
Abstract
We have found a novel nonsense mutation in the C-terminus of HERG in a four-generation Chinese family with long QT syndrome and investigated the molecular mechanism of this mutation in vitro. Six family members, including the proband, were clinically affected. Syncope and ventricular tachycardia of torsades de pointes were triggered by startling or emotional stress, and β-adrenergic blockade treatment was ineffective. Haplotype analysis showed that only LQT2 markers cosegregated with the disease, and sequence analysis revealed a substitution of T with C at nucleotide position 2770 of the HERG gene (U04270), which creates a stop codon at amino acid position 863 (R863X) of the HERG protein, leading to a deletion of 296 amino acids. Whole cell patch clamp studies showed that the R863X HERG could not induce time-dependent current. Coexpression of R863X with wild-type HERG showed reduced current densities and accelerated voltage-dependent inactivation of HERG channels. Subcellular localization of R863X-EGFP revealed that the mutant did not traffic to the cell surface. These data suggest that R863X failed to form functional HERG channels, contributing to a prolongation of the QT interval and long QT syndrome with a dominant phenotype. These findings provide new insights into the structure-function relationships of the HERG C-terminus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Familial long QT syndrome (LQTS) is an inherited cardiac disorder characterized by delayed cardiac repolarization, ventricular arrhythmia, syncope, and sudden death [1]. Although there is a variable spectrum of cardiac electrical abnormalities and penetrance, the majority of LQTS pedigrees show dominant transmission.
Six LQTS-associated genes have been hitherto identified [2, 3]. The human ether-a-go-go related gene (HERG) is responsible for one form of long QT syndrome (LQT2) and encodes the pore-forming subunit of the rapidly activating, delayed rectifier current I kr, which plays an important role in the repolarization of the cardiac action potential [4, 5]. Although the role of other factors contributing to I kr current, such as HERG splice variants and interacting β-subunits, is still under investigation [6, 7, 8, 9], mutations in HERG are one of the most common causes for congenital LQTS [10].
So far, more than 100 mutations in the HERG gene have been identified in LQT2 patients [10]. Various mechanisms underlying the dysfunction of the HERG channel in these mutants have been suggested, including abnormal channel processing [11, 12, 13], generation of nonfunctional channels [14, 15] and altered channel gating [16, 17, 18, 19].
From the primary HERG structure point of view, there are several sites where mutations have occurred that could result in HERG channel dysfunction. For example, mutations in the N-terminus usually accelerate HERG channel deactivation [19], while mutations in the pore region affect HERG channel inactivation or ion selectivity [15, 17, 18]. More recently, mutations in LQT2 have been mapped to the C-terminus which contains regions essential for I kr function [20], but the mechanisms underlying HERG channel dysfunction in association with C-terminal mutations have not been fully characterized yet.
In this study, a novel mutation, R863X, of HERG was identified in a Chinese LQTS family, leading to a deletion of 296 amino acids in the C-terminus of HERG protein. The functional consequences of this mutation were characterized in vitro.
Materials and methods
Subjects
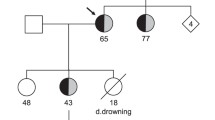
This study was conducted in a four-generation Chinese family with LQTS (Fig. 1A). Informed consent was obtained from the family members in accordance with the study protocols approved by the Review Board of Center for Molecular Cardiology, Chinese Academic of Medical Sciences. Detailed medical histories, physical examinations and 12-lead electrocardiography (ECG) and a 24-h Holter recording were obtained from 26 familial members. Diagnosis of LQTS was made on the basis of symptoms and corrected QT interval (QTc) on ECG according to the criteria of Schwartz et al. [21].

Pedigree of the LQTS family and their genotypes obtained with HERG microsatellite markers. Squares indicate men, circles women, solid symbols affected individuals, slashed symbols deceased individuals, and half-solid symbols individuals with an uncertain phenotypic status. B Sequence identification of a C to T transition of one HERG allele in an affected individual (both alleles shown), causing replacement of arginine 863 by a stop codon (R863X)
One hundred normal controls without heart or other systemic diseases were recruited based on medical history, physical examination, echocardiography, ECG and serum biochemical analysis. Peripheral blood was collected from all participants.
Haplotype analysis
Genomic DNA was extracted from blood samples by standard procedures. Polymorphic microsatellite markers covering HERG, KCNQ1, SCN5A, KCNE1 and KCNE2 loci were amplified by polymerase chain reaction (PCR) as described (http://www.gdb.org), including D3S1298 and D3S1767 for SCN5A; D7S483, D7S636, D7S1824 and D7S2461 for HERG; D11S1323, D11S2362 and D11S1318 for KCNQ1; and D21S2049 and D21S1435 for KCNE1 and KCNE2. These sequences were based on the data in the genome databases. After PCR, aliquots of 2 μl reaction mixture were denatured and separated on 7% or 10% polyacrylamide gels including 7 M urea. The gels were then silver-stained and dried. Two independent investigators with no knowledge of each individual’s status scored the alleles. Genotypes were grouped into the most likely paternal and maternal haplotypes. Haplotype analysis was performed to confirm the cosegregation of the haplotypes with the disease.
Mutation screening
The DNA fragments encoding the HERG gene were amplified using the primer sets as described [22] on a GeneAmp PCR System 9700 Thermal Cycler (Perkin Elmer, Foster City, Calif.) in the presence of 1 U Taq DNA polymerase, 0.2 mM deoxyribonucleotides, 1.5 mM MgCl2, 100 ng forward and reverse primers and 0.2 μg genomic DNA. PCR fragments were subsequently screened with single strand conformation polymorphism assays (SSCP) according to standard procedures at 4°C and the gels were silver-stained. If abnormal SSCP patterns were found, the same fragments (all family members and the 100 unrelated normal controls) were reamplified and subjected to dye-terminator sequencing using an ABI 377 automatic sequencer. The primer sequences used to detect the HERG gene in exon 10 were 5′ AGTTCTCCGACCACTTCT 3′ and 5′ TCAATGTCACAC AGCAAA 3′.
Construction of wild-type HERG (WT) and R863X HERG
WT HERG cDNA cloned into the pSP64 vector was a kind gift from Dr. Michael Sanguinetti (University of Utah) and was subcloned into pcDNA3.1 (Invitrogen, Carlsbad, Calif.) through the HindIII and BamHI sites. The pEGFP-HERG plasmid was a kind gift from Dr. Thomas V. McDonald (Albert Einstein College of Medicine, N.Y.)
R863X-HERG was constructed by PCR based-mutagenesis. The sequence was amplified using the forward primer 5′-CCCTGTCCTCTC CATGGCCT-3′ and the reverse primer 5′-TGGATCCACAGGTTGAAGGTG-3′ with Pfx DNA polymerase (Gibco, Rockville, Md.). The resultant PCR products were verified by sequencing and subcloned into pcDNA3.1 vector through the HindIII and BamHI sites and the pEGFP-C2 vector through the EcoRI and BamHI sites.
Cell culture and transfection
CHO cells were maintained in MEM-media supplemented with 10% fetal calf serum (Gibco-BRL, Rockville, Md.) and penicillin/streptomycin at 37°C under 5% CO2. Gene transfer was performed using 1–5 μg of Qiagen Midiprep purified plasmid DNA (QIAGEN,Germany). The cells were transfected using LipofectAMINE according to the manufacturer’s instructions (Gibco-BRL). The pcDNA3/GFP plasmid encoding green fluorescent protein was cotransfected with the test plasmid and the target cells were identified by fluorescence microscopy. Patch clamp recordings were conducted 24–48 h after transfection.
Electrophysiologic experiments and data analysis
Whole cell patch clamp recordings were performed using an EPC-9 amplifier (HEKA Elektronik, Lambrecht). The pulse software 8.0 (HEKA Elektronik) was used for the generation of voltage pulse protocols and data acquisition. All experiments were conducted at room temperature. The extracellular solution contained 150 mM NaCl, 1.8 mM CaCl2, 2 mM KCl, 1 mM MgCl2, 5 mM glucose, 10 mM HEPES and 10 mM sucrose, pH 7.4. The pipette solution consisted of 120 mM KCl, 2 mM MgSO4, 5 mM EGTA, 0.5 mM CaCl2, 10 mM HEPES, pH 7.2. Series resistance was actively compensated and was below 5 Mω. The holding potential was −80 mV in all experiments.
The voltage protocols are presented in the figure legends. The current densities were calculated by dividing the maximal peak current by the capacities of the cells tested. Voltage activation data were plotted as peak tail current amplitudes against the test potential values and were fitted to a Boltzmann function, I/I max=1/(1+exp[(V 1/2−V)/k]), where I is the measured tail current, V is the applied membrane voltage,V 1/2 is the voltage at half-maximal activation, and k is the slope factor. Activation, deactivation, and inactivation kinetics were fitted with a single or double exponential function. Data analysis and drawings were performed by using IGOR software (WaveMetrics, Lake Oswego, Ore.) and kaleidagraph 3.5 software (Synergy, Reading, Pa.). All data were expressed as means±SEM. Statistical differences were assessed by Student’s t-test, P<0.05 was considered significant.
Confocal imaging
CHO cells were cultivated as described in the previous section and transiently transfected using LipofectAMINE with pEGFP-HERG or pEGFP-R863X. After 24–48 h, the transfected cells were replated on chambers with glass coverslips and allowed to attach for at least 12 h prior to imaging. The confocal images were obtained using a Leica TCS NT laser scanning microscope (Leica, Bensheim). Image analysis was performed by using Imagepro and Photoshop 7.0 software.
Results
Clinical presentation
In this family, six members were clinically diagnosed as LQTS. The proband was a 57-year-old woman, who experienced her first syncope with seizure at age 27, with ECG documented torsade de pointes ventricular tachycardia (Tdp). The ECG revealed a prolonged QTc interval (580 ms at rest and 480 ms after exercise) and a notched T wave or a T-U-wave complex in several leads. The Tdp attacks were easily and repeatedly provoked by startling or emotional stress. The proband was treated with atenolol or propranolol but remained symptomatic.
Molecular genetics
Microsatellite markers for KCNQ1, SCN5A, KCNE1 and KCNE2 were used to identify the affected individuals by haplotype analysis.The most likely haplotypes obtained with these HERG microsatellite markers indicated that the haplotype 2-2-3-4 cosegregated with the disease, suggesting that the disease gene in the family is linked to this locus Fig. 1A. The family was then screened for mutations by PCR-SSCP, with intronic PCR primers covering the coding regions of the HERG gene. The resultant PCR products generated from HERG exon 10 had an aberrant pattern in the patient, and were not observed in DNA samples from 100 control subjects. DNA sequencing revealed a heterozygous C to T transversion at nucleotide position 2770 of the HERG gene (GenBank accession U04270), causing the substitution of a conserved arginine residue at position 863 for a stop codon (R863X) with a deletion of 296 amino acids in the HERG C-terminus (Fig. 1B). All clinically affected family members (diagnosed by ECG) were found to be heterozygous for R863X. The proband’s niece (III-3) had a normal QTc, but carried this mutation. The mutation was not identified in other unaffected family members nor in 100 normal controls.

Steady state activation kinetics of WT and R863X/WT. A Typical current traces in CHO cells transfected with the constructs indicated. The currents were elicited with 30 sec pulses to potentials ranging from −70 to 30 mV and the tail currents were measured on return of the voltage to −40 mV. B The relationship of tail current density and test pulses. C I-V relationships of the tail currents in CHO cells transfected with WT and R863X/WT. Amplitudes of tail currents were normalized and plotted as a function of the test potential and were fitted to a Boltzmann function
R863X HERG alone could not form functional channels
To investigate the functional consequence of this mutation, we measured the whole cell K+ currents from CHO cells transiently transfected with either WT or R863X HERG plasmids. Typical whole cell current traces are seen in Fig. 2A. The mutation could not produce voltage-gated K+ current when expressed alone. This indicates that R863X HERG alone could not form functional channels.
Coexpression of WT with R863X HERG reduced the current densities of HERG channels
To assess the interaction between WT and the mutant proteins, we coexpressed equal amounts (1.5 μg) of WT and R863X HERG in CHO cells and characterized the currents, making quantitative analysis feasible. Typical whole cell current traces are seen in Fig. 2A. The tail currents were measured at −40 mV after a depolarizing pulse from −70 mV to +30 mV and the peak tail current density was 52.47±6.73 pA/pF (n=25) for WT, and 35.44±3.16 pA/pF (n=24) for R863X/WT (P<0.01) (Fig. 2B). Coexpression of WT and R863X HERG reduced HERG current densities about 30%. The apparent degree of HERG current reduction, however, indicated that co-expression of WT with R863X HERG did not exert a dominant negative effect on WT HERG expression. In addition, the maximal tail current amplitudes were obtained at the peak of the third pulse in the inactivation protocol, when almost all of the channels were active (Fig. 3). The results also demonstrated that the current densities of R863X/WT HERG were lower than that of WT (WT, 357±78 pA/pF, n=8; R863X/WT 256±82 pA/pF, n=8, P<0.05).

Time courses of inactivation of expressed currents in CHO cells transfected with WT or R863X/WT. The inset illustrates the voltage protocol. A Representative current recordings were those in CHO cells transfected with WT or R863X/WT. Inactivation time constants (τ) were measured by fitting inactivating currents during test pulses at each potential with a single-exponential function. B τvalues representing inactivation time constants for expressed currents in CHO cells transfected with WT and R863X/WT were plotted as a function of test potential (VT). *P<0.05 for time constant between R863X/WT and WT
Coexpression of WT with R863X HERG also modified the gating property of HERG channels
Figure 2C represents the normalized I-V relationships for amplitudes of tail currents for WT and R863X/WT. The voltage at which the current was half-activated (V 1/2)for R863X/WT was not much different from that for WT, but the slope factor k for R863X/WT (7.29±1.1) was increased slightly compared with that for WT (5.97±0.65, P<0.05).
To analyze the deactivation time course accurately long hyperpolarizing test pulses were applied after a depolarizing conditioning pulse. Deactivating currents during test pulses could be fitted to a double exponential function. At all test potentials, the fast and slow time constants for R863X/WT were not different from that for WT (data not shown).
The inactivation time course of expressed currents was analyzed by applying brief hyperpolarizing pulses to allow the HERG channel to recover from inactivation after an initial long depolarizing pulse, and then depolarizing test pulses were applied to record inactivating currents. The time course of inactivating currents could be fitted by a single-exponential function. The inactivation time constants of R863X/WT channels (10.97±1.28 ms at −50 mV, and 7.12±0.47 ms at 10 mV, n=10) were decreased slightly compared with WT channels (13.88±1.52 ms at −50 mV, P=0.001 and 8.08±0.40 ms at 10 mV, n=10, P<0.01) (Fig. 3). The results revealed that R863X HERG could accelerate the channel inactivation when coexpressed with WT.
R863X HERG does not translocate to the plasma membrane
Several LQT2 mutations have been found to cause reduced I kr due to defective transport of the mutant protein to the plasma membrane or through enhanced degradation of the protein. To examine whether the R863X mutant proteins could be translocated to the plasma membrane, we visualized the location of HERG protein in CHO cells using GFP fusion proteins for both WT and R863X HERG. The GFP signal of the R863X mutant protein was located mainly in the cytoplasm and was not found in the cell plasma membrane, whereas the GFP signal of the WT protein was distributed both in the cytoplasm and in the plasma membrane (Fig. 4), indicating that the R863X protein could not be effectively translocated to the plasma membrane.

Localization of WT and R863X mutant HERG in living cells by GFP fluorescence. A The GFP signal of WT protein was located in the cytoplasm and the plasma membrane. The unstained region within the cell is the nucleus. B The GFP signal of R863X mutant protein was located in the cytoplasm, but not in the plasma membrane
Discussion
In this study, a novel mutation, R863X, in the HERG C-terminus was identified in a Chinese LQTS family. Carriers of this mutation have a high risk of developing lethal arrhythmias. In comparsion with other LQT2 C terminus mutations (Fig. 5), R863X exhibited unique electrophysiological properties. R863X HERG alone failed to form functional HERG channels, consistent with other reported C-terminus mutations [23, 24, 25]. However, coexpression of R863X HERG with WT reduced the current densities and accelerated the inactivation of WT-HERG channels, which was not the case for mutants in the cyclic nucleotide binding domain (CNBD) and Y667X. The latter has no influence on the current amplitudes and the properties of HERG channels when coexpressed with WT, whereas coexpression of CNBD mutants with WT has not been found to alter current amplitudes significantly, butchanged the half voltage of activation (V 1/2). The R863X mutation is positioned just downstream of the CNBD region with a deletion of 296 amino acids (residues 863–1159) in the HERG C-terminus. Interestingly, it has been reported that the residues 863–1159 in the HERG C-terminus may contain a domain involving the inactivation process of the channel.

The C-terminal mutations of the HERG protein
Failure of cell surface expression has now been identified as a mechanism of ion channel dysfunction in several human familial arrhythmias, including LQTS and Brugada syndrome [11, 12, 13, 26]. Many of the mutant HERG proteins fail to generate HERG current because of trafficking problems [11, 12, 13]. Aydar and Palmer [27] have reported that HERG proteins with a C-terminal truncation of 311 or more amino acid residues cannot form functional channels. Moreover, Akhavan et al. [28] have recently identified a region between residues 860–899 that is critical for trafficking, and have shown that truncations or deletion of residues 860–899 result in a decreased expression level and an absence of the mature glycosylated form of the HERG protein. Our study revealed that the mutant R863X alone failed to form functional channels due to ineffective trafficking to the plasma membrane. These results demonstrated that C-terminus of the HERG protein may play an important role in HERG trafficking and channel maturation.
Coexpression of R863X HERG with WT reduced the current densities of HERG channels, and speeded up their inactivation slightly. This faster inactivation contributed to a lower current density. However, the maximal current densities of R863X/WT channels were lower than those of WT channels when all of the channels were open, which suggested that R863X may also interfere with WT protein trafficking and/or accelerate its degradation. The decrease in current density by R863X may result in the longer QT intervals documented in patients with this mutation.
A recent study has showed that the 14-3-3 protein family, interact with HERG channels and alter the effect of β-adrenergic signaling upon HERG activity [29]. This may provide a mechanism for plasticity in cardiac electrophysiological response to stress. Members of the 14-3-3 family are highly conserved and are present in all eukaryotic organisms. HERG associates with 14-3-3ε and potentiates the cAMP/PKA effect upon HERG. 14-3-3 accelerates and enhances HERG activation, an effect that requires phosphorylation of HERG at S283 and S1137. The mutant R863X lacks the S1137 PKA site. Therefore, the R863X/WT HERG channel can not efficiently interact with 14-3-3, which may weaken cAMP/PKA effects on HERG, leading to an increased risk for fatal cardiac arrhythmias in the face of stress. This could be the mechanism for the clinical events seen in this family.
In summary, we have identified a novel mutation, R863X, in the HERG gene. The mutation results in a deletion of 296 amino acids. The R863X mutant can not translocate normally to the cell surface. Coexpression of WT and R863X HERG showed reduced current densities and faster inactivation. These may contribute to a prolongation of QT intervals and thus to LQT2. The results indicate that these residues are necessary for the expression of functional HERG channels.
References
Priori SG, Barhanin J, Hauer RN, Haverkamp W, Jongsma HJ, Kleber AG, McKenna WJ, Roden DM, Rudy Y, Schwartz K, Schwartz PJ, Towbin JA, Wilde AM (1999) Genetic and molecular basis of cardiac arrhythmias: impact on clinical management parts I and II. Circulation 99:518–528
Keating MT, Sanguinetti MC (2001) Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104:569–580
Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V (2003) Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421:634–639
Sanguinetti MC, Jiang C, Curran ME, Keating MT (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81:299–307
Trudeau MC, Warmke JW, Ganetzky B, Robertson GA (1995) HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269:92–95
Kupershmidt S, Snyders DJ, Raes A, Roden DM (1998) A K+ channel splice variant common in human heart lacks a C-terminal domain required for expression of rapidly activating delayed rectifier current. J Biol Chem 273:27231–27235
London B, Trudeau MC, Newton KP, Beyer AK, Copeland NG, Gilbert DJ, Jenkins NA, Satler NA, Robertson GA (1997) Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K+ current. Circ Res 81:870–878
Lees-Miller JP, Kondo C, Wang L, Duff HJ (1997) Electrophysiological characterization of an alternatively processed ERG K+ channel in mouse and human hearts. Circ Res 81:719–726
Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SAN (1999) MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97:175–187
Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT (2000) Spectrum of mutations in long-QT syndrome genes KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102:1178–1185
Paulussen A, Raes A, Matthijs G, Snyders DJ, Cohen N, Aerssens J (2002) A novel mutation (T65P) in the PAS domain of the human potassium channel HERG results in the long QT syndrome by trafficking deficiency. J Biol Chem 277:48610–48616
Furutani M, Trudeau MC, Hagiwara N, Seki A, Gong Q, Zhou Z, Imamura S, Nagashima H, Kasanuki H, Takao A, Momma K, January CT, Robertson GA, Matsuoka R (1999) Novel mechanism associated with an inherited cardiac arrhythmia: defective protein trafficking by the mutant HERG (G601S) potassium channel. Circulation 99:2290–2294
Zhou Z, Gong Q, Epstein ML, January CT (1998) HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem 273:21061–21066
Nakajima T, Kurabayashi M, Ohyama Y, Kaneko Y, Furukawa T, Itoh T, Taniguchi Y, Tanaka T, Nakamura Y, Hiraoka M, Nagai R (2000) Characterization of S818L mutation in HERG C-terminus in LQT2. Modification of activation-deactivation gating properties. FEBS Lett 481:197–203
Huang FD, Chen J, Lin M, Keating MT, Sanguinetti MC (2001) Long-QT syndrome-associated missense mutations in the pore helix of the HERG potassium channel. Circulation 104:1071–1075
Nakajima T, Furukawa T, Hirano Y, Tanaka T, Sakurada H, Takahashi T, Nagai R, Itoh T, Katayama Y, Nakamura Y, Hiraoka M (1999) Voltage-shift of the current activation in HERG S4 mutation (R534C) in LQT2. Cardiovasc Res 44:283–293
Lees-Miller JP, Duan Y, Teng GQ, Thorstad K, Duff HJ (2000) Novel gain-of-function mechanism in K(+) channel-related long-QT syndrome: altered gating and selectivity in the HERG1 N629D mutant. Circ Res 86:507–513
Nakajima T, Furukawa T, Tanaka T, Katayama Y, Nagai R, Nakamura Y, Hiraoka M (1998) Novel mechanism of HERG current suppression in LQT2: shift in voltage dependence of HERG inactivation. Circ Res 83:415–422
Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC (1999) Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J Biol Chem 274:10113–10118
Berthet M, Denjoy I, Donger C, Demay L, Hammoude H, Klug D, Schulze-Bahr E, Richard P, Funke H, Schwartz K, Coumel P, Hainque B, Guicheney P (1999) C-terminal HERG mutations: the role of hypokalemia and a KCNQ1-associated mutation in cardiac event occurrence. Circulation 99:1464–1470
Schwartz PJ, Moss AJ, Vincent GM, Crampton RS (1993) Diagnostic criteria for the long QT syndrome: an update. Circulation 88:782–784
Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH, Keating MT (1998) Genomic structure of three long QT syndrome genes: KCNQ1, HERG and KCNE1. Genomics 51:86–97
Nakajima T, Kurabayashi M, Ohyama Y, Kaneko Y, Furukawa T, Itoh T, Taniguchi Y, Tanaka T, Nakamura Y, Hiraoka M, Nagai R (2000) Characterization of S818L mutation in HERG C-terminus in LQT2. Modification of activation-deactivation gating properties. FEBS Lett 481:197–203
Cui J, Kagan A, Qin D, Mathew J, Melman YF, McDonald TV (2001) Analysis of the cyclic nucleotide binding domain of the HERG potassium channel and interactions with KCNE2. J Biol Chem 276:17244–17251
Paulussen A, Yang P, Pangalos M, Verhasselt P, Marrannes R, Verfaille C, Vandenberk I, Crabbe R, Konings F, Luyten W, Armstrong M (2000) Analysis of the human KCNH2(HERG) gene: identification and characterization of a novel mutation Y667X associated with long QT syndrome and a non-pathological 9 bp insertion. Hum Mutat 15:483
Baroudi G, Pouliot V, Denjoy I, Guicheney, P (2001) Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G). Circ Res 88:E78–83
Aydar E, Palmer C (2001) Functional characterization of the C-terminus of the human ether-a-go-go-related gene K+ channel (HERG). J Physiol 534:1–14
Akhavan A, Atanasiu R, Shrier A (2003) Identification of a C-terminal segment involved in maturation and stability of HERG potassium channel. J Biol Chem (in press)
Kagan A, Melman YF, Krumerman A, McDonald TV (2002) 14-3-3 amplifies and prolongs adrenergic stimulation of HERG K+ channel activity. EMBO J 21:1889–1898
Acknowledgements
We are grateful to the family members because this study would be impossible without their enthusiastic participation, and to Professors Michael C.Sanguinetti and Thomas V. McDonald for their gifts of plasmids, and Drs Jielin Pu and Dirk Isbrandt for their advice. This study was financially supported by the International Cooperation Department, the Ministry of Science and Technology, China (to R.H.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Teng, S., Ma, L., Dong, Y. et al. Clinical and electrophysiological characterization of a novel mutation R863X in HERG C-terminus associated with long QT syndrome. J Mol Med 82, 189–196 (2004). https://doi.org/10.1007/s00109-003-0504-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-003-0504-1