
Abstract
The thioester homocysteine (Hcy)-thiolactone, product of an error-editing reaction in protein biosynthesis, forms when Hcy is mistakenly selected by methionyl-tRNA synthetase. Accumulating evidence suggests that Hcy-thiolactone plays an important role in atherothrombosis. The thioester chemistry of Hcy-thiolactone underlies its ability to form isopeptide bonds with protein lysine residues, which impairs or alters protein function and has pathophysiological consequences including activation of an autoimmune response and enhanced thrombosis. Mammalian organisms, including human, have evolved the ability to eliminate Hcy-thiolactone. One such mechanism involves paraoxonase 1 (PON1), which has the ability to hydrolyze Hcy-thiolactone. This article outlines Hcy-thiolactone pathobiology and reviews evidence documenting the role of PON1 in minimizing Hcy-thiolactone and N-Hcy-protein accumulation.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
- Autoantibodies
- Atherosclerosis
- CBS
- Fibrinogen
- Hyperhomocysteinemia
- Homocysteine thiolactone
- Immune activation
- MTHFR
- Paraoxonase/thiolactonase
- Protein N-homocysteinylation
- Thrombosis
1 Homocysteine Metabolism
In mammals, including humans, homocysteine (Hcy) is formed from methionine (Met) as a result of cellular methylation reactions (Mudd et al., 2001). In this pathway Met is first activated by ATP to yield S-adenosylmethionine (AdoMet). As a result of the transfer of its methyl group to an acceptor, AdoMet is converted to S-adenosylhomocysteine (AdoHcy). Enzymatic hydrolysis of AdoHcy is the only known source of Hcy in the human body. Levels of Hcy are regulated by remethylation to Met, catalyzed by Met synthase (MS), and transsulfuration to cysteine, the first step of which is catalyzed by cystathionine β-synthase (CBS). The remethylation requires vitamin B12 and 5,10-methyl-tetrahydrofolate (CH3-THF), generated by 5,10-methylene-THF reductase (MTHFR). The transsulfuration requires vitamin B6 (Fig. 1).

The metabolism and pathophysiology of Hcy-thiolactone. N-Hcy-Fbg, N-Hcy-LDL – N-homocysteinylated forms of fibrinogen and low density lipoprotein, respectively (adapted from (Chwatko et al., 2007)). See text for discussion
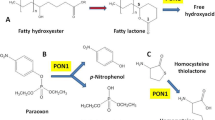
Hcy is also metabolized to the thioester Hcy-thiolactone in an error-editing reaction in protein biosynthesis when Hcy is mistakenly selected in place of Met by methionyl-tRNA synthetase (MetRS) (Fig. 2) (Jakubowski, 2005c, 2000a, 2001a, b, c, 2004, 2005a, Jakubowski and Goldman, 1993). The flow through the Hcy-thiolactone pathway increases when the remethylation or transsulfuration reaction is impaired by genetic alterations of enzymes, such as CBS (Jakubowski, 1991, 1997; 2002a; 2007), MS (Jakubowski, 1991, 2002a), and MTHFR (Jakubowski, 2006), or by inadequate supply of co-factors, e.g. CH3-THF (Jakubowski, 1997, 2000; Jakubowski et al., 2000). As will be discussed in a greater detail elsewhere in this chapter, Hcy-thioactone is hydrolyzed to Hcy by paraoxonase 1 (PON1) (Fig. 1).

The formation of Hcy-thiolactone catalyzed by MetRS. During protein biosynthesis Hcy is often mistakenly selected in place of Met by methionyl-tRNA synthetase (MetRS) and activated with ATP to form Hcy-AMP (upper panel). The misactivated Hcy is not transferred to tRNA but converted to Hcy-thiolactone in an error-editing reaction (lower panel) (adapted from (Jakubowski, 2001))
2 Pathophysiology of Hyperhomocysteinemia
Among pathophysiological manifestations of genetic hyperhomocysteinemia, which include mental retardation, ectopia lentis, and osteoporosis, vascular complications, including increased thrombosis, remain the major cause of morbidity and mortality in untreated patients (Kluijtmans et al., 1999; Mudd et al., 1985; Rosenblatt and Fenton, 2001; Yap et al., 2001). McCully observed advanced arterial lesions in children with inborn errors in Hcy metabolism and proposed a hypothesis that Hcy causes vascular disease (McCully, 1969). Even mild hyperhomocysteinemia, quite prevalent in the general population, is associated with an increased risk of vascular events (Clarke et al., 2007; Shishehbor et al., 2008).
Patients suffering from hyperhomocysteinemia improve upon vitamin B therapy, which lowers plasma Hcy levels. For example, Hcy lowering by vitamin B supplementation improves vascular outcomes in CBS-deficient patients, which suggests that Hcy plays a causal role in atherothrombosis. Specifically, untreated CBS-deficient patients suffer one vascular event per 25 patient-years (Mudd et al., 1985) while treated CBS-deficient patients suffer only one vascular event per 263 patient-years (relative risk 0.091, p < 0.001) (Yap et al., 2001). Hcy-lowering therapy started early in life also prevents brain disease from severe MTHFR deficiency (Rosenblatt and Fenton, 2001; Strauss et al., 2007).
Furthermore, lowering plasma Hcy by vitamin B supplementation also improves cognitive function in the general population (Durga et al., 2007). High-risk stroke (Lonn et al., 2006; Spence et al., 2005) but not myocardial infarction patients (Bonaa et al., 2006; Lonn et al., 2006) benefit from lowering of plasma Hcy by vitamin B supplementation. These findings suggest that Hcy plays a greater role in stroke than in myocardial infarction, a suggestion consistent with the observations that in untreated CBS-deficient patients cerebrovascular incidents are eight times more frequent than myocardial infarctions (Mudd et al., 1985). These findings also suggest that Hcy plays a more pronounced role in brain compared with heart pathophysiology. Studies of genetic and nutritional hyperhomocysteinemia in animal models provide additional support for a causal role of Hcy in atherothrombosis (Lentz, 2005).
3 The Hcy-Thiolactone Hypothesis
A preponderance of biochemical and genetic data suggest that elevated Hcy promotes a proatherothrombotic phenotype. Proposed mechanisms underlying Hcy pathobiology include protein modification by Hcy-thiolactone, oxidative stress, inflammation and autoimmune response, endothelial dysfunction, and thrombosis (Jakubowski, 2004, 2007; Lentz, 2005).
The Hcy-thiolactone hypothesis (Jakubowski, 1997) states that a pathway initiated by metabolic conversion of Hcy to Hcy-thiolactone catalyzed by methionyl-tRNS synthetase (Fig. 2) (Jakubowski and Goldman, 1993) contributes to Hcy pathobiology (Fig. 1) (Jakubowski, 2004, 2006, 2007). Hcy-thiolactone is a reactive metabolite that causes protein N-homocysteinylation through the formation of amide bonds with protein lysine residues (Fig. 3), which impairs or alters the protein’s function (Jakubowski, 1997, 1999). Plasma Hcy-thiolactone and N-linked protein Hcy(N-Hcy-protein), originally discovered in vitro in human fibroblasts and endothelial cells (Jakubowski, 1997, 1999, 2000; Jakubowski et al., 2000), occur in the human body (Chwatko and Jakubowski, 2005a, b; Jakubowski, 2000; Jakubowski et al., 2000), and are greatly elevated under conditions predisposing to atherothrombosis, such as hyperhomocysteinemia caused by mutations in CBS or MTHFR gene in humans or a high-Met diet in mice (Chwatko et al., 2007; Glowacki and Jakubowski, 2004; Jakubowski, 2001, 2002b; Jakubowski et al., 2008). N-Hcy-protein accumulates in atherosclerotic lesions in ApoE-deficient mice, and the accumulation increases in animals fed a high methionine diet (Perla-Kajan et al., 2008).

N-Hcy-protein forms in a reaction of Hcy-thiolactone with a protein lysine residue
4 Toxicity of Hcy-Thiolactone
Accumulating evidence from human, animal, and tissue culture studies shows that Hcy-thiolactone is involved in pathophysiology (reviewed in (Jakubowski, 2004, 2005, 2006, 2007)). Chronic treatments of animals with Hcy-thiolactone cause pathophysiological changes similar to those observed in human genetic hyperhomocysteinemia. For instance, Hcy-thiolactone infusions or Hcy-thiolactone-supplemented diet produce atherosclerosis in baboons (Harker et al., 1974) or rats (Endo et al., 2006) while treatments with Hcy-thiolactone cause developmental abnormalities in chick embryos, including optic lens dislocation (Maestro de las Casas et al., 2003), characteristic of CBS-deficient human patients (Mudd et al., 2001; 1985). A recent study shows that plasma Hcy-thiolactone levels are associated with the development and progression of diabetic macrovasculopathy (Gu et al., 2008).
Hcy-thiolactone induces apoptotic death in cultured human vascular endothelial (Kerkeni et al., 2006; Mercie et al., 2000) and promyeloid cells (Huang et al., 2001) and placental trophoblasts (Kamudhamas et al., 2004), and inhibits insulin signaling in rat hepatoma cells (Najib and Sanchez-Margalet, 2005). Hcy-thiolactone also induces endoplasmic reticulum (ER) stress and unfolded protein response (UPR) in retinal epithelial cells (Roybal et al., 2004). Furthermore, Hcy-thiolactone is more toxic to cultured cells than Hcy itself (Huang et al., 2001; Kamudhamas et al., 2004; Kerkeni et al., 2006; Mercie et al., 2000; Roybal et al., 2004).
5 Consequences of Protein N -Homocysteinylation by Hcy-Thiolactone
Cellular physiology can be impacted by Hcy-thiolactone-mediated protein modification, which changes the primary protein sequence, disrupts protein folding, and creates altered proteins with newly acquired interactions. Small changes in amino acid sequence caused by Hcy incorporation have the potential to create misfolded protein aggregates. Indeed, N-Hcy-proteins do have a propensity to form protein aggregates (Jakubowski, 1999). The appearance of misfolded/aggregated proteins in the ER activates a signaling pathway, the UPR, that, when overwhelmed, leads to cell death via apoptosis (Lawrence de Koning et al., 2003; Lentz, 2005). These pathways are induced by treatments of cultured cells and mice with excess Hcy (Hossain et al., 2003), which is metabolized to Hcy-thiolactone (Jakubowski, 1997, 2007; Jakubowski et al., 2000). Hcy-thiolactone is more effective than Hcy in inducing ER and UPR (Roybal et al., 2004). In this scenario the formation of N-Hcy-proteins leads to the UPR and induction of the apoptotic pathway. In humans, Hcy incorporation into proteins triggers an autoimmune response (Jakubowski, 2005b) and increases vascular inflammation (Bogdanski et al., 2007)(Fig. 2), known modulators of atherogenesis (Libby, 2006).
Hcy incorporation is detrimental to protein function. For example, lysine oxidase (Liu et al., 1997), trypsin (Jakubowski, 1999), MetRS (Jakubowski, 1999), and PON1 (Ferretti et al., 2003) are inactivated by N-homocysteinylation with Hcy-thiolactone. N-Homocysteinylation of albumin (Glowacki and Jakubowski, 2004) and cytochrome c (Perla-Kajan et al., 2007) impairs their redox function. N-Hcy-proteins (Jakubowski, 1999, 2000), including N-Hcy-LDL (Naruszewicz et al., 1994), tend to form aggregates in vitro. N-Hcy-LDL, but not native LDL, induces cell death in human endothelial cells (Ferretti et al., 2004), a finding consistent with the inherent toxicity of protein aggregates (Stefani, 2004).
Plasma N-linked Hcy protein is correlated with plasma total Hcy in humans (Jakubowski, 2000, 2002b; Jakubowski et al., 2008). Hcy-thiolactone and N-Hcy-protein levels are greatly elevated by CBS or MTHFR deficiency in humans (Chwatko et al., 2007; Jakubowski et al., 2008). In cultured human cells, CBS deficiency orantifolate drugs such as aminopterin (Jakubowski, 1997) increase the accumulation of Hcy-thiolactone and N-Hcy-protein, whereas supplementation with folic acid decreases the levels of these metabolites (Jakubowski et al., 2000). As will be discussed in greater detail in the final section of this chapter, the accumulation of N-Hcy-protein is also inhibited by the Hcy-thiolactonase activity of PON1 (Fig. 1) (Jakubowski, 2000; Jakubowski et al., 2000).
6 Prothrombotic Properties of N -Hcy-Fibrinogen
Fibrinogen undergoes facile N-homocysteinylation by Hcy-thiolactone in vitro (Jakubowski, 1999, 2000) and in vivo in humans (Jakubowski, 2002b; Jakubowski et al., 2008). Sauls et al. (Sauls et al., 2006) showed that clots formed from Hcy-thiolactone-treated normal human plasma or fibrinogen lyse slower than clots from untreated controls. Some of the lysine residues susceptible to N-homocysteinylation are close to tissue plasminogen activator and plasminogen binding, or plasmin cleavage, sites, which can explain abnormal characteristics of clots formed from N-Hcy-fibrinogen (Sauls et al., 2006). The detrimental effects of elevated plasma tHcy on clot permeability and resistance to lysis in humans are consistent with a mechanism involving fibrinogen modification by Hcy-thiolactone (Undas et al., 2006). Furthermore, CBS-deficient patients have significantly elevated plasma levels of prothrombotic N-Hcy-fibrinogen (Jakubowski et al., 2008), which explains increased atherothrombosis observed in these patients (Mudd et al., 1985).
7 Autoimmunogenicity of N -Hcy-Protein
Atherosclerosis is now widly recognized as a chronic inflammatory disease that involves both innate and adaptive immunity (Libby, 2006). Like other modified proteins, N-Hcy-proteins elicit an autoimmune response in humans, manifested by the induction of IgG autoantibodies directed against Nε-Hcy-Lys epitopes. This response is enhanced in stroke and coronary artery disease (CAD) patients, suggesting that it is a general feature of atherosclerosis (Undas et al., 2005, 2004, 2006). Elevated levels of anti-N-Hcy-protein IgG autoantibodies are a consequence of elevated levels of N-Hcy-protein observed in CAD patients (Yang et al., 2006).
The involvement of an autoimmune response against N-Hcy-protein in CAD is supported by the findings that lowering plasma Hcy by folic acid supplementation lowers anti-N-Hcy-protein autoantibodies levels in control subjects but not in patients with CAD (Undas et al., 2006). These findings suggest that, once accumulated, the antigens causing the antibody response, i.e. N-Hcy proteins, persist and that chronic protein damage caused by N-homocysteinylation cannot be easily reversed in CAD patients. Furthermore, these findings also suggest that while primary Hcy-lowering intervention by vitamin supplementation is beneficial, secondary intervention may be ineffective, and may explain at least in part the failure of vitamin therapy to lower cardiovascular events in myocardial infarction patients (Bonaa et al., 2006; Lonn et al., 2006).
8 The Role of PON1 in Elimination of Hcy-Thiolactone
Because Hcy-thiolactone is linked to human pathophysiology (Fig. 1), it is not surprising that the human body evolved the ability to eliminate Hcy-thiolactone. Indeed, we found that an high-density lipoprotein (HDL)-associated enzyme, Hcy-thiolactonase/paraoxonase 1 (PON1) is able to hydrolyze the toxic metabolite Hcy-thiolactone in human serum (Domagała et al. 2006; Jakubowski, 2000; Jakubowski et al., 2001; Lacinski et al., 2004). More recently, Hcy-thiolactonase/bleomycin hydrolase (BLH) was found to hydrolyze Hcy-thiolactone intracellularly (Zimny et al., 2006). Another mechanism of Hcy-thiolactone elimination involves clearance by the kidney (Chwatko and Jakubowski, 2005).
PON1 is synthesized exclusively in the liver and carried on HDL in the circulation. Owing to its ability to detoxify organophosphate insecticides and nerve gases, PON1 has been studied in the field of toxicology since the 1960 s. More recent studies have implicated PON1 in the pathogenesis of cardiovascular disease. For example, PON1-deficient mice are more susceptible to a high-fat diet-induced atherosclerosis than wild-type littermates (however, the animals do not develop atherosclerosis on a normal chow diet) (Shih et al., 1998). PON1 transgenic mice (carrying three copies of the human PON1) are less susceptible to atherosclerosis (Tward et al., 2002). In vitro studies indicate that HDL from PON1-deficient animals does not prevent LDL oxidation, whereas HDL from PON1 transgenic animals protects LDL against oxidation more effectively than HDL from wild-type mice. However, the biochemical basis for the putative antioxidative function of PON1 is unclear and its pathophysiologically relevant lipid-related substrate is not known (Vos, 2008).
Hcy-thiolactone, ubiquitously present in living organisms, including humans, is a natural substrate of PON1, which therefore should be more appropriately called Hcy-thiolactonase (HTase) (Jakubowski, 2000). The conclusion that PON1 is an HTase is based on proteomic and genetic evidence. For example, purified human HTase has a molecular weight and N-terminal amino acid sequence identical to those of human PON1 protein. Sera from PON1-deficient mice are also deficient in HTase activity (Table 1) (Jakubowski, 2000, 2001). HTase activity is absent in serum from chicken (Jakubowski, 2000, 2001), in which the PON1 gene is known to be absent. HTase activity of PON1 requires calcium for activity and stability, and is inhibited by isoleucine and the antiarthritic drug D-penicillamine (Jakubowski, 2000). Human PON1 has genetic polymorphisms, e.g. PON1-M55L, PON1-R192Q (Jarvik et al., 2000), which are responsible for about 10-fold inter-individual variation in HTase activity (Domagała et al. 2006; Jakubowski et al., 2001; Lacinski et al., 2004). High HTase activity is associated with L55 and R192 alleles more frequent in blacks than in whites, whereas low HTase activity is associated with M55 and Q192 alleles, more frequent in whites than in blacks (Jakubowski et al., 2001).
In vitro studies show that HTase activity of PON1 prevents Hcy-thiolactone and N-Hcy-protein accumulation in cultures of human endothelial cells (Jakubowski et al., 2001, 2000) (Table 2). The high HTase activity form of PON1 affords better protection against protein N-homocysteinylation than the low activity form in human serum (Jakubowski et al., 2001) (Fig. 4). High Hcy-thiolactonase activity in rabbit serum protects serum proteins against N-homocysteinylation (Fig. 4) (Jakubowski et al., 2001) and could account for the observation that infusions with Hcy-thiolactone failed to produce atherosclerosis in these animals (Donahue et al., 1974). In humans, HTase activity of PON1 is negatively correlated with plasma total Hcy (Lacinski et al., 2004) and predicts cardiovascular disease (Domagała et al. 2006). In mice, Hcy is a negative regulator of PON1 expression (Robert et al., 2003).

High HTase activity protects against N-Hcy-protein accumulation in human and animal sera. In humans, L55 and R192 are high HTase activity, whereas M55 and Q192 are low HTase activity PON1 alleles (adapted from (Jakubowski et al., 2001))
Recent development of highly sensitive HPLC-based assays facilitated examination of Hcy-thiolactone (Chwatko et al., 2007; Chwatko and Jakubowski, 2005, 2005) and N-Hcy-protein (Jakubowski, 2008) metabolism in vivo both in humans and in mice. These assays also allowed examination of the role of PON1 in Hcy-thiolactone and N-Hcy-protein metabolism. We found that in C57BL/6 J mice fed a normal diet (n = 8), plasma and urinary Hcy-thiolactone levels were 3.7 ± 2.1 nM and 136 ± 22 nM (Table 2), respectively. These levels are similar to plasma and urinary Hcy-thiolactone levels in humans (Chwatko et al., 2007).
In mice fed a high-Met diet (n = 14), Hcy-thiolactone concentrations increased 4- to 25-fold, to 13.8.0 ± 4.8 and 3490 ± 3780 nM, in plasma and urine, respectively. Plasma and urinary tHcy levels increased 17.3-fold (to 51.8 ± 22.7 μM) and 30-fold (to 1360 ± 840 μM) (Table 3), respectively, compared to mice fed a normal chow diet (Chwatko et al., 2007).
We then measured levels of plasma Hcy-thiolactone and plasma N-Hcy-protein in PON1–/– and PON1+/+ mice fed a normal chow diet or a high-Met diet for up to 18 weeks. The levels of Hcy-thiolactone and N-Hcy were then normalized to Hcy levels. We found that the normalized plasma Hcy-thiolactone levels (Hcy-thiolactone/Hcy ratios) were similar in PON1–/–and PON1+/+ mice fed a normal chow diet (Table 4). In animals fed a high-Met diet for 2, 8, and 18 weeks, which increases Hcy-thiolactone and Hcy accumulation (Table 3), the Hcy-thiolactone/Hcy ratios did not differ between PON1–/– and PON1+/+ mice.
Because the bulk of Hcy-thiolactone is excreted in urine (Chwatko et al., 2007; Chwatko and Jakubowski, 2005), we also measured urinary Hcy-thioactone in PON1–/– and PON1+/+ mice fed a normal diet or a high-Met diet. We found that urinary Hcy-thiolactone/Hcy ratios were significantly elevated in PON1–/– mice relative to PON1+/+ mice fed a normal chow diet (Table 5). The Hcy-thiolactone/Hcy ratios also increased in PON1–/– relative to PON1+/+ mice fed a high-Met diet, but the difference did not reach statistical significance.
The reaction of Hcy-thiolactone with proteins (Jakubowski, 1997, 1999; Jakubowski et al., 2000) could lead to an apparent lack of Hcy-thiolactone accumulation in mice fed a high-Met diet. To examine this possibility, we measured plasma N-Hcy-protein levels in WT and PON1–/– mice. Plasma N-Hcy-protein levels were normalized to plasma Hcy levels (N-Hcy-protein/Hcy ratio). We found that there was no difference in N-Hcy-protein/Hcy ratios between PON1–/– and PON1+/+ mice fed a normal chow diet (0.28 ± 0.21 vs. 0.33 ± 0.21). However, the N-Hcy-protein/Hcy ratios were significantly higher in PON1–/– mice compared with PON1+/+ mice fed a high-Met diet for 2 weeks (1.21 ± 1.19 vs. 0.25 ± 0.11, p = 0.027) and 8 weeks (0.98 ± 0.20 vs. 0.72 ± 0.19, p = 0.022) (Table 6). The differences between PON1–/– and PON1+/+ were the highest at 2 weeks, diminished at 8 weeks, and became insignificant at 18 weeks (0.16 ± 0.07 vs. 0.13 ± 0.03) (Table 6). Taken together, these results show that PON1 controls the accumulation of Hcy-thiolactone in mice and that PON1 protects against transient elevation of N-Hcy-protein in mice fed a hyperhomocysteinemic high-Met diet.
Leptin administration in male Wistar rats lowers plasma activities of PON1 (Beltowski et al., 2003), including HTase activity (Beltowski et al., 2008), and increases plasma N-Hcy-protein levels, but has no effect on plasma Hcy levels (Beltowski et al., 2008). Co-treatment of rats with a synthetic agonist to liver X receptor, T0901317, prevents leptin-induced decrease in HTase activity of PON1 and increase in N-Hcy-protein accumulation in rat serum. Control experiments show that T0901317 has no effect on HTase activity and N-Hcy-protein accumulation in animals not receiving leptin. These results suggest that PON1 controls N-Hcy-protein accumulation in rats (Beltowski et al., 2008).
9 Conclusion
Despite advances in our understanding of cardiovascular disease, coronary heart disease is still the major cause of mortality in industrial nations. Traditional risk factors such as hypertension, diabetes, hyperlipidemia, and smoking do not accurately predict cardiovascular events. Thus identification of novel risk factors and their mechanisms of action has important public health implications. Hcy is a novel risk factor for the development of cardiovascular disease (Clarke et al., 2007; Shishehbor et al., 2008). Studies of severe genetic hyperhomocysteinemia in humans and genetic and nutritional hyperhomocysteinemia in animal models show that Hcy plays a causal role in atherothrombosis. Metabolic conversion of Hcy to Hcy-thiolactone and inadvertent protein modification by Hcy-thiolactone (which induces pathophysiological responses, such as an autoimmune activation and increased thrombosis) contribute to proatherogenic changes in the cardiovascular system. Chronic activation of these processes in nutritional or genetic deficiencies in Hcy metabolism can cause vascular disease. The ability to hydrolyze Hcy-thiolactone and minimize the accumulation of N-Hcy-protein is likely to contribute to the cardioprotective function of PON1.
References
Beltowski J, Wojcicka G, Jamroz A. Leptin decreases plasma paraoxonase 1 (PON1) activity and induces oxidative stress: the possible novel mechanism for proatherogenic effect of chronic hyperleptinemia. Atherosclerosis. 2003 Sep;170(1):21–29.
Beltowski J, Wojcicka G, Jamroz-Wisniewska A, Marciniak A. Liver X receptor agonist, T0901317, normalizes plasma PON1 activity and reduces protein homocysteinylation in rats with experimental hyperleptinemia. 3rd International Conference on Paraoxonases; 2008; Los Angeles, CA. 2008.
Bogdanski P, Pupek-Musialik D, Dytfeld J, Lacinski M, Jablecka A, Jakubowski H. Plasma homocysteine is a determinant of tissue necrosis factor-alpha in hypertensive patients. Biomed Pharmacother. 2007 Dec 3;62:360–365
Bonaa KH, Njolstad I, Ueland PM, Schirmer H, Tverdal A, Steigen T, et al. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med. 2006 Apr 13;354(15):1578–1588.
Chwatko G, Boers GH, Strauss KA, Shih DM, Jakubowski H. Mutations in methylenetetrahydrofolate reductase or cystathionine beta-synthase gene, or a high-methionine diet, increase homocysteine thiolactone levels in humans and mice. Faseb J. 2007 Jun;21(8):1707–1713.
Chwatko G, Jakubowski H. The determination of homocysteine-thiolactone in human plasma. Anal Biochem. 2005 Feb 15;337(2):271–277.
Chwatko G, Jakubowski H. Urinary excretion of homocysteine-thiolactone in humans. Clin Chem. 2005 Feb;51(2):408–415.
Clarke R, Lewington S, Sherliker P, Armitage J. Effects of B-vitamins on plasma homocysteine concentrations and on risk of cardiovascular disease and dementia. Curr Opin Clin Nutr Metab Care. 2007 Jan;10(1):32–39.
Domagała TB, Łacinski M, Trzeciak WH, Mackness B, Mackness MI, Jakubowski H. The correlation of homocysteine-thiolactonase activity of the paraoxonase (PON1) protein with coronary heart disease status. Cell Mol Biol (Noisy-le-grand). 2006;52(5):4–10.
Donahue S, Struman JA, Gaull G. Arteriosclerosis due to homocyst(e)inemia. Failure to reproduce the model in weanling rabbits. Am J Pathol. 1974 Nov;77(2):167–163.
Durga J, van Boxtel MP, Schouten EG, Kok FJ, Jolles J, Katan MB, et al. Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet. 2007 Jan 20;369(9557):208–216.
Endo N, Nishiyama K, Otsuka A, Kanouchi H, Taga M, Oka T. Antioxidant activity of vitamin B6 delays homocysteine-induced atherosclerosis in rats. Br J Nutr. 2006 Jun;95(6):1088–1093.
Ferretti G, Bacchetti T, Marotti E, Curatola G. Effect of homocysteinylation on human high-density lipoproteins: a correlation with paraoxonase activity. Metabolism. 2003 Feb;52(2):146–151.
Ferretti G, Bacchetti T, Moroni C, Vignini A, Nanetti L, Curatola G. Effect of homocysteinylation of low density lipoproteins on lipid peroxidation of human endothelial cells. J Cell Biochem. 2004 May 15;92(2):351–360.
Glowacki R, Jakubowski H. Cross-talk between Cys34 and lysine residues in human serum albumin revealed by N-homocysteinylation. J Biol Chem. 2004 Mar 19;279(12):10864–10871.
Gu W, Lu J, Yang G, Dou J, Mu Y, Meng J, et al. Plasma homocysteine thiolactone associated with risk of macrovasculopathy in Chinese patients with type 2 diabetes mellitus. Adv Ther. 2008 Sep;25(9):914–924.
Harker LA, Slichter SJ, Scott CR, Ross R. Homocystinemia. Vascular injury and arterial thrombosis. N Engl J Med. 1974 Sep 12;291(11):537–543.
Hossain GS, van Thienen JV, Werstuck GH, Zhou J, Sood SK, Dickhout JG, et al. TDAG51 is induced by homocysteine, promotes detachment-mediated programmed cell death, and contributes to the development of atherosclerosis in hyperhomocysteinemia. J Biol Chem. 2003 Aug 8;278(32):30317–30327.
Huang RF, Huang SM, Lin BS, Wei JS, Liu TZ. Homocysteine thiolactone induces apoptotic DNA damage mediated by increased intracellular hydrogen peroxide and caspase 3 activation in HL-60 cells. Life Sci. 2001 May 11;68(25):2799–2811.
Jakubowski H. Proofreading in vivo: editing of homocysteine by methionyl-tRNA synthetase in the yeast Saccharomyces cerevisiae. Embo J. 1991 Mar;10(3):593–598.
Jakubowski H. Metabolism of homocysteine thiolactone in human cell cultures. Possible mechanism for pathological consequences of elevated homocysteine levels. J Biol Chem. 1997 Jan 17;272(3):1935–1942.
Jakubowski H. Protein homocysteinylation: possible mechanism underlying pathological consequences of elevated homocysteine levels. Faseb J. 1999 Dec;13(15):2277–2283.
Jakubowski H. Homocysteine thiolactone: metabolic origin and protein homocysteinylation in humans. J Nutr. 2000 Feb;130(2S Suppl):377S–381S.
Jakubowski H. Calcium-dependent human serum homocysteine thiolactone hydrolase. A protective mechanism against protein N-homocysteinylation. J Biol Chem. 2000 Feb 11;275(6):3957–3962.
Jakubowski H. Biosynthesis and reactions of homocysteine thiolactone. In: Jacobson D, Carmel R (eds.). Homocysteine in Health and Disease. Cambridge, UK: Cambridge University Press; 2001. 21–31.
Jakubowski H. Translational accuracy of aminoacyl-tRNA synthetases: implications for atherosclerosis. J Nutr. 2001 Nov;131(11):2983S–2987S.
Jakubowski H. Protein N-homocysteinylation: implications for atherosclerosis. Biomed Pharmacother. 2001 Oct;55(8):443–447.
Jakubowski H. The determination of homocysteine-thiolactone in biological samples. Anal Biochem. 2002 Sep 1;308(1):112–119.
Jakubowski H. Homocysteine is a protein amino acid in humans. Implications for homocysteine-linked disease. J Biol Chem. 2002 Aug 23;277(34):30425–30428.
Jakubowski H. Molecular basis of homocysteine toxicity in humans. Cell Mol Life Sci. 2004 Feb;61(4):470–487.
Jakubowski H. Accuracy of aminoacyl-tRNA synthetases: Proofreading of amino acids. In: Ibba M, Francklyn C, Cusack S (eds.). The Aminoacyl-tRNA Synthetases. Georgetown, TX: Landes Bioscience/Eurekah.com 2005. 384–396.
Jakubowski H. Anti-N-homocysteinylated protein autoantibodies and cardiovascular disease. Clin Chem Lab Med. 2005;43(10):1011–1014.
Jakubowski H. tRNA Synthetase Editing of Amino Acids. Encyclopedia of Life Sciences. Chichester, UK: John Wiley & Sons, Ltd; 2005. p. http://www.els.net/doi:10.1038/npg.els.0003933.
Jakubowski H. Pathophysiological consequences of homocysteine excess. J Nutr. 2006 Jun;136(6 Suppl):1741S–1749S.
Jakubowski H. The molecular basis of homocysteine thiolactone-mediated vascular disease. Clin Chem Lab Med. 2007;45(12):1704–1716.
Jakubowski H. New method for the determination of protein N-linked homocysteine. Anal Biochem. 2008 Sep 15;380(2):257–261.
Jakubowski H, Ambrosius WT, Pratt JH. Genetic determinants of homocysteine thiolactonase activity in humans: implications for atherosclerosis. FEBS Lett. 2001 Feb 23;491(1–2):35–39.
Jakubowski H, Boers GH, Strauss KA. Mutations in cystathionine beta-synthase or methylenetetrahydrofolate reductase gene increase N-homocysteinylated protein levels in humans. FASEB J. 2008;22:4071–4076.
Jakubowski H, Goldman E. Synthesis of homocysteine thiolactone by methionyl-tRNA synthetase in cultured mammalian cells. FEBS Lett. 1993 Feb 15;317(3):237–240.
Jakubowski H, Zhang L, Bardeguez A, Aviv A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: implications for atherosclerosis. Circ Res. 2000 Jul 7;87(1):45–51.
Jarvik GP, Rozek LS, Brophy VH, Hatsukami TS, Richter RJ, Schellenberg GD, et al. Paraoxonase (PON1) phenotype is a better predictor of vascular disease than is PON1(192) or PON1(55) genotype. Arterioscler Thromb Vasc Biol. 2000 Nov;20(11):2441–2447.
Kamudhamas A, Pang L, Smith SD, Sadovsky Y, Nelson DM. Homocysteine thiolactone induces apoptosis in cultured human trophoblasts: a mechanism for homocysteine-mediated placental dysfunction? Am J Obstet Gynecol. 2004 Aug;191(2):563–571.
Kerkeni M, Tnani M, Chuniaud L, Miled A, Maaroufi K, Trivin F. Comparative study on in vitro effects of homocysteine thiolactone and homocysteine on HUVEC cells: evidence for a stronger proapoptotic and proinflammative homocysteine thiolactone. Mol Cell Biochem. 2006 Oct;291(1-2):119–126.
Kluijtmans LA, Boers GH, Kraus JP, van den Heuvel LP, Cruysberg JR, Trijbels FJ, et al. The molecular basis of cystathionine beta-synthase deficiency in Dutch patients with homocystinuria: effect of CBS genotype on biochemical and clinical phenotype and on response to treatment. Am J Hum Genet. 1999 Jul;65(1):59–67.
Lacinski M, Skorupski W, Cieslinski A, Sokolowska J, Trzeciak WH, Jakubowski H. Determinants of homocysteine-thiolactonase activity of the paraoxonase-1 (PON1) protein in humans. Cell Mol Biol (Noisy-le-grand). 2004 Dec;50(8):885–893.
Lawrence de Koning AB, Werstuck GH, Zhou J, Austin RC. Hyperhomocysteinemia and its role in the development of atherosclerosis. Clin Biochem. 2003 Sep;36(6):431–441.
Lentz SR. Mechanisms of homocysteine-induced atherothrombosis. J Thromb Haemost. 2005 Aug;3(8):1646–1654.
Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006 Feb;83(2):456S–4560S.
Liu G, Nellaiappan K, Kagan HM. Irreversible inhibition of lysyl oxidase by homocysteine thiolactone and its selenium and oxygen analogues. Implications for homocystinuria. J Biol Chem. 1997 Dec 19;272(51):32370–32377.
Lonn E, Yusuf S, Arnold MJ, Sheridan P, Pogue J, Micks M, et al. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med. 2006 Apr 13;354(15):1567–1577.
Maestro de las Casas C, Epeldegui M, Tudela C, Varela-Moreiras G, Perez-Miguelsanz J. High exogenous homocysteine modifies eye development in early chick embryos. Birth Defects Res A Clin Mol Teratol. 2003 Jan;67(1):35–40.
McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969 Jul;56(1):111–128.
Mercie P, Garnier O, Lascoste L, Renard M, Closse C, Durrieu F, et al. Homocysteine-thiolactone induces caspase-independent vascular endothelial cell death with apoptotic features. Apoptosis. 2000 Nov;5(5):403–411.
Mudd SH, Levy HL, Krauss JP. Disorders of transsulfuration. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW et al. (eds.). The metabolic and molecular bases of inherited disease. 8th ed. New York: Mc Graw-Hill; 2001. 2007–2056.
Mudd SH, Skovby F, Levy HL, Pettigrew KD, Wilcken B, Pyeritz RE, et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet. 1985 Jan;37(1):1–31.
Najib S, Sanchez-Margalet V. Homocysteine thiolactone inhibits insulin-stimulated DNA and protein synthesis: possible role of mitogen-activated protein kinase (MAPK), glycogen synthase kinase-3 (GSK-3) and p70 S6K phosphorylation. J Mol Endocrinol. 2005 Feb;34(1):119–126.
Naruszewicz M, Olszewski AJ, Mirkiewicz E, McCully KS. Thiolation of low density lipoproteins by homocysteine thiolactone causes increased aggregation and altered interaction with cultured macrophages. Nutr Metab Cardiovasc Dis. 1994;4:70–77.
Perla-Kajan J, Marczak L, Kajan L, Skowronek P, Twardowski T, Jakubowski H. Modification by homocysteine thiolactone affects redox status of cytochrome c. Biochemistry. 2007 May 29;46(21):6225–6231.
Perla-Kajan J, Stanger O, Luczak M, Ziolkowska A, Malendowicz LK, Twardowski T, et al. Immunohistochemical detection of N-homocysteinylated proteins in humans and mice. Biomed Pharmacother. 2008 May 2;62(7):473–479.
Robert K, Chasse JF, Santiard-Baron D, Vayssettes C, Chabli A, Aupetit J, et al. Altered gene expression in liver from a murine model of hyperhomocysteinemia. J Biol Chem. 2003 Aug 22;278(34):31504–31511.
Rosenblatt D, Fenton W. Disorders of transsulfuration. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, et al. (eds.). The metabolic and molecular bases of inherited disease. 8th ed. New York: Mc Graw-Hill; 2001. 2007–2056.
Roybal CN, Yang S, Sun CW, Hurtado D, Vander Jagt DL, Townes TM, et al. Homocysteine increases the expression of vascular endothelial growth factor by a mechanism involving endoplasmic reticulum stress and transcription factor ATF4. J Biol Chem. 2004 Apr 9;279(15):14844–14852.
Sauls DL, Lockhart E, Warren ME, Lenkowski A, Wilhelm SE, Hoffman M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: a potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry. 2006 Feb 28;45(8):2480–2487.
Shih DM, Gu L, Xia YR, Navab M, Li WF, Hama S, et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature. 1998 Jul 16;394(6690):284–287.
Shishehbor MH, Oliveira LP, Lauer MS, Sprecher DL, Wolski K, Cho L, et al. Emerging cardiovascular risk factors that account for a significant portion of attributable mortality risk in chronic kidney disease. Am J Cardiol. 2008 Jun 15;101(12):1741–1746.
Spence JD, Bang H, Chambless LE, Stampfer MJ. Vitamin Intervention For Stroke Prevention trial: an efficacy analysis. Stroke. 2005 Nov;36(11):2404–2409.
Stefani M. Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim Biophys Acta. 2004 Dec 24;1739(1):5–25.
Strauss KA, Morton DH, Puffenberger EG, Hendrickson C, Robinson DL, Wagner C, et al. Prevention of brain disease from severe 5,10-methylenetetrahydrofolate reductase deficiency. Mol Genet Metab. 2007 Jun;91(2):165–175.
Tward A, Xia YR, Wang XP, Shi YS, Park C, Castellani LW, et al. Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation. 2002 Jul 23;106(4):484–490.
Undas A, Brozek J, Jankowski M, Siudak Z, Szczeklik A, Jakubowski H. Plasma homocysteine affects fibrin clot permeability and resistance to lysis in human subjects. Arterioscler Thromb Vasc Biol. 2006 Jun;26(6):1397–1404.
Undas A, Jankowski M, Twardowska M, Padjas A, Jakubowski H, Szczeklik A. Antibodies to N-homocysteinylated albumin as a marker for early-onset coronary artery disease in men. Thromb Haemost. 2005 Feb;93(2):346–350.
Undas A, Perla J, Lacinski M, Trzeciak W, Kazmierski R, Jakubowski H. Autoantibodies against N-homocysteinylated proteins in humans: implications for atherosclerosis. Stroke. 2004 Jun;35(6):1299–1304.
Undas A, Stepien E, Glowacki R, Tisonczyk J, Tracz W, Jakubowski H. Folic acid administration and antibodies against homocysteinylated proteins in subjects with hyperhomocysteinemia. Thromb Haemost. 2006 Sep;96(3):342–347.
Vos E. Homocysteine levels, paraoxonase 1 (PON1) activity, and cardiovascular risk. JAMA. 2008 Jul 9;300(2):168–169; author reply 9.
Yang X, Gao Y, Zhou J, Zhen Y, Yang Y, Wang J, et al. Plasma homocysteine thiolactone adducts associated with risk of coronary heart disease. Clin Chim Acta. 2006 Feb;364(1–2):230–234.
Yap S, Boers GH, Wilcken B, Wilcken DE, Brenton DP, Lee PJ, et al. Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol. 2001 Dec;21(12):2080–2085.
Zimny J, Sikora M, Guranowski A, Jakubowski H. Protective mechanisms against homocysteine toxicity: the role of bleomycin hydrolase. J Biol Chem. 2006 Aug 11;281(32):22485–22492.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Humana Press, a part of Springer Science+Business Media, LLC
About this paper
Cite this paper
Jakubowski, H. (2010). The Role of Paraoxonase 1 in the Detoxification of Homocysteine Thiolactone. In: Reddy, S. (eds) Paraoxonases in Inflammation, Infection, and Toxicology. Advances in Experimental Medicine and Biology, vol 660. Humana Press. https://doi.org/10.1007/978-1-60761-350-3_11
Download citation
DOI: https://doi.org/10.1007/978-1-60761-350-3_11
Published:
Publisher Name: Humana Press
Print ISBN: 978-1-60761-349-7
Online ISBN: 978-1-60761-350-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)