000000000359.
Bethapudi S, Ritchie DA, Macduff E, Straiton J. Imaging in osteofibrous dysplasia, osteofibrous dysplasia-like adamantinoma, and classic adamantinoma. Clin Radiol. 2014;69(2):200–8. https://doi.org/10.1016/j.crad.2013.09.011 (Epub 2013Nov 5).
Khanna M, Delaney D, Tirabosco R, Saifuddin A. Osteofibrous dysplasia, osteofibrous dysplasia-like adamantinoma and adamantinoma: correlation of radiological imaging features with surgical histology and assessment of the use of radiology in contributing to needle biopsy diagnosis. Skeletal Radiol. 2008;37(12):1077–84. https://doi.org/10.1007/s00256-008-0553-1.
Scholfield DW, Sadozai Z, Ghali C, Sumathi V, Douis H, Gaston L, et al. Does osteofibrous dysplasia progress to adamantinoma and how should they be treated? Bone Joint J. 2017;99-B(3):409–16. https://doi.org/10.1302/0301-620X.99B3.38050.
Inwards CY, Czerniak B, Dei, Tos AP. Undifferentiated pleomorphic sarcoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:480–482.
Gulati N, Allen CE. Langerhans cell histiocytosis: Version 2021. Hematol Oncol. 2021;39(Suppl 1):15–23. https://doi.org/10.1002/hon.2857.
Pileri SA, Cheuk W, Picarsic J. Langerhans cell histiocytosis. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:492–494
Rajakulasingam R, Siddiqui M, Michelagnoli M, Saifuddin A. Skeletal staging in Langerhans cell histiocytosis: a multimodality imaging review. Skeletal Radiol. 2021;50(6):1081–93. https://doi.org/10.1007/s00256-020-03670-y.
Azouz EM, Saigal G, Rodriguez MM, Podda A. Langerhans’ cell histiocytosis: pathology, imaging, and treatment of skeletal involvement. Pediatr Radiol. 2005;35(2):103–15. https://doi.org/10.1007/s00247-004-1262-0.
Emile JF, Haroche J, Picarsic J, Tirabosco R. Erdheim-Chester disease. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:495–497.
Choraria A, Andrei V, Rajakulasingam R, Saifuddin A. Musculoskeletal imaging features of non-Langerhans cell histiocytoses. Skeletal Radiol. 2021;50(10):1921–40. https://doi.org/10.1007/s00256-021-03765-0.
Emile JF, Cohen-Aubart F, Collin M, Fraitag S, Idbaih A, Abdel-Wahab O, et al. Histiocytosis Lancet. 2021;398(10295):157–70. https://doi.org/10.1016/S0140-6736(21)00311-1.
Rosenberg AE, Demicco EG. Rosai-Dorfman disease. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:498–499.
Garcia RA, DiCarlo EF. Rosai-Dorfman Disease of Bone and Soft Tissue. Arch Pathol Lab Med. 2022;146(1):40–6. https://doi.org/10.5858/arpa.2021-0116-RA.
de Álava E, Lessnick SL, Stamenkovic I. Ewing sarcoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:323–325.
Antonescu CR, Yoshida A. CIC-rearranged sarcoma. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:330–332.
Antonescu CR, Puls F, Tirode F. Sarcoma with BCOR genetic alterations. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:333–335.
Le Loarer F, Szuhai K, Tirode F. Round cell sarcoma with EWSR1–non-ETS fusions. In: Lokuhetty D, White V, Cree I, eds. WHO classification of tumours, soft tissue and bone tumors. 5th ed. Lyon Cedex, France: International Agency for Research on Cancer (IARC); 2020:326–329.
Kallen ME, Hornick JL. From the ashes of “Ewing-like” sarcoma: A contemporary update of the classification, immunohistochemistry, and molecular genetics of round cell sarcomas. Semin Diagn Pathol. 2022;39(1):29–37. https://doi.org/10.1053/j.semdp.2021.10.002.
Brady EJ, Hameed M, Tap WD, Hwang S. Imaging features and clinical course of undifferentiated round cell sarcomas with CIC-DUX4 and BCOR-CCNB3translocations. Skeletal Radiol. 2021;50(3):521–9. https://doi.org/10.1007/s00256-020-03589-4.
Sirisena UDN, Rajakulasingam R, Saifuddin A. Imaging of bone and soft tissue BCOR-rearranged sarcoma. Skeletal Radiol. 2021;50(7):1291–301. https://doi.org/10.1007/s00256-020-03683-7.
Funding
National Cancer Center,P30 CA008748
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
• Chondroblastoma, chondromyxoid fibroma, and aneurysmal bone cyst are classified as benign. Osteofibrous dysplasia-like adamantinoma and synovial chondromatosis are now categorized as intermediate (locally aggressive).
• The designation of atypical cartilaginous tumors is reserved for the appendicular skeleton and the histologically identical locally aggressive hyaline cartilage tumor is termed chondrosarcoma grade 1 in the axial skeleton and flat bones.
• Erdheim-Chester Erdheim-Chester disease is no longer considered an intermediate locally aggressive tumor due to its unfavorable clinical outcomes by its multiorgan involvement and is now classified as a hematopoietic neoplasm of bone.
• Ewing sarcoma is no longer classified as a bone tumor and is now addressed in a new category, titled “undifferentiated small round cell sarcomas of bone and soft tissue.”
Rights and permissions
About this article

Cite this article
Hwang, S., Hameed, M. & Kransdorf, M. The 2020 World Health Organization classification of bone tumors: what radiologists should know. Skeletal Radiol 52, 329–348 (2023). https://doi.org/10.1007/s00256-022-04093-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00256-022-04093-7