
Abstract
Breast cancer represents the major cause of death in female cancer patients. New efficient treatments are desperately needed, particularly especially for patients suffering from advanced stages and metastases, or those who are no longer responding to the clinically established drugs such as cisplatin or carboplatin. New promising therapy regimens and platinum complexes have emerged over the last few years that displayed efficacy in advanced platinum- and/or drug-resistant breast tumors and metastases. This chapter provides an overview of the latest developments in the field of platinum-based drugs against advanced and resistant breast cancers since 2013.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Platinum complexes
- Anticancer agents
- Breast cancer
- Metastasis
- Multidrug resistance (MDR)
- Triple-negative breast cancer (TNBC)
13.1 Introduction
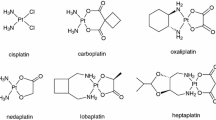
Rosenberg and coworkers discovered the anticancer activity of the platinum(II) complex cisplatin in 1969, and after its approval by the FDA about 10 years later cisplatin became a salient drug in the therapy of solid tumors (Fig. 13.1) [1]. DNA is the main cellular target of cisplatin which binds to it via metal coordination to the N-7 atom of purine bases such as guanine in exchange for its chlorido ligands. The resulting intra- and interstrand links lead to morphological changes of the platinated DNA eventually evoking apoptosis of the affected cells [2, 3]. Platinum therapy, however, comes at a price. Toxicity, severe side-effects and intrinsic or acquired platinum resistance confine the clinical applicability of platinum complexes [3,4,5,6]. This is true also for the second and third generation drugs carboplatin and oxaliplatin that are clinically approved in the USA and the EU (Fig. 13.1) [2,3,4]. Fortunately, the renaissance of interest in the medicinal chemistry of platinum opens a way out of this predicament. A plethora of new promising platinum complexes was disclosed that harness novel structural motifs, oxidation states, and conjugates with other drugs to overcome the eminent drawbacks of cisplatin, carboplatin and oxaliplatin [7,8,9,10]. Typical such examples are trans-Pt complexes, Pt(IV) complexes, heteronuclear complexes, and N-heterocyclic carbene complexes.

Platinum complexes in advanced stages of investigation as breast cancer therapeutics
Breast cancer still represents the major cause of death among all female cancer patients (more than 40,000 deaths per year alone in the USA), although a reduction of the mortality rates by 36% was observed since 1989 [11, 12]. Surgery and chemotherapy as well as hormone therapy for estrogen receptor positive breast cancer represent the main treatment options for breast cancer patients. Platinum complexes such as cisplatin and its less toxic congener carboplatin appear to be promising agents against particularly aggressive triple-negative breast cancers (TNBC) [13]. The efficacy of cisplatin and carboplatin treatment against breast cancer cells seems to be regulated epigenetically and tightly correlated with certain miRNA expression profiles including tumor suppressor miRNAs and oncogenic miRNAs (oncomirs) [14, 15]. Several new platinum complexes were disclosed that proved particularly active against aggressive, metastatic and/or drug-resistant breast cancers [8, 16]. In the following, an overview is presented of platinum–based anticancer agents for the targeted treatment of drug-resistant breast cancer and breast cancer metastases, published over the last 4 years, with a focus on those breast cancers that are associated with a poor prognosis.
13.2 Platinum Complexes in Advanced Stages of Investigation for the Treatment of Aggressive and Resistant Breast Cancers
Clinically approved platinum complexes such as cisplatin and carboplatin already represent valuable options for the treatment of advanced breast cancer diseases either alone or in combination with other drugs [17]. About 15–20% of all patients are diagnosed with triple-negative breast cancer (TNBC) which lacks expression of estrogen receptor (ER) and progesterone receptor (PR) and doesn’t overexpress human epidermal growth factor receptor 2 (HER2) [18]. TNBC is often associated with the development of brain and lung metastases and, thus, with a reduced overall survival rate and poor prognosis [18]. Platinum complexes have gained importance concerning the therapy of TNBC because of their DNA-damaging properties [13]. In a recent phase II trial, several patients suffering from metastatic TNBC responded well to platinum-based therapy (either cisplatin or carboplatin). In particular, patients with either germline BRCA1/2 mutations or with otherwise induced homologous recombination (HR) deficiency associated with ineffective DNA damage repair exhibited therapy response (Table 13.1) [19]. Indeed, HR deficiency on the basis of loss of heterozygosity (LOH), telomeric allelic imbalance (TAI) and large-scale state transition (LST) was discovered as a prognostic factor for BRCA1/2-mutated and sporadic TNBC response to treatment with platinum drugs [20]. In line with this finding, it was shown that the HR-repair inhibitor triapine augmented cisplatin activity in BRCA wild-type cancer cells [21]. A recent phase III trial of cisplatin in combination with gemcitabine for the treatment of metastatic TNBC revealed very promising results (median PFS/progression free survival = 7.73 months) when compared with paclitaxel plus gemcitabine (median PFS = 6.57 months), and cisplatin plus gemcitabine was suggested as the preferred first-line chemotherapy for this tumor disease in the future (Table 13.1) [22]. Another study revealed a median PFS of 4 months in patients with heavily pre-treated metastatic breast cancer who received cisplatin plus ifosfamide as salvage treatment (Table 13.1) [23]. The combination of cisplatin with the bisphosphonate zoledronic acid (a clinically approved drug for the treatment of cancer-mediated bone diseases) exhibited synergistic effects in TNBC cells (MDA-MB-231) which was associated with suppressed Mcl-1 expression and inhibition of mTOR signalling [24].
The less toxic cisplatin congener carboplatin was investigated in combination with the mTOR inhibitor everolimus in 25 metastatic TNBC patients in a phase 2 trial (Table 13.1) [25]. Carboplatin treatment was well tolerated by the patients and one complete response, six partial responses, and seven stable diseases were observed while eight patients showed progressing disease [25]. In a study with TNBC intracranial models, the combination of carboplatin with the PARP inhibitor ABT888 showed improved survival in the BRCA-mutant intracranial TNBC models and might be a suitable therapy option for BRCA-mutant TNBC patients with brain metastases in future clinical trials [26].
The second generation platinum complex nedaplatin is a close analog of carboplatin and its activity against advanced breast cancer was evaluated and compared with cisplatin [27]. Indeed, nedaplatin-based chemotherapy (in combination with paclitaxel or docetaxel, and gemcitabine or navelbine) showed distinctly longer time-to-treatment failure (TTF) = 13.87 months and overall survival (OS) times = 31.53 months in advanced breast cancer patients when compared with cisplatin-based chemotherapy (TTF = 8.7 months, OS = 24.87 months) (Table 13.1) [27].
Oxaliplatin with the characteristic trans-(1R,2R)-DACH (diaminocyclohexane) ligand is approved for the therapy of colorectal cancer because it lacks cross-resistance to cisplatin and carboplatin. Thus, the effects of oxaliplatin against TNBC were evaluated in a phase 2 trial as well [28]. The biweekly administered combination of oxaliplatin and vinorelbine against pre-treated second- or third-line metastatic TNBC revealed a median PFS of 4.3 months, an OS of 12.6 months and it was characterized by a good safety profile that warrants a phase 3 study of this therapy regimen (Table 13.1) [28].
13.3 New Platinum Complexes for the Treatment of Advanced and Resistant Breast Cancers
As a suitable model for TNBC, the MDA-MB-231 breast carcinoma cell line was frequently employed in order to study the effects of new platinum complexes at the pre-clinical stage [16]. The cis-diphenyl pyridineamine platinum(II) complex 1 inhibited MDA-MB-231 TNBC cell growth at much lower doses (IC50 = 1.0 μM) than cisplatin (IC50 = 10 μM) (Fig. 13.2) [29]. In addition to its DNA-binding and apoptosis induction, complex 1 also suppressed the migration of MDA-MB-231 cells and, thus, has potential as an anti-metastatic agent [29]. The trans-2-phenylindole platinum complex 2a and the cis-derivative 2b revealed distinct growth inhibition of MDA-MB-231 cells (IC50 = 4.3–4.4 μM) (Fig. 13.2) [30]. While 2a caused changes in the tertiary structure of treated plasmid DNA, 2b exerted no effects on plasmid DNA [30]. The di-n-butyl-DACH platinum(II) complex 3 proved more strongly growth inhibitory (IC50 = 13.79 μM) against MDA-MB-231 cells than oxaliplatin (IC50 = 26.82 μM) and cisplatin (IC50 = 18.27 μM) [31]. In addition, complex 3 bound more slowly to DNA when compared with cisplatin due to sterical hindrance by the dibutyl-DACH ligand which suggests a mode of action different from cisplatin (Fig. 13.2) [31]. The Schiff base platinum(II) complex 4 was tested against MDA-MB-231 cells and showed an IC50 value of 6.6 μM (Fig. 13.2) [32]. Complex 4 induced cell cycle arrest (G1-phase) and apoptosis while DNA interaction proceeded via intercalation [32]. Another Schiff base (N-octyl-salicylimine)(cis-cyclooctene)platinum(II) complex 5 was found to be a strong inducer of apoptosis in MDA-MB-231 cells (Fig. 13.2) [33]. The diiodido complex 6 inhibited MDA-MB-231 cell growth (IC50 = 6.6 μM) much more strongly than cisplatin (IC50 = 21.9 μM) due to its increased accumulation in cancer cells and to an increased DNA binding (Fig. 13.2) [34]. The 2-hydroxybenzimidazole oxalatoplatinum(II) complex 7 showed growth inhibition in MDA-MB-231 cells similar to cisplatin and greater than carboplatin (Fig. 13.2) [35]. Complex 7 changed the tertiary structure of plasmid DNA like cisplatin and efficiently protected plasmid DNA from digestion by a restriction enzyme [35]. The cycloplatinated benzophenone imine 8 also inhibited MDA-MB-231 tumor cell growth (IC50 = 5.0 μM), it showed antioxidant activity, and it bound to plasmid DNA leading to changes in its tertiary structure (Fig. 13.2) [36]. The ferrocene-platinum(II) complexes 9a and 9b strongly inhibited growth of MDA-MB-231 cells (IC50 = 1.4 μM) (Fig. 13.2) [37]. While 9a initiated distinct changes in the tertiary structure of plasmid DNA, complex 9b showed no such effects at all, which disproves DNA binding being a major aspect of the mode of action of these novel anticancer platinum complexes [37]. The 1,10-phenanthroline 2-(2′-hydroxy-5′-methylphenyl)-benzotriazole platinum complex 10 also showed significant growth inhibitory activity against MDA-MB-231 TNBC cells (IC50 = 5.2 μM) (Fig. 13.2) [38]. The triphenylphosphino chloroquine complex 11 was antiproliferative in MDA-MB-231 cells at similar concentrations (IC50 = 5.5 μM) (Fig. 13.2) [39]. Complex 11 bound to DNA and bovine serum albumin (BSA). When reacted with guanosine complex 11 underwent a Pt coordination to guanosine via the N7 atom [39]. The luminescent platinum(II) complex 12 featuring a pincer ligand led to a distinct growth inhibition of MDA-MB-231 cells growth inhibition (IC50 = 1.6 μM) when compared with cisplatin (IC50 = 25 μM) and it accumulated in the cancer cell lysosomes leading to an increased lysosomal membrane permeability and eventually to cell death (Fig. 13.2) [40]. The cationic platinum(II) complex phenanthriplatin (13) was of similar antiproliferative activity in MDA-MB-231 cells (IC50 = 3.1 μM) (Fig. 13.2) [41]. Its conjugation to tobacco mosaic virus (TMV) as a nano-carrier system gave a conjugate 13-TMV with even higher activity (IC50 = 2.2 μM). It also led to a distinct tumor growth reduction in MDA-MB-231 tumor xenograft models at doses of 1.0 mg/kg (weekly i.v. injection) with the 13-TMV nanoparticles accumulating in the tumor tissue [41]. The acridine-platinum(II) complex conjugate 14 inhibited MDA-MB-231 cell growth completely at doses between 5 and 10 μg/mL after 72 h (Fig. 13.2) [42]. Increased accumulation of 14 in MDA-MB-231 cells was achieved by coating of multi-walled carbon nanotubes with 14 (14-MWCNT) which also induced S-phase arrest and non-apoptotic cell death in MDA-MB-231 breast cancer cells [42]. The trans-1,2-diaminocyclopentane platinum(II) complex 15 and its conjugate with a fructose-based glyco-methacrylate-copolymer carrier (15-FMA) revealed potent activity against MDA-MB-231 cells (IC50 = 5.1 μM for 15, IC50 = 4.8 μM for 15-FMA) and the conjugate was readily taken up by breast cancer cells probably via the GLUT-5 receptor (Fig. 13.2) [43]. Reaction of trans-(1S,2S)-diaminocyclohexane-dichloridoplatinum(II) with 1,10-phenanthroline gave the bis-cationic complex 16 which was very active against MDA-MB-231 cells (IC50 = 0.64 μM) (Fig. 13.2) [44]. Intercalation of the cationic complex 16 into montmorillonite clay as a drug vehicle only slightly reduced the activity against MDA-MB-231 cells (IC50 = 0.9 μM) [44].

Platinum(II) complexes with activity against the TNBC model MDA-MB-231
The dinuclear berenil-platinum(II) complex 17a with isopropylamino ligands showed distinct tumor cell growth inhibition (IC50 = 18 μM) of MDA-MB-231 cells in contrast to cisplatin (IC50 = 96 μM) (Fig. 13.2) [45]. Complex 17a increased ROS levels in MDA-MB-231 cells and decreased the cellular concentrations of antioxidants such as GSH and vitamin E [45]. The analogous berenil-complex 17b with 3-butylpyridine ligands disclosed improved growth inhibitory activity in MDA-MB-231 cells (IC50 = 11 μM) when compared with complex 17a [46]. Complex 17b induced apoptosis in MDA-MB-231 cells in a caspase-dependent way via mitochondrial damage [46]. The new dinuclear berenil 4-ethylpyridine platinum(II) complex 17c also exhibited stronger growth inhibition of MDA-MB-231 cells (IC50 = 18 μM) when compared with cisplatin (IC50 = 92 μM) [47]. Complex 17c showed a more pronounced apoptosis induction in MDA-MB-231 cells (38%, 10 μM 17c) than cisplatin (11%, 10 μM cisplatin), and the activity of 17c was augmented by combination with anti-MUC1 antibodies (58% apoptotic cells, 10 μM 17c and 10 μg/mL anti-MUC1) which was associated with increased levels of caspases-8, −9, and −3, and of the pro-apoptotic Bax protein [48]. More recently, another potent dinuclear berenil-platinum(II) complex 17d with 3,4-dimethylpyridine ligands was disclosed (IC50 = 12 μM, MDA-MB-231 cells) which induced apoptosis both by mitochondrial damage and by the external pathway [49]. A micelles-forming carboxy-functionalized polymer was reacted with cisplatin in order to obtain the diammineplatinum(II) functionalized multinuclear polymer 18 for improved drug delivery (Fig. 13.2) [50]. Though the growth inhibitory activity of 18 (IC50 ca. 10 μg/mL after 48 h) was reduced in MDA-MB-231 cells when compared with cisplatin, increased platinum release was observed from the polymer micelles 18 at lower pH values (pH 5) [50].
Platinum(IV) complexes are usually more inert than platinum(II) complexes and they need to get activated by reduction to cytotoxic platinum(II) species in the hypoxic tumor environment. A prominent example is the orally applicable Pt(IV) complex satraplatin (Fig. 13.3) that had entered advanced clinical trials [51]. However, a phase 2 trial of satraplatin for the treatment of metastatic breast cancer patients dating from 2009 revealed only limited activity of satraplatin as a single agent (2 PRs, 18 SDs, from a total number of 31 metastatic breast cancer patients) [52]. A more focussed clinical study with patients suffering from advanced breast cancer characterized by HR repair deficiency would probably lead to better results for satraplatin treatment as it was the case for cisplatin and carboplatin [19, 20]. Due to the octahedral structure of Pt(IV) complexes, two more ligands can be introduced which may be applied for the fine-tuning of the biological and pharmacological properties. Inorganic chemists already took advantage of this option in designing new potent anticancer active Pt(IV) complexes [53].

Platinum(IV) complexes with activity against TNBC cells
The lipophilic ibuprofen platinum(IV) complex 19 revealed excellent growth inhibitory activity in MDA-MB-231 cells (IC50 = 0.05 μM) and was much more active than the platinum(II) complex cisplatin (IC50 = 20 μM) as a consequence of a much higher accumulation in the cancer cells (Fig. 13.3) [54]. LA-12 (20), a close adamantylamine analog of satraplatin, was also distinctly inhibiting the growth of MDA-MB-231 cells (IC50 = 2.4 μM) (Fig. 13.3) [55]. Interestingly, its formulation as a tumor-targeted folate-cyclodextrin conjugate augmented its activity against MDA-MB-231 cells significantly (IC50 = 0.7 μM) [55]. Since the FPR1/2 formyl peptide receptor is overexpressed in immune cells as well as in metastases, the Pt(IV) complex 21 was conjugated to a FPR1/2-targeting peptide (WKYMVm) in order to achieve synergy effects [56]. While 21 exhibited growth inhibitory activity against MDA-MB-231 cells in the range of cisplatin, 21 led to an enhanced secretion of TNF-α and IFN-γ in peripheral blood mononuclear cells (PBMC) when compared to cisplatin [56]. The fact that PBMCs activated by 21 efficiently inhibited MDA-MB-231 cell growth renders this complex a promising potential immunomodulating drug candidate [56]. Another Pt(IV) complex 22, comprising an aggregation-induced emission luminogen and the integrin-targeting moiety cRGD (cyclic arginine-glycine-aspartate), was used for the study of bio-reduction of the Pt(IV) moiety [57]. The αvβ3 integrin-expressing MDA-MB-231 cells responded much better to 22a (IC50 = 30.2 μM) than MCF-7 breast cancer cells with only low integrin expression (no response up to 50 μM) [57]. Following this, a similar cRGD-Pt(IV) complex 22b linked to a photosensitizer with AIE characteristics was prepared, and irradiation with light strongly enhanced the growth inhibitory activity of 22b in MDA-MB-231 cells (IC50 = 4.2 μM) when compared with its efficacy in the dark (IC50 = 37.1 μM) and with that of cisplatin (IC50 = 33.4 μM) [58]. Human serum albumin (HSA) was linked to Pt(IV) via a succinate to give complex 23 which served as starting material for the preparation of calcium phosphate(CaP)-23 nanoparticles that release the platinum drug under acidic and hypoxic conditions [59]. Indeed, CaP-23 exhibited better activity against MDA-MB-231 cells (IC50 = 1.36 μM) than cisplatin (IC50 = 2.66 μM) [59]. Another potent Pt(IV) complex is the bis-benzoyl complex 24 which was highly active against MDA-MB-231 cells (IC50 = 0.59 μM) [60]. Incorporation of 24 into silk fibroin nanoparticles (SNF) even augmented the activity of 24 slightly (IC50 = 0.39 μM) and increased its tumor selectivity [60]. MDA-MB-468 is another TNBC cell line that was applied to study the anticancer effects of mitaplatin 25 [61]. Complex 25 (1 mg/kg) inhibited the in vivo growth of MDA-MB-468 mouse xenograft tumors distinctly (tumor volume ca. 200 mm3 for 25 vs. ca. 900 mm3 for the control mice after 24 days) [61]. Encapsulation of 25 into polymer nanoparticles led to a similar tumor growth inhibition and to a prolonged drug circulation in the blood system of the treated mice while the accumulation in the kidneys was reduced [61].
ER-positive T47D breast carcinoma cells are less responsive to cisplatin than ER-positive MCF-7 breast carcinoma cells due to an enhanced glutathione-S-transferase (GST)-mediated drug resistance [62, 63]. However, the triazolopyrimidine diacetatoplatinum(II) complex 26 (Fig. 13.4) showed excellent and tumor selective activity against T47D breast cancer cells (IC50 = 0.26 μM) and it exceeded the activity both of cisplatin (IC50 = 14.4 μM) and of oxaliplatin (IC50 = 18.3 μM) by far [64]. A similar malonatoplatinum(II) complex 27 exhibited distinct growth inhibition of T47D cells (IC50 = 3.4 μM) while non-malignant cells were affected less (IC50 = 55.8 μM) [65]. A new platinum(II) conjugate 28 bearing a steroidal 7-azaindole ligand also showed increased activity against T47D cells (IC50 = 13 μM) when compared with cisplatin (IC50 = 33 μM) (Fig. 13.4) [66]. Complex 28 was also accumulated to a greater extend in the T47D cancer cells than cisplatin, and it displaced the intercalator ethidium bromide from plasmid DNA and inhibited cathepsin B [66]. The analogous tri-(p-trifluoromethylphenyl)-phosphinoplatinum(II) complex 29 inhibited the growth of T47D cells much more strongly (IC50 = 1.84 μM) than cisplatin (IC50 = 30 μM) [67]. Complex 29 arrested the cancer cell cycle in the G0/G1 phase and it inhibited cathepsin B (IC50 = 8.1 μM) [67]. The trans-dichloridoplatinum(II) complex 30 featuring a ferrocene-based ligand was also a stronger inhibitor of the growth of T47D cells (IC50 = 2.4 μM) than cisplatin (IC50 = 15 μM) [68].

Platinum complexes with improved activity against cisplatin-resistant T47D breast cancer cells
HER2 epidermal growth factor receptors are overexpressed in many aggressive tumors including breast cancer. Trastuzumab is a clinically approved monoclonal antibody that targets HER2, and a trastuzumab-platinum(IV) conjugate 31 was prepared as a tumor-targeted drug (Fig. 13.5) [69]. Complex 31 bound to HER2 and was much more active against HER2-positive SK-BR-3 breast carcinoma cells when compared with HER2-negative cell lines and it inhibited the growth of SK-BR-3 cells (IC50 = 21.3 μM) as effectively as cisplatin (IC50 = 20.7 μM) [69]. A platinum(II) conjugate 32 (Fig. 13.5) of the HER2-targeting antibody herceptin showed similar results (IC50 = 19.7 μM in SK-BR-3 cells) and an activity better than that of oxaliplatin (IC50 = 31.0 μM) [70]. In addition, a HER2-targeting affibody (= small peptidic antibody mimics) was conjugated to cisplatin-loaded liposomes, and the resulting affisome showed increased cytotoxicity and cellular accumulation in SK-BR-3 cells and it exhibited distinct tumor growth inhibition of HER2-positive TUBO breast cancer xenograft models [71].

Platinum complexes with distinct activity against various advanced or resistant breast cancers
Another study disclosed that epithelial breast cancer cells were 16-times more sensitive to complex 33 (IC50 = 5.3 μM) than to cisplatin (IC50 = 94.7 μM) (Fig. 13.5) [72]. Complex 33 reduced the expression of the anti-apoptotic Bcl-2 protein and augmented pro-apoptotic Bax expression leading to the efficient induction of apoptosis by 33 in cisplatin-resistant epithelial breast cancer cells [72].
ER-positive MCF-7 breast cancer cells under hypoxic conditions showed reduced sensitivity to cisplatin. The tetrachloridoplatinum(IV) complex 34 (Fig. 13.5) containing the alkylating nitrogen mustard motif exhibited higher activity against MCF-7 cells both under normoxic (IC50 = 11.4 μM) and hypoxic conditions (IC50 = 8.6 μM) than cisplatin (IC50 = 14.1 μM under normoxic, 18.7 μM under hypoxic conditions) [73]. In addition, 34 was more efficacious in MCF-7 cells supplemented with the cisplatin-resistance factor glutathione (GSH) (IC50 = 12.9 μM under normoxic, 11.2 μM under hypoxic conditions) when compared with cisplatin (IC50 = 27.8 μM under normoxic, 29.0 μM under hypoxic conditions). 34 also showed an increased accumulation in MCF-7 cells (more than twice as high than that for cisplatin) [73]. In addition, 34 induced apoptosis and reduced the motility of MCF-7 cells [73]. The new water-soluble oxaliplatin/carboplatin analogue 35 showed growth inhibitory activity (IC50 = 15.0 μM) comparable with oxaliplatin (IC50 = 10.4 μM) and much better than carboplatin (IC50 = 154 μM) in multidrug-resistant MCF-7/ADR breast cancer cells (Fig. 13.5) [74]. The in vivo anticancer activity of 35 was evaluated in KM mice bearing Sarcoma 180. Complex 35 led to a greater inhibition of the tumor growth (53.2% inhibition) than oxaliplatin (32.5% inhibition) [74].
In addition, various N-heterocyclic carbene platinum complexes were recently investigated for their effect on multidrug-resistant MCF-7/Topo breast cancer cells which overexpress the BCRP transporter [75,76,77]. Complex 36 (Fig. 13.5) showed excellent and selective growth inhibition (IC50 = 0.15 μM) in MCF-7/Topo cells when compared with cisplatin (IC50 = 10.6 μM) [75]. Although 36 did not bind covalently to DNA, this complex induced DNA aggregation in addition to cell cycle arrest in the G1 phase. It also led to the disruption of blood vessels [75]. Similar DNA aggregation effects were observed for biscarbene complex 37 (Fig. 13.5), which also showed strong MCF-7/Topo cell growth inhibition (IC50 = 0.52 μM) [76]. Another trans-diiodidoplatinum(II) NHC complex 38 featuring a histidine-derived NHC-ligand also strongly inhibited the growth of MCF-7/Topo cells (IC50 = 1.6 μM) (Fig. 13.5) [77]. Complex 38 induced morphological changes in plasmid DNA and caused vascular disruption [77]. Its in vivo activity was evaluated in mice with cisplatin-resistant A2780cis ovarian tumors. Complex 38 (30 mg/kg, i.p.) was roughly as effective a tumor growth inhibitor as cisplatin (6 mg/kg, i.p.), yet showed a superior toxicity profile with treated mice regaining their normal weight far more quickly [77]. Hence, complex 38 is likely applicable in much higher doses than cisplatin to the effect of a significantly better tumor mass reduction.
In order to reduce the systemic toxicity of platinum complexes, a tumor-selective Pt(IV) complex conjugate 39 comprising a short self-assembling peptide sequence was prepared (Fig. 13.6) [78]. Alkaline-phosphatase (AP)-catalyzed cleavage of the phosphate group of 39 led to self-assembly and bioreduction to active platinum species in the tumor (high levels of AP are found in the environment of many tumors). Increased tumor cell accumulation as well as reduced liver and kidney toxicity were observed for 4T1-breast carcinoma xenograft models treated with 39 while the in vivo 4T1 tumor growth was inhibited by 39 similarly to cisplatin [78].

Self-assembling Pt(IV) complex prodrug 39 for 4T1-breast cancer targeting
13.4 Conclusions
The platinum complex cisplatin has been and still is a mainstay in the therapy of solid tumors. However, meanwhile more platinum complexes have passed clinical trials and quite a few of them were found active against drug-resistant and advanced breast cancers. HR-repair deficient triple-negative breast cancers appeared to be especially sensitive to platinum drugs. Their chemical tuning in terms of structure, redox chemistry, and synergistic effects of ligands and co-conjugates has led to a plethora of new complexes with enhanced activity against and selectivity for drug-resistant and/or aggressive/metastatic breast cancers. In addition, novel delivery systems for the targeted therapy of breast cancers with platinum complexes have overcome the notorious drawbacks of the first- and second-generation platinum complexes. Taken together, there are distinct glimpses of hope that new therapies with platinum complexes will prevent or overcome drug resistance, improve prognosis and survival, reduce side-effects, and increase the quality of life of breast cancer patients in a not too distant future.
References
Rosenberg B, VanCamp L, Trosko JE, Mansour VH (1969) Platinum compounds: a new class of potent antitumour agents. Nature 222:385–386
Jamieson ER, Lippard SJ (1999) Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev 99:2467–2498
Wang D, Lippard SJ (2005) Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 4:307–320
Kartalou M, Essigmann JM (2001) Mechanisms of resistance to cisplatin. Mutat Res Fundam Mol Mech Mutagen 478:23–43
Oh GS, Kim HJ, Shen A, Lee SB, Khadka D, Pandit A, So HS (2014) Cisplatin-induced kidney dysfunction and perspectives on improving treatment strategies. Electrolyte Blood Pressure 12:55–65
Dasari S, Tchounwou PB (2014) Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740:364–378
Schobert R, Biersack B, Dietrich A, Grotemeier A, Müller T, Kalinowski B, Knauer S, Voigt W, Paschke R (2007) Monoterpenes as drug shuttles: cytotoxic (6-aminomethylnicotinate) dichloridoplatinum(II) complexes with potential to overcome cisplatin resistance. J Med Chem 50:1288–1293
Zoldakova M, Biersack B, Kostrhunova K, Ahmad A, Padhye S, Sarkar FH, Schobert R, Brabec V (2011) (Carboxydiamine)Pt(II) complexes of a combretastatin A-4 analogous chalcone: the influence of the diamine ligand on DNA binding and anticancer effects. Med Chem Commun 2:493–499
Najajreh Y, Perez JM, Navarro-Ranninger C, Gibson D (2002) Novel soluble cationic trans-diaminedichloroplatinum(II) complexes that are active against cisplatin resistant ovarian cancer cell lines. J Med Chem 45:5189–5195
Liu W, Gust R (2013) Metal N-heterocyclic carbene complexes as potential antitumor metallodrugs. Chem Soc Rev 42:755–773
Ferlay J, Soerjomataram I, Dikshit R, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386
DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A (2016) Breast cancer statistics, 2015: convergence of incidence rates between black and white women. CA Cancer J Clin 66:31–42
Petrelli F, Coinu A, Borgonovo K, Cabiddu M, Ghilardi M, Lonati V, Barni S (2014) The value of platinum agents as neoadjuvant chemotherapy in triple-negative breast cancers: a systemic review and meta-analysis. Breast Cancer Res Treat 144:223–232
Chen X, Lu P, Wu Y, Wang D, Zhou S, Yang S, Shen H, Zhang X, Zhao J, Tang T (2016) MiRNAs-mediated cisplatin resistance in breast cancer. Tumor Biol 37:12905–12913
Biersack B (2017) Interactions between anticancer active platinum complexes and non-coding RNAs/microRNAs. Non-Coding RNA Res 2:1–17
Biersack B, Schobert R (2013) Platinum and ruthenium complexes for the therapy of breast cancer diseases. In: Ahmad A (ed) Breast cancer metastasis and drug resistance. Springer Science+Business Media, New York
Cobleigh MA (2011) Other options in the treatment of advanced breast cancer. Semin Oncol Suppl 2:S11–S16
Lin NU, Vanderplas A, Hughes ME, Theriault RL, Edge SB, Wong YN, Blayney DW, Niland JC, Winter EP, Weeks JC (2012) Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the national comprehensive cancer network. Cancer 118:5463–5472
Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, Rugo HS, Liu MC, Stearns V, Come SE, Timms KM, Hartman A-R, Borger DR, Finkelstein DM, Garber JE, Ryan PD, Winer EP, Goss PE, Ellisen LW (2015) TBCRC009: a multicenter phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J Clin Oncol 33:1902–1909
Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, Isakoff SJ, Ryan PD, Greene-Colozzi A, Gutin A, Sangale Z, Iliev D, Neff C, Abkevich V, Jones JT, Lanchbury JS, Hartman A-R, Garber JE, Ford JM, Silver DP, Richardson AL (2016) Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res 22:3764–3773
Ratner ES, Zhu Y-L, Penketh PG, Berenblum J, Whicker ME, Huang PH, Lee Y, Ishiguro K, Zhu R, Sartorelli AC, Lin ZP (2016) Triapine potentiates platinum-based combination therapy by disruption of homologous recombination repair. Br J Cancer 114:777–786
Hu X-C, Zhang J, Xu B-H, Cai L, Ragaz J, Wang Z-H, Wang B-Y, Teng Y-E, Tong Z-S, Pan Y-Y, Yin Y-M, Wu C-P, Jiang Z-F, Wang X-J, Lou G-Y, Liu D-G, Feng J-F, Luo J-F, Sun K, Gu Y-J, Wu J, Shao Z-M (2015) Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): a randomized, open-label, multicenter, phase 3 trial. Lancet Oncol 16:436–446
Habbel P, Kurreck A, Schulz C-O, Regierer AC, Kaul D, Scholz CW, Neumann C, Possinger K, Eucker J (2015) Cisplatin plus ifosfamide with/without etoposide as salvage therapy in heavily-pre-treated patients with metastatic breast cancer. Anticancer Res 35:5091–5096
Ibrahim T, Liverani C, Mercatali L, Sacanna E, Zanoni M, Fabbri F, Zoli W, Amadori D (2013) Cisplatin in combination with zoledronic acid: a synergistic effect in triple-negative breast cancer cell lines. Int J Oncol 42:1263–1270
Singh JC, Novik Y, Stein S, Volm M, Meyers M, Smith J, Omene C, Speyer J, Schneider R, Jhaveri K, Formenti S, Kyriakou V, Joseph B, Goldberg JD, Li X, Adams S, Tiersten A (2014) Phase 2 trial of everolimus and carboplatin combination in patients with triple negative metastatic breast cancer. Breast Cancer Res 16:R32
Karginova O, Siegel MB, Van Swearingen AED, Deal AM, Adamo B, Sambade MJ, Bazyar S, Nikolaishvili-Feinberg N, Bash R, O’Neal S, Sandison K, Parker JS, Santos C, Darr D, Zamboni W, Lee YZ, Miller CR, Anders CK (2015) Efficacy of carboplatin alone or in combination with ABT888 in intracranial murine models of BRCA-mutated and BRCA-wild-type triple-negative breast cancer. Mol Cancer 14:920–930
Pang H, Feng T, Lu H, Meng Q, Chen X, Shen Q, Dong X, Cai L (2016) Efficacy and safety of nedaplatin in advanced breast cancer therapy. Cancer Investig 34:167–172
Zhang J, Wang L, Wang Z, Hu X, Wang B, Cao J, Lv F, Zhen C, Zhang S, Shao Z (2015) A phase II trial of biweekly vinorelbine and oxaliplatin in second- or third-line metastatic triple-negative breast cancer. Cancer Biol Ther 16:225–232
Varela JG, Chatterjee AD, Guevara P, Ramirez V, Metta-Magana AJ, Villagrán D, Varela-Ramirez A, Das S, Nunez JE (2014) Synthesis, characterization, and evaluation of cis-diphenyl pyridineamine platinum(II) complexes as potential anti-breast cancer agents. J Biol Inorg Chem 19:967–979
Tomé M, López C, González A, Ozay B, Quirante J, Font-Bardía M, Calvet T, Calvis C, Messeguer R, Baldomá L, Badía J (2013) Trans- and cis-2-phenylindole platinum(II) complexes as cytotoxic agents against human breast adenocarcinoma cell lines. J Mol Struct 1048:88–97
Zhang H, Gou S, Zhao J, Chen F, Xu G, Liu X (2015) Cytotoxicity profile of novel sterically hindered platinum(II) complexes with (1R,2R)-N1,N2-dibutyl-1,2-diaminocyclohexane. Eur J Med Chem 96:187–195
Peng Y, Zhong H, Chen Z-F, Liu Y-C, Zhang G-H, Qin Q-P, Liang H (2014) A planar Schiff base platinum(II) complex: crystal structure, cytotoxicity and interaction with DNA. Chem Pharm Bull 62:221–228
Jean S, Cormier K, Patterson AE, Vogels CM, Decken A, Robichaud GA, Turcotte S, Westcott SA (2015) Synthesis, characterization, and anticancer properties of organometallic Schiff base platinum complexes. Can J Chem 93:1140–1146
Savic A, Filipovic L, Arandelovic S, Dojcinovic B, Radulovic S, Sabo TJ, Grguric-Sipka S (2014) Synthesis, characterization and cytotoxic activity of novel platinum(II) iodide complexes. Eur J Med Chem 82:372–384
Utku S, Ozcelik AB, Gümüs F, Yilmaz S, Arsoy T, Acik L, Keskin AC (2014) Synthesis, in-vitro cytotoxic activity and DNA interactions of new dicarboxylatoplatinum(II) complexes with 2-hydroxymethylbenzimidazole as carrier ligands. J Pharm Pharmacol 66:1593–1605
Albert J, D’Andrea L, Granell J, Pla-Vilanova P, Quirante J, Khosa MK, Calvis C, Messeguer R, Badía J, Baldomà L, Font-Bardia M, Calvet T (2014) Cyclopalladated and cycloplatinated benzophenone imines: antitumor, antibacterial and antioxidant activities, DNA interaction and cathepsin B inhibition. J Inorg Biochem 140:80–88
Talancón D, López C, Font-Bardía M, Calvet T, Quirante J, Calvis C, Messeguer R, Cortés R, Cascante M, Baldomà L, Badia H (2013) Diastereomerically pure platinum(II) complexes as antitumoral agents. The influence of the mode of binding {(N), (N,O)− or (C,N)}− of (1S,2R)-[(η5 –C5H5)Fe{(η5 –C5H4)-CH=N-CH(Me)-CH(OH)-C6H5}] and the arrangement of the auxiliary ligands. J Inorg Biochem 118:1–12
El-Asmy HA, Butler IS, Mouhri ZS, Jean-Claude BJ, Emmam MS, Mostafa SI (2014) Zinc(II), ruthenium(II), rhodium(III), palladium(II), silver(I), platinum(II) and MoO22+ complexes of 2-(2′-hydroxy-5′-methylphenyl)-benzotriazole as simple or primary ligand and 2,2′-bipyridyl, 9,10-phenanthroline or triphenylphosphine as secondary ligands: structure and anticancer activity. J Mol Struct 1059:193–201
Villareal W, Colina-Vegas L, de Oliveira CR, Tenorio JC, Ellena J, Gozzo FC, Cominetti MR, Ferreira AG, Ferreira MAB, Navarro M, Batista AA (2015) Chiral platinum(II) complexes featuring phosphine and chloroquine ligands as cytotoxic and monofunctional DNA-binding agents. Inorg Chem 54:11709–11720
Tsai JL-L, Zou T, Liu J, Chen T, Chan AO-Y, Yang C, Lok C-N, Che C-M (2015) Luminescent platinum(II) complexes with self-assembly and anti-cancer properties: hydrogel, pH dependent emission color and sustained-release properties under physiological conditions. Chem Sci 6:3823–3830
Czapar AE, Zheng Y-R, Riddell IA, Shukla S, Awuah SG, Lippard SJ, Steinmetz NF (2016) Tobacco mosaic virus delivery of phenanthriplatin for cancer therapy. ACS Nano 10:4119–4126
Fahrenholtz CD, Ding S, Bernish BW, Wright ML, Zheng Y, Yang M, Yao X, Donati GL, Gross MD, Bierbach U, Singh R (2016) Design and cellular studies of a carbon nanotube-based delivery system for a hybrid platinum-acridine anticancer agent. J Inorg Biochem 165:170–180
Dag A, Callari M, Lu H, Stenzel MH (2016) Modulating the cellular uptake of platinum drugs with glycopolymers. Polym Chem 7:1031–1036
Apps MG, Ammit AJ, Gu A, Wheate NJ (2014) Analysis of montmorillonite clay as a vehicle in platinum anticancer drug delivery. Inorg Chim Acta 421:513–518
Gegotek A, Cyunczyk M, Luczaj W, Bielawska A, Bielawski K, Skrzydlewska E (2014) The redox status of human breast cancer cell lines (MCF-7 and MDA-MB231) treated with novel dinuclear berenil-platinum(II) complexes. Pharmazie 69:923–928
Bielawski K, Czarnomysy R, Muszynska A, Bielawska A, Poplawska B (2013) Cytotoxicity and induction of apoptosis of human breast cancer cells by novel platinum(II) complexes. Environ Toxicol Pharmacol 35:254–264
Gornowicz A, Kaluza Z, Bielawska A, Gabryel-Porowska H, Czarnomysy R, Bielawski K (2014) Cytotoxic efficacy of a novel dinuclear platinum(II) complex used with anti-MUC1 in human breast cancer cells. Mol Cell Biochem 392:161–174
Gornowicz A, Bielawska A, Czarnomysy R, Gabryel-Porowska H, Muszynska A, Bielawski K (2015) The combined treatment with novel platinum(II) complex and anti-MUC1 increases apoptotic response in MDA-MB-231 breast cancer cells. Mol Cell Biochem 408:103–113
Czarnomysy R, Bielawski K, Muszynska A, Bielawska A, Gornowicz A (2016) Biological evaluation of dimethylpyridine-platinum complexes with potent antiproliferative activity. J Enzyme Inhib Med Chem 31:150–165
Shahin M, Safaei-Nikouei N, Lavasanifar A (2014) Polymeric micelles for pH-responsive delivery of cisplatin. J Drug Target 22:629–637
Voigt W, Dietrich A, Schmoll H-J (2006) Cisplatin und seine Analoga. Pharm Unserer Zeit 35:134–143
Smith JW III, McIntyre KJ, Avecedo PV, Encarnacion CA, Tedesco KL, Wang Y, Asmar L, O’Shaughnessy (2009) Results of a phase II open-label, non-randomized trial of oral satraplatin in patients with metastatic breast cancer. Breast Cancer Res Treat 118:361–367
Wilson JJ, Lippard SJ (2014) Synthetic methods for the preparation of platinum anticancer complexes. Chem Rev 114:4470–4495
Neumann W, Crews BC, Sárosi MB, Daniel CM, Ghebreselasie K, Scholz MS, Marnett LJ, Hey-Hawkins E (2015) Conjugation of cisplatin analogues and cyclooxygenase inhibitors to overcome cisplatin resistance. Chem Med Chem 10:183–192
Giglio V, Oliveri V, Viale M, Gangemi R, Natile G, Intini FP, Vecchio G (2015) Folate-cyclodextrin conjugates as carriers of the platinum(IV) complex LA-12. Chem Plus Chem 80:536–543
Wong DYQ, Yeo CHF, Ang WH (2014) Immuno-chemotherapeutic platinum(IV) prodrugs of cisplatin as multimodal anticancer agents. Angew Chem Int Ed 53:6752–6756
Yuan Y, Chen Y, Tang BZ, Liu B (2014) A targeted theranostic platinum(IV) prodrug containing a luminogen with aggregation-induced emission (AIE) characteristics for in situ monitoring of drug activation. Chem Commun 50:3868–3870
Yuan Y, Zhang C-J, Liu B (2015) A platinum prodrug conjugated with a photosensitizer with aggregation-induced emission (AIE) characteristics for drug activation monitoring and combinatorial photodynamic-chemotherapy against cisplatin resistant cancer cells. Chem Commun 51:8626–8629
Shi H, Cheng Q, Yuan S, Ding X, Liu Y (2015) Human serum albumin conjugated nanoparticles for pH and redox-responsive delivery of a prodrug of cisplatin. Chem Eur J 21:16547–16554
Lozano-Pérez AA, Gil AL, Pérez SA, Cutillas N, Meyer H, Pedreno M, Aznar-Cervantes SD, Janiak C, Cenis JL, Ruiz J (2015) Antitumor properties of platinum(IV) prodrug-loaded silk fibroin nanoparticles. Dalton Trans 44:13513–13521
Johnstone TC, Kulak N, Pridgen EM, Farokhzad OC, Langer R, Lippard SJ (2013) Nanoparticle encapsulation of mitaplatin and the effect thereof on in vivo properties. ACS Nano 7:5675–5683
Ang WH, Khalaila I, Allardyce CS, Juillerat-Jeanneret L, Dyson PJ (2005) Rational design of platinum(IV) compounds to overcome glutathione-S-transferase mediated drug resistance. J Am Chem Soc 127:1382–1383
LaPensee EW, Schwemberger SJ, LaPensee CR, Bahassi EM, Afton SE, Ben-Jonathan N (2009) Prolactin confers resistance against cisplatin in breast cancer cells by activating glutathione-S-transferase. Carcinogenesis 30:1298–1304
Hoffmann K, Lakomska I, Wisniewska J, Kaczmarek-Kedziera A, Wietrzyk J (2015) Acetate platinum(II) compound with 5,7-ditertbutyl-1,2,4-triazolo[1,5-a]pyrimidine that overcomes cisplatin resistance: structural characterization, in vitro cytotoxicity, and kinetic studies. J Coord Chem 68:3193–3208
Lakomska I, Hoffmann K, Wojtczak A, Sitkowski J, Maj E, Wietrzyk J (2014) Cytotoxic malonate platinum(II) complexes with 1,2,4-triazolo[1,5-a]pyrimidine derivatives: structural characterization and mechanism of the suppression of tumor cell growth. J Inorg Biochem 141:188–197
Zamora A, Rodríguez V, Cutillas N, Yellol GS, Espinosa A, Samper K, Capdevila M, Palacios O, Ruiz J (2013) New steroidal 7-azaindole platinum(II) antitumor complexes. J Inorg Biochem 128:48–56
Cutillas N, Mart’nez A, Yellol GS, Rodríguez V, Zamora A, Pedreno M, Donaire A, Janiak C, Ruiz J (2013) Anticancer C,N-cycloplatinated(II) complexes containing fluorinated phosphine ligands: synthesis, structural characterization, and biological activity. Inorg Chem 52:13529–13535
Nieto D, Bruna S, González-Vadillo AM, Perles J, Carillo-Hermosilla F, Antinolo A, Padrón JM, Plata GB, Cuadrado I (2015) Catalytically generated ferrocene-containing guanidines as efficient precursors for new redox-active heterometallic platinum(II) complexes with anticancer activity. Organometallics 34:5407–5417
Huang R, Wang Q, Zhang X, Zhu J, Sun B (2015) Trastuzumab-cisplatin conjugates for targeted delivery of cisplatin to HER2-overexpressing cancer cells. Biomed Pharmacother 72:17–23
Huang R, Sun Y, Zhang X, Sun B, Wang Q, Zhu J (2015) Biological evaluation of a novel Herceptin-platinum(II) conjugate for efficient and cancer cell specific delivery. Biomed Pharmacother 73:116–122
Alavizadeh SH, Akhtari J, Badiee A, Golmohammadzadeh S, Jaafari MR (2015) Improved therapeutic activity of HER2 affibody-targeted cisplatin liposomes in HER2-expressing breast tumor models. Exp Opin Drug Deliv 13:325–336
Muscella A, Vetrugno C, Fanizzi FP, Manca C, De Pascali SA, Marsigliante S (2013) A new platinum(II) compound anticancer drug candidate with selective cytotoxicity for breast cancer cells. Cell Death Dis 4:e796
Karmakar S, Chatterjee S, Purkait K, Mukherjee A (2016) Anticancer activity of a chelating nitrogen mustard bearing tetrachloroplatinum(IV) complex: better stability yet equipotent to the Pt(II) analogue. Dalton Trans 45:11710–11722
Liu W, Ye Q, Jiang J, Lou L, Xu Y, Xie C, Xie M (2013) cis-[PtII(1R,2R-DACH)(3-acetoxy-1,1-cyclobutanedicarboxylato)], a water-soluble, oxalate-free and stable analogue of oxaliplatin: synthesis, characterization, and biological evaluations. Chem Med Chem 8:1465–1467
Muenzner JK, Rehm T, Biersack B, Casini A, de Graaf IAM, Worawutputtapong P, Noor A, Kempe R, Brabec V, Kasparkova J, Schobert R (2015) Adjusting the DNA interaction and anticancer activity of Pt(II) N-heterocyclic carbene complexes by steric shielding of the trans leaving group. J Med Chem 58:6283–6292
Rehm T, Rothemund M, Muenzner JK, Noor A, Kempe R, Schobert R (2016) Novel cis-[(NHC)1(NHC)2(L)Cl]platinum(II) complexes – synthesis, structures, and anticancer activities. Dalton Trans 45:15390–15398
Schmitt F, Donnelly K, Muenzner JK, Rehm T, Novohradsky V, Brabec V, Kasparkova J, Albrecht M, Schobert R, Mueller T (2016) Effects of histidine-2-ylidene vs. imidazole-2-ylidene ligands on the anticancer and antivascular activity of complexes of ruthenium, iridium, platinum, and gold. J Inorg Biochem 163:221–228
Liu H, Li Y, Lyu Z, Wan Y, Li X, Chen H, Chen H, Li X (2014) Enzyme-triggered supramolecular self-assembly of platinum prodrug with enhanced tumor-selective accumulation and reduced systemic toxicity. J Mater Chem B 2:83038309
Acknowledgments
Own work referenced in this chapter was supported by grants from the Deutsche Forschungsgemeinschaft (Scho 402/8 and Scho 402/12).
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Biersack, B., Schobert, R. (2019). Current State of Platinum Complexes for the Treatment of Advanced and Drug-Resistant Breast Cancers. In: Ahmad, A. (eds) Breast Cancer Metastasis and Drug Resistance. Advances in Experimental Medicine and Biology, vol 1152. Springer, Cham. https://doi.org/10.1007/978-3-030-20301-6_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-20301-6_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-20300-9
Online ISBN: 978-3-030-20301-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)