
Abstract
Vitamin D plays an essential role in calcium and inorganic phosphate (Pi) homeostasis, maintaining their optimal levels to assure adequate bone mineralization. Vitamin D, as calcitriol (1,25(OH)2D), not only increases intestinal calcium and phosphate absorption but also facilitates their renal reabsorption, leading to elevated serum calcium and phosphate levels. The interaction of 1,25(OH)2D with its receptor (VDR) increases the efficiency of intestinal absorption of calcium to 30–40% and phosphate to nearly 80%. Serum phosphate levels can also influence 1,25(OH)2D and fibroblast growth factor 23 (FGF23) levels, i.e., higher phosphate concentrations suppress vitamin D activation and stimulate parathyroid hormone (PTH) release, while a high FGF23 serum level leads to reduced vitamin D synthesis. In the vitamin D-deficient state, the intestinal calcium absorption decreases and the secretion of PTH increases, which in turn causes the stimulation of 1,25(OH)2D production, resulting in excessive urinary phosphate loss. Maintenance of phosphate homeostasis is essential as hyperphosphatemia is a risk factor of cardiovascular calcification, chronic kidney diseases (CKD), and premature aging, while hypophosphatemia is usually associated with rickets and osteomalacia. This chapter elaborates on the possible interactions between vitamin D and phosphate in health and disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Introduction
Vitamin D research has more than 100 years of history since McCollum and Davis’s discovered the “growth-promoting fat-soluble vitamin” that was found in cod liver oil [20]. The effect of this growth-promoting factor in the treatment of rickets was so effective that cod liver oil was regarded as a panacea and gave a powerful impetus to further research on vitamin D throughout the world [71]. In the last 20 years, it has been shown that vitamin D‘s biological activities extend far beyond its involvement in calcium metabolism. Along with proven efficacy in pathological conditions and diseases such as rickets, bone loss, and osteomalacia, some novel effects of vitamin D on very diverse physiological processes have been well established [8, 39]. Vitamin D deficiency remains a critical health issue worldwide, and it has been estimated that around one billion people suffer from various vitamin D-related disorders [35].
The biological effects of 1,25(OH)2D can be divided into two types: skeletal (primarily related to calcemic and phosphatemic activities) and non-skeletal, typically not associated with mineral metabolism [15]. The homeostasis of serum phosphate mediated by vitamin D is of paramount importance for adequate bone mineralization, muscle contraction, nerve conduction, and many other vital functions [26]. This brief chapter reviews our understanding of vitamin D-mediated regulation of phosphate homeostasis in health and diseases.
5.2 Physiological Regulation of Phosphate Homeostasis
Phosphorus is the sixth most abundant chemical element in the body [34]. In nature it mainly exists as phosphates, the form most suitable for living organisms [14]. In mammals, the phosphate group is primarily concentrated (~85%) in bones and teeth as hydroxyapatite. The remaining ~15% are distributed in the other tissues as intracellular ortho- and pyrophosphate groups, either free (“inorganic”) or as a part of nucleotides, coenzymes, and high-energy phosphate compounds. (referred to as “organophosphates”). Inorganic phosphates exist in two forms: monovalent dihydrogen phosphate (H2PO4−) and divalent hydrogen phosphate (HPO42−). In the cytosol dihydrogen phosphate is contributing bulk amounts (62% of all cytosolic phosphates).
The extracellular fluid contains only <1% of the whole pool of body’s inorganic phosphates [27, 33]. Interestingly, compared to the cytosol, the proportion H2PO4−/ HPO42− is inverted, so that the major component is now hydrogen phosphate (61% of all phosphates). In general, a 70-kilogram adult with 25% body fat content would have total body phosphorus of approximately 630 g (~21 mol) [34].
Due to its unique chemical structure, various phosphate groups (especially as nucleoside triphosphates) are key players in cellular energy metabolism, in genetic information storage, in signaling pathways, and as phospholipid components of the cell membranes [37]. Inorganic phosphates, together with bicarbonate and protein buffer systems, constitute the basis of the acid-base homeostasis of the body [42].
A healthy adult consumes 1000 mg on average of dietary phosphate per day (Fig. 5.1). Of this amount 700 mg. is absorbed in the small intestine through passive and active pathways [97]. The unabsorbed phosphate is excreted in the feces. Approximately 150 mg. phosphate is secreted into the gut in the saliva, intestinal and pancreatic secretions, while some of it is reabsorbed [47]. Although dietary phosphate intake differs from day to day, principally, phosphate homeostasis is adjusted by intestinal absorption, renal reabsorption, and skeletal resorption. The average serum phosphate concentration in healthy adults is 2.5–4.9 mg/dl [67].

The kidneys filter about 9000 mg. of phosphate daily, 80–90% of which is reabsorbed mainly in the proximal tubule [68]. At least three distinct cotransporters are involved for phosphate transcellular reabsorption in the proximal tubule, namely NaPi-IIa (SLC34A1), NaPi-IIc (SLC34A3), and PiT-2 (SLC20A2) [7] (Fig. 5.2). Phosphate reabsorption is coupled with sodium-dependent (Na+) transport. Type NaPi II cotransporters are capable of transporting both H2PO4− and HPO42− across brush border membrane (BBM) of the proximal tubules [90]. In contrast, in the small intestine, phosphate is absorbed by both transcellular (active) and paracellular (passive) processes, with the active transport being mainly mediated by NaPi-IIb [55].

Main transcellular phosphate traffic mechanisms
Given the generally acknowledged role of phosphate in almost every molecular and cellular function, altered phosphate balance can lead to untoward effects. The serum phosphate homeostasis is firmly regulated by endocrine communication among parathyroid hormone (PTH), calcitriol (1,25(OH)2D), and fibroblast growth factor 23 (FGF-23) [5, 11].
5.2.1 Parathyroid Hormone (PTH)
PTH, a polypeptide containing 84 amino acids with MW 9500 Da, is secreted by chief cells of parathyroid glands [92]. Extracellular calcium concentration is the main modulator of PTH secretion [60]. PTH stimulates calcium resorption from bone tissue, increases calcium reabsorption in the renal tubules, facilitates hydroxylation of 25(OH)D to 1,25(OH)2D in the kidneys, and induces renal excretion of phosphate [50, 69].
In bone tissue, PTH at a permissive level of 1,25(OH)2D promotes calcium resorption by activating osteoclasts [93]. In the intestine, PTH increases the reabsorption of calcium and phosphate by enhancing 1,25(OH)2D synthesis [69]. High serum PTH levels and hypophosphatemia lead to activation of vitamin D-activating enzyme 1α-hydroxylase [57]. 1,25(OH)2D facilitates absorption of calcium and phosphate for bone mineralization and homeostatic metabolism, preventing low serum levels of these elements [43]. PTH also stimulates the synthesis of vitamin D in the kidneys [52].
The effect of PTH on the renal tubules leads to decreased phosphate reabsorption and its increased renal excretion due to the lowered NaPi cotransporters. In general increased PTH secretion results in a decrease in serum phosphate levels [30]. The main role of 1,25(OH)2D is to determine the availability of calcium and phosphate to form new bone and prevent the development of hypocalcemia and hypophosphatemia [3, 30]. This hormone increases intestinal phosphate absorption elevating its serum concentration.
Secretion PTH by the parathyroid glands is mainly triggered by low extracellular calcium by acting on Ca-sensing receptors (CaSR) [85]. Stimulation of CaSR (they belong to the class of G-protein-coupled receptors) activates multiple heterotrimeric G-proteins, in turn passing the signal to mitogen-activated protein kinase (MAPK) pathways. This cascade of reactions ultimately leads to the suppression of PTH secretion by a negative feedback loop. It has been shown that 1,25(OH)2D upregulates the transcription of the gene encoding the CaSR in the parathyroid gland [13]. Additionally, a low level of calcium indirectly induces parathyroid hyperplasia [23]. However, there is also evidence of the opposite effect of stimulation of parathyroid cell proliferation in response to a high calcium concentration [81].
Interestingly, high serum phosphate levels (hyperphosphatemia) also increase PTH secretion independently of shifts in extracellular calcium [41, 86]. The further secretion of PTH is directly suppressed by 1,25(OH)2D, acting on VDR of parathyroid glands [79].
5.2.2 Vitamin D (Calcitriol)
From a biological point of view, vitamin D is a steroid hormone, as it is synthesized in the body and has a highly specific receptor (VDR). Most vitamin D (90–95%) is formed in the skin under the influence of UVB light, and only a minor fraction of it is obtained from dietary sources [8].
Vitamin D is stored mainly in the liver with a half-life of approximately 14 days. When a larger amount of vitamin D is absorbed, its excess is stored mainly in adipose tissue [1]. Furthermore, vitamin D in association with the vitamin D-binding protein (VBP) is transferred to the liver, where it is hydroxylated to form 25(OH)D, which subsequently undergoes 1α-hydroxylation in the renal tubules, turning into 1,25(OH)2D. This biologically active form of vitamin D is under control by serum PTH, phosphate, and FGF23 concentrations. The synthesis of 1,25(OH)2D is stimulated by low serum phosphate levels and high PTH concentrations [78].
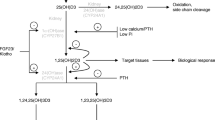
Vitamin D promotes the intestinal absorption of calcium and phosphate, significantly increases their renal reabsorption, and also inhibits the PTH secretion [40] (Fig. 5.3). Thus the major effects of 1,25(OH)2D are to augment the intestinal absorption of both calcium and phosphate for proper bone mineral matrix formation [40]. In the intestine and kidneys, 1,25(OH)2D increases the formation of calcium-binding proteins (calbidins), which promote transmembrane calcium transport to control homeostasis [2]. In bone, 1,25(OH)2D potentiates the effects of PTH, stimulates bone resorption by osteoclasts, and promotes maturation of monocytes into osteoclasts [70, 84]. In parathyroid glands, 1,25(OH)2D binds to the VDR, resulting in the suppression of PTH production [96]. The optimal level of serum phosphate is maintained by the interaction of hormones; lowering serum phosphate level by PTH and FGF23, while, increasing serum phosphate level by elevating its absorption in the intestine (1,25(OH)2D) and its resorption from bones (PTH, 1.25(OH)2D) [37]. PTH directly activates osteoclasts and causes phosphate resorption, and indirectly enhances intestinal phosphate absorption by stimulating 1,25(OH)2D production [44].

Possible regulation of phosphate homeostasis by vitamin D
Activation of the VDR is a potent and rapid modulator of FGF23 expression, thus forming a “classical” endocrine negative feedback loop between FGF23 and vitamin D [17]. In addition, 1,25(OH)2D is a potent suppressor of PTH gene expression [9].
5.2.3 Fibroblast Growth Factor 23 (FGF23)
FGF23, secreted in bone (osteocytes, osteoblasts, and odontoblasts), is an around 32 kDa glycoprotein, which can be converted in its inactive form through cleavage by a proconvertase-type enzyme into two smaller fragments: 18 kDa (amino fragment) and 12 kDa (carboxy fragment) [32].
FGF23, like PTH, reduces renal phosphate reabsorption, which leads to a drop-in plasma phosphate levels [18]. This hormone also suppresses the secretion of PTH and inhibits the 1α-hydroxylase activity of the kidneys, thus reducing the synthesis of 1,25(OH)2D [46, 51]. FGF23 acts by stimulating its receptors, for the normal function of which a cofactor is needed, i.e. the Klotho protein, synthesized, mostly in the kidneys [87]. The transmembrane Klotho protein is essential for FGF23 to exert its phosphaturic effects in the kidney [72,73,74, 89].
A decrease in serum phosphate under the FGF23 is achieved by inhibiting phosphate reabsorption in the renal tubules, as well as by stimulating PTH secretion and suppressing 1,25(OH)2D synthesis [12, 51, 56, 72, 91]. In contrast, calcitonin, is another hormone produced by the thyroid gland, slightly lowers serum calcium due to inhibition of renal and intestinal calcium reabsorption, reducing calcium and phosphate resorption from bones [36]. Plasma calcium is regulated by a complex system involving PTH and 1,25(OH)2D on the intestine, bones, and kidneys. As mentioned, parathyroid gland cells respond to serum calcium concentration via CaSR. A high level of calcium in extracellular fluid stimulates CaSR receptors and activates cellular mechanisms, which ultimately leads to inhibition of PTH release [6].
Imbalance of calcium and phosphate is manifested as a shift in the calcium, phosphate levels in serum and the levels of serum hormones [PTH and 25(ОН)D], as well as the development of bone pathology and cardiovascular calcification with soft anomalies [76, 88]. The exact etiology and pathogenesis of serum phosphate derangements (hyperphosphatemia and hypophosphatemia) will need further studies.
5.3 Hyperphosphatemia
Renal failure is the most common cause of hyperphosphatemia [80]. The decline in estimated glomerular filtration rate disrupts phosphate homeostasis: when it falls below 30 mL/min/1.73 m2, the reabsorption of phosphate is maximally suppressed and fractional excretion markedly reduced. As a result, the serum level of phosphate increases [16, 21]. A primary increase in tubular reabsorption of phosphate is less common and can be observed in hypoparathyroidism, acromegaly, and tumoral calcification [38].
Excessive phosphate can be released from the intracellular compartment, which is observed in acute tumor lysis syndrome, rhabdomyolysis, hemolysis, hyperthermia, profound catabolic stress, and acute leukemia. Tumor lysis syndrome is commonly observed in malignant hematological patients, particularly non-Hodgkin’s lymphoma and acute leukemia, following chemotherapy [4]. Risk factors for developing the syndrome include impaired renal function, increased levels of lactate dehydrogenase, and hyperuricemia [95]. The latter is caused by the disturbances in FGF23-mediated phosphate regulation in the proximal tubule of the kidney [10]. Increased intestinal phosphate absorption is mainly caused either by the use of phosphate-containing oral laxative, or by vitamin D overdoses [59].
5.4 Hypophosphatemia
Hypophosphatemia may be a consequence of the decreased intestinal absorption, internal redistribution, and increased urinary loss of phosphate [31]. The acute shift of phosphate from the extracellular to the intracellular compartment is most often caused by respiratory alkalosis and refeeding syndrome in hospitalized patients [19, 54]. Respiratory alkalosis causes an increase in intracellular pH, which stimulates phosphofructokinase, leading to severe hypophosphatemia with plasma phosphate of >0.32 mmol/L [82]. The intracellular shift of phosphate is also observed in the treatment of diabetic ketoacidosis and hungry bone syndrome, which occurs after parathyroidectomy performed for patients with long-standing hyperparathyroidism [31]. At the same time, in the postoperative period, serum calcium and phosphate concentrations significantly decrease.
Low phosphate intake rarely causes hypophosphatemia, probably because the phosphate content in the diet almost always exceeds the phosphate loss through the gastrointestinal tract, and the kidneys can reabsorb nearly all of the filtered phosphate [24]. Excessive urinary loss of phosphate is observed in both primary and secondary hyperparathyroidism caused by impaired vitamin D metabolism, Fanconi syndrome, diuretics, and tumor-induced osteomalacia (TIO) [31, 48]. TIO is a rare paraneoplastic syndrome characterized by hypophosphatemia, phosphaturia, decreased 1,25(OH)2D level, normal 25(OH)D levels, and osteomalacia [29]. Overproduction of FGF23 caused by TIO reduces tubular phosphate reabsorption and 1,25(OH)2D production [58].
5.5 Genetic Disorders Associated with Hypophosphatemia
Several inherited abnormalities are characterized by phosphate-wasting syndromes, commonly mediated by FGF23. These diseases, resulted by impaired FGF23 metabolism, include autosomal dominant hypophosphatemic rickets (ADHR), X-linked hypophosphatemic rickets (XLHR), and autosomal recessive hypophosphatemic rickets (ARHR) [94].
ADHR (OMIM 193100) is produced by FGF23 gain-of-function mutation, which causes the resistance of the mutant FGF23 to proteolytic degradation [22]. ADHR manifests as a defect in renal phosphate transport, associated with decreased 1,25(OH)2D levels, while the PTH levels remain normal. ADHR is characterized by hypophosphatemia, renal phosphate loss, short stature, and bone disorders [25].
ARHR (OMIM 241520) is caused by mutations in the DMP1 gene (located on chromosome locus 4q21). Patients with ARHR suffer from decreased renal phosphate reabsorption and typically display hyperphosphaturia, hypophosphatemia, reduced 1,25(OH)2D concentration, with PTH values remaining normal [28, 49].
XLHR (OMIM 307800) appears as a result of mutations inactivating PHEX (phosphate-regulating gene with homologies to endopeptidases located on the X-chromosome). The PHEX gene encodes a zinc-dependent metalloproteinase, and is strongly expressed in osteoblasts, osteocytes, and odontoblasts [53]. The XLHR symptoms include growth retardation, hypophosphatemia, osteomalacia, and defective renal phosphate reabsorption. The diseased state is resistant to phosphate and vitamin D therapy [63].
5.6 Conclusions
Serum phosphate levels are tightly regulated by hormonal and metabolic factors mainly related to the triad “vitamin D-PTH-FGF23” as well as dietary phosphate. Experimental studies have convincingly shown that disorders and disturbances in phosphate regulation can lead to serious systemic complications [45, 61, 62, 64, 65, 75, 77]. Particular attention should be placed on the central activity of vitamin D in phosphate metabolism, as 1,25(OH)2D both, directly and indirectly, impact serum phosphate levels. However, despite the well-studied pivotal roles of vitamin D in phosphate homeostasis, many aspects remain unclear. For instance, what are the underlying mechanisms by which vitamin D acts on renal phosphate reabsorption, and how exactly do calcium and vitamin D modulate FGF23 production? A better understanding of these processes and interactions would help to develop more efficient strategies for the treatment of phosphate-related disorders.
References
Abbas MA (2017) Physiological functions of Vitamin D in adipose tissue. J Steroid Biochem Mol Biol 165:369–381
Armbrecht HJ, Boltz MA, Christakos S, Bruns ME (1998) Capacity of 1,25-dihydroxyvitamin D to stimulate expression of calbindin D changes with age in the rat. Arch Biochem Biophys 352:159–164
Avcil S, Uysal P, Yilmaz M, Erge D, Demirkaya SK, Eren E (2017) Vitamin D deficiency and a blunted parathyroid hormone response in children with attention-deficit/hyperactivity disorder. Clin Lab 63:435–443
Belay Y, Yirdaw K, Enawgaw B (2017) Tumor lysis syndrome in patients with hematological malignancies. J Oncol 2017:9684909
Bergwitz C, Jüppner H (2010) Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med 61:91–104
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529
Biber J, Hernando N, Forster I, Murer H (2009) Regulation of phosphate transport in proximal tubules. Pflugers Arch 458:39–52
Bikle DD (2014) Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol 21:319–329
Bohnert BN, Daniel C, Amann K, Voelkl J, Alesutan I, Lang F, Heyne N, Häring H-U, Artunc F (2015) Impact of phosphorus restriction and vitamin D-substitution on secondary hyperparathyroidism in a proteinuric mouse model. Kidney Blood Press Res 40:153–165
Boyce AM, Lee AE, Roszko KL, Gafni RI (2020) Hyperphosphatemic Tumoral calcinosis: pathogenesis, clinical presentation, and challenges in management. Front Endocrinol (Lausanne) 11:293
Brown RB, Haq A, Stanford CF, Razzaque MS (2015) Vitamin D, phosphate, and vasculotoxicity. Can J Physiol Pharmacol 93:1077–1082
Buchanan S, Combet E, Stenvinkel P, Shiels PG (2020) Klotho, aging, and the failing kidney. Front Endocrinol (Lausanne) 11:560
Canaff L, Hendy GN (2002) Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J Biol Chem 277:30337–30350
Chapuy MC, Schott AM, Garnero P, Hans D, Delmas PD, Meunier PJ (1996) Healthy elderly French women living at home have secondary hyperparathyroidism and high bone turnover in winter. EPIDOS Study Group. J Clin Endocrinol Metab 81:1129–1133
Charoenngam N, Shirvani A, Holick MF (2019) Vitamin D for skeletal and non-skeletal health: what we should know. J Clin Orthop Trauma 10:1082–1093
Chen W, Bushinsky D (2017) Chronic kidney disease–mineral and bone disorder. In: Nissenson, A.R., Fine, R.N.B.T. Handbook of Dialysis Therapy, 5th edn. Elsevier, pp 685–697.e1
Clinkenbeard EL, White KE (2016) Systemic control of bone homeostasis by FGF23 signaling. Curr Mol Biol Rep 2:62–71
Coyac BR, Hoac B, Chafey P, Falgayrac G, Slimani L, Rowe PS, Penel G, Linglart A, McKee MD, Chaussain C, Bardet C (2018) Defective mineralization in X-linked hypophosphatemia dental pulp cell cultures. J Dent Res 97:184–191
De Marchi S, Cecchin E, Basile A, Bertotti A, Nardini R, Bartoli E (1993) Renal tubular dysfunction in chronic alcohol abuse–effects of abstinence. N Engl J Med 329:1927–1934
Deluca HF (2014) History of the discovery of vitamin D and its active metabolites. Bonekey Rep 3:479
Diez C, Mohr P, Koch D, Silber R-E, Schmid C, Hofmann H-S (2009) Age- and gender-specific values of estimated glomerular filtration rate among 6232 patients undergoing cardiac surgery. Interact Cardiovasc Thorac Surg 9:593–597
Drezner MK, Whyte MP (2018) Chapter 40: Heritable renal phosphate wasting disorders. In: Thakker RV, Whyte MP, Eisman JA, Igarashi TBT (eds) Genetics of bone biology and skeletal disease, 2nd edn. Academic, pp 761–782
Drüeke TB (2001) Parathyroid gland hyperplasia in uremia. Kidney Int 59:1182–1183
Dvm SDF, Moreland KJ (1989) Hypophosphatemia. J Vet Intern Med 3:149–159
Econs MJ (2005) Chapter 70: Disorders of phosphate metabolism: autosomal dominant hypophosphatemic rickets, tumor induced osteomalacia, fibrous dysplasia, and the pathophysiological relevance of FGF23. In: Feldman DBT (ed) Vitamin D, 2nd edn. Academic, Burlington, pp 1189–1195
Erem S, Razzaque MS (2018) Dietary phosphate toxicity: an emerging global health concern. Histochem Cell Biol 150:711–719
Farrow EG, White KE (2010) Recent advances in renal phosphate handling. Nat Rev Nephrol 6:207–217
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Florenzano P, Gafni RI, Collins MT (2017) Tumor-induced osteomalacia. Bone Rep 7:90–97
Fukumoto S (2014) Phosphate metabolism and vitamin D. Bonekey Rep 3:497
Gaasbeek A, Meinders AE (2005) Hypophosphatemia: an update on its etiology and treatment. Am J Med 118:1094–1101
Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, Kuro-o M, Mohammadi M (2010) Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A 107:407–412
Goretti Penido M, Alon US (2012) Phosphate homeostasis and its role in bone health. Pediatr Nephrol 27:2039–2048
Heaney RP (2011) Chapter 34: Vitamin D: role in the calcium and phosphorus economies. In: Feldman D, Pike JW, Adams JSBT (eds) Vitamin D, 3rd edn. Academic, San Diego, pp 607–624
Holick MF (2007) Vitamin D deficiency. N Engl J Med 357:266–281
Ikegame M, Ejiri S, Ozawa H (2004) Calcitonin-induced change in serum calcium levels and its relationship to osteoclast morphology and number of calcitonin receptors. Bone 35:27–33
Jacquillet G, Unwin RJ (2019) Physiological regulation of phosphate by vitamin D, parathyroid hormone (PTH) and phosphate (Pi). Pflugers Arch 471:83–98
Kamenický P, Blanchard A, Gauci C, Salenave S, Letierce A, Lombès M, Brailly-Tabard S, Azizi M, Prié D, Souberbielle J-C, Chanson P (2012) Pathophysiology of renal calcium handling in acromegaly: what lies behind hypercalciuria? J Clin Endocrinol Metab 97:2124–2133
Khammissa RAG, Fourie J, Motswaledi MH, Ballyram R, Lemmer J, Feller L (2018) The biological activities of vitamin D and its receptor in relation to calcium and bone homeostasis, cancer, immune and cardiovascular systems, skin biology, and Oral health. Biomed Res Int 2018:9276380
Khazai N, Judd SE, Tangpricha V (2008) Calcium and vitamin D: skeletal and extraskeletal health. Curr Rheumatol Rep 10:110–117
Kilav R, Silver J, Naveh-Many T (1995) Parathyroid hormone gene expression in hypophosphatemic rats. J Clin Invest 96:327–333
Klumpp S, Krieglstein J (2002) Phosphorylation and dephosphorylation of histidine residues in proteins. Eur J Biochem 269:1067–1071
Korvala J, Hartikka H, Pihlajamäki H, Solovieva S, Ruohola J-P, Sahi T, Barral S, Ott J, Ala-Kokko L, Männikkö M (2010) Genetic predisposition for femoral neck stress fractures in military conscripts. BMC Genet 11:95
Koumakis E, Cormier C, Roux C, Briot K (2021) The causes of hypo- and hyperphosphatemia in humans. Calcif Tissue Int 108:41–73
Lanske B, Razzaque MS (2007) Premature aging in klotho mutant mice: cause or consequence? Ageing Res Rev 6:73–79
Lederer E (2014) Regulation of serum phosphate. J Physiol 592:3985–3995
Leung J, Crook M (2019) Disorders of phosphate metabolism. J Clin Pathol 72:741–747
Levi M (2001) Post-transplant hypophosphatemia. Kidney Int 59:2377–2387
Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, Parvari R (2010) Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet 86:273–278
Lips P (2006) Vitamin D physiology. Prog Biophys Mol Biol 92:4–8
Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD (2006) Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol 17:1305–1315
Lofrese JJ, Basit H, Lappin SL (2021) Physiology, parathyroid. Treasure Island (FL)
Magen D, Zelikovic I (2012) Chapter 27: Hereditary tubular disorders of mineral handling. In: Glorieux FH, Pettifor JM, Jüppner HBT (eds) Pediatric bone, 2nd edn. Academic, San Diego, pp 727–770
Marik PE, Bedigian MK (1996) Refeeding hypophosphatemia in critically ill patients in an intensive care unit. A prospective study. Arch Surg 131:1043–1047
Marks J (2019) The role of SLC34A2 in intestinal phosphate absorption and phosphate homeostasis. Pflugers Arch 471:165–173
Marthi A, Donovan K, Haynes R, Wheeler DC, Baigent C, Rooney CM, Landray MJ, Moe SM, Yang J, Holland L, di Giuseppe R, Bouma-de Krijger A, Mihaylova B, Herrington WG (2018) Fibroblast growth Factor-23 and risks of cardiovascular and noncardiovascular diseases: a meta-analysis. J Am Soc Nephrol 29:2015–2027
Miller WL (2017) Genetic disorders of vitamin D biosynthesis and degradation. J Steroid Biochem Mol Biol 165:101–108
Minisola S, Peacock M, Fukumoto S, Cipriani C, Pepe J, Tella SH, Collins MT (2017) Tumour-induced osteomalacia. Nat Rev Dis Prim 3:17044
Moe SM, Daoud JR (2014) Chapter 11: Disorders of mineral metabolism: calcium, phosphorus, and magnesium. In: Gilbert SJ, Weiner DE (eds) National Kidney Foundation Primer on Kidney Diseases, 6th edn, Philadelphia, pp 100–112
Muresan Z, MacGregor RR (1994) The release of parathyroid hormone and the exocytosis of a proteoglycan are modulated by extracellular Ca2+ in a similar manner. Mol Biol Cell 5:725–737
Nakatani T, Ohnishi M, Razzaque MS (2009a) Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J 23:3702–3711
Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS (2009b) In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. FASEB J 23:433–441
Neves RL, Chiarantin GMD, Nascimento FD, Pesquero JB, Nader HB, Tersariol ILS, McKee MD, Carmona AK, Barros NMT (2016) Expression and inactivation of osteopontin-degrading PHEX enzyme in squamous cell carcinoma. Int J Biochem Cell Biol 77:155–164
Ohnishi M, Razzaque MS (2010) Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J 24:3562–3571
Ohnishi M, Nakatani T, Lanske B, Razzaque MS (2009) In vivo genetic evidence for suppressing vascular and soft-tissue calcification through the reduction of serum phosphate levels, even in the presence of high serum calcium and 1,25-dihydroxyvitamin d levels. Circ Cardiovasc Genet 2:583–590
Osuka S, Razzaque MS (2012) Can features of phosphate toxicity appear in normophosphatemia? J Bone Miner Metab 30:10–18
Peacock M (2015) Chapter 31: Primary hyperparathyroidism and the kidney. In: Bilezikian JPBT (ed) The parathyroids, 3rd edn. Academic, San Diego, pp 455–467
Prasad N, Bhadauria D (2013) Renal phosphate handling: physiology. Indian J Endocrinol Metab 17:620–627
Pu F, Chen N, Xue S (2016) Calcium intake, calcium homeostasis and health. Food Sci Hum Wellness 5:8–16
Quinn JM, Fujikawa Y, McGee JO, Athanasou NA (1997) Rodent osteoblast-like cells support osteoclastic differentiation of human cord blood monocytes in the presence of M-CSF and 1,25 dihydroxyvitamin D3. Int J Biochem Cell Biol 29:173–179
Rajakumar K (2003) Vitamin D, cod-liver oil, sunlight, and rickets: a historical perspective. Pediatrics 112:e132–e135
Razzaque MS (2009a) FGF23-mediated regulation of systemic phosphate homeostasis: is klotho an essential player? Am J Physiol Renal Physiol 296:F470–F476
Razzaque MS (2009b) Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol Dial Transplant 24:4–7
Razzaque MS (2014) Bone-kidney axis in systemic phosphate turnover. Arch Biochem Biophys 561:154–158
Razzaque MS, Lanske B (2006) Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med 12:298–305
Razzaque MS, St-Arnaud R, Taguchi T, Lanske B (2005) FGF-23, vitamin D and calcification: the unholy triad. Nephrol Dial Transplant 20:2032–2035
Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B (2006) Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J 20:720–722
Rigo J, Pieltain C, Viellevoye R, Bagnoli F (2018) Calcium and phosphorus homeostasis: pathophysiology. In: Buonocore G, Bracci R, Weindling M (eds) Neonatology: a practical approach to neonatal diseases. Springer International Publishing, Cham, pp 639–668
Ritter CS, Armbrecht HJ, Slatopolsky E, Brown AJ (2006) 25-Hydroxyvitamin D3 suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int 70:654–659
Ritz E, Gross M-L (2005) Hyperphosphatemia in renal failure. Blood Purif 23:6–9
Roussanne MC, Lieberherr M, Souberbielle JC, Sarfati E, Drüeke T, Bourdeau A (2001) Human parathyroid cell proliferation in response to calcium, NPS R-467, calcitriol and phosphate. Eur J Clin Investig 31:610–616
Rudolph EH, Gonin JM (2012) Chapter 79: Disorders of phosphorus metabolism. In: Lerma EV, Nissenson ARBT (eds) Nephrology secrets, 3rd edn. Mosby, Saint Louis, pp 551–559
Schiavi SC, Kumar R (2004) The phosphatonin pathway: new insights in phosphate homeostasis. Kidney Int 65:1–14
Schmitt A, Ehnert S, Schyschka L, Buschner P, Kühnl A, Döbele S, Siebenlist S, Lucke M, Stöckle U, Nussler AK (2012) Monocytes do not transdifferentiate into proper osteoblasts. Sci World J 2012:384936
Shoback DM, Bilezikian JP, Turner SA, McCary LC, Guo MD, Peacock M (2003) The calcimimetic cinacalcet normalizes serum calcium in subjects with primary hyperparathyroidism. J Clin Endocrinol Metab 88:5644–5649
Slatopolsky E, Brown A, Dusso A (2001) Role of phosphorus in the pathogenesis of secondary hyperparathyroidism. Am J Kidney Dis 37:S54–S57
Stefanopoulos D, Nasiri-Ansari N, Dontas I, Vryonidou A, Galanos A, Psaridi L, Fatouros IG, Mastorakos G, Papavassiliou AG, Kassi E, Tournis S (2020) Fibroblast growth factor 23 (FGF23) and klotho protein in Beta-thalassemia. Horm Metab Res – Horm und Stoffwechselforsch 52:194–201
Sun M, Wu X, Yu Y, Wang L, Xie D, Zhang Z, Chen L, Lu A, Zhang G, Li F (2020) Disorders of calcium and phosphorus metabolism and the proteomics/metabolomics-based research. Front Cell Dev Biol 8:576110
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–774
Villa-Bellosta R, Ravera S, Sorribas V, Stange G, Levi M, Murer H, Biber J, Forster IC (2009) The Na+-Pi cotransporter PiT-2 (SLC20A2) is expressed in the apical membrane of rat renal proximal tubules and regulated by dietary Pi. Am J Physiol Renal Physiol 296:F691–F699
Vogt I, Haffner D, Leifheit-Nestler M (2019) FGF23 and phosphate-cardiovascular toxins in CKD. Toxins (Basel) 11:647
Voinescu A, Martin KJ (2013) Chapter 19: Calcium, phosphate, PTH, Vitamin D and FGF-23 in chronic kidney disease. In: Kopple JD, Massry SG, Kalantar-Zadeh KBT (eds) Nutritional management of renal disease. Academic, pp 263–283
Wald A, Narasimhan S, Nieves-Arriba L, Waggoner S (2009) Prolonged hypercalcemia following resection of dysgerminoma: a case report. Obstet Gynecol Int 2009:956935
White KE, Evans WE, O’Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Will A, Tholouli E (2011) The clinical management of tumour lysis syndrome in haematological malignancies. Br J Haematol 154:3–13
Xiao Z, Dallas M, Qiu N, Nicolella D, Cao L, Johnson M, Bonewald L, Quarles LD (2011) Conditional deletion of Pkd1 in osteocytes disrupts skeletal mechanosensing in mice. FASEB J 25:2418–2432
Yamaguchi T, Sugimoto T, Chihara K (2002) Intestinal absorption of phosphate. In: Morii H, Nishizawa Y, Massry SG (eds) Calcium in internal medicine. Springer, London, pp 123–135
Acknowledgement
The authors would like to thank Dr. Margo Wolfe for carefully reading the manuscript and providing useful suggestions.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Akimbekov, N.S., Digel, I., Sherelkhan, D.K., Razzaque, M.S. (2022). Vitamin D and Phosphate Interactions in Health and Disease. In: Razzaque, M.S. (eds) Phosphate Metabolism . Advances in Experimental Medicine and Biology, vol 1362. Springer, Cham. https://doi.org/10.1007/978-3-030-91623-7_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-91623-7_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-91621-3
Online ISBN: 978-3-030-91623-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)