
Abstract
The implications of a tumor microenvironment in cancer initiation and progression have drawn interest in recent years. Within the tumor stroma, fibroblasts represent a predominant cell type and are responsible for the majority of extracellular components within the tumor microenvironment, such as matrix and soluble factors. A switch from quiescent fibroblasts to cancer-associated fibroblasts triggers a large variety of pro-tumorigenic signals that support tumor progression and shape the surrounding pathological stroma, with the remodeling of tissue architecture and repression of the local immune response. The heterogeneous nature of cancer-associated fibroblasts and their multiple functions are subject of active research as they could represent promising targets for cutting-edge therapeutic approaches to cancer and the tumor microenvironment.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cancer-associated fibroblasts
- Epithelial-to-mesenchymal transition
- Tumor microenvironment
- Pro-tumorigenic cytokines
- Extracellular matrix remodeling
- Tumor neoangiogenesis
- Immunosuppression
- Cancer-stroma crosstalk
- Chemoresistance
- Targeted therapy
- Cancer treatment
2.1 Switching the Focus from Tumor to Tumor Microenvironment
The biological implications of the tumor microenvironment (TME) on cancer progression and its spreading have begun to be suggested over the past few years. Several studies have demonstrated that TME is not just a silent bystander, but rather an active promoter of cancer progression. A milieu of immunosuppressive T-reg lymphocytes, tumor-associated macrophages, fibroblasts, and adipocytes makes up the TME, providing a real sanctuary for cancer [48]. In particular, cancer-associated fibroblasts (CAF) are key components of the TME, closely supporting cancer by secreting mitogenic growth factors such as a fibroblast growth factor (FGF) or the insulin-like growth factor 1 (IGF-1) [4]. Furthermore, CAF are centrally involved in the NF-kB inflammatory signaling pathway which promotes tumor progression, also stimulating neo-angiogenesis. The transforming growth factor beta (TGF-β) deriving from CAF induces the epithelial-to-mesenchymal transition (EMT), which is considered the key process in cancer invasion and distant spread due to the acquisition of mesenchymal stem cell features. Not only are CAF involved in such a complex crosstalk between cancer cells and TME, they are also structurally fundamental for a cancer-supporting TME. Indeed, CAF produce extracellular matrix (ECM) proteins , which are responsible for the desmoplastic reaction at the edges of a tumor and within cancer cells, protecting them from antitumor immune responses and chemotherapeutics. Recently, the interest of researchers and clinicians has focused on CAF, considered to be key mediators of cancer-stroma crosstalk, and a promising target for novel therapeutic approaches toward TME in cancer treatment.
2.2 The Heterogeneous Nature of Cancer-Associated Fibroblasts
2.2.1 Origins and Functions of CAF
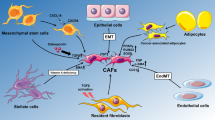
Fibroblasts are the most abundant stromal cells in the TME , accounting for up to 80% of the tumor mass in certain solid tumors characterized by a desmoplastic reaction [120]. They are particularly important because of their continuous and complex crosstalk with cancer cells [51, 91]. From a quiescent state, fibroblasts can be reversibly or irreversibly activated to form myofibroblasts in response to different inputs (Fig. 2.1). Myofibroblasts, induced by TGF-β-mediated signaling, proliferate, gain contractile properties, and unleash an injury response to repair the cellular damage and to restore tissue homeostasis [23, 26, 66, 111]. When fibroblast activation persists even in absence of the initial injury (e.g., in chronic tissue damage or fibrosis), a pathological remodeling occurs, partly depending on epigenetic regulation [121], and tumor initiation is promoted [27, 29, 98], so that tumors are considered “wounds that do not heal” [30]. By supporting tumorigenesis and by interacting with cancer cells at all tumor stages, hyperactivated fibroblasts gain enhanced proliferative properties and become a functionally diverse population, called cancer-associated fibroblasts (CAF) [52]. CAF may derive from a variety of cells, including normal fibroblasts, but also surrounding endothelial cells, pericytes, stellate cells, bone marrow-derived mesenchymal cells, and adipocytes [52, 60, 92]. Depending on their origin, the function of such activated fibroblasts can be diverse and unique (Fig. 2.2). Mediators for CAF transformation are growth factors, cytokines, and micro-RNAs soaked in the tissue milieu that can modulate the cellular response through a variety of molecular mechanisms [34]. In the early stages of neoplasia, the pathological tissue remodeling may initiate tumor-promoting functions in fibroblasts through the secretion of pro-inflammatory cues, such as interleukin (IL)-1β by immune cells [36]. Later, as the tumor grows, most of the CAF-transforming factors, including TGF-β, platelet-derived growth factors (PDGF), and FGF2, derive from direct secretion by cancer or stroma cells, either as soluble factors or transported by exosomes [3, 10, 34, 63, 65]. Moreover, matrix stiffness and solid stress in the TME constitute additional physical factors that cause sustained activation of CAF, through a feedback loop involving YAP activation and Rho-associated protein kinase (ROCK) signaling pathways [14].

Activation of fibroblasts in the tumor microenvironment , from quiescent resident fibroblasts to activated myofibroblasts to hyperactivated CAF, with sequential acquisition of key phenotypical and functional features

CAF origins in the tumor microenvironment. The acquisition of a CAF phenotype is associated with the expression of a variety of CAF-related markers
Multifaceted bio-functions of CAF aim to orchestrate the TME and manage the tumor-stroma interface via intercellular contacts, secretion of a number of factors, modification of the ECM, and promotion of malignant transformation of epithelial cells [67, 82]. CAF contribute to hypoxia-dependent tumor neo-angiogenesis and are key actors in the restricted penetration of drugs and nanodrugs in the tumor tissue, thus modifying tumor responsiveness and therapeutic efficacy of several drugs [52, 61]. Additionally, there is evidence that CAF promote cytotoxic T cell exclusion from the tumor and hinder antitumor immune responses [57].
2.2.2 Coexisting CAF Subsets
Unlike normal fibroblasts, CAF are characterized by an increased expression of certain biomarkers, which have been recently studied as potential targets for innovative therapeutics [16, 18, 112]. Depending on tumor type and origin, CAF express high levels of alpha-smooth muscle actin (α-SMA), fibroblast activation protein (FAP), fibroblast specific protein 1 (FSP1 or S100A4), vimentin, and platelet-derived growth factor receptor (PDGFR)-α and β [53, 85, 86, 107, 118]. Leucine-rich repeat containing 15 (LRRC15) membrane protein, CD10, and G protein-coupled receptor 77 (GPR77) were also found highly expressed in CAF in many solid tumors [24, 59, 89, 108]. Unfortunately, none of the identified markers are currently able to select CAF with a high degree of specificity, because of a high-grade heterogeneity characterizing this cell population [2]. As an example, the loss of caveolin 1 (CAV1) expression in breast tumor cases defines fibroblasts with pro-tumorigenic functions [102]; however, a high expression of CAV1 in CAF could also facilitate tumor invasion via ECM remodeling [41]. Thus, nowadays, it is becoming increasingly recognized that CAF represent a heterogeneous cell population of multiple origins [49]. Researchers have demonstrated the existence of distinct subsets of CAF with different localization within the tumor mass and specificity per tumor type [79, 109]. The existence of four CAF subsets has been demonstrated in triple-negative breast cancer (S1–4) and pancreatic ductal adenocarcinoma (subsets A-D). All subtypes have unique properties and expression profiles, as assessed by marker analysis and transcriptomic investigation [6]. Of note, a specific CAF phenotype corresponds to a prognostic impact. In breast cancer, S1-CAF are associated with immunosuppressive TME by promotion of T cell differentiation into T-reg, while S4-CAF are associated with high CD8+ T cell infiltration into the tumor [21].
2.2.3 Friend or Foe?
In many tumors CAF accumulation in the TME is often correlated with poor prognosis [7, 118]. Indeed, their presence is an effective predictor of tumor reoccurrence in colorectal cancer patients and has been highlighted as a significant prognostic factor in a number of other tumor types [12, 13]. At the same time, the functional role of CAF in cancer progression and metastasis is emerging as being complex and bimodal, with both cancer-promoting and cancer-restraining actions. Recent studies have suggested that CAF can restrain pancreatic ductal adenocarcinoma (PDAC) by reducing fibrosis and hypoxia [95]. Also, patients with high desmoplasia can have improved prognosis and overall survival in PDAC, breast cancer, and lung cancer, as demonstrated by correlation studies between CAF markers and disease outcome [38, 84]. CAF have also been suggested to play a tumor-suppressive role via the I kappa B kinase/NF-kB pathway, lowering hepatocyte growth factor (HGF) secretion and reducing tumor size and metastasis [79]. Keeping all of this in mind, CAF are not a unique population, but rather an updated description of CAF requires taking into consideration their dynamic state, with epigenetic changes and variable gene expression and functions.
2.3 Fibroblasts and Tumor Progression: A Key Role in Tumor Architecture Remodeling and Desmoplasia
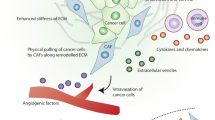
Over time, researchers have progressively realized that initiation, proliferation, invasion, and metastases of tumors do not rely on tumor cells properties alone, but they are influenced by the pathological stroma. From the “seed and soil” hypothesis, it has been recognized that the dynamic crosstalk between cancer cells (“seed”) and TME (“soil”) has a pivotal relevance in a variety of processes such as proliferation, migration, invasion, survival, angiogenesis, and EMT [83]. Through EMT, cancer cells gradually lose their epithelial hallmarks and acquire mesenchymal properties related to invasiveness and the remodeling of surrounding ECM [58]. The final result of EMT is the capability of cancer cells to reach blood circulation and metastasize at distant sites, making cancer progress from an in situ lesion to an invasive disease [46]. CAF have been shown to support cancer cell growth and metastatic dissemination in several ways [11, 51, 97] (Fig. 2.3). Their effects are mediated through both paracrine and autocrine stimulation by a variety of growth factors and cytokines, including TGF-β, bFGF, vascular endothelial growth factor (VEGF), PDGF, and interleukins (IL) [75]. TGF-β/TGF-βR signaling is required for advanced carcinogenesis via EMT induction, angiogenesis, and the modification of the stromal compartment [22, 42]. CAF-derived TGF-β1 was identified as a central molecular regulator of mesenchymal stem cells as well as a tumor-promoting factor in prostate cancer and other types of carcinoma [68, 71, 96]. Other important cues that drive the gaining of mesenchymal traits include HGF, stromal-derived factor-1α (SDF-1), osteopontin (OPN) and key cytokines released by CAF able to reprogram cancer cells through activation of the Wnt/β-catenin signaling pathway, which fosters migration and metastasis [110]. HGF and IL-6 are also considered drivers of tumor initiation and progression, through their interaction with MAPK, PI3K/Akt, and JAK/STAT signaling pathways, along with the subsequent induction of c-MET expression as positive feedback regulation [25, 116]. The coordination of these pathways controls tumorigenic progression in response to CAF’s paracrine activity. CAF-derived SDF-1, also known as CXCL12, is also able to induce an angiogenic response in synergy with the chemokine ligand CXCL -8 and enhances the invasiveness of pancreatic cancer cells [69, 81]. A gene knockdown assay and gain- and loss-of-function assays revealed that CAF secrete TGF-β and SDF-1, which promote the formation of capillary-like structures, participate in vascular endothelial cells migration, tube formation, and angiogenesis via interaction with TGF-βR1 and CXCR-4 in tumor cells [37, 119].

Multivalent activity of CAF and their secretome for shaping the tumor microenvironment
The ability to control the local remodeling of ECM is another critical function of CAF and a feature of paramount importance during the desmoplastic reaction occurring in many carcinomas. Activated fibroblasts are an important source of ECM-degrading proteases, including matrix metalloproteinases (MMP), zinc-dependent endopeptidases that facilitate cancer cell migration across ECM [101, 105, 114]. MMP-3, produced by CAF, promotes EMT by cleavage of E-cadherin and induces invasiveness of cancer cells [64, 104]. MMP-13 promotes angiogenesis by releasing VEGF and increasing the invasive capabilities of squamous cell carcinoma cells [56]. Additionally, other stromal MMP, such as MMP-1, MMP-9, and MMP-14, are able to induce cancer invasiveness, and their expression has been associated with tumor progression in several carcinomas [8, 106].
2.4 Cancer-Associated Fibroblasts in Immunosuppression and Chemoresistance
Generally, CAF are known to promote an immunosuppressive TME. Fibroblasts are a significant source of immunomodulatory cytokines and chemokines, notably interferon-γ, IL-6, CCL2, and tumor-necrosis factor-α, which can influence the mobilization of cytotoxic T lymphocytes, natural killer cells, and macrophages [43, 93, 100] (Fig. 2.3). Paracrine CAF–immune cell signaling may induce differentiation of immunosuppressive myeloid cells and affect macrophage recruitment to the tumor [55, 72, 113]. T cell recruitment and activation also involves cytokines that are found in the CAF secretome, such as CXCL9, CXCL10, and SDF-1 [5]. A recent study has shown that programmed cell death 1 ligand (PDL) 1 and 2 in a subset of CAF derived from patients with lung cancer may carry an immunosuppressive effect on T cell activation ex vivo [77, 87].
Beyond CAF secretome which switches off anticancer immunity , CAF-orchestrated ECM plays a crucial role in restricting access of immune cells to cancer, by generating a physical barrier to tumor infiltration and unmasking cryptic binding sites that could promote immune cell adhesion [33, 50]. In orthotopic tumor grafts, targeting FAP+ CAF with a DNA vaccine showed antitumor effects via suppression of collagen synthesis and intratumoral recruitment of CD8+ T cells, with the subsequent immuno-control of tumor growth [62, 80]. CAF distribution at the interface between blood vessels and tumor cells contributes to increasing the tumor interstitial fluid pressure, which represents a physical barrier to several drugs [7]. Moreover, dynamic ECM alterations may induce tissue stiffening and increased tension, which have been associated with poor outcome in patients with many solid tumors [14]. The immunosuppressive and poorly accessible TME drastically limits the potential of effective therapeutics, which have raised new hopes for the treatment of several malignant tumors. Therefore, favoring ECM remodeling and overcoming immunosuppression in the tumor is of fundamental importance for effective anticancer treatment .
2.5 Targeting Cancer-Associated Fibroblasts: Current Clinical Evidence
Considering the central role of CAF in cancer progression and diffusion, it is quite surprising that TME-targeted treatments have been so poorly explored in clinical trials to date. A main reason for the lack in clinical data is the relatively difficult specific targeting of CAF. A promising candidate for CAF targeting is FAP, a cell surface glycoprotein expressed in over 90% of these stromal cells while normally not expressed in most healthy tissues. In 1994 a first phase I study evaluated the clinical use of a monoclonal antibody toward FAP for imaging purposes, labeling it with iodine 131, to detect liver metastases from colorectal cancer [115]. In accrued patients, iodine 131-labeled anti-FAP antibodies were administered 1 week before liver surgery or regional chemotherapy, demonstrating a high accumulation within liver metastases but not in liver normal parenchyma, and no significant toxicities. Therefore, a first proof-of-concept on selective overexpression of FAP in metastatic colorectal cancer was provided, together with the usefulness of focusing on TME for clinical purposes. Subsequently, the anti-FAP antibody named sibrotuzumab was clinically assessed for anticancer efficacy in further trials. First, a phase I clinical study evaluated sibrotruzumab in FAP+ metastatic colorectal and non-small cell lung cancer [99]. After 12 weeks, treatment with sibrotuzumab showed no significant toxicities and was overall well tolerated. However, on the other hand, cancer progression was observed in all included patients, and no objective tumor response was reported. In another phase II trial, sibrotuzumab was administered in metastatic colorectal cancer patients: unfortunately, all patients still experienced cancer progression except for 2 cases, where a stable disease was observed [44]. Despite, yet again, the fact that no significant toxicities were reported, the trial failed to provide a benefit from sibrotuzumab and it was terminated. Furthermore, although no severe adverse events were reported, it should be noted that FAP is overexpressed also in bone marrow, further making clinical translation difficult. The discouraging findings from the above-mentioned trials have resulted into a long-lasting abandonment of the interest toward sibrotuzumab ,; however it has also produced a number of lines of thought. Targeting the TME could probably be a winning strategy in preventing reactivation and progression of dormant metastatic tumor cells, rather than arresting the metastatic storm once the TME has elicited its promoting activity [19]. Indeed, once cancer progression has started and metastatic disease occurs, a large amount of cancer-promoting forces are activated, making it difficult to be effectively counteracted by targeting tumor stroma only. Targeted therapy for TME might therefore be preferred as an ancillary treatment to support conventional chemotherapy in the first-line therapy of cancer, since its anticancer efficacy as a stand-alone treatment is limited, as demonstrated in preclinical studies on FAP inhibition [20] or in clinical trials targeting other TME actors, such as metalloproteinases [78]. More recently, another approach to target CAF activity has been proposed, based on inhibition of FAP enzymatic activity rather than targeting FAP itself. In a phase II clinical trial, Talabostat , an orally available amino boronic dipeptide which competitively inhibits the dipeptidyl peptidase activity, has been administered as a stand-alone therapy in metastatic colorectal patients previously treated with conventional chemotherapy [76]. Although 21% of patients maintained a stable disease for up to 25 weeks, no objective responses were observed, demonstrating a minimal clinical activity of Talabostat. However, since it was tolerated well by patients, Talabostat was further assessed in non-small cell lung cancer patients in combination with docetaxel; however, only 3 patients out of 42 reported an objective response [31]. Since Talabostat has been related to increased production of cytokines leading to enhanced antitumor immunity [1], this FAP inhibitor represented a hope for new treatment approaches in highly immunogenic malignancies, such as melanoma. Inspired by the intriguing discovery that Talabostat with cisplatin makes mice resistant to rechallenge with melanoma cells, a phase II trial evaluating Talabostat and cisplatin as a second-line therapy for metastatic melanoma was conducted [32]. A partial response was observed in less than 10% of included patients, similarly to treatment with cisplatin alone: thus, Talabostat added no clinical benefit. Furthermore, about one-third of patients experienced severe side effects related to the use of Talabostat , mainly anemia, thrombocytopenia, and neutrophilia. Regarding the minimal clinical effect, it should be noted that Talabostat acts by inhibiting the peptidase activity of FAP only; however, it was recently demonstrated that FAP promotes cancer growth and progression also through non-enzymatic activities, such as stimulating ECM remodeling by MMP-9 [47]. Phase III clinical trials on Talabostat combined with docetaxel or pemetrexed for treatment of late-stage non-small cell lung cancer were initiated, but these studies were prematurely stopped at the interim evaluation due to the observation of a lower survival rate in the Talabostat group compared to the placebo group [9]. The current difficulty in targeting FAP or in inhibiting its enzymatic activity has not decreased the great interest in implementing an effective strategy toward TME in cancer management. Indeed, while the targeting of cancer cells must follow their evolving wide heterogeneity with frequent onset of resistance, TME and interactions between cancer and TME are much more universal and common to different types of cancer, making targeting TME a promising approach. Therefore, an innovative strategy was proposed which focuses on growth factors deriving from CAF, such as FGF. Nintedanib is a pan-tyrosin kinase inhibitor, acting toward receptors for FGF, VEGF, or PDGF, overexpressed in cancer cells. By inhibiting the activity of the above-mentioned growth factors, the downstream support from CAF to cancer cells could theoretically be reduced or abolished, avoiding stimulation of tumor proliferation, migration, and survival. In 2010 Nintedanib was evaluated in a phase I clinical trial on 61 patients affected by advanced solid malignancies, demonstrating to be limited by G3-G4 reversible liver enzyme elevation but substantially showing a decent level of tolerability on behalf of patients. Despite the fact that only 3 clinical responses were reported, in 55% of patients, a significant reduction in tumor blood flow was observed, suggesting that targeting a CAF-derived growth factor may significantly impact on TME and its neoangiogenesis [74]. A further clinical trial of Nintedanib in advanced or metastatic relapsed non-small cell lung cancer administration achieved disease stabilization in 46% of patients, with a median progression-free survival of 6.9 weeks [94]. These encouraging findings warranted further clinical exploration of this strategy, and after the finding that Nintedanib in addition to docetaxel improves the overall survival rate, it is currently an established second-line treatment for non-small cell lung cancer [88]. Under the new perspective of targeting the signaling network of CAF, a monoclonal antibody toward TGF-β has been developed and named Fresolimumab . Recent clinical trials have evaluated Fresolimumab in previously treated melanoma, renal cell cancer [73], or metastatic breast cancer [39], but a limited clinical response was conjugated with the occurrence of secondary cutaneous malignancies, stopping any further clinical trial with TGF-β antagonists. Indeed, TGF-β may stimulate cancer in advanced stages making its inhibition a potential anticancer treatment; on the other hand TGF-β could mediate inhibition of cancer development in normal tissues [35].
2.6 Future Trends for Cancer Therapy Through Fibroblasts
2.6.1 CAF Reprograming
As suggested by the major concerns emerged from tout-court CAF-inhibiting strategies, TME might play several different roles in cancer progression, including both cancer-promoting and cancer-suppressing pathways. TME was classically depicted as a stable and universal feature of cancer, while it is increasingly recognized that it is highly heterogeneous. The coexistence of different subpopulations of CAF has been proposed, ranging from cancer-inhibiting to cancer-enhancing fibroblasts [51]. Therefore, a precision medicine approach should also be preferred in targeting CAF, and turning CAF from a cancer-enhancing profile to one that is cancer-inhibiting might be a more suitable strategy than the total depletion of CAF. Two recently proposed specific surface biomarkers of tumor-enhancing CAF are CD10 and GPR77, and a monoclonal antibody toward the latter receptor has shown reduced chemoresistance in a patient-derived breast cancer xenograft [108]. Beyond a precise targeting of cancer-supporting CAF, the main challenge is how to reprogram them in order to convert an immunosuppressive into an immune-permissive TME. An interesting approach has been proposed to block those signals fueling fibroblast activity, such as the angiotensin II-angiotensin II receptor type-1 axis. Indeed, angiotensin II transforms quiescent fibroblasts into CAF; therefore angiotensin receptor blockers (ARBs) should hypothetically reverse the process and reprogram CAF. A clinical concern is represented by the potent antihypertensive effects of ARBs, making them useless as anticancer treatment in clinical practice. However, ARBs have been recently nano-conjugated with pH-dependent degradable polymers in order to selectively direct ARBs into the acidic TME in a murine model of metastatic breast cancer [15]. Intriguingly, this strategy allowed for the reprogramming of CAF without hypotensive effects, deleting the immunosuppression promoted by TME and improving the T lymphocyte antitumor response, thus extending survival of mice with concurrent administration of immune checkpoint blockers. Another original approach for CAF reprograming is based on epigenetic regulation. The use of a selective inhibitor of histone deacetylases (HDACs) has been successfully used to interfere with TGF-β-mediated CAF differentiation, thus reversing CAF activation and delaying cancer growth [54].
2.6.2 Immunotherapy
Following the increasing interest toward antitumor immunity and strategies based on enhancement of T cell responses to cancer cells, a similar approach may be translated as anti-CAF treatment. In particular, combined treatments toward cancer cells and CAF are particularly promising. A specifically engineered T-cell engager for both FAP and human CD3 has been inserted into an oncolytic virus: the binding with CD3+ effector T lymphocytes and with FAP-expressing CAF lead to T cells activation and cytotoxicity toward CAF, while the oncolytic activity of the viral vector exerted its well-known anticancer effect [103]. This oncolytic approach not only results in CAF depletion, but it may also mediate a reversal of TME from immunosuppressive to immune-permissive, as shown by the repolarization of M2 macrophages toward a proinflammatory profile [40] in fresh prostate cancer tissue derived from biopsy samples. In other words, tumor-infiltrating lymphocytes could be reeducated to kill CAF, leading to TME remodeling and cancer suppression. Beyond oncolytic viruses, an elegant solution for priming the natural intratumoral immune response toward CAF is the use of specific vaccines. Tolerance toward FAP can be broken by specific DNA vaccines to exploit the cytotoxic activity of CD8+ and CD4+ T lymphocytes toward CAF. Interestingly, the T cell-mediated CAF depletion also decreased macrophage infiltration and increased intratumoral lymphocytes; furthermore this strategy was improved by adding tumor-specific DNA vaccines in different cancer models [28]. As previously stated, targeting TME as a stand-alone therapy might be ineffective, especially in aggressive cancers or where metastatic spread has already occurred. A combination strategy toward both cancer and TME could maximize the outcome. Therefore, other DNA vaccines to prime cytotoxic T lymphocytes toward FAP-positive CAF have been developed and tested in combination with chemotherapeutics with immunomodulatory activity, such as cyclophosphamide [117], demonstrating enhanced anticancer efficacy. An original sort of FAP-specific vaccination has been proposed by fusing dendritic cells, which normally present antigens to start the immune response, with CAF [90]. The resulting hybrid cells effectively activated T cells to generate a specific cytotoxic immune response toward CAF, inhibiting cancer growth.
2.6.3 Nano-strategies to Target CAF
Nanoparticles have been profoundly explored as an excellent drug delivery system in tumors, first exploiting their natural intratumoral delivery due to extravasation from leaky vasculature (the so-called enhanced permeability and retention effect, EPR). Then, by conjugation with specific antibodies, nanoparticles have been increasingly evaluated for actively targeting cancer. In both cases a high anticancer efficacy combined with a significantly lower toxicity have been reported, thanks to the specific action of drugs loaded inside cancer cells, thus avoiding off-target adverse effects in healthy tissues. Despite nanomedicine demonstrating great potential for cancer treatment, its clinical translation is a slow process, due to production costs and safety concerns. A special interest in nanomedicine has recently been developed also for targeting TME. Nano-liposomes conjugated with a peptide recognizing tenascin C, overexpressed in CAF, have been demonstrated to adequately address the anti-apoptotic drug Navitoclax in TME [17]. As a consequence, downregulation of ECM deposition, decreased interstitial fluid pressure, and increased blood perfusion with a subsequent improvement in chemotherapeutics penetration have been observed. The reduction in the high intratumoral interstitial pressure due to TME has been observed also with gold nanoparticles in xenograft of colorectal cancer [123]. Interestingly, after treatment with naked gold nanoparticles, CAF and pro-fibrotic signals decreased as well as TME stiffness, leading to increased penetrance and activity of cisplatin, which was subsequently administered. Similar findings were reported also for ovarian cancer , where gold nanoparticles were demonstrated to affect the VEGF signaling, thus blocking neoangiogenesis by disrupting the cancer cell-TME crosstalk [122]. The innate capability of untargeted gold nanoparticles to inhibit the interaction between cancer cells and TME has been more deeply studied: not only do they act on AKT pathways and VEGF signaling, they also modulate cancer cell secretome to reduce the desmoplastic feature in pancreatic cancers [70]. A more intriguing feature of gold nanoparticles might explain their natural anti-TME effects not only affecting cell crosstalk but also finely modulating the CAF profile. As recently demonstrated, gold nanoparticles increase lipid intracellular content by inducing an expression of lipogenesis genes in CAF, which use endogenously synthetized lipids to convert into quiescent fibroblasts [45]. Also, actively targeted nanoparticles toward CAF have been evaluated. A biocompatible ferritin-based nanocage has been engineered with a FAP-specific single-chain variable fragment to provide a prompt targeting and internalization into CAF, for subsequent photoirradiation exploiting the photosensitizing feature of ferritin [124]. By this nano-based photoimmunotherapy, CAF were efficiently depleted, enhancing T cell infiltration and tumor suppression in immunocompetent mice, again providing a proof-of-concept on the usefulness of targeting TME to increase antitumor immunity.
An increased interest toward implementation of anti-TME treatments for cancer therapy is expected over the next years. After the failure of clinical trials to demonstrate a significant benefit provided by anti-FAP monoclonal antibodies, it appeared clearer that, beyond merely killing CAF, other strategies aiming at reeducating CAF to modulate TME merit further exploration. Promising therapies in reaching this goal are selective inhibitors of CAF signaling, DNA vaccines toward CAF, and targeted nanodrugs; however, further characterization of CAF molecular biomarkers is needed in order to exploit suitable targets and thus avoid a tout-court action on all fibroblasts, including those providing anti-cancer activity, and avoid off-target toxicities. Finally, a selective modulation of TME could be an optimal treatment to prevent the invasive features of primary cancer or, in the best case, to prevent metastatic cancer cells in distant niches, but its potential efficacy for advanced/metastatic cancers is much less clear and combination strategies with cytotoxic drugs could maximize the outcome in these cases.
References
Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP (2004) PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res 64:5471–5480
Alkasalias T, Moyano-Galceran L, Arsenian-Henriksson M, Lehti K (2018) Fibroblasts in the tumor microenvironment: shield or spear? Int J Mol Sci 19
Aoyagi Y, Oda T, Kinoshita T, Nakahashi C, Hasebe T, Ohkohchi N, Ochiai A (2004) Overexpression of TGF-beta by infiltrated granulocytes correlates with the expression of collagen mRNA in pancreatic cancer. Br J Cancer 91:1316–1326
Balkwill FR, Capasso M, Hagemann T (2012) The tumor microenvironment at a glance. J Cell Sci 125:5591–5596
Barnas JL, Simpson-Abelson MR, Yokota SJ, Kelleher RJ, Bankert RB (2010) T cells and stromal fibroblasts in human tumor microenvironments represent potential therapeutic targets. Cancer Microenviron 3:29–47
Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, Madsen CD, Lindgren D, Pekar G, Karlsson G et al (2018) Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun 9:5150
Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, Dray C, Guiet R, Maridonneau-Parini I, Le Gonidec S et al (2013) Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res 73:5657–5668
Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120:303–313
Brennen WN, Isaacs JT, Denmeade SR (2012) Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Mol Cancer Ther 11:257–266
Bronzert DA, Pantazis P, Antoniades HN, Kasid A, Davidson N, Dickson RB, Lippman ME (1987) Synthesis and secretion of platelet-derived growth factor by human breast cancer cell lines. Proc Natl Acad Sci U S A 84:5763–5767
Bruzzese F, Hägglöf C, Leone A, Sjöberg E, Roca MS, Kiflemariam S, Sjöblom T, Hammarsten P, Egevad L, Bergh A et al (2014) Local and systemic protumorigenic effects of cancer-associated fibroblast-derived GDF15. Cancer Res 74:3408–3417
Calon A, Espinet E, Palomo-Ponce S, Tauriello DVF, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung P, Zhang XH-F et al (2012) Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 22:571–584
Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DVF, Byrom D et al (2015) Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet 47:320–329
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G et al (2013) Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 15:637–646
Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P et al (2013) Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun 4:2516
Chen X, Song E (2019) Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov 18(2):99–115
Chen B, Wang Z, Sun J, Song Q, He B, Zhang H, Wang X, Dai W, Zhang Q (2016) A tenascin C targeted nanoliposome with navitoclax for specifically eradicating of cancer-associated fibroblasts. Nanomedicine 12:131–141
Chen Q, Liu G, Liu S, Su H, Wang Y, Li J, Luo C (2018) Remodeling the tumor microenvironment with emerging Nanotherapeutics. Trends Pharmacol Sci 39:59–74
Cheng JD, Weiner LM (2003) Tumors and their microenvironments: tilling the soil. Commentary re: A. M. Scott et al., A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res., 9: 1639–1647, 2003. Clin Cancer Res 9:1590–1595
Cheng JD, Dunbrack RL, Valianou M, Rogatko A, Alpaugh RK, Weiner LM (2002) Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res 62:4767–4772
Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, Sirven P, Magagna I, Fuhrmann L, Bernard C et al (2018) Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 33:463–479.e10
Cruz-Bermúdez A, Laza-Briviesca R, Vicente-Blanco RJ, García-Grande A, Coronado MJ, Laine-Menéndez S, Alfaro C, Sanchez JC, Franco F, Calvo V et al (2019) Cancer-associated fibroblasts modify lung cancer metabolism involving ROS and TGF-β signaling. Free Radic Biol Med 130:163–173
Darby IA, Laverdet B, Bonté F, Desmoulière A (2014) Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol 7:301–311
De Francesco EM, Sims AH, Maggiolini M, Sotgia F, Lisanti MP, Clarke RB (2017) GPER mediates the angiocrine actions induced by IGF1 through the HIF-1α/VEGF pathway in the breast tumor microenvironment. Breast Cancer Res 19:129
Ding X, Ji J, Jiang J, Cai Q, Wang C, Shi M, Yu Y, Zhu Z, Zhang J (2018) HGF-mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis 9:867
Driskell RR, Watt FM (2015) Understanding fibroblast heterogeneity in the skin. Trends Cell Biol 25:92–99
Dumont N, Liu B, Defilippis RA, Chang H, Rabban JT, Karnezis AN, Tjoe JA, Marx J, Parvin B, Tlsty TD (2013) Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia 15:249–262
Duperret EK, Trautz A, Ammons D, Perales-Puchalt A, Wise MC, Yan J, Reed C, Weiner DB (2018) Alteration of the tumor stroma using a consensus DNA vaccine targeting fibroblast activation protein (FAP) synergizes with antitumor vaccine therapy in mice. Clin Cancer Res 24:1190–1201
Durning P, Schor SL, Sellwood RA (1984) Fibroblasts from patients with breast cancer show abnormal migratory behaviour in vitro. Lancet 2:890–892
Dvorak HF (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315:1650–1659
Eager RM, Cunningham CC, Senzer N, Richards DA, Raju RN, Jones B, Uprichard M, Nemunaitis J (2009a) Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin Oncol (R Coll Radiol) 21:464–472
Eager RM, Cunningham CC, Senzer NN, Stephenson J, Anthony SP, O’Day SJ, Frenette G, Pavlick AC, Jones B, Uprichard M et al (2009b) Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma. BMC Cancer 9:263
Egeblad M, Werb Z (2002) New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2:161–174
Elenbaas B, Weinberg RA (2001) Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res 264:169–184
Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T (1999) Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res 59:3379–3386
Erez N, Truitt M, Olson P, Arron ST, Hanahan D (2010) Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 17:135–147
Fang D, Sun L, Lin S, Zhou L, Su N, Yuan S, Yu B (2012) Vinorelbine inhibits angiogenesis and 95D migration via reducing hypoxic fibroblast stromal cell-derived factor 1 secretion. Exp Biol Med (Maywood) 237:1045–1055
Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A et al (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14:518–527
Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, Ratikan JA, Felix C, Hwang L, Faull KF et al (2018) Focal irradiation and systemic TGFβ blockade in metastatic breast cancer. Clin Cancer Res 24:2493–2504
Freedman JD, Duffy MR, Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, Campo L, Bryant RJ, Verrill C, Lambert A et al (2018) An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res 78:6852–6865
Goetz JG, Minguet S, Navarro-Lérida I, Lazcano JJ, Samaniego R, Calvo E, Tello M, Osteso-Ibáñez T, Pellinen T, Echarri A et al (2011) Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 146:148–163
Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, Andò S, Martinez-Outschoorn U et al (2012) Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 11:3019–3035
Harper J, Sainson RCA (2014) Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol 25:69–77
Hofheinz R-D, al-Batran S-E, Hartmann F, Hartung G, Jäger D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H et al (2003) Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 26:44–48
Hossen MN, Rao G, Dey A, Robertson JD, Bhattacharya R, Mukherjee P (2019) Gold nanoparticle transforms activated cancer-associated fibroblasts to quiescence. ACS Appl Mater Interfaces 11:26060–26068
Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y et al (2008) Regulation of in situ to invasive breast carcinoma transition. Cancer Cell 13:394–406
Huang Y, Simms AE, Mazur A, Wang S, León NR, Jones B, Aziz N, Kelly T (2011) Fibroblast activation protein-α promotes tumor growth and invasion of breast cancer cells through non-enzymatic functions. Clin Exp Metastasis 28:567–579
Hui L, Chen Y (2015) Tumor microenvironment: sanctuary of the devil. Cancer Lett 368:7–13
Ishii G, Ochiai A, Neri S (2016) Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev 99:186–196
Kalluri R (2003) Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer 3:422–433
Kalluri R (2016) The biology and function of fibroblasts in cancer. Nat Rev Cancer 16:582–598
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401
Kelly T (2005) Fibroblast activation protein-alpha and dipeptidyl peptidase IV (CD26): cell-surface proteases that activate cell signaling and are potential targets for cancer therapy. Drug Resist Updat 8:51–58
Kim DJ, Dunleavey JM, Xiao L, Ollila DW, Troester MA, Otey CA, Li W, Barker TH, Dudley AC (2018) Suppression of TGFβ-mediated conversion of endothelial cells and fibroblasts into cancer associated (myo)fibroblasts via HDAC inhibition. Br J Cancer 118:1359–1368
Kim JH, Oh S-H, Kim E-J, Park SJ, Hong SP, Cheon JH, Kim TI, Kim WH (2012) The role of myofibroblasts in upregulation of S100A8 and S100A9 and the differentiation of myeloid cells in the colorectal cancer microenvironment. Biochem Biophys Res Commun 423:60–66
Kudo Y, Iizuka S, Yoshida M, Tsunematsu T, Kondo T, Subarnbhesaj A, Deraz EM, Siriwardena SBSM, Tahara H, Ishimaru N et al (2012) Matrix metalloproteinase-13 (MMP-13) directly and indirectly promotes tumor angiogenesis. J Biol Chem 287:38716–38728
Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD (2018) Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T cells to protect tumour cells. Nat Commun 9:948
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15:178–196
LeBien TW, McCormack RT (1989) The common acute lymphoblastic leukemia antigen (CD10) – emancipation from a functional enigma. Blood 73:625–635
LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R (2013) Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19:1047–1053
Li X-Y, Hu S-Q, Xiao L (2015) The cancer-associated fibroblasts and drug resistance. Eur Rev Med Pharmacol Sci 19:2112–2119
Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA (2009) Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One 4:e7965
Lippman ME, Dickson RB, Gelmann EP, Rosen N, Knabbe C, Bates S, Bronzert D, Huff K, Kasid A (1988) Growth regulatory peptide production by human breast carcinoma cells. J Steroid Biochem 30:53–61
Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ (1997) Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol 139:1861–1872
Löhr M, Schmidt C, Ringel J, Kluth M, Müller P, Nizze H, Jesnowski R (2001) Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res 61:550–555
Lynch MD, Watt FM (2018) Fibroblast heterogeneity: implications for human disease. J Clin Invest 128:26–35
Marsh T, Pietras K, McAllister SS (2013) Fibroblasts as architects of cancer pathogenesis. Biochim Biophys Acta 1832:1070–1078
Massagué J (2012) TGFβ signalling in context. Nat Rev Mol Cell Biol 13:616–630
Matsuo Y, Ochi N, Sawai H, Yasuda A, Takahashi H, Funahashi H, Takeyama H, Tong Z, Guha S (2009) CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int J Cancer 124:853–861
Melamed JR, Riley RS, Valcourt DM, Day ES (2016) Using gold nanoparticles to disrupt the tumor microenvironment: an emerging therapeutic strategy. ACS Nano 10:10631–10635
Meulmeester E, Ten Dijke P (2011) The dynamic roles of TGF-β in cancer. J Pathol 223:205–218
Mishra P, Banerjee D, Ben-Baruch A (2011) Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. J Leukoc Biol 89:31–39
Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, Hsu FJ, Berzofsky JA, Lawrence DP (2014) Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One 9:e90353
Mross K, Stefanic M, Gmehling D, Frost A, Baas F, Unger C, Strecker R, Henning J, Gaschler-Markefski B, Stopfer P et al (2010) Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res 16:311–319
Mueller MM, Fusenig NE (2004) Friends or foes – bipolar effects of the tumour stroma in cancer. Nat Rev Cancer 4:839–849
Narra K, Mullins SR, Lee H-O, Strzemkowski-Brun B, Magalong K, Christiansen VJ, McKee PA, Egleston B, Cohen SJ, Weiner LM et al (2007) Phase II trial of single agent Val-boroPro (Talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biol Ther 6:1691–1699
Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ, Yokota SJ, Bankert RB (2007) Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol 178:5552–5562
Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM (2000) Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol 18:1135–1149
Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E (2019) In search of definitions: cancer-associated fibroblasts and their markers. Int J Cancer. https://doi.org/10.1002/ijc.32193
Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, Daigo Y, Ogasawara K (2015) Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci 106:134–142
Orimo A, Weinberg RA (2006) Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 5:1597–1601
Ostman A, Augsten M (2009) Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr Opin Genet Dev 19:67–73
Paget S (1989) The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev 8:98–101
Paulsson J, Micke P (2014) Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol 25:61–68
Paulsson J, Ehnman M, Östman A (2014) PDGF receptors in tumor biology: prognostic and predictive potential. Future Oncol 10:1695–1708
Pietras K, Pahler J, Bergers G, Hanahan D (2008) Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med 5:e19
Pinchuk IV, Saada JI, Beswick EJ, Boya G, Qiu SM, Mifflin RC, Raju GS, Reyes VE, Powell DW (2008) PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology 135:1228–1237, 1237.e1-2
Popat S, Mellemgaard A, Fahrbach K, Martin A, Rizzo M, Kaiser R, Griebsch I, Reck M (2015) Nintedanib plus docetaxel as second-line therapy in patients with non-small-cell lung cancer: a network meta-analysis. Future Oncol 11:409–420
Purcell JW, Tanlimco SG, Hickson J, Fox M, Sho M, Durkin L, Uziel T, Powers R, Foster K, McGonigal T et al (2018) LRRC15 is a novel mesenchymal protein and stromal target for antibody-drug conjugates. Cancer Res 78:4059–4072
Qian L, Tang Z, Yin S, Mo F, Yang X, Hou X, Liu A, Lu X (2018) Fusion of dendritic cells and cancer-associated fibroblasts for activation of anti-tumor cytotoxic T lymphocytes. J Biomed Nanotechnol 14:1826–1835
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19:1423–1437
Quante M, Tu SP, Tomita H, Gonda T, Wang SSW, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS et al (2011) Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19:257–272
Raffaghello L, Dazzi F (2015) Classification and biology of tumour associated stromal cells. Immunol Lett 168:175–182
Reck M, Kaiser R, Eschbach C, Stefanic M, Love J, Gatzemeier U, Stopfer P, von Pawel J (2011) A phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancer. Ann Oncol 22:1374–1381
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW et al (2014) Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25:735–747
Sánchez-Elsner T, Botella LM, Velasco B, Corbí A, Attisano L, Bernabéu C (2001) Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem 276:38527–38535
Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, Dias-Santagata D, Koeva M, Stemmer SM, Whitesell L et al (2014) The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 158:564–578
Schor SL, Schor AM, Grey AM, Rushton G (1988) Foetal and cancer patient fibroblasts produce an autocrine migration-stimulating factor not made by normal adult cells. J Cell Sci 90(Pt 3):391–399
Scott AM, Wiseman G, Welt S, Adjei A, Lee F-T, Hopkins W, Divgi CR, Hanson LH, Mitchell P, Gansen DN et al (2003) A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res 9:1639–1647
Silzle T, Randolph GJ, Kreutz M, Kunz-Schughart LA (2004) The fibroblast: sentinel cell and local immune modulator in tumor tissue. Int J Cancer 108:173–180
Simian M, Hirai Y, Navre M, Werb Z, Lochter A, Bissell MJ (2001) The interplay of matrix metalloproteinases, morphogens and growth factors is necessary for branching of mammary epithelial cells. Development 128:3117–3131
Simpkins SA, Hanby AM, Holliday DL, Speirs V (2012) Clinical and functional significance of loss of caveolin-1 expression in breast cancer-associated fibroblasts. J Pathol 227:490–498
de Sostoa J, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R (2019) Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer 7:19
Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z (1999) The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 98:137–146
Stetler-Stevenson WG, Aznavoorian S, Liotta LA (1993a) Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol 9:541–573
Stetler-Stevenson WG, Liotta LA, Kleiner DE (1993b) Extracellular matrix 6: role of matrix metalloproteinases in tumor invasion and metastasis. FASEB J 7:1434–1441
Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG (1995) Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 130:393–405
Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F et al (2018) CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172:841–856.e16
Sugimoto H, Mundel TM, Kieran MW, Kalluri R (2006) Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther 5:1640–1646
Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, Apuzzo T, Sperduti I, Volpe S, Cocorullo G et al (2014) CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 14:342–356
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3:349–363
Truffi M, Mazzucchelli S, Bonizzi A, Sorrentino L, Allevi R, Vanna R, Morasso C, Corsi F (2019) Nano-strategies to target breast cancer-associated fibroblasts: rearranging the tumor microenvironment to achieve antitumor efficacy. Int J Mol Sci 20(6)
Van Linthout S, Miteva K, Tschöpe C (2014) Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res 102:258–269
Vosseler S, Lederle W, Airola K, Obermueller E, Fusenig NE, Mueller MM (2009) Distinct progression-associated expression of tumor and stromal MMPs in HaCaT skin SCCs correlates with onset of invasion. Int J Cancer 125:2296–2306
Welt S, Divgi CR, Scott AM, Garin-Chesa P, Finn RD, Graham M, Carswell EA, Cohen A, Larson SM, Old LJ (1994) Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J Clin Oncol 12:1193–1203
Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X, Li J, Li C, Yan M, Zhu Z et al (2017) IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 8:20741–20750
Xia Q, Zhang F-F, Geng F, Liu C-L, Wang Y-Q, Xu P, Lu Z-Z, Xie Y, Wu H, Chen Y et al (2016) Improvement of anti-tumor immunity of fibroblast activation protein α based vaccines by combination with cyclophosphamide in a murine model of breast cancer. Cell Immunol 310:89–98
Yamashita M, Ogawa T, Zhang X, Hanamura N, Kashikura Y, Takamura M, Yoneda M, Shiraishi T (2012) Role of stromal myofibroblasts in invasive breast cancer: stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 19:170–176
Yang J, Lu Y, Lin Y-Y, Zheng Z-Y, Fang J-H, He S, Zhuang S-M (2016) Vascular mimicry formation is promoted by paracrine TGF-β and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett 383:18–27
Yu M, Tannock IF (2012) Targeting tumor architecture to favor drug penetration: a new weapon to combat chemoresistance in pancreatic cancer? Cancer Cell 21:327–329
Zeisberg EM, Zeisberg M (2013) The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J Pathol 229:264–273
Zhang Y, Xiong X, Huai Y, Dey A, Hossen MN, Roy RV, Elechalawar CK, Rao G, Bhattacharya R, Mukherjee P (2019) Gold nanoparticles disrupt tumor microenvironment - endothelial cell cross talk to inhibit Angiogenic phenotypes in vitro. Bioconjug Chem 30:1724
Zhao X, Pan J, Li W, Yang W, Qin L, Pan Y (2018) Gold nanoparticles enhance cisplatin delivery and potentiate chemotherapy by decompressing colorectal cancer vessels. Int J Nanomedicine 13:6207–6221
Zhen Z, Tang W, Wang M, Zhou S, Wang H, Wu Z, Hao Z, Li Z, Liu L, Xie J (2017) Protein Nanocage mediated fibroblast-activation protein targeted Photoimmunotherapy to enhance cytotoxic T cell infiltration and tumor control. Nano Lett 17:862–869
Acknowledgment
The authors thank Associazione Italiana per la Ricerca sul Cancro for research support (AIRC IG 20172 to F.C.)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Truffi, M., Sorrentino, L., Corsi, F. (2020). Fibroblasts in the Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1234. Springer, Cham. https://doi.org/10.1007/978-3-030-37184-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-37184-5_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-37183-8
Online ISBN: 978-3-030-37184-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)