
Abstract
The different forms of lymphoid organization that coexist in our bodies appeared at distinct time points during the evolution of the animal kingdom. Some of these forms are constitutive, either in fully dedicated organs, such as lymph nodes, or in tissue interfacing with the external environment, such as mucosal-associated lymphoid tissues. Others, known as tertiary lymphoid structures (TLS), are selectively induced in response to inflammation in any peripheral tissues and organs. In this chapter, we discuss the functional interest of each of these lymphoid organizations under different physiopathological conditions. In the context of cancer, recent findings have identified TLS formation as a hallmark of active T- and B-cell immune responses against tumors. TLS are thus a powerful prognostic factor in nearly all solid cancers, which must be taken into account along with the tumor microenvironment. The presence of TLS also predicts the response to immunotherapy including immune checkpoint blockade. With tumor-associated TLS now a key target for the next generation of immunotherapy, this chapter discusses their potential therapeutic manipulations in oncology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Tertiary lymphoid structure
- Ectopic lymphoid organ
- Lymphoid aggregate
- Tumor microenvironment
- T cell
- B cell
- Dendritic cell
- Regulatory T cell
- Solid tumor
- Prognostic factor
- Predictive factor
- Secondary lymphoid organ
- High endothelial venule
- Immunotherapy
- Immune checkpoint
1 Introduction
The tumor microenvironment is an essential source of information for understanding the heterogeneity of tumors and their evolution over time. Among the approaches used to investigate this complexity microenvironment, immunohistochemistry is remarkable in its ability both to measure the density of tumor-infiltrating cells and to locate them and their positions relative to each other within a complex architecture. This approach made it possible to describe the organization of inflammatory cells into tertiary lymphoid structures (TLS) two decades ago in autoimmune diseases [1] and much more recently in solid cancers [2]. The literature refers to these structures by many names besides TLS: tertiary lymphoid organ/tissue/aggregate, tertiary lymphatic organ, ectopic lymphoid organ/structure/tissue, lymph node-like structures/aggregate/organization, lymphoid nodule/aggregate, ectopic germinal center, ectopic follicular structure, ectopic reactive lymphoid tissue, extranodal lymphoid reaction, Crohn’s-like lymphoid reaction, nodular lymphatic infiltrate, and induced bronchus-associated lymphoid tissue (in lung). This de novo organization of immune cells is not an organ or a tissue but rather emerges within them. Thus, TLS are induced in response to inflammation in nonlymphoid sites and disappear with its resolution. Their presence in association with tumors raises the question of their immune function within the tumor microenvironment and consequently of their potential manipulation for therapeutic purposes. In this chapter, we describe several concepts, address issues related to TLS that are still debated in different pathological contexts, and conclude by discussing their potential therapeutic manipulation in oncology.
2 TLS Versus SLO: Which Came First in Animal Evolution?
The similarity in structure and cellular organization of TLS and secondary lymphoid organs (SLO) is striking, with TLS lacking only the natural killer (NK) cells found in SLO [3]. TLS are composed of a T-cell-rich area with mainly T lymphocytes and mature dendritic cells (DC), adjacent to a B-cell zone consisting of B cells, follicular helper T cells, macrophages, and follicular dendritic cells (FDC) [4]. Like canonical SLO, TLS are key sites for the initiation of adaptive immune responses. It is therefore legitimate to wonder why such ectopic lymphoid structures develop at the effector site when humans are born with a spleen, around 600 lymph nodes distributed throughout the entire body, and numerous mucosa-associated lymphoid tissues, known as MALT (e.g., G(gut)ALT, N(nasal)ALT, B(bronchus)ALT, C(conjunctival)ALT, L(larynx)ALT, S(skin)ALT, V(vulvo-vaginal)ALT).
Part of the answer may come from an analysis of the immune system throughout the evolution of animals. This analysis may enable us to determine whether these two types of lymphoid organization, TLS and SLO, coevolved over time or, if instead, the survival of various species required immune systems to become increasingly sophisticated, by the emergence of TLS and then SLO, in response to pathological situations. Infectious diseases were likely at the forefront of the selective pressure on the evolution of the immune system in animals for millions of years. This is exemplified on a shorter scale in humans, among whom aging-related diseases, such as many cancers and metabolic illnesses, have only recently become leading causes of death. Before then, recurrent epidemics of infectious diseases due to pathogens decimated millions until the implementation of vaccination, due to the pioneering work of Dr. Edward Jenner in 1796 against smallpox and Louis Pasteur in 1885 [5] against rabies.
The route by which pathogens enter the human body may explain the critical need for the establishment of immune defense at animals’ mucosal sites. Analysis of the evolution of the animal kingdom over time shows that TLS and GALT appeared long before lymph nodes [6]. TLS appeared in lower vertebrates, such as jawless fish, about 500 million years ago during the Cambrian period, whereas lymph nodes appeared much later, 200 million years ago, at the boundary between the Triassic and Jurassic periods, in mammals and rare birds. MALT and TLS are the first barrier of immune defense against pathogens and play a major role in preventing—or at least limiting—the spread of infectious agents throughout the body. Constitutively present, MALT are key sentinels in mucosa, already organized and functional at any moment. The inducible nature of TLS means that they can selectively develop at a pathogen’s entry site, regardless of the tissue, with the ability to mobilize innate and adaptive immune cells locally within a few hours. In contrast to GALTs, TLS can also develop in nonmucosal tissues and organs. They therefore offer a more sophisticated level of immune defense to preserve the integrity of the body in diseases that can emerge in any tissue or organ, such as cancer or infectious diseases. A study of postmortem specimens of human fetal and infant lungs [7] demonstrated that TLS (then called BALT) are detected in all cases with a diagnosed infection. TLS have been observed in fetal lungs as early as 16 weeks of gestation, thus earlier than MALT formation, which has been detected around week 20. The capacity of TLS to be generated in any tissue is underlined by their presence in the brain of patients with multiple sclerosis, for this organ has long regarded as an immunological sanctuary. In this case, the presence of TLS may be related to the pathological process [8].
As the immune system has adapted to many different pathological contexts from the time of the earliest animals by developing ever more sophisticated structures, different lymphoid organizations now coexist in all mammals including humans, each of them with its own specificities.
3 TLS and SLO: Is There Redundancy in the Immune System?
The strong similarities between TLS and SLO can make it tempting to speculate that their immune functions are identical. Several observations nonetheless challenge this assumption. Studies in mice lacking SLO (i.e., without a spleen, lymph nodes, and Peyer’s patches) have shown that these animals can develop TLS in the lungs and mount effective primary and secondary immune responses after infection by the influenza virus [9, 10]. These findings suggest that TLS alone are sufficient to protect animals against this virus. Functional complementarity between TLS and SLO during disease development cannot be formally excluded. Since TLS return to normal size after resolution of the acute phase of inflammation, SLO may be secondary immune hubs where the adaptive immune response initially mounted in TLS can be maintained, amplified, and then disseminated to other immune organs. In cancer, the migration of memory B and T lymphocytes from TLS (induced by the chronic inflammation associated with tumor growth) to the draining lymph node, and then to other lymph nodes, is likely to be essential in fighting metastases through the development of systemic immune surveillance. We can accordingly presume that the main role of SLO is as a niche for the homing of memory adaptive immune cells that have been differentiated within TLS. Cell tracking experiments in animals will be helpful in confirming or disproving this hypothesis.
Moreover, it is important to note that cancer behaves markedly differently in TLS and in SLO: tumor metastases occur in SLO, but not in TLS (except for a rare subtype of HCV+ non-alcohol-dependent patients with hepatocellular carcinoma, HCC) [11]. Thus, TLS, contrary to SLO, likely harbor specific cellular and molecular features that prevent their invasion by tumor cells. Although they emerge in a tumor, TLS are mostly localized in the invasive margin of the tumor. This observation reflects their dynamics and ability to escape tumor invasion. This finding again raises the question of what consequences these differences between TLS and SLO might have on patients’ clinical outcome.
Taken together, the enormous plasticity of TLS over time (as transient structures) and in space (through their potential presence in any organ or tissue), capable of avoiding functional disruption by tumor cell invasion, makes them a powerful immune hub for the initiation of adaptive immunity at the effector site, eventually relayed to SLO. As discussed below, this central role of TLS in setting the adaptive immune response in motion has major consequences in the control of diseases, especially cancers.
4 TLS: a Powerful Prognostic Biomarker in Cancers
4.1 Prognostic Factor Across Human Cancer Types
The similarity between TLS and SLO provides evidence supporting the involvement of TLS in the establishment of adaptive immune responses. One way to measure their involvement in antitumor immunity is to assess their correlation with the clinical outcome of cancer patients. TLS were first observed in solid tumors among patients with non-small cell lung carcinoma (NSCLC), and a high density of these structures was associated in this report with patients’ long-term survival [2] (Table 1).
Nearly all subsequent studies of TLS took place in carcinomas, the most commonly diagnosed cancer, which originates in organs and glands. Until now, TLS have been shown to be strongly prognostic of and an independent factor favoring survival in most primary carcinomas (i.e., breast, colorectal, gastric, Merkel cell, oral, ovarian, pancreatic, renal cell, HCC, urothelial bladder, and biliary tract cancers, together with melanomas) (Table 1) as well as in metastatic lesions (Table 2). Interestingly, the tumor’s immunogenicity, which depends, at least in part, on its neoepitope load and on the presence of immunosuppressive factors, has no incidence on the predictive value of TLS. TLS neogenesis occurs both for highly immunogenic tumors (i.e., NSCLC and melanoma) and for those that are poorly immunogenic (i.e., pancreatic and ovarian carcinomas), and high densities of these structures are associated with a favorable clinical outcome. The presence of TLS appears to shape the local immune microenvironment. In NSCLC, TLShigh tumors are extensively infiltrated by CD8+ T cells, whereas the reverse is not true [12], that is, a high density of CD8+ T lymphocytes is not associated with a favorable outcome in TLSlow tumors in either NSCLC [12] or ovarian cancer [13]. This finding indicates that the presence of TLS is critical for the local education of effector T cells against endogenous tumor-associated antigens in hot as well as cold tumors. Similarly, no difference in patient outcome has been reported for tumors induced—or not—by an oncogenic virus as long as TLS density is high. This finding suggests that the composition of the tumor microenvironment is far more important than the presence of viral antigens in the formation and immune function of TLS.
Two studies, on the other hand, have reported negative effects by TLS on the survival of cancer patients (Table 1). The first recently showed that high TLS densities are correlated with poor outcome for patients with primary breast cancer although they correlate with long-term survival in its metastatic lesions [14]. Other studies have also described a positive influence by TLS in patients with other metastatic tumors (Table 2). A possible explanation for these paradoxical observations is that, in contrast to newly emerging metastases, the microenvironment of the primary tumor has changed considerably by the most advanced stage of the disease; immunosuppressive mechanisms have been established, some of them affecting the antitumor immune function of TLS. A deeper analysis of tumor-infiltrating immunoregulatory cells and expression of immune checkpoint molecules on individual TLS during tumor progression would be very helpful for understanding these paradoxical observations. The second study also associated TLS with a poor prognosis in a rare subset of HCV− non-alcohol-dependent patients with HCC, most probably because TLS provide microniches for tumor progenitor cells in this disease [11]. More investigation is needed to know whether this surprising observation is restricted to this rare HCC subtype. Thus far, tumor-invaded TLS have not been observed in other types of cancer, including NSCLC (personal data). Finally, only a few studies have investigated the prognostic value of TLS in sarcomas, a type of cancer affecting soft and connective tissues such as bones, blood vessels, nerves, fat, and muscles. TLS have been described in retroperitoneal liposarcoma, and high densities were associated with a trend toward a favorable clinical outcome [15, 16]. Analysis of datasets from two pooled cohorts (TCGA SARC and GSE21050) and a retrospective cohort of patients with sarcomas has confirmed this point [17].
A few studies have also reported TLS in nontumoral tissue but close to the invasive margin of the tumor (these TLS are called “peritumoral TLS” in contrast to the TLS found within the invasive margin). In breast cancer, a high density of peritumoral TLS is correlated with the worst outcome [18] (Table 1). In that study, however, the authors neither investigated the prognostic impact of the intratumor TLS nor compared the impact of the TLS in these two different areas with the clinical outcome in their cohort. In patients with HCC, the positive prognostic impact of TLS disappears when the density of adjacent nontumoral TLS is compared to that of tumor-associated TLS [19]. More investigations are clearly necessary to improve our understanding of the role of peritumoral TLS, compared with TLS in the invasive margin. Animal models should certainly offer some key answers and provide a dynamic view of the immune function of TLS in different tumor areas.
4.2 Methods for TLS Quantification
Two different approaches—i.e., immunohistochemistry on paraffin-embedded tumor sections and gene expression (TLS signature) on frozen tumor samples—have been used for TLS quantification and produce generally similar results. Nonetheless, discrepancies in a few studies most likely reflect key methodological differences (Table 1). The number of patients enrolled is critical to a study’s statistical power (i.e., Kaplan-Meier curves, log-rank test), particularly when it includes several histological types, various TNM stages, and treatment by radiotherapy and/or chemotherapy. The selection of the tumor sample (biopsy or surgical excision) for immunohistochemistry is also determinant, as TLS are mainly detected in the tumor’s invasive margin. Most studies use immunohistochemistry (single or multiple immunostainings), but others prefer to quantify TLS by histology (counterstaining) of paraffin-embedded tumor sections: hematoxylin and eosin counterstaining, which has the advantages of easy and rapid performance, together with low cost. This technique does not, however, enable an assessment of the level of TLS organization that can unambiguously discriminate between lymphoid aggregates and fully mature TLS, characterized by the presence of DC and T and B lymphocytes. Consequently, hematoxylin and eosin counterstaining may be less accurate than multiple immunostainings for quantifying fully organized TLS.
Another key issue for stratifying tumors by TLS density involves the choice of the threshold value. Some studies have used as a cutoff value the absence of TLS versus the presence of at least one, while others have used the median value of the number of TLS. Still others have used a mathematical method called “optimal cutoff P value” based on Altman’s formula [20]. This method determines the cutoff value that best distinguishes patients according to their clinical outcome.
It is clearly now time to normalize these criteria and standardize the method of TLS evaluation and quantification. Some guidelines can be established (Table 3): (1) select a block of tumor containing the invasive margin and the highest immune infiltrate (most objective criteria, highly reproducible); (2) include at least 20 patients in the smallest group (i.e., the minimum number of patients needed to achieve sufficient statistical power); and (3) use Altman’s unbiased methods to stratify patients into high versus low groups according to TLS density.
Taken together, these guidelines will improve the standardization of TLS studies in humans, standardization necessary for these structures that are likely to serve as a powerful biomarker in a number of cancers.
5 TLS: A Key Target for Next-generation Immunotherapy
The association of TLS with a better prognosis in the vast majority of solid cancers opens new avenues for the development of innovative immunotherapeutic protocols. Formidable challenges remain to be addressed. It is still unclear how TLS can be induced in situ and which cancer patients should be treated with such a TLS-inducing therapy.
TLS can be described as a remarkable “antitumor school” where the initiation and/or reactivation of the adaptive immune response can take place [21, 22]. That is, within TLS, tumor-associated antigens are continuously sampled and processed into peptides that are presented to T cells by mature DC and B cells, thus triggering the development of an adaptive antitumor immune response. This ability of TLS to continuously process tumor antigens and mount adaptive immune responses holds major interest for the development of new approaches to immunotherapy. Targeting the epitopes and neoepitopes of tumor-associated antigens is one of the most important and elusive challenges in immunotherapeutic clinical trials aimed either at triggering adaptive antitumor responses in different ways (vaccination or ICP use) or at using effector cells with defined epitope specificities (CAR-T or cytotoxic T cells). Thus, strategies focused on the induction of functional TLS have a huge advantage: they do not need to start by defining which tumor-associated antigens (or derived peptides) to target since endogenous processing of antigens occurs in TLS. In addition, these strategies might be applicable to all cancer patients, each of them setting up his or her own “antitumor school.”
Targeting TLS, however, requires the identification of molecular inducers, which is itself a challenge. Several studies have investigated the molecular and cellular mechanisms leading to TLS neogenesis to discover key TLS inducers. They have underlined that the TLS formation program shares many similarities with the SLO organogenesis program, including the lymphoid chemokine (i.e., CCL19, CCL21, and CXCL13) and lymphotoxin-β (LTβ)/LIGHT axis. The cross-talk in the lymph nodes between DC and endothelial cells is critical for the differentiation and maturation of the latter into high endothelial venules (HEV); it is mediated through the LTβ signaling pathway [23]. HEV, because they are always located in close proximity to TLS in tumors, have been suggested as the major gateway by which circulating immune cells are recruited into TLS. Strategies aimed at inducing de novo HEV in tumors may be an interesting way to boost TLS neogenesis. An anti-VEGFR2/anti-PD-L1 antibody combination has been used to test this approach [24]. Activation of LTβR signaling induced HEV formation and thereby enhanced CD8+ T-cell recruitment in PyMT breast cancer and RT2-PNET pancreatic neuroendocrine tumors but not in glioblastoma. Interestingly, in this study, agonist anti-LTβR antibody facilitated HEV formation and CTL activity even in glioblastoma. Nonetheless, the failure of the immune cells to organize into fully mature TLS indicated that another signal downstream from the LTβ/LTβR axis or through another pathway is required for TLS formation. Consistent with this observation, CD8+ T cells and NK cells can induce the expression of PNAd, a marker of HEV, in an LTβR-independent manner via the engagement of soluble LTα3 and TNF-α [25, 26]. This finding shows that LTα3 and TNF-α are alternative triggers for the differentiation of HEV, which can, in turn, induce TLS neogenesis.
Another strategy targets the LIGHT axis. Fu et al. have engineered an anti-EGFR human/mouse LIGHT fusion protein to activate LIGHT signaling in close proximity to EGFR+ tumor cells [27]. This fusion protein controlled tumor growth and initiated adaptive immune responses in an LTβR-dependent manner in various murine tumor models and xenograft human tumor models. More importantly, this engineered molecule was able to overcome tumor resistance to treatment by anti-PD-1 antibody through the de novo infiltration of immune cells that organized into TLS. This work suggests that combining a TLS inducer with an immune checkpoint (ICP) blockade could be proposed to patients who do not respond to ICP treatment alone.
Another study that also focused on the LIGHT axis used a mouse LIGHT-vascular targeting peptide (VTP) fusion protein to target tumor vasculature in a pancreatic tumor model [28]. Despite significant side effects, LIGHT-VTP normalized blood vessels (i.e., the remodeling and alignment of endothelial cells surrounded by pericytes) and induced TLS via an LTβR-independent HVEM-dependent mechanism. Interestingly, two subsets of pericytes have been reported [29]. Each has been observed around large blood vessel walls and small capillaries, but only type-2 pericytes promote tumor angiogenesis. Given that the phenotype of pericytes matures after LIGHT-VTP treatment [30], it will be important to decipher which subset is associated with—and may contribute to—TLS neogenesis.
In contrast to the previous study, associating LIGHT-VTP with one ICP molecule (CTLA-4 or PD-1) produced no observable survival benefit. Inversely, the combination of LIGHT-VTP with anti-CTLA-4 and anti-PD-1 antibodies inhibited tumor growth, enhanced survival rates, and generated both cytotoxic and memory antitumor T cells. It thus demonstrated the efficacy of targeting TLS at the tumor site and its synergistic effect when combined with ICP therapy.
Interestingly, a HEV density has been negatively correlated with the Treg/CD3+ T cell ratio in patients with breast cancer [31]. Tregs can home into distinct tumor areas including TLS in primary tumors (i.e., breast, prostate, and lung squamous cell carcinomas [32,33,34,35]) and in pulmonary metastases from colorectal cancer [36]. The association of a decreased Treg/Th1 T cell ratio in TLS with spontaneous human prostate cancer regression [34] suggests that increasing this ratio within TLS might inhibit an ongoing immune response. In line with this observation, Treg depletion can boost HEV and TLS formation and cause immune-mediated tumor eradication [37, 38]. Finally, another elegant strategy provoked TLS neogenesis directly by injecting stromal cells that act as lymphoid tissue organizing (LTo) cells [39]. This approach, besides inducing TLS in the tumor microenvironment, also contributed to de novo lymphocyte recruitment, most probably through the secretion of chemokines (e.g., CCL2, CCL3, CCL4) and decreased PD-1 and TIM-3 expression on tumor-infiltrating CD8+ T cells. These findings were accompanied by the suppression of tumor growth.
Evidently, these findings leave numerous questions about the patients who can be treated by TLS inducers. The types of cancers, disease stages, and route of infusion are among the many issues to be solved, along with whether cold tumors can be manipulated. Moreover, the combination of TLS inducer-based therapy with some current clinical practices might be deleterious for the induction of TLS in an inflammatory milieu. This is exemplified in corticosteroid-treated SCC patients who show fewer TLS than untreated patients [40]. Clearly, further investigation is required to identify TLS inducers with adequate safety and efficacy profiles.
6 Conclusion and Perspectives
TLS can be analogized to a powerful antitumor school where many tumor-associated antigens (i.e., antigens that are overexpressed, mutated, or truncated, as well as neoantigens) are continuously sampled and presented by DC and B cells to T lymphocytes within the tumor. This process develops an individual antitumor adaptive immune response in each patient. Analysis of the specificity of TLS-derived T and B cells might therefore allow the identification of new targets in long-term survivors as well as among those receiving immune or other therapy.
TLS can serve as a powerful prognostic biomarker in nearly all solid tumors. As discussed above, despite some pending technical issues about TLS quantification, it is now basically feasible to evaluate TLS density in tumor samples and thus to identify patients at high risk of relapse. Patients with high TLS density may benefit from ICP blockade, according to recent reports that TLS presence can also predict response to ICP therapy in patients with melanoma, ccRCC, and sarcoma [17, 41, 42]. The key question now is how to induce TLS in cancer patients lacking these structures or with low TLS density or in inoperable patients to force a switch from TLSlow to TLShigh status. Different strategies have proved effective in inducing TLS in several preclinical models. The success of this next-generation immunotherapy lies in our ability to control the selectivity of the targeting in the desired organ and to manage possible side effects. We can also hope that the modification of the tumor microenvironment that follows TLS induction also triggers a positive feedback loop allowing the maintenance of these lymphoid structures and their antitumor function in the long term.
Now more than ever, TLS appear to be a key ingredient in next-generation immunotherapy, offering promise for boosting endogenous TLS-dependent antitumor immunity and for optimizing current anticancer therapy—immune and others (Fig. 1).

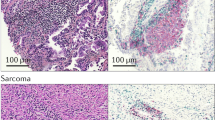
Organization of tumor-infiltrating immune cells into TLS in NSCLC patient
Multiple stainings on FFPE lung tumor sections. (a–b) TLS-CD20 (red)+ B cells are mainly detected in the invasive margin of the tumor (hematoxylin counterstaining). (c–b) TLS-T cell zone consists of CD3 (blue)+ T cells and DC-Lamp (red, arrow)+ mature DC. (c, inserts) Both mature DC and pneumocytes type II (Pn) are positive for DC-lamp, but they can be distinguished by the morphology of the staining (dot staining for mature DC, cytoplasmic staining for pneumocyte type II). Abbreviations: mDC mature DC, Pn pneumocyte. Original magnifications: A,C: ×6; B,D: ×150
References
Pipi E, Nayar S, Gardner DH et al (2018) Tertiary lymphoid structures: autoimmunity goes local. Front Immunol 9:1952. https://doi.org/10.3389/fimmu.2018.01952
Dieu-Nosjean M-C, Antoine M, Danel C et al (2008) Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol 26:4410–4417. https://doi.org/10.1200/JCO.2007.15.0284
Platonova S, Cherfils-Vicini J, Damotte D et al (2011) Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res 71(16):5412–5422. https://doi.org/10.1158/0008-5472.CAN-10-4179
Germain C, Gnjatic S, Tamzalit F et al (2014) Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med 189:832–844. https://doi.org/10.1164/rccm.201309-1611OC
De Gregorio E, Rappuoli R (2014) From empiricism to rational design: a personal perspective of the evolution of vaccine development. Nat Rev Immunol 14(7):505–514. https://doi.org/10.1038/nri3694
Hofmann J, Greter M, Du Pasquier L, Becher B (2010) B-cells need a proper house, whereas T-cells are happy in a cave: the dependence of lymphocytes on secondary lymphoid tissues during evolution. Trends Immunol 31(4):144–153. https://doi.org/10.1016/j.it.2010.01.003
Gould SJ, Isaacson PG (1993) Bronchus-associated lymphoid tissue (BALT) in human fetal and infant lung. J Pathol 169(2):229–234. https://doi.org/10.1002/path.1711690209
Louveau A (2018) Meningeal immunity, drainage, and tertiary lymphoid structure formation. Methods Mol Biol 1845:31–45. https://doi.org/10.1007/978-1-4939-8709-2_3
Moyron-Quiroz JE, Rangel-Moreno J, Kusser K et al (2004) Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med 10(9):927–934. https://doi.org/10.1038/nm1091
Moyron-Quiroz JE, Rangel-Moreno J, Hartson L et al (2006) Persistence and responsiveness of immunologic memory in the absence of secondary lymphoid organs. Immunity 25(4):643–654
Finkin S, Yuan D, Stein I et al (2015) Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol 16:1235–1244. https://doi.org/10.1038/ni.3290
Goc J, Germain C, Vo-Bourgais TKD et al (2014) Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res 74:705–715. https://doi.org/10.1158/0008-5472.CAN-13-1342
Truxova I, Kasikova L, Hensler M et al (2018) Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J Immunother Cancer 6(1):139. https://doi.org/10.1186/s40425-018-0446-3
Lee M, Heo SH, Song IH et al (2019) Presence of tertiary lymphoid structures determines the level of tumor-infiltrating lymphocytes in primary breast cancer and metastasis. Mod Pathol 32(1):70–80. https://doi.org/10.1038/s41379-018-0113-8
Tseng WW, Malu S, Zhang M et al (2015) Analysis of the intratumoral adaptive immune response in well differentiated and dedifferentiated retroperitoneal liposarcoma. Sarcoma 2015:547460. https://doi.org/10.1155/2015/547460
Yan L, Wang Z, Cui C et al (2019) Comprehensive immune characterization and T-cell receptor repertoire heterogeneity of retroperitoneal liposarcoma. Cancer Sci 110(10):3038–3048. https://doi.org/10.1111/cas.14161
Petitprez F, de Reyniès A, Keung EZ et al (2020) B cells are associated with survival and immunotherapy response in sarcoma. Nature 577(7791):556–560. https://doi.org/10.1038/s41586-019-1906-8
Sofopoulos M, Fortis SP, Vaxevanis CK et al (2019) The prognostic significance of peritumoral tertiary lymphoid structures in breast cancer. Cancer Immunol Immunother 68(11):1733–1745. https://doi.org/10.1007/s00262-019-02407-8
Calderaro J, Petitprez F, Becht E et al (2019) Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol 70(1):58–65. https://doi.org/10.1016/j.jhep.2018.09.003
Altman DG, Lausen B, Sauerbrei W, Schumacher M (1994) Dangers of using “optimal” cutpoints in the evaluation of prognostic factors. J Natl Cancer Inst 86(11):829–835
Dieu-Nosjean MC, Giraldo NA, Kaplon H et al (2016) Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev 271(1):260–275. https://doi.org/10.1111/imr.12405
Teillaud J-L, Dieu-Nosjean M-C (2017) Tertiary lymphoid structures: An anti-tumor school for adaptive immune cells and an antibody factory to fight cancer? Front Immunol 8:830. https://doi.org/10.3389/fimmu.2017.00830
Moussion C, Girard JP (2011) Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature 479:542–546. https://doi.org/10.1038/nature10540
Allen E, Jabouille A, Rivera LB et al (2017) Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med 9(385):pii: eaak9679. https://doi.org/10.1126/scitranslmed.aak9679
Peske JD, Thompson ED, Gemta L, Baylis RA, Fu YX, Engelhard VH (2015) Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun 6:7114. https://doi.org/10.1038/ncomms8114
Colbeck EJ, Jones E, Hindley JP et al (2017) Treg depletion licenses T cell-driven HEV neogenesis and promotes tumor destruction. Cancer Immunol Res 5(11):1005–1015. https://doi.org/10.1158/2326-6066.CIR-17-0131
Tang H, Wang Y, Chlewicki LK et al (2016) Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell 29(3):285–296. https://doi.org/10.1016/j.ccell.2016.08.011
Johansson-Percival A, He B, Li ZJ, Kjellen A, Russell K, Li J et al (2017) De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat Immunol 18:1207–1217. https://doi.org/10.1038/ni.3836
Birbrair A, Zhang T, Wang ZM et al (2014) Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol 307(1):C25–C38. https://doi.org/10.1152/ajpcell.00084.2014
He B, Jabouille A, Steri V et al (2018) Vascular targeting of LIGHT normalizes blood vessels in primary brain cancer and induces intratumoural high endothelial venules. J Pathol 245(2):209–221. https://doi.org/10.1002/path.5080
Martinet L, Filleron T, Le Guellec S et al (2013) High endothelial venule blood vessels for tumor-infiltrating lymphocytes are associated with lymphotoxin β-producing dendritic cells in human breast cancer. J Immunol 191(4):2001–2008. https://doi.org/10.4049/jimmunol.1300872
Gobert M, Treilleux I, Bendriss-Vermare N et al (2009) Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res 69(5):2000–2009. https://doi.org/10.1158/0008-5472.CAN-08-2360
Gu-Trantien C, Migliori E, Buisseret L et al (2017) CXCL13-producing TFH cells link immune suppression and adaptive memory in human breast cancer. JCI Insight 2(11):pii: 91487. https://doi.org/10.1172/jci.insight.91487
García-Hernández ML, Uribe-Uribe NO, Espinosa-González R et al (2017) A unique cellular and molecular microenvironment is present in tertiary lymphoid organs of patients with spontaneous prostate cancer regression. Front Immunol 8:563. https://doi.org/10.3389/fimmu.2017.00563
Nishihira M, Nakazato Y, Maeda S et al (2019) Impact of tumor infiltrating lymphocytes and lymphoid follicle formation on patient survival following surgery for lung squamous cell carcinoma. Thorac Cancer 10(2):219–225. https://doi.org/10.1111/1759-7714.12935
Schweiger T, Berghoff AS, Glogner C et al (2016) Tumor-infiltrating lymphocyte subsets and tertiary lymphoid structures in pulmonary metastases from colorectal cancer. Clin Exp Metastasis 33(7):727–739. https://doi.org/10.1007/s10585-016-9813-y
Hindley JP, Jones E, Smart K, Bridgeman H, Lauder SN, Ondondo B et al (2012) T-cell trafficking facilitated by high endothelial venules is required for tumor control after regulatory T-cell depletion. Cancer Res 72:5473–5482. https://doi.org/10.1158/0008-5472.CAN-12-1912
Joshi NS, Akama-Garren EH, Lu Y et al (2015) Regulatory T cells in tumor-associated tertiarylymphoid structures suppress anti-tumor T cellresponses. Immunity 43:579–590. https://doi.org/10.1016/j.immuni.2015.08.006
Zhu G, Nemoto S, Mailloux AW et al (2018) Induction of tertiary lymphoid structures with antitumor function by a lymph node-derived stromal cell line. Front Immunol 9:1609. https://doi.org/10.3389/fimmu.2018.01609
Silina K, Soltermann A, Movahedian Attar F, Casanova R, Uckeley ZM, Thut H, Wandres M, Isajevs S, Cheng PF, Curioni Fontecedro A, Foukas P, Levesque MP, Moch H, Linē A, van den Broek M (2018) Germinal centers determine the prognostic relevance of tertiary lymphoid structures and are impaired by corticosteroids in lung squamous cell carcinoma. Cancer Res 78:1308–1320. https://doi.org/10.1158/0008-5472.CAN-17-1987
Helmink BA, Reddy SM, Gao J et al (2020) B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577(7791):549–555. https://doi.org/10.1038/s41586-019-1922-8
Cabrita R, Lauss M, Sanna A et al (2020) Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577(7791):561–565. https://doi.org/10.1038/s41586-019-1914-8
Pfannstiel C, Strissel PL, Chiappinelli KB et al (2019) The tumor immune microenvironment drives a prognostic relevance that correlates with bladder cancer subtypes. Cancer Immunol Res 7(6):923–938. https://doi.org/10.1158/2326-6066.CIR-18-0758
Goeppert B, Frauenschuh L, Zucknick M et al (2013) Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br J Cancer 109(10):2665–2674. https://doi.org/10.1038/bjc.2013.610
Martinet L, Garrido I, Filleron T et al (2011) Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res 71:5678–5687. https://doi.org/10.1158/0008-5472.CAN-11-0431
Gu-Trantien C, Loi S, Garaud S et al (2013) CD4+ follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest 123:2873–2892. https://doi.org/10.1172/JCI67428
Lee HJ, Park IA, Song IH et al (2016) Tertiary lymphoid structures: prognostic significance and relationship with tumour-infiltrating lymphocytes in triple-negative breast cancer. J Clin Pathol 69(5):422–430. https://doi.org/10.1136/jclinpath-2015-203089
Prabhakaran S, Rizk VT, Ma Z et al (2017) Evaluation of invasive breast cancer samples using a 12-chemokine gene expression score: correlation with clinical outcomes. Breast Cancer Res 19(1):71. https://doi.org/10.1186/s13058-017-0864-z
Liu X, Tsang JYS, Hlaing T et al (2017) Distinct tertiary lymphoid structure associations and their prognostic relevance in HER2 positive and negative breast cancers. Oncologist 22(11):1316–1324. https://doi.org/10.1634/theoncologist.2017-0029
Ogino S, Nosho K, Irahara N et al (2009) Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res 15(20):6412–6420. https://doi.org/10.1158/1078-0432.CCR-09-1438
Väyrynen JP, Sajanti SA, Klintrup K et al (2014) Characteristics and significance of colorectal cancer associated lymphoid reaction. Int J Cancer 134(9):2126–2135. https://doi.org/10.1002/ijc.28533
Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A et al (2014) Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin Cancer Res 20:2147–2158. https://doi.org/10.1158/1078-0432.CCR-13-2590
Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A et al (2011) Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am J Pathol 179:37–45. https://doi.org/10.1016/j.ajpath.2011.03.007
Bindea G, Mlecnik B, Tosolini M et al (2013) Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39(4):782–795. https://doi.org/10.1016/j.immuni.2013.10.003
Giraldo NA, Becht E, Pagès F et al (2015) Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clin Cancer Res 21(13):3031–3040. https://doi.org/10.1158/1078-0432.CCR-14-2926
Remark R, Lupo A, Alifano M et al (2016) Immune contexture and histological response after neoadjuvant chemotherapy predict clinical outcome of lung cancer patients. Oncoimmunology 5(12):e1255394. https://doi.org/10.1080/2162402X.2016.1255394
Wirsing AM, Rikardsen OG, Steigen SE et al (2014) Characterisation and prognostic value of tertiary lymphoid structures in oral squamous cell carcinoma. BMC Clin Pathol 14:38. https://doi.org/10.1186/1472-6890-14-38
Li K, Guo Q, Zhang X et al (2020) Oral cancer-associated tertiary lymphoid structures: gene expression profile and prognostic value. Clin Exp Immunol 199(2):172–181. https://doi.org/10.1111/cei.13389
Hiraoka N, Ino Y, Yamazaki-Itoh R et al (2015) Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer 112(11):1782–1790. https://doi.org/10.1038/bjc.2015.145
Castino GF, Cortese N, Capretti G et al (2015) Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology 5(4):e1085147
Kuwabara S, Tsuchikawa T, Nakamura T et al (2019) Prognostic relevance of tertiary lymphoid organs following neoadjuvant chemoradiotherapy in pancreatic ductal adenocarcinoma. Cancer Sci 110(6):1853–1862. https://doi.org/10.1111/cas.14023
McMullen TP, Lai R, Dabbagh L et al (2010) Survival in rectal cancer is predicted by T cell infiltration of tumour-associated lymphoid nodules. Clin Exp Immunol 161(1):81–88. https://doi.org/10.1111/j.1365-2249.2010.04147.x
Messina JL, Fenstermacher DA, Eschrich S et al (2012) 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep 2:765. https://doi.org/10.1038/srep00765
Behr DS, Peitsch WK, Hametner C et al (2014) Prognostic value of immune cell infiltration, tertiary lymphoid structures and PD-L1 expression in Merkel cell carcinomas. Int J Clin Exp Pathol 7(11):7610–7621
Hennequin A, Derangère V, Boidot R et al (2015) Tumor infiltration by Tbet+ effector T cells and CD20+ B cells is associated with survival in gastric cancer patients. Oncoimmunology 5(2):e1054598. https://doi.org/10.1080/2162402X.2015.1054598
Hill DG, Yu L, Gao H et al (2018) Hyperactive gp130/STAT3-driven gastric tumourigenesis promotes submucosal tertiary lymphoid structure development. Int J Cancer 143(1):167–178. https://doi.org/10.1002/ijc.31298
Meshcheryakova A, Tamandl D, Bajna E et al (2014) B cells and ectopic follicular structures: novel players in anti-tumor programming with prognostic power for patients with metastatic colorectal cancer. PLoS One 9(6):e99008. https://doi.org/10.1371/journal.pone.0099008
Remark R, Alifano M, Cremer I et al (2013) Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin Cancer Res 19:4079–4091. https://doi.org/10.1158/1078-0432.CCR-12-3847
Acknowledgments
I am grateful to Dr. Jean-Luc Teillaud for helpful scientific discussions. I also thank Drs. Jérémy Goc, Claire Germain, Hélène Kaplon, Myriam Lawand, and Priyanka Devi as well as Mrs. Claudia Gutierrez-Chavez and Samantha Knockaert, for their contribution to the research work discussed here. We also thank Drs. S. Hammond and K. Steele (MedImmune, Gaithersburg, MD, USA) and Dr. S. Gnjatic (Mount Sinai Institute, Tisch Cancer Institute, New York, USA) for fruitful collaborations. I am also grateful to all clinicians and NSCLC patients involved in the studies performed in my laboratory and discussed here.
Funding
Part of this work was supported by Institut National de la Santé et de la Recherche Médicale (INSERM), Sorbonne Université, Université de Paris, Institut National du Cancer (INCa), MedImmune, the Fondation ARC pour la Recherche sur le Cancer, and La Ligue Nationale contre le Cancer.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Dieu-Nosjean, MC. (2021). Tumor-Associated Tertiary Lymphoid Structures: A Cancer Biomarker and a Target for Next-generation Immunotherapy. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1329. Springer, Cham. https://doi.org/10.1007/978-3-030-73119-9_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-73119-9_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-73118-2
Online ISBN: 978-3-030-73119-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)