
Abstract
Sequence-specific nucleases (SSNs), such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspersed short palindromic repeats (CRISPR)/CRISPR-associated protein 9 nuclease (Cas9) system, are powerful tools for understanding gene function and for developing novel traits in plants. In plant species for which transformation and regeneration systems using protoplasts are not yet established, direct delivery to nuclei of SSNs either in the form of RNA or protein is difficult. Thus, Agrobacterium-mediated transformation of SSN expression constructs in cultured cells is a practical means of delivering targeted mutagenesis in some plant species including rice. Because targeted mutagenesis occurs stochastically in transgenic cells and SSN-mediated targeted mutagenesis often leads to no selectable phenotype, identification of highly mutated cell lines is a critical step in obtaining regenerated plants with desired mutations.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key word
1 Introduction
The TALENs and CRISPR/Cas9 systems both rely on endonucleases that initiate DNA double-strand breaks (DSBs) at virtually any genomic target sequence and both are used for targeted mutagenesis [1]. While both technologies are popular, it is better to select one or the other as appropriate depending on the purpose. The TALENs system has the advantage of little limitation in target sequence because TALEs contain multiple 33- to 35-amino acid repeat domains that each recognize a single base pair (Fig. 1a) [2, 3]. On the other hand, when Streptococcus pyogenes Cas9 (SpCas9) and guide RNAs (gRNAs) are used, CRISPR/Cas9 targets must immediately precede an NGG site protospacer adjacent motif (PAM) sequence recognized by Cas9 (Fig. 1b) [4–7]; it is usually not difficult to locate NGG sites for knockouts, but this constraint sometimes causes problems for other applications. Nevertheless, CRISPR/Cas9 has the advantage of being able to knock out multiple genes at the same time because the target specificity of CRISPR/Cas9 relies not on protein/DNA recognition but rather on ribonucleotide complex formation; thus, mutations can be introduced in multiple genes when multiple guide RNAs (gRNAs) determining different target sequences are expressed together with Cas9. In contrast to TALENs, which rely on protein domains to confer DNA-binding specificity, Cas9 forms a complex with a small guide RNA (gRNA) that directs the enzyme to its DNA target via Watson-Crick base pairing. Consequently, the system is simple and fast to design for each application and requires only the production of a short oligo RNA to direct DNA binding to the target locus.

Schematic representation of TALENs- and CRISPR/Cas9 -mediated DNA cleavage. (a) Schematic diagram of TALENs binding to target DNA. The TALEN consists of N- and C-terminal regions containing nuclear localization signals, a central DNA-binding domain (TAL effector repeats) with typically 34 amino acid repeats, and a DNA cleavage domain with the restriction enzyme Fok I. The amino acid sequences of each TAL effector repeat are highly conserved, but the 12th and 13th amino acids are variable and recognize a single base pair (NI = A, NG = T, NN = G or A and HD = C). (b) Schematic diagram of Cas9 and gRNA complex binding to target DNA. A limitation of the CRISPR/Cas9 system is the 5′-NGG PAM sequence on the genomic DNA. The 20-bp target sequence preceding a PAM sequence must exist on the gRNA. Note that the PAM sequence required for target recognition by Cas9 is never present as part of the gRNA itself
2 Materials
2.1 Vector Construction
-
1.
Several types of assembly kit for TALENs (https://www.addgene.org/talen/) and various CRISPR/Cas9 plasmids for use in plants are available from Addgene (https://www.addgene.org/crispr/plant/). When pU6gRNA and pZH_gYSA_MMCas9 [8], (see Note 1 ) are used for construction of Cas9, gRNA , selection marker all-in-one vector, materials 2–5 are needed.
-
2.
Restriction enzymes: Bbs I, Asc I, Pac I.
-
3.
Gel Extraction Kit (e.g., QIAquick, Qiagen Germany).
-
4.
Ligation Kit.
-
5.
E. coli DH5α competent cells.
2.2 Agrobacterium-Mediated Transformation
2.2.1 Plant and Agrobacterium Materials
-
1.
Mature rice seeds (Oryza sativa L. cv. Nipponbare).
-
2.
Agrobacterium tumefaciens strain EHA105 [9].
2.2.2 Media for Agrobacterium-Mediated Transformation
Autoclaved media are poured as 50 mL in each dish (9 cm in diameter, 2 cm in depth). All media are stored at 4 °C. Media are incubated at room temperature and the lids are opened to dry water drops on the surface of the medium in a clean bench just before use.
-
2.
2,4-Dichlorophenoxy acetic acid (2,4-D), naphtalene acetic acid (NAA), kinetin, acetosyringone, meropenem, hygromycin B, G418: See ref. 10 and Nishizawa-Yokoi et al., Chapter 10.
2.3 Selection and Regeneration of TALENs or Cas9- and gRNA -Transformed Calli
2.3.1 Media for Selection of Transgenic Calli
-
1.
Selection medium (1L): After autoclaving N6D medium, add 1 mL of 25 mg/mL meropenem and 50 mg/L hygromycin B.
-
2.
Regeneration (ReIII) medium (1L): After autoclaving, add 1 mL of 12.5 mg/mL meropenem and 50 mg/L hygromycin B.
-
3.
Hormone-free (HF) medium (1L): After autoclaving, add 1 mL of 12.5 mg/mL meropenem.
2.4 Detection of Mutations
2.4.1 DNA Extraction
-
1.
Agencourt chloropure (Beckman Coulter, USA).
2.4.2 Polymerase Chain Reaction
-
1.
For PCR amplification, 2 μL of 5× PCR reaction buffer , 0.8 μL of 2.5 mM dNPTs, 0.25 μL of each 10 μM primer, 1 μL of 1/100 diluted genomic DNA, and 0.25 units of Taq DNA polymerase (e.g., PrimeSTAR GXL DNA polymerase, Takara Bio Inc.) are mixed with distilled water to give a final volume of 10 μL.
2.4.3 Cleaved Amplified Polymeric Sequences (CAPS) Analysis
-
1.
PCR product (500 bp to 1 kb).
-
2.
Restriction enzymes.
-
3.
1 or 2 % agarose gel.
-
4.
Digital gel imaging system (e.g., Chemi Doc imaging system, BioRad, USA).
2.4.4 Analysis of Mutation Patterns in Calli by Sequencing
-
1.
Gel Extraction Kit (e.g., QIAquick, Qiagen).
-
2.
PCR Cloning Kit (e.g., Zero Blunt TOPO, Thermo Fisher Scientific, USA).
-
3.
E. coli DH5α Competent Cells.
-
4.
BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific).
-
5.
Sequencer (e.g., ABI PRISM 2100 Genetic Analyzer, Applied Biosystems, USA).
2.4.5 Guide-It Resolvase Assay
-
1.
PCR product (500 bp to 1 kb).
-
2.
Guide-it Mutation Detection Kit (Takara Bio Inc.).
2.4.6 Heteroduplex Mobility Shift Assay (HMA)
-
1.
PCR product (100–200 bp).
-
2.
Polyacrylamide gel: 20 % (w/v) polyacrylamide (a mixture of 99 % acrylamide and 1 % methylenebisacrylamide) in 1× TBE, 0.5 % (w/v) ammonium persulfate (APS), 0.01 % (v/v) N,N,N′,N′ Tetramethylethylene diamine (TEMED).
-
3.
30 % Acrylamide: for 100 ml, 29.7 g of acrylamide, 0.3 g of methylenebisacrylamide.
-
4.
10× TBE: for 1 L, combine 108 g of Tris base, 55 g of boric acid, 40 mL of 0.5 % EDTA (pH 8.0). Autoclave for 20 min.
3 Methods
3.1 Selecting Target Sequence
3.1.1 TALENs
In nature, the target sites of TAL effectors derived from Xanthomonas start mostly from T at the 5′-end. Thus, a T at the 5′-end is required to select the target site of the TAL effector DNA-binding domain in many cases. The TAL effector repeat arrays of the DNA-binding domain can be designed easily using software such as TAL effector Nucleotide Targeter (TALE-NT 2.0, https://tale-nt.cac.cornell.edu/, 11) and Scoring Algorithm for Predicting TALE(N) Activity (SAPTA, http://bao.rice.edu/Research/BioinformaticTools/TAL_targeter.html, 12).
3.1.2 CRISPR/Cas9
When Streptococcus pyogenes Cas9 (SpCas9) is used, a requirement for the guide sequence is the 20-bp target sequence preceding a 5′-NGG protospacer adjacent motif (PAM) sequence recognized by Cas9 (Fig. 1b). SpCas9 cuts 3-nt upstream of the PAM site. If the expected cleavage site is on the restriction enzyme recognition site, mutation can be detected by CAPS analysis. If the Guide-it resolvase assay or HMA is used, restriction enzyme sites do not need to be considered.
3.2 Vector Construction
3.2.1 TALENs
Several platforms, including Golden Gate assembly technology [13], REAL [14] and FLASH [15], have been developed for the construction of TAL effector repeat arrays. Among them, Golden Gate assembly technology has been used widely and validated in multiple organisms. Using this system, TAL effector repeat arrays are constructed by digestion with a type IIS restriction enzyme and ligation of each TAL repeat and are subsequently cloned into a vector carrying the TAL backbone and catalytic domain of the Fok I endonuclease. For induction of TALEN-mediated target mutagenesis in rice, expression vectors carrying expression cassettes of TALEN pairs driven by mono- or polycistronic expression constructs (for details, see ref. 16).
3.2.2 CRISPR/Cas9
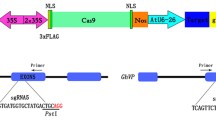
Details of the construction method differ depending on the vectors used for expressing Cas9 and gRNA . Examples of vector construction strategies used in [8] are as follows: only the 20-nt target sequence of the gRNA needs to be replaced for targeting different genomic sites while the scaffold sequence of the gRNA remains the same. Therefore, the gRNA cloning vector allows the target sequence to be swapped easily. For example, pU6gRNA [8] contains two Bbs I sites in place of the gRNA target sequence such that digestion leaves overhangs complementary to the annealed oligo overhang (Fig. 2a). Because the cleavage site of Bbs I, 5′-GAAGAC(N)2/-3′ is outside of its recognition sequence, we can avoid the addition of unnecessary sequence in this step. Once a target 20 bp is determined, forward and reverse oligos consisting of the 20-nt target site with 5′ and 3′ overhangs complementary to the digested gRNA cloning vector are annealed and cloned into the gRNA cloning vector. Because the OsU6 RNA polymerase III promoter prefers to start transcripts with a guanine (G) nucleotide, a G is included immediately 5′ of the target for inserts cloned in the pU6gRNA vector (Fig. 2a).

Strategy for CRISPR/Cas9 vector construction. (a) Schematic representation of cloning the target sequence into a plasmid containing the OsU6 promoter and the gRNA scaffold. The annealed target sequence oligos contain overhangs to allow ligation into the pair of Bbs I sites in the sgRNA vector, pU6gRNA. (b) Schematic representation of cloning the gRNA expression construct into a binary vector containing Cas9 and HPT expression constructs. I-Sce I or combination of Asc I and Pac I can be used for this cloning
For stable rice transformation , the gRNA expression construct OsU6pro::gRNA::polyT completed on pU6gRNA must be placed in a binary vector containing the Cas9 expression construct and selection marker. A restriction enzyme reaction (I-Sce I only or the combination of Asc I and Pac I) can be used to replace the gRNA expression construct when pZH_gRNA_MMCas9 (Fig. 2b, [8]) is used as an all-in-one binary vector expressing Cas9, gRNA and the selection marker.
Details:
-
1.
Combine 1 μL of each oligo DNA (100 μM) and 48 μL of ddH2O. Boil 5 min and leave at room temperature for 20 min to anneal the oligos.
-
2.
Digest the gRNA cloning plasmid, pU6gRNA as follows: combine 2 μg of pU6gRNA plasmid DNA, 5 μL of 10× NEB cut smart buffer, 1 μL of Bbs I enzyme (5 unit/μL) and ddH2O to 50 μL. Incubate at 37 °C for 2–16 h. Run the digested products on 1 % (w/v) agarose gel in TAE buffer and purify the linearized vector using a QIAquick PCR purification kit.
-
3.
Ligate the annealed gRNA oligos into the digestion vector using a Rapid DNA ligation kit as follows: combine 4 μL of digested pU6gRNA (10 ng/μL), 4 μL of annealed oligo, 2 μL of 5× DNA dilution buffer, 10 μL of 2× T4 DNA ligation buffer, and 1 μL of T4 DNA ligase. Incubate at room temperature for 5 min and transform the ligation reaction into competent E. coli cells according to the manufacturer’s instructions.
-
4.
Isolate plasmid DNA and sequence the plasmid using primer OsU6-2F (5′-TGCTGGAATTGCCCTTGGATCATGAACCAA-3′) to verify that the clones harbor the corrected gRNA.
-
5.
Digest the all-in-one Cas9, gRNA , and selection marker vector (pZH_gYSA_MMCas9) and the completed gRNA cloning vector (from step 4) with Asc I and Pac I and purify the resulting 17,257 bp (pZH_gYSA_MMCas9) and 486 bp (pU6gRNA) fragments.
-
6.
Ligate Asc I-, Pac I-digested all-in-one vector and gRNA expression construct (OsU6pro::gRNA) (from step 6) to complete the Cas9, gRNA, selection marker all-in-one binary vector.
3.3 Agrobacterium-Mediated Transformation Using Primary Callus of Rice
-
1.
Agrobacterium strain EHA105 harboring an all-in-one binary vector (from Subheading 3.2.2) containing Cas9, gRNA and selection marker expression cassettes is used for transformation. Transformation and selection steps basically follow the method published by [10].
-
2.
Transgenic calli grown on selection medium are transferred onto regeneration medium (ReIII) containing 25 mg/L meropenem and the appropriate concentration of selection agents, and grown at 28 °C under constant light for 10–14 days (see Note 2 ).
-
3.
Shoots arising from callus on ReIII medium are transferred to HF medium containing 25 mg/L meropenem and grown at 28 °C under constant light for 2 weeks.
3.4 Detection of Mutation in Rice Calli (See Note 3 )
3.4.1 DNA Isolation
Extract DNA from transgenic calli using Agencourt chloropure according to the manufacturer’s instructions.
3.4.2 Amplification of the Targeted Sequence
Design PCR primers that should amplify a 0.5–2 kb fragment including the gRNA -targeted sequence and set up the following PCR. When CAPS assay or surveyor nuclease is to be used for detecting mutations, the targeted sequence is better to be located near the center of the PCR products. This facilitates separation of mutated and non-mutated PCR products on agarose gels (see Note 4 ).
Genomic DNA | 1 μL |
5× PCR reaction buffer | 2 μL |
2.5 mM dNTPs | 0.8 μL |
10 μM forward primer | 0.25 μL |
10 μM reverse primer | 0.25 μL |
ddH2O | 5.45 μL |
Taq DNA polymerase (e.g., PrimeStar GXL) | 0.25 μL |
Total | 10 μL |
PCR conditions
Cycle 1, 95 °C for 2 min; Cycles 2, 95 °C for 15 s and 68 °C for 1 min, repeat 35 times; Cycle 3, 68 °C for 7 min.
3.4.3 CAPS Assay
When an appropriate restriction enzyme recognition sequence is located on the expected cleavage site, PCR and subsequent restriction enzyme reactions can be used to detect the mutation of interest (Fig. 3). Use the PCR product from Subheading 3.4.2 to set up the following restriction enzyme reaction.

Example of CAPS analysis using transgenic calli of Cas9 and gRNA. First, genomic DNAs from Cas9, gRNA-transformed calli, or regenerated plants are amplified by PCR. PCR products are then digested with restriction enzyme, which recognizes the wild-type target sequence. Mutated DNAs are resistant to the restriction enzyme reaction because of loss of the restriction enzyme recognition sequence and are revealed as uncleaved bands (indicated by black arrowhead) in agarose gels. Mutation frequency can be measured from the relative intensity of cleaved and uncleaved bands
PCR product | 5 μL |
10× buffer | 2 μL |
ddH2O | 12.5 μL |
Restriction enzyme | 0.5 μL |
Total | 20 μL |
Mix the reaction well and spin down. Incubate the reaction at the appropriate temperature for 2 h. Run the digested DNA on an agarose gel. Uncleaved bands detected in Cas9-, gRNA -transformed cells are proof of mutation.
3.4.4 Estimation of the Precise Mutation Frequency
An approximate mutation frequency can be estimated by measuring the intensity of the PCR amplicon and cleaved bands with gel quantification software.
Mutation (%) = A/(A + B + C) × 100 (A, intensity of the non-digested PCR product; B and C, intensity of the digested PCR product).
3.4.5 Analysis of Mutation Patterns in Calli by Sequencing
Purify the undigested bands using a gel purification kit according to the manufacturer’s instructions and clone these fragments into pENTR-D TOPO using a Zero Blunt TOPO PCR cloning kit. Identify the mutations by sequencing of plasmid DNA isolated from overnight cultures of single colonies.
3.5 Guide-It Resolvase Assay
This mutation detection method is based on a mismatch-specific DNA endonuclease (Fig. 4). All steps are performed according to the manufacturer’s instructions provided in the Guide-it Mutation Detection Kit.

Schematic representation of Guide-it resolvase assay. First, genomic DNAs from Cas9-, gRNA -transformed calli, or regenerated plants are amplified by PCR. PCR products are then denatured and re-annealed using a thermal cycler. Guide-it resolvase digests heteroduplex DNA at mismatches and an extrahelical loop is formed by single or multiple nucleotides
3.6 Heteroduplex Mobility Shift Assay (HMA)
HMA is based on the denaturing and annealing of PCR-amplified nucleotide strands that are not fully complementary and therefore generate homo- and hetero-duplexes. Heteroduplexes can be separated from homoduplexes by polyacrylamide gel electrophoresis because heteroduplex migrates more slowly due to an opened single-strand configuration surrounding the mismatched region (Fig. 5) (for details, see ref. 17). PCR amplifications (100–200 bp) are mixed with loading dye and electrophoresed on 20 % acrylamide gels in 1× TBE running buffer at 200 V for 1 h. After electrophoresis, gels are stained with ethidium bromide and images captured using a gel imaging system.

Schematic representation of HMA. First, genomic DNAs from Cas9-, gRNA -transformed calli or regenerated plants are amplified by PCR. PCR products are then denatured and re-annealed using a thermal cycler. Heteroduplex and homoduplex forms are separated by polyacrylamide gel electrophoresis
4 Notes
-
1.
These plasmids are not deposited to Addgene yet but available to academic investigators for non-commercial research purposes. Please contact to Seiichi Toki (stoki@affrc.go.jp) or Masaki Endo (mendo@affrc.go.jp) for the request .
-
2.
G418 often inhibits regeneration. Thus, it is better to eliminate G418 from ReIII medium when transgenic calli are fully isolated on selection medium.
-
3.
Because mutation efficiency sometimes differs between independent transgenic callus clones due to different expression levels of Cas9 and gRNA , we recommend to check mutation frequency in the clone before going to the regeneration step.
-
4.
Use wild-type genomic DNA as a negative control.
References
Gaj T, Gersbach CA, Barbas CF III (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405
Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL (2012) The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335:716–719
Deng D, Yan C, Pan X, Mahfouz M, Wang J, Zhu JK, Shi Y, Yan N (2012) Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335:720–723
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Garneau JE, Dupuis MÈ, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71
Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V (2011) The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39:9275–9282
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67
Mikami M, Toki S, Endo M (2015) Comparison of CRISPR/Cas9 expression constructs for efficient targeted mutagenesis in rice. Plant Mol Biol 88:561–572
Hood EE, Gelvin SB, Melchers LS, Hoekema A (1993) New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res 2:208–218
Saika H, Onodera H, Toki S (2012) Visual selection in rice: a strategy for the efficient identification of transgenic calli accumulating transgene products. Methods Mol Biol 847:67–74
Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, VanDyk JK, Bogdanove AJ (2012) TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res 40(Web Server issue):W117–W122
Lin Y, Fine EJ, Zheng Z, Antico CJ, Voit RA, Porteus MH, Cradick TJ, Bao G (2014) SAPTA: a new design tool for improving TALE nuclease activity. Nucleic Acids Res 42:e47
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39:e82
Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK, Yeh JR (2011) Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol 29:697–698
Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK (2012) FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30:460–465
Cermak T, Starker CG, Voytas DF (2015) Efficient design and assembly of custom TALENs using the Golden Gate platform. Methods Mol Biol 1239:133–159
Ota S, Hisano Y, Muraki M, Hoshijima K, Dahlem TJ, Grunwald DJ, Okada Y, Kawahara A (2013) Efficient identification of TALEN-mediated genome modifications using heteroduplex mobility assays. Genes Cells 18:450–458
Acknowledgments
We thank M. Mikami and K. Abe for technical assistance. This research was supported by a grant from the Ministry of Agriculture, Forestry and Fisheries of Japan (Genomics for Agricultural Innovation PGE1001), the Council for Science, Technology and Innovation (CSTI), Cross-ministerial Strategic Innovation Promotion Program (SIP), and the NIAS Strategic Research Fund.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Endo, M., Nishizawa-Yokoi, A., Toki, S. (2016). Targeted Mutagenesis in Rice Using TALENs and the CRISPR/Cas9 System. In: Murata, M. (eds) Chromosome and Genomic Engineering in Plants. Methods in Molecular Biology, vol 1469. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-4931-1_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-4931-1_9
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-4929-8
Online ISBN: 978-1-4939-4931-1
eBook Packages: Springer Protocols