
Abstract
Fungi are known for their ability to produce a vast array of valuable chemical compounds, which are utilized in biotechnology and the pharmaceutical industry. In this chapter, we outline recent advances in the development of genomic and bioinformatics resources for the discovery of genes involved in natural product biosynthesis, focusing on the major natural product classes: polyketides, nonribosomal peptides, and terpenes. These groups of compounds are all produced from simple, scaffold molecules that are further modified to create a diverse portfolio of bioactive products. Biosynthetic clusters of genes responsible for the production of these compounds are prolific in fungal systems and typically include key scaffold-producing enzymes and a multitude of modification enzymes. This chapter highlights how the recent explosion in bioinformatics tools and genomic resources can be applied to mining fungal genomes for the novel natural product biosynthetic pathways, providing key examples of well-studied fungal biosynthetic gene clusters.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
The fungal kingdom represents a vast and largely untapped resource for the discovery of new natural products and their biosynthetic pathways . It is estimated that the number of fungal species (~ 1.5 million species) exceeds that of land plants by a ratio of 10:1. Only a fraction of this diversity (~ 100,000 species) has been described. Ascomycota (filamentous fungi) and Basidiomycota (including mushroom forming fungi) make up the vast majority of this diversity [1, 2]. However, despite this remarkable species diversity, relatively few fungi have been studied and even fewer species investigated for their ability to make natural products. Such studies are hindered by the complex life cycles of fungi, unknown or difficult to reproduce conditions for growth and natural products production, and genetic intractability of the majority of fungi [3]. Despite these challenges, natural products made by filamentous fungi such as Aspergillus, Penicillium have been used clinically as antibiotics, antifungals, immunosuppressants, and cholesterol-lowering agents (Fig. 4.1) [4–6]. Compared to filamentous fungi, Basidiomycota are a largely uncharted territory for natural products discovery, with only a very small fraction of the nearly 30,000 described species examined for their ability to produce secondary metabolites [7–10]. Even fewer studies have focused on the biosynthetic genes responsible for natural product production in this fascinating class of organisms [11–24].

Natural products from fungi have very complex structures and a wide range of biological activities. Many of these compounds have been used to make synthetic analogs with improved therapeutic applications. Some bioactive compounds isolated from fungi and representing different natural product classes include, a the immunosuppressant cyclosporine A (a nonribosomal peptide) [139], b the immunosuppressant sirolimus (rapamycin) (hybrid nonribosmal peptide and polyketide) [139], c the antimicrobial fumagillin [140] (meroterpenoid, hybrid terpenoid-polyketide), and d the antitumor compound illudin S (sesquiterpenoid). [141]
Genome sequencing initiatives such as the US Department of Energy’s Fungal Genomics Program [25] have resulted in a massive influx of fungal DNA sequence data over the course of the last 10 years. Driven by an interest in fungi as sources for lingocellulose-degrading enzymes [26] and affordable next-generation sequencing technologies [27], the number of Basidiomycota genomes alone has ballooned from less than 5 a few years ago to currently over 100 draft genomes listed at the Joint Genome Institute’s (JGI) MycoCosm genome portal (http://genome.jgi.doe.gov/programs/fungi/index.jsf), of which only a select few have been annotated in detail [28–36]. About twice as many Ascomycota genome sequences are listed at this portal, and many more are deposited at the National Center for Biotechnology Information (NCBI). The large number of fungal genome data provides a tremendous opportunity for bioinformatics-guided approaches to assess and access the natural products potential of fungi independent from whether their biosynthetic pathways can be induced under laboratory conditions.
One of the hallmarks of fungal genetic organization, similar to bacteria, is the physical clustering of core secondary metabolic genes of natural products pathways in the genome. Genomic clustering is believed to facilitate efficient regulation of natural product biosynthesis through transcription factors and epigenetics [37–44]. Physical linkage between biosynthetic genes in a given pathway greatly aids in the characterization, discovery, and biotechnological exploitation of fungal natural biosynthetic pathways . Inexpensive DNA synthesis and the development of sophisticated synthetic biology approaches for heterologous biosynthetic pathway assembly will eventually enable scientists to bypass current challenges such as finding conditions for growth and natural products biosynthesis, and tools for genetic manipulations.
In this chapter, we will provide an overview of fungal natural products biosynthesis with an emphasis on genomic- and bioinformatic-driven pathway discovery. Regulation of secondary metabolic pathways (including silent and cryptic pathways) will be discussed elsewhere in this book and will not be a focus of this chapter. We will begin by examining the different bioinformatics tools available for genome mining for biosynthetic gene clusters with an emphasis on open-source accessible algorithms and software. We will then discuss elucidation and characterization of gene clusters responsible for the biosynthesis of major fungal secondary metabolite classes, polyketides (PK)/nonribosomal peptides (NRP), and terpenes . Apart from representing major fungal natural product classes that have been characterized in some detail, differences in PK/NRP and terpenoid biosynthetic cluster abundance in Ascomycota and Basidiomycota genomes provide some insights into the natural products repertoire of these two fungal groups. Each section will provide key examples of the workflow required to identify and characterize the genes responsible for synthesizing the biologically active compounds introduced above.
The Bioinformatic Tools of Genome Mining for Natural Products in Fungi
The physical clustering of biosynthetic genes for a given natural product provides an elegant means of elucidating fungal biosynthetic pathways . Unlike in plants, whose genomes are more complex and lack clearly delineated biosynthetic gene clusters [37], in fungi , the identification of a gene encoding a key enzyme in a biosynthetic pathway may lead directly to most of the remaining genes in the pathway. This can be particularly important for the discovery and characterization of natural product scaffold-activating cytochrome P450 enzymes. Like in plants, this enzyme family has undergone extensive gene duplication in fungi, particularly in Basidiomycota [45]. This makes it difficult to determine specific P450 functions based on sequence homology alone, especially for novel, multifunctional fungal P450 gene families [46, 47]. The identification of biosynthetic gene clusters has led to the characterization of fungal P450s involved in statin [48], PK [49], and alkaloid biosynthesis [50, 51]. With the increase of genomic sequence data, initially for filamentous fungi and more recently for Basidiomycota, much effort has, therefore, been invested in the development of bioinformatics tools to mine fungal genomes for these clusters.
The identification of biosynthetic gene clusters has typically begun with an mRNA or genomic “anchor sequence” based on one or more known biosynthetic genes in a pathway. Such an “anchor gene”, typically (but not always, [52]) encodes the first key enzyme in the biosynthetic pathway—for example, a polyketide synthase (PKS), nonribosomal peptide synthase (NRPS) [18, 24], terpene synthase (TPS), or other enzyme—depending on the type of natural product scaffold formed [12, 53–55]. Traditionally, clusters were identified through molecular genetic techniques. For example, the creation and sequencing of a cosmid library led to the identification of the first gene cluster responsible for the production of trichothecene, a sequiterpenoid mycotoxin, in Fusarium graminearum F15 [56]. In addition, when the anchor gene was cloned and sequenced, fungal biosynthetic gene clusters were identified through subsequent genome walking [54, 55, 57]. These molecular techniques, however, have only allowed for the identification of clusters up to a certain size (up to ~ 20 kb) and with the anchor gene fully sequenced. The more recent availability of fungal whole-genome sequence data has allowed for the identification of biosynthetic clusters of any size, provided the genome assembly data include sufficiently large scaffolds. Basic Local Alignment Search Tool (BLAST) searches of fungal genome sequences for classes of enzymes, such as PKS, NRPS, and TPS genes, typically yield several potential candidate genes for a target enzyme. Gene prediction algorithms, such as Augustus [58], can then be applied to predict open reading frames (ORFs) and encoding cDNAs of the putative anchor genes and of upstream and/or downstream located additional biosynthetic genes. Such an approach led to the discovery of multiple terpenoid biosynthetic gene clusters in Coprinus cinereus and Omphalotus olearius by our group [11, 12]. However, manual gene prediction and identification of cluster genes is tedious, and cluster boundaries can be hard to pin down.
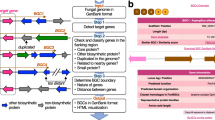
As a result, several tools have been developed to more easily identify the putative fungal biosynthetic clusters (reviewed in [59–61]). The most extensive software tool available for the identification of biosynthetic gene clusters is antibiotics and Secondary Metabolite Analysis SHell (antiSMASH) [61]. When given a bacterial or fungal genome sequence data, antiSMASH identifies putative gene clusters by comparing all predicted gene products to a hand-curated set of models corresponding to gene families common to the two dozen types of secondary metabolic pathways it recognizes. Putative clusters are then further analyzed through sequence homology to identify types of multidomain enzymes such as PKSs or NRPSs, enzyme specificities, and potentially even a core structure of the natural product [61]. Originally released in 2011 [60] and updated in 2013 [61], antiSMASH now has the capability to analyze raw contig assemblies of entire genomes, although it should be noted that the assemblies must be relatively clean to avoid redundant identifications. The output visualizes cluster predictions (Fig. 4.2a), which shows homologs of clusters in other microbial species (i.e., fungi) (Fig. 4.2b), and allows for easy sequence extraction (Fig. 4.2c).

Sample output using antiSMASH 2.0 [61] to analyze the genome of the basidiomycete O. olearius. The raw scaffolds were uploaded to the antiSMASH server and analyzed for secondary metabolic genes/clusters. a The initial output lists all putative clusters and attempts to classify them by type. b Upon clicking on a cluster the view is expanded, and individual genes are shown within their genomic context. Additionally, homologous clusters from other fungi are shown below. c Each putative protein sequence can be quickly accessed and used to perform BLAST searches. (This figure was adapted from the output of antiSMASH 2.0)
While antiSMASH is extremely powerful, we found that it has some notable limitations in its ability to correctly predict genes and their encoded proteins and identify full biosynthetic pathways . First, individual biosynthetic genes may be easily missed by antiSMASH. In O. olearius, our group identified 11 terpene synthases through manual BLAST searches, while antiSMASH was only able to identify four of those genes. Second, the rules for defining the boundaries of secondary metabolic clusters are not, as of yet, entirely clear. While the “core” cluster may be predicted with ease, individual biosynthetic genes some distance away (10–20 kilobases) that may still be involved in late-pathway modifications are frequently missed. In some cases, satellite clusters of biosynthetic genes are located at distinct loci in the genome. For example, while most trichothecene biosynthetic genes in Fusarium sporotrichioides and F. graminearum exist in the tri5 core gene cluster, two late pathway genes are clustered elsewhere [62]. Finally, accurate structural annotation of genome sequences is crucial for the identification of biosynthetic genes; unfortunately common gene prediction algorithms (such as Augustus [58]) trained on other eukaryotic genomes typically perform poorly for prediction in Basidiomycota, whose genomes are rich in small intron and exons. Furthermore, genes may be closely spaced and neighboring gene models may or may not be fused together and cryptic alternative splicing is not uncommon [63–67].
Our experience has shown that the gene prediction models used by most algorithms often lead to incorrect structural gene annotation in Basidiomycota, frequently requiring manual re-annotation using protein sequence alignment-guided identification of the most likely correct splice isoforms together with tedious attempts at obtaining correctly spliced cDNAs encoding functional proteins. Efforts to uncover the first biosynthetic gene of a biosynthetic pathway such as a terpenoid pathway can be arduous [68]. The presence of introns, some of which can be unusually small, complicates polymerase chain reaction (PCR) amplification [67]. In our experience, many splice variants can be amplified from a given cDNA pool, though only one splice variant has ever been confirmed to produce an active enzyme after transcription. Even when a good splicing model is predicted, and the expected gene is amplified, the resultant protein may still be inactive, as was the case with Cop5, a sesquiterpene synthase our laboratory cloned and attempted to characterize from Coprinus cinereus [12]. We appear to be just beginning to understand the complex splicing, transcriptional regulation , and possibly posttranslational regulation that leads to active secondary metabolic genes.
In the future, deep RNA sequencing will be a key to improving computational prediction of fungal biosynthetic genes and gene clusters. Already, RNAseq data has been shown to improve the accuracy of gene prediction models [29, 65, 69, 70] and has shed light on differential splicing [71]. In addition, transcriptomic data has also been useful in the delineation of gene cluster boundaries [24]. Presently only some of the more recently sequenced Basidiomycota genomes have associated deep RNA sequence data [29, 70] useful for biosynthetic pathway identification. Advances in HT-RNA sequencing and continued cost reductions for sequencing now allow affordable rapid and deep profiling transcriptome analysis under a variety of conditions or for diverse genotypes to collect large data sets for one species. Such data can be used to create gene coexpression networks built on physical distance to a seed natural products biosynthetic gene (e.g., TPS, NRPS, PKS, P450s) (guilt by association) as a powerful tool for pathway discovery. The fact that NP pathway genes are generally co-regulated through levels of shared transcriptional control elements (e.g., transcription factors and upstream intergenic gene regions) [72] represents yet another approach for network analysis within and also across species. Significant advances have been made in understanding the regulatory control elements of NP pathways in filamentous fungi , including the velvet family of regulatory proteins that are conserved among Ascomycota and Basidiomycota [39, 73, 74]. Genome analysis of Ganoderma and Schizophyllum [29, 31, 75] suggests high conservation of regulatory networks among mushroom forming fungi, which can be exploited for network building. Yet, gene coexpression network analysis so far has been largely applied for the discovery of natural products genes in plant [76, 77]. Guilt by association-based analysis based on DNA expression arrays was only recently applied to natural product biosynthetic gene cluster analysis in A. nidulans [78].
Polyketide Synthases (PKS) and Nonribosomal Peptide Synthases (NRPS)
Polyketides (PK) and nonribosomal peptides (NRPs) are major, structurally diverse classes of natural products known to be produced by numerous filamentous fungi [79–81] and bacteria [82]. From an ecological perspective, these polyketide- and peptide-based secondary metabolites afford the host organism a wide array of largely cytotoxic or general antibiotic compounds, which effectively restrict the growth and development of organisms that may compete for space and nutrients. Synthesis of these metabolites is achieved by a simple and highly conserved general mechanism involving the iterative elongation of either amino acid or carboxylic acid building blocks, for nonribosomal peptides and polyketides, respectively. In a similar manner as fatty acid synthases (FAS), the relevant enzymes that coordinate the production of these varied metabolites are multifunctional, mostly iterative enzymes, with a predictable set of core domains that repeatedly utilize the same active site to elongate peptide or polyketide chains. For PKSs, these include ketoacyl synthase (KS), acyltransferase (AT), acyl carrier protein (ACP), and thioesterase (TE) domains [83]. Utilizing this core domain set, the condensation of activated acetate units produces a polyketide scaffold, upon which a vast array of modifications can be imposed by one or more sparsely conserved ketoreductase (KR), dehydratase (DH), methyltransferase (MT), and enoyl reductase (ER) domains. The relative reduction status and product profile of a given PKS is linked to the presence or absence of these domains, with so-called “nonreducing PKSs” lacking these domains entirely and having a relatively limited product profile, while highly reducing PKSs contain all three of these domains and drastically alter the scaffold molecule into a wide array of alcohols, ketones, and other interesting chemical variants (reviewed in [79]).
Conservation of domains and reaction mechanisms suggest that PKSs and NRPSs share ancestral origins. It is, therefore, not surprising that NRPSs also contain a core set of domains similar to those in PKSs, including the peptidyl carrier protein (PCP), adenylation (A), condensation (C), and the thioesterase (TE) domains [84–86], which are required for the production of a scaffold peptide. This basal molecule can then be modified to varying degrees by a number of ancillary domains, including an epimerization or dual/epimerization domain (E and D/E, respectively), a reductase domain (R), and others involved in oxidation, cyclization, and methylation [85]. Additionally, genomic resources have revealed that many fungal PKSs and NRPSs cluster with cytochrome P450s (for example [87]) and that modifications of PKS and NRPS-derived scaffolds molecules by P450s have been implicated in the production of relevant secondary metabolites including mycophenolic acid, a grisan scaffold, tenellin, and the antifungal pneumocandin [88–91]. The propensity of fungi to cluster an array of modification enzymes such as P450s around core scaffold-producing enzymes such as PKSs and NRPS s is a widely conserved means to create vast libraries of products from simple PK, NRP, and terpenoid (discussed in next section) building blocks.
From an engineering perspective, a great deal of information has already been accumulated regarding the structure and function of PKS/NRPS enzymes, with numerous recent reports highlighting the potential for engineering PKSs and also NRPSs from Ascomycetes such as Fusarium and Aspergillus [92, 93] and Basidiomycetes such as Ustilago maydis and Suillus grevillei [15, 17]. Ma et al. [92] conducted a detailed characterization of the lovastatin nonaketide synthase LovB; a highly reducing PKS catalyzing the production of dihydromonacolin L. Extensive in vitro analyses, as well as production from Saccharomyces cerevisiae and substrate feeding experiments provided the authors with a detailed understanding of LovB structure and function. The production of lovastatin from dihydromonacolin L is known to require LovB and the enoyl reductase (ER) domain of its partner enzyme, LovC [92]. Interestingly, in vitro experiments with LovB, LovC, and all required cofactors failed to release dihydromonacolin L, indicating that the action of another domain might be required. The authors successfully released dihydromonacolin L after coexpression of heterologous thioesterase-containing enzymes from Gibberella zeae, supporting the aforementioned claim. Moreover, the same can be accomplished with the ER domain protein LovC. Complementation of dihydromonacolin L release can be achieved via the heterologous expression of MlcG; an analogous ER domain containing protein from the compactin biosynthetic cluster of Penicillium citrinum [92]. These reports support the claim that the function of these enzymatic partners is more promiscuous than once believed and that engineering of designer pathways by swapping analogous domains from related PKSs and NRPSs is a viable strategy for production of novel chemistry as explained later in this chapter.
Despite the vast potential for isolation and production of valuable compounds from these metabolic clusters, there are significant gaps in our current level of understanding of NRP and PK biosynthesis in fungal systems. Indeed, a brief survey of the SciFinder returns fewer than 100 PKS- or NRPS-derived compounds from Basidiomycota, although recent work highlighting PKS diversity in Basidiomycota suggests that the number of biosynthetic genes grossly exceeds the number of reported PKs from these organisms [94]. This discrepancy most likely reflects a lack in the characterization of compounds produced by these enzymes.
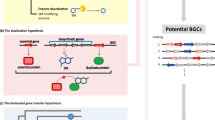
A number of very recent reports have utilized multifaceted approaches to mine fungal genomes and increase our understanding of PK and NRP diversity. For example, a study by Lackner et al. [94] aimed to identify new PKSs in Basidiomycota probing the aforementioned genome resource at JGI with the KS domain of AflC of Aspergillus parasiticus and a selected group of related sequences. Thirty-five Basidiomycota genomes were queried, yielding more than 100 putative PKS genes [94], thus supporting the claim that the myriad of domain architectures presented by fungal PKSs represent an “in silico gold mine” for the discovery of new enzymes and possibly enzymes with variant domains. A similar approach has also been used with the well-conserved PCP, A, C, and TE domains of an NRPS to infer a great deal regarding the phylogeny and functional diversity of NRPSs [85] (reviewed in [84, 86]). Combining the information accrued from these reports, with greatly expanded genomic resources and the knowledge that a great deal of variation exists within the domain structure of PKSs, NRPSs, and also hybrid PK-NRPSs [94], it seems plausible to apply more advanced computational strategies for the isolation of novel metabolites produced from common scaffolds. In this scenario, known domain elements isolated from PKSs and/or NRPSs of interest could be used as queries against fungal genomic databases to isolate novel variants from a wide range of diverse genera (Fig. 4.3). From an engineering perspective, these variant domains represent modules that could be swapped interchangeably, yielding chimeric enzymes with the potential for novel chemistries (Fig. 4.3). This strategy has been implemented, albeit on a small scale, to engineer novel chemistry from an engineered hybrid PKS combining the asperfuranone and sterigmatocystin biosynthetic pathways of A. nidulans [95].

Scheme for developing novel PKS- and NRPS-derived chemistries. Known domain elements of NRPSs (green boxes; PCP, A, C, and TE) as well as downstream, modification domains (purple boxes; E D/E, R, and ME) can be used to query vast fungal databases to isolate variants in domain of interest. Variants isolated in this way for the epimerization and methylation domains (E D/E and ME) are shown as examples in dashed boxes. Subsequent domain shuffling experiments would allow for the construction of novel, chimeric enzymes with the potential for producing novel metabolites. An NRPS is shown as an example, but the same scheme could be applied to PKSs and also hybrid PK-NRPSs
In addition to genome mining targeting PKSs and NRPSs themselves, another potential strategy for identifying biosynthetic gene clusters is to elicit and examine their transcription. Many biosynthetic clusters are transcriptionally inactive (a.k.a. cryptic gene clusters) under normal cultivation conditions, particularly in endophytic fungi reliant on small molecule signaling from another organism [96]. One way to combat this transcriptional repression is to alter the expression of transcriptional activators/repressors through global epigenetic regulators or with genetic knockouts. An excellent review of the function of LaeA and the velvet complex mentions the implication of these global regulators of secondary metabolism in more than half of the PKS and NRPS genes in Trichoderma reesei [41]. Both knockouts and overexpression of global regulators like LaeA and its homologs [38] (or other related machinery, such as histone acetyltransferases [97]) can allow for more detailed genome mining when examining RNA-sequencing datasets and searching for areas of the genome in which transcription is directly affected. A comprehensive discussion of fungal secondary metabolic pathway regulation is provided elsewhere in this book.
Terpene Synthases and Terpenoid Biosynthetic Clusters
Terpenoids are all derived from the five-carbon isoprene units isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP). Prenyldiphosphate synthases then catalyze the head to tail condensation of two, three or four of these five-carbon units to produce 10-, 15- or 20-carbon (C10, C15, C20) isoprenoid diphosphate molecules, which serve as the substrates for Class I terpene synthases that are dependent on the ionization of the allylic diphosphate to form a reactive carbocation and triggering a cascade of cyclization and rearrangement reactions in the enzymes active site [98]. Depending on their chain-length specificity, this class of terpene synthases utilize the 10-carbon substrate geranyl diphosphate (C10, GPP) to form monoterpenes, farnesyl diphosphate (C15, FPP) to generate sesquiterpenes, or geranylgeranyl diphosphate (C20, GGPP) to synthesize diterpenes [99]. Terpenoids with more than 20 carbons are typically formed by the head to head condensation of two FPP or GGPP molecules, yielding longer isoprene chains that are then modified into various C30 (sterols) and C40 (carotenoids) terpenoid structures. For example, sterols are formed by Class II terpene synthases that rely on a protonation-initiated cyclization mechanism that yields the scaffolds of bioactive triterpenoids isolated from many fungi [100, 101].
Identification of the first terpene synthases from filamentous fungi , such as the sesquiterpene synthases aristolochene [102, 103] and trichodiene synthase [53] required laborious efforts. Advances in sequencing and the increasing availability of sequences data has led to the discovery and characterization of a suite of novel fungal terpenoid biosynthetic enzymes in the past few years. For example, in the past 5 years more than two dozen new fungal sesquiterpene synthases have been cloned and characterized [104], [52], [12], [11, 105] [105–107]. Genome mining efforts not only enable the discovery of new types of terpene synthases and enzymes with new cyclization activities as discussed later but also comparative analysis of the natural production potential encoded by the genomes of the two major fungal phyla.
While polyketides and nonribosomal peptides are the major class of secondary metabolites discovered in filamentous fungi , terpenoids appear to be a predominant class of secondary metabolites in Basidiomycota. In contrast to the number of PKS and NRPS genes mentioned previously, Basidiomycota genomes contain large collections of sesquiterpenoid biosynthetic genes. In 2012 we identified more than 500 putative sesquiterpene synthases (TPS, see later) in only 40 Basidiomycota genomes , and many of the putative TPS appear to be part of a biosynthetic gene cluster [11]. As the number of publicly available genome datasets has doubled, so has the number of putative TPS genes, totaling nearly 1000. As a comparison, only 179 putative TPS genes were identified in close to 2000 bacterial genomes [108], and we identified only ~ 250 TPS genes in 80 Ascomycota genomes examined.
Conspicuously absent in sequenced Basidiomycota genomes are genes that could encode diterpene synthases, although some mushroom-forming fungi (including the pleuromutilin antibiotic producing fungus Clitiopilus passeckerianus [109]) have been reported to produce diterpenoids [110]. Ascomycota on the other hand are known to be prolific producers of bioactive diterpenoids and several biosynthetic gene clusters have been characterized. These include the well-studied gibberellin pathways found in several fungi that use a bifunctional diterpene synthase that combines the domains of a class I and class II terpene synthases (which are separate enzymes in plant diterpene biosynthesis) to cyclize GGPP [111–113]. Mining of Ascomycota genomes followed by gene deletion studies and stepwise heterologous coexpression of pathway genes in Aspergillus has led to the elucidation of additional gene clusters involved in the biosynthesis of the diterpene compounds fusicoccin, brassicicene, aphidicolin and phomopsene [54, 55, 114–120], and a sesterterppenoid (C25) [121]. Intriguingly, these terpenoid scaffolds are built by a novel-type of chimeric terpene synthases that combines the domains of a class I terpene synthase and a prenyldiphosphate synthase that provides the C25 isoprene substrate for the cyclase [55].
Genome sequences of several Aspergillus strains have recently enabled the discovery and heterologous reconstitution of a series of biosynthetic gene clusters involved in the biosynthesis of medicinally important meroterpenoids (e.g., pyripyropene [122], terretonin [123–125], fumagillin [126] (Fig. 4.1c), austinol, and dehydroaustinol [127]), which are polyketide-terpenoid hybrid compounds. Except for the fumagillin cluster, all of these biosynthetic pathways involve an iterative, nonreducing PKS and an aromatic prenyltransferase [125] that attaches a prenyl chain (typically C15) to the polyketide moiety. The attached prenyl chain is then epoxidated by a flavin-dependent monooxygase to allow for cyclization by a novel type of membrane-bound Class II terpene synthase [122] [123–125] [127]. In fumagallin biosynthesis [126], however, a novel membrane-bound Class I TPS first generates the cyclized terpenoid scaffold that is then attached to a polyketide chain. Yet another novel membrane-bound type II terpene synthase has recently been proposed to catalyze the cyclization of the geranylgeranyl chain attached to the indole moiety in indole-diterpene biosynthesis [128]. Several indole-diterpene biosynthetic clusters have been identified in filamentous fungi [128–132]. Common to all clusters are genes that encode a putative GGPP synthase, aromatic prenyltransferase and a flavin monoxygenase and cyclase proposed to catalyze epoxidation and cyclization, respectively, of the GGPP chain[128–132].
The aforementioned studies illustrate that even with a genome sequence at hand, it may not be possible to assign function to biosynthetic gene clusters solely based on homology to known biosynthetic enzymes. The identification in filamentous fungi of different types of chimeric biosynthetic pathways and of novel enzyme folds that catalyze similar reactions as in the case of terpene cyclization indicates that we may have only just begun to scratch the surface of the fungal secondary metabolome.
While the abundance of putative TPS genes may appear daunting to characterize biochemically, we found that when focusing on sesquiterpene synthases in Basidiomycota a relationship between sequence and function could be uncovered. Specifically, we examined sequence conservation as it relates to the first committed bond-forming step in the cyclization of the terpene molecule. We discovered that, despite relatively poor automatic gene prediction in the publicly available databases, the TPS genes partitioned to five clades, which appear to segregate based on their cyclization mechanism (Fig. 4.4) [11]. These clades represent the four initial cyclizations of FPP known to be catalyzed by TPS. With this information it is now possible to sort through the large set of TPS sequences based on the initial cyclization believed to lead to the desired product. Our group recently carried out a study focused on validating the predictive framework by examining sesquiterpene biosynthesis in the crust fungus Stereum hirsutum. Not only did this fungus possess a large repertoire of terpene synthases, it also has been studied for its production of bioactive natural products. As many of the sesquiterpenoids reported are derived from a humulyl cation, we chose to target protoilludene synthase homologs, which we expected to go through the same cyclization mechanism. Using the framework described previously, it was possible to clone and characterize three novel protoilludene synthases [119]. While our work only focused on sesquiterpenes, similar studies can be carried out to examine other classes of terpenoids in order to provide the same genome mining roadmap.

Schematic representation of an unrooted neighbor-joining phylogenetic tree of sesquiterpene synthases identified in Basidiomycota genomes. We surveyed 42 Basidiomycota genomes and found 542 putative terpene synthase sequences, of which 392 were built into a phylogenetic tree to establish a link between sequence and function in this class of enzymes. By applying context to the tree through the inclusion of biochemical data, conservation of the initial cyclization reaction of farnesyl diphosphate (FPP) was identified. [11]
Terpenoid biosynthetic gene clusters may be extremely difficult to characterize due to the complex nature of the clusters themselves. Clusters can range in size from only two genes (e.g., a terpene synthase and a P450 monooxygenase) to greater than a dozen. Complicating the matter further is the propensity for separate biosynthetic clusters to work together to form the same types of products. For example, in searching for the enzymes responsible for illudin biosynthesis in the Jack O’Lantern fungus Omphalotus olearius, two protoilludene (the precursor to illudin compounds) synthases were identified (Omp6 and Omp7). Both were part of biosynthetic clusters, though one contained only three genes [11]. Varying kinetic values for the two terpene synthases indicate a possible mechanism for overcoming rate-limiting steps in the biosynthesis of illudins, though this has yet to be experimentally validated. A parallel example from the fungus F. sporotrichioides shows two clusters and an independent gene responsible for trichothecene biosynthesis. The larger cluster contains 12 genes, while the second smaller cluster and the independent gene are responsible for late-pathway reactions [62]. Additionally, very similar strains may contain orthologous biosynthetic genes, but some may be pseudogenes, inactivated by the accumulation of mutations [62, 113]. This sort of genomic segregation implies a need for tight transcriptional control on late-pathway genes, perhaps to minimize toxicity/reactivity of intermediates. This sort of variation in the genetic organization of biosynthetic clusters makes it particularly challenging to find all of the genes responsible for any given product.
After cluster gene identification, the next step is to characterize the function of individual genes. For a genetically tractable fungus, gene function can be determined in part by knockout and complementation studies. Additionally, if the clusters appear to be transcriptionally inactive, background mutations can be made to alter the level of transcription. In order to study the biosynthesis of botridal in B. cinerea, a knockout strain was engineered for the bcg1 gene responsible for downregulation of the pathway [52]. In another example studying tricothecene production in F. graminearum, the expression of the cluster proteins was too low, so a strain was engineered to contain an overexpression cassette with the FgTri6 gene responsible for regulating the biosynthetic cluster [56].
When no genetic tools are available for the cluster’s source organism, the biosynthetic cluster must be heterologously characterized. As expected, Escherichia coliis often the first prokaryotic chassis used for characterization of biosynthetic genes. While some enzymes, such as the terpene synthases discussed previously, express well and have high activity in E. coli, many of the other pathway enzymes prove difficult to characterize in this host [62]. This is, in part, due to the prevalence of P450 monooxgenases in many biosynthetic clusters, which are associated with the cell membrane and tend to express poorly in E. coli. Recent work, however, suggests different strategies to accommodate these enzymes, though the applicability across many different P450 homologs is unknown [133]. Several genes from the trichothecene biosynthetic cluster have been successfully characterized in E. coli, despite the difficulties described previously [62].
A more commonly used chassis for the expression and characterization of late-pathway biosynthetic enzymes is S. cerevisiae. Our laboratory characterized a terpene synthase and two associated P450 monooxgenases through standard plasmid-based expression [12]. While this is feasible for a small number of genes for characterization purposes, the assembly of an entire pathway for stable high-level production, as shown by Keasling’s group in their efforts to produce artemisinin, is very laborious and requires extensive strain engineering [134]. Another consideration when producing secondary metabolites is their inherent toxicity and the absence of the machinery required to protect the host organism. For this reason and others stated previously, the development of genetically tractable and easy-to-manipulate fungal strains is the next step in studying biosynthetic pathways. Additionally, many fungal systems have developed transporters designed specifically to export or compartmentalize the toxic intermediates and products in order to maintain high levels of biosynthesis. Perhaps the most well-understood system is the “aflatoxisomes” in Aspergillus , which are responsible for the compartmentalization of aflatoxin in vesicles before export from the cell [135, 136]. Another analogous system in Fusarium graminearium involves the use of toxisomes to compartmentalize toxic compounds in tricothecene biosynthesis . Using colocalization experiments with GFP/RFP-tagged pathway enzymes, two P450s (P450-Tri1, P450-Tri4) and the HMG-CoA reductase (the rate-controlling enzyme of the mevalonate precursor pathway) were found to localize to toxisomes that interact with smaller vesicles. The smaller vesicles contain the MFS transporter Tri12 and are believed to accumulate toxic pathway products that are compartmentalized and then eliminated by fusion with the vacuole and plasma membrane. Interestingly, many biosynthetic clusters contain transporters believed to be similar in function to Tri12 (Fig. 4.5) [137, 138].

Secondary metabolic clusters from three different fungi. Shown here are sesquiterpenoid biosynthetic cluster from a Omphalotus olearius [11]. b Stereum hirsutum [107], and c Fusarium graminearium [56]. Predicted ORFs are colored according to their putative function, with gray arrows with dotted outlines representing P450 enzymes, white arrows with solid outlines representing enzymes with predicted roles in sesquiterpene scaffold modification, light gray arrows with dotted outlines representing a transporter, and white arrows with dotted outlines representing the respective sesquiterpene synthases in each cluster. Black ORFs indicate hypothetical/unknown proteins and known transcriptional regulators are gray arrows with solid outlines in the trichothecene (Tri) biosynthetic cluster [142]
With the massive amount of sequence data available we are now limited by the speed by which we can biochemically characterize terpenoid biosynthetic genes for their function. The next step, after sufficient biochemical data has been collected, is to use this secondary metabolic enzyme toolbox to generate products not found in nature. Synthetic biology and metabolic engineering provide us with the tools to do something at a much faster rate than evolution. By combining interesting enzymes across many different fungi we will likely be able to generate terpenoids never seen before in nature (Fig. 4.6). For example, we may find multiple clusters in which the first committed step is for a specific sesquiterpene. While that step shows very little variance, the next step—the modification of that terpene scaffold—presents an opportunity for metabolic engineering of nonnatural pathways. These new pathways may contain P450 enzymes from a number of different fungi know to produce the precursor compound of interest, and known to modify that precursor to for a final product with interesting biological activity. These new compounds may have slightly different biological activities and serve as new starting compounds for useful pharmaceutical compounds.

Strategy for the biosynthesis of nonnatural products through combinatorial approaches. In the example shown here, a sesquiterpene synthase converts its substrate, farnesyl diphosphate to a sesquiterpene hydrocarbon scaffold. Such a hydrocarbon scaffold is typically activated through oxygenation catalyzed by P450 monooxygenases. In this example, P450s from three different fungal sources and sesquiterpene biosynthesis pathways are used to differentially oxygenate the product to different final sesquiterpenoid products. Further extension of these pathways with additional combinations of modifying enzymes could lead to a range of novel products
Conclusion
Researchers have only begun to scratch the surface of the myriad of natural products and biosynthetic pathways that can be discovered through mining the genomes of higher fungi. The influx of fungal genome sequencing data in recent years, along with the development of bioinformatics tools, from BLAST to Augustus to antiSMASH, have allowed us to probe rapidly into the biosynthetic gene clusters abundant in this class of organisms. However, additional bioinformatic and genomic approaches need to be developed or adapted for improved gene prediction and functional annotation, and especially for the identification of biosynthetic clusters made up by novel types of enzymes, delineating cluster boundaries, and for finding satellite and chimeric clusters.
So far, sequencing data only exists for a tiny percentage of identified fungal species. As the amount of fungal genome sequencing data increases, fungi are bound to become a much greater source of new natural products and their biosynthetic enzymes. In addition to the development of genomic data and bioinformatic resources, a critical component of natural product discovery is the detailed characterization of the product and pathway enzymes. At this point in time, the biochemical characterization of biosynthetic gene clusters and heterologous refactoring of pathways is equally if not more challenging than their identification. To fully exploit the natural products potential of fungi, significant efforts are required that aim at developing synthetic biology approaches for high-throughput heterologous fungal pathway assembly; ideally facilitating direct translation of sequence information encoded in fungal genomes into biosynthetic output by a heterologous expression and production platform. The final, and arguably most interesting step, will be combinatorial biosynthetic approaches through the creation of novel biosynthetic assemblies for the production of an even greater diversity of potentially bioactive compounds.
References
Stajich JE, Wilke SK, Ahren D, Au CH, Birren BW, Borodovsky M et al (2010 Jun 29) Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc Natl Acad Sci USA 107(26):11889–11894
Blackwell M (2011 Mar) The fungi: 1, 2, 3.. 5.1 million species? Am J Bot 98(3):426–438
Elisashvili V (2012) Submerged cultivation of medicinal mushrooms: bioprocesses and products (review). Int J Med Mushrooms 14(3):211–239
Hu F, Liu J, Du G, Hua Z, Zhou J, Chen J (2012 Aug) Key cytomembrane abc transporters of Saccharomyces cerevisiae fail to improve the tolerance to d-limonene. Biotechnol Lett 34(8):1505–1509
Kang J, Park J, Choi H, Burla B, Kretzschmar T, Lee Y et al (2011) Plant ABC transporters. Arabidopsis Book 9:e0153
Brase S, Encinas A, Keck J, Nising CF (2009 Sep) Chemistry and biology of mycotoxins and related fungal metabolites. Chem Rev 109(9):3903–3990
Zhong JJ, Xiao JH (2009) Secondary metabolites from higher fungi: discovery, bioactivity, and bioproduction. Adv Biochem Eng Biotechnol 113:79–150
Lindequist U, Niedermeyer THJ, Julich WD (2005) The pharmacological potential of mushrooms. Evid Based Complement Alternat Med 2(3):285–299
Alves MJ, Ferreira IC, Dias J, Teixeira V, Martins A, Pintado M (2012 Nov) A review on antimicrobial activity of mushroom (basidiomycetes) extracts and isolated compounds. Planta Med 78(16):1707–1718
Zjawiony JK (2004 Feb) Biologically active compounds from Aphyllophorales (polypore) fungi. J Nat Prod 67(2):300–310
Wawrzyn GT, Quin MB, Choudhary S, Lopez-Gallego F, Schmidt-Dannert C (2012 Jun 22) Draft genome of omphalotus olearius provides a predictive framework for sesquiterpenoid natural product biosynthesis in basidiomycota. Chem Biol 19(6):772–783
Agger S, Lopez-Gallego F, Schmidt-Dannert C (2009 Jun) Diversity of sesquiterpene synthases in the basidiomycete Coprinus cinereus. Mol Microbiol 72(5):1181–1195
Lopez-Gallego F, Wawrzyn GT, Schmidt-Dannert C (2010 Dec) Selectivity of fungal sesquiterpene synthases: role of the active site’s h-1 a loop in catalysis. Appl Environ Microbiol 76(23):7723–7733
Lopez-Gallego F, Agger SA, Abate-Pella D, Distefano MD, Schmidt-Dannert C (2010 May) Sesquiterpene synthases cop4 and cop6 from Coprinus cinereus: catalytic promiscuity and cyclization of farnesyl pyrophosphate geometric isomers. Chembiochem [Article] 11(8):1093–1106
Wackler B, Lackner G, Chooi YH, Hoffmeister D (2012 Aug 13) Characterization of the Suillus grevillei quinone synthetase grea supports a nonribosomal code for aromatic alpha-keto acids. Chembiochem 13(12):1798–1804
Schneider P, Bouhired S, Hoffmeister D (2008 Nov) Characterization of the atromentin biosynthesis genes and enzymes in the homobasidiomycete Tapinella panuoides. Fungal Genet Biol 45(11):1487–1496
Winterberg B, Uhlmann S, Linne U, Lessing F, Marahiel MA, Eichhorn H et al (2010 Mar) Elucidation of the complete ferrichrome a biosynthetic pathway in Ustilago maydis. Mol Microbiol 75(5):1260–1271
Welzel K, Eisfeld K, Antelo L, Anke T, Anke H (2005 Aug 1) Characterization of the ferrichrome a biosynthetic gene cluster in the homobasidiomycete Omphalotus olearius. FEMS Microbiol Lett 249(1):157–163
Quin MB, Wawrzyn G, Schmidt-Dannert C (2013 May) Purification, crystallization and preliminary x-ray diffraction analysis of omp6, a protoilludene synthase from Omphalotus olearius. Acta Crystallogr Sect F Struct Biol Cryst Commun 69(5):574–577
Bushley KE, Ripoll DR, Turgeon BG (2008 Dec 3) Module evolution and substrate specificity of fungal nonribosomal peptide synthetases involved in siderophore biosynthesis. BMC Evol Biol 8(328). doi:10.1186/1471-2148-8-328
Bushley KE, Turgeon BG (2010 Jan 26) Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol Biol 10(26). doi:10.1186/1471-2148-10-26
Condon BJ, Leng YQ, Wu DL, Bushley KE, Ohm RA, Otillar R et al (2013 Jan) Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. Plos Genet 9(1):e1003233
Turgeon BG, Oide S, Bushley K (2008 Feb) Creating and screening Cochliobolus heterostrophus non-ribosomal peptide synthetase mutants. Mycol Res 112(Pt 2):200–206
Bushley KE, Raja R, Jaiswal P, Cumbie JS, Nonogaki M, Boyd AE et al (2013) The genome of Tolypocladium inflatum: evolution, organization and expression of the cyclosporin biosynthetic gene cluster. PLoS Genet 9(6):e1003496
Martin F, Cullen D, Hibbett D, Pisabarro A, Spatafora JW, Baker SE et al (2011 Jun) Sequencing the fungal tree of life. New Phytol 190(4):818–821
Gibson DM, King BC, Hayes ML, Bergstrom GC (2011 Jun) Plant pathogens as a source of diverse enzymes for lignocellulose digestion. Curr Opin Microbiol 14(3):264–270
Shendure J, Lieberman Aiden E (2012 Nov) The expanding scope of DNA sequencing. Nat Biotechnol 30(11):1084–1094
Morin E, Kohler A, Baker AR, Foulongne-Oriol M, Lombard V, Nagy LG et al (2012 Oct 23) Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc Natl Acad Sci USA 109(43):17501–17506
Chen S, Xu J, Liu C, Zhu Y, Nelson DR, Zhou S et al (2012) Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat Commun 3:913
Nishida H, Nagatsuka Y, Sugiyama J (2011) Draft genome sequencing of the enigmatic basidiomycete Mixia osmundae. J Gen Appl Microbiol 57(1):63–67
Ohm RA, de Jong JF, Lugones LG, Aerts A, Kothe E, Stajich JE et al (2010 Sep) Genome sequence of the model mushroom schizophyllum commune. Nat Biotechnol 28(9):957–963
Martinez D, Challacombe J, Morgenstern I, Hibbett D, Schmoll M, Kubicek CP et al (2009 Feb 10) Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc Natl Acad Sci USA 106(6):1954–1959
Martin F, Aerts A, Ahren D, Brun A, Danchin EG, Duchaussoy F et al (2008 Mar 6) The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 452(7183):88–92
Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B et al (2012 Jun 29) The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336(6089):1715–1719
Fernandez-Fueyo E, Ruiz-Duenas FJ, Ferreira P, Floudas D, Hibbett DS, Canessa P et al (2012 Apr 3) Comparative genomics of Ceriporiopsis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis. Proc Natl Acad Sci USA 109(14):5458–5463
Bao D, Gong M, Zheng H, Chen M, Zhang L, Wang H et al (2013) Sequencing and comparative analysis of the straw mushroom (Volvariella volvacea) genome. PLoS ONE 8(3):e58294
Osbourn A (2010 Oct) Secondary metabolic gene clusters: evolutionary toolkits for chemical innovation. Trends Genet 26(10):449–457
Brakhage AA, Schroeckh V (2011 Jan) Fungal secondary metabolites—strategies to activate silent gene clusters. Fungal Gen Biol 48(1):15–22
Brakhage AA (2013 Jan) Regulation of fungal secondary metabolism. Nat Rev Microbiol 11(1):21–32
Yazaki K (2005 Jun) Transporters of secondary metabolites. Curr Opin Plant Biol 8(3):301–307
Prasad R, Goffeau A (2012) Yeast ATP-binding cassette transporters conferring multidrug resistance. Annu Rev Microbiol 66:39–63
Luzhetskyy A, Vente A, Bechthold A (2005 Jul) Glycosyltransferases involved in the biosynthesis of biologically active natural products that contain oligosaccharides. Mol Biosyst 1(2):117–126
Singh S, Phillips GN Jr, Thorson JS (2012 Oct) The structural biology of enzymes involved in natural product glycosylation. Nat Prod Rep 29(10):1201–1237
Lim FY, Sanchez JF, Wang CC, Keller NP (2012) Toward awakening cryptic secondary metabolite gene clusters in filamentous fungi. Meth Enzymol 517:303–324
Cresnar B, Petric S (2011 Jan) Cytochrome p450 enzymes in the fungal kingdom. Biochim Biophys Acta 1814(1):29–35
Podust LM, Sherman DH (2012 Oct) Diversity of P450 enzymes in the biosynthesis of natural products. Nat Prod Rep [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review] 29(10):1251–1266
Kelly DE, Krasevec N, Mullins J, Nelson DR (2009 Mar) The cypome (cytochrome P450 complement) of Aspergillus nidulans. Fungal Gen Biol [Research Support, Non-U.S. Gov’t] 46(Suppl 1):53–61
Barriuso J, Nguyen DT, Li JW, Roberts JN, MacNevin G, Chaytor JL et al (2011 Jun 1) Double oxidation of the cyclic nonaketide dihydromonacolin l to monacolin j by a single cytochrome P450 monooxygenase, LovA. J Am Chem Soc [Research Support Non-US Gov’t] 133(21):8078–8081
Artigot MP, Loiseau N, Laffitte J, Mas-Reguieg L, Tadrist S, Oswald IP et al (2009 May) Molecular cloning and functional characterization of two cyp619 cytochrome P450s involved in biosynthesis of patulin in Aspergillus clavatus. Microbiology [Research Support NIH Extramural Research Support Non-US Gov’t] 155(5):1738–1747
Haarmann T, Ortel I, Tudzynski P, Keller U (2006 Apr) Identification of the cytochrome p450 monooxygenase that bridges the clavine and ergoline alkaloid pathways. Chembiochem [Research Support Non-US Gov’t] 7(4):645–652
Kelly SL, Kelly DE (2013 Feb 19) Microbial cytochromes P450: biodiversity and biotechnology. Where do cytochromes P450 come from, what do they do and what can they do for us? Philos Trans R Soc Lond B Biol Sci [Research Support NIH Extramural Research Support Non-US Govt Review] 368(1612):20120476
Pinedo C, Wang CM, Pradier JM, Dalmais B, Choquer M, Le Pecheur P et al (2008 Dec 19) Sesquiterpene synthase from the botrydial biosynthetic gene cluster of the phytopathogen Botrytis cinerea. ACS Chem Biol 3(12):791–801
Hohn TM, Beremand PD (1989 Jun) Isolation and nucleotide sequence of a sesquiterpene cyclase gene from the trichothecene producing fungus Fusarium sporotrichoides. Gene [Article] 79(1):131–138
Toyomasu T, Nakaminami K, Toshima H, Mie T, Watanabe K, Ito H et al (2004 Jan) Cloning of a gene cluster responsible for the biosynthesis of diterpene aphidicolin, a specific inhibitor of DNA polymerase alpha. Biosci Biotechnol Biochem [Article] 68(1):146–152
Toyomasu T, Tsukahara M, Kaneko A, Niida R, Mitsuhashi W, Dairi T et al (2007 Feb) Fusicoccins are biosynthesized by an unusual chimera diterpene synthase in fungi. Proc Natl Acad Sci USA [Article] 104(9):3084–3088
Kimura M, Tokai T, O'Donnell K, Ward TJ, Fujimura M, Hamamoto H et al (2003 Mar 27) The trichothecene biosynthesis gene cluster of Fusarium graminearum F15 contains a limited number of essential pathway genes and expressed non-essential genes. FEBS Lett 539(1–3):105–110
Toyomasu T, Tsukahara M, Kenmoku H, Anada M, Nitta H, Ohkanda J et al (2009 Jul 16) Transannular proton transfer in the cyclization of geranylgeranyl diphosphate to fusicoccadiene, a biosynthetic intermediate of fusicoccins. Org Lett 11(14):3044–3047
Stanke M, Steinkamp R, Waack S, Morgenstern B (2004 Jul) Augustus: a web server for gene finding in eukaryotes. Nucleic Acids Res [Article] 32:W309–W312
Chen B, Ling H, Chang MW (2013) Transporter engineering for improved tolerance against alkane biofuels in Saccharomyces cerevisiae. Biotechnol Biofuels 6(1):21
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA et al (2011 Jul) antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39(Web Server issue):W339–346
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E et al (2013 June 3) antiSMASH 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41:W204–12
Kimura M, Tokai T, Takahashi-Ando N, Ohsato S, Fujimura M (2007 Sep) Molecular and genetic studies of Fusarium trichothecene biosynthesis: pathways, genes, and evolution. Biosci Biotechnol Biochem 71(9):2105–2123
Csuros M, Rogozin IB, Koonin EV (2011 Sep) A detailed history of intron-rich eukaryotic ancestors inferred from a global survey of 100 complete genomes. PLoS Comput Biol 7(9):e1002150
Freitag J, Ast J, Bolker M (2012 May 24) Cryptic peroxisomal targeting via alternative splicing and stop codon read-through in fungi. Nature 485(7399):522–525
Larsen PE, Trivedi G, Sreedasyam A, Lu V, Podila GK, Collart FR (2010) Using deep RNA sequencing for the structural annotation of the Laccaria bicolor mycorrhizal transcriptome. PLoS ONE 5(7):e9780
Misiek M, Braesel J, Hoffmeister D (2011 Aug) Characterisation of the ArmA adenylation domain implies a more diverse secondary metabolism in the genus Armillaria. Fungal Biol 115(8):775–781
Misiek M, Hoffmeister D (2008 Feb) Processing sites involved in intron splicing of Armillaria natural product genes. Mycol Res [Article] 112:216–224
Wawrzyn GT, Bloch SE, Schmidt-Dannert C (2012) Discovery and characterization of terpenoid biosynthetic pathways of fungi. Meth Enzymol 515:83–105
Keller O, Kollmar M, Stanke M, Waack S (2011 Mar 15) A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics [Research Support Non-US Gov’t] 27(6):757–763
Yu GJ, Wang M, Huang J, Yin YL, Chen YJ, Jiang S et al (2012) Deep insight into the Ganoderma lucidum by comprehensive analysis of its transcriptome. PLoS ONE 7(8):e44031
McGettigan PA (2013 Feb) Transcriptomics in the RNA-seq era. Curr Opin Chem Biol [Research Support Non-US Govt Review] 17(1):4–11
Noble LM, Andrianopoulos A (2013 May 21) Fungal genes in context: genome architecture reflects regulatory complexity and function. Genome Biol Evol 5(7):1336–52
Yin W, Keller NP (2011 Jun) Transcriptional regulatory elements in fungal secondary metabolism. J Microbiol 49(3):329–339
Bayram O, Braus GH (2012 Jan) Coordination of secondary metabolism and development in fungi: the velvet family of regulatory proteins. FEMS Microbiol Rev 36(1):1–24
Ohm RA, de Jong JF, de Bekker C, Wosten HA, Lugones LG (2011 Sep) Transcription factor genes of Schizophyllum commune involved in regulation of mushroom formation. Mol Microbiol 81(6):1433–1445
Hur M, Campbell AA, Almeida-de-Macedo M, Li L, Ransom N, Jose A et al (2013) A global approach to analysis and interpretation of metabolic data for plant natural product discovery. Nat Product Rep 30(4):565–583
Higashi Y, Saito K (2013 Jan 21) Network analysis for gene discovery in plant-specialized metabolism. Plant Cell Environ 36(9):1597–606
Andersen MR, Nielsen JB, Klitgaard A, Petersen LM, Zachariasen M, Hansen TJ et al (2013 Jan 2) Accurate prediction of secondary metabolite gene clusters in filamentous fungi. Proc Natl Acad Sci USA 110(1):E99–E107
Chooi YH, Tang Y (2012 Nov 16) Navigating the fungal polyketide chemical space: from genes to molecules. J Org Chem 77(22):9933–9953
Evans BS, Robinson SJ, Kelleher NL (2011 Jan) Surveys of non-ribosomal peptide and polyketide assembly lines in fungi and prospects for their analysis in vitro and in vivo. Fungal Genet Biol 48(1):49–61
Boettger D, Hertweck C (2013 Jan 2) Molecular diversity sculpted by fungal PKS-NRPS hybrids. Chembiochem 14(1):28–42
Donadio S, Monciardini P, Sosio M (2007 Oct) Polyketide synthases and nonribosomal peptide synthetases: the emerging view from bacterial genomics. Nat Prod Rep 24(5):1073–1109
Paumi CM, Chuk M, Snider J, Stagljar I, Michaelis S (2009 Dec) ABC transporters in Saccharomyces cerevisiae and their interactors: new technology advances the biology of the ABCC (MRP) subfamily. Microbiol Mol Biol Rev 73(4):577–593
Wang X (2009 Oct 20) Structure, mechanism and engineering of plant natural product glycosyltransferases. FEBS Lett 583(20):3303–3309
Crouzet J, Roland J, Peeters E, Trombik T, Ducos E, Nader J et al (2013 May) Ntpdr1, a plasma membrane ABC transporter from Nicotiana tabacum, is involved in diterpene transport. Plant Mol Biol 82(1–2):181–192
Gupta RP, Kueppers P, Schmitt L, Ernst R (2011 Jan) The multidrug transporter Pdr5: a molecular diode? Biol Chem 392(1–2):53–60
Syed K, Yadav JS (2012 Nov) P450 monooxygenases (P450ome) of the model white rot fungus Phanerochaete chrysosporium. Crit Rev Microbiol 38(4):339–363
Hansen FT, Sorensen JL, Giese H, Sondergaard TE, Frandsen RJ (2012 Apr 16) Quick guide to polyketide synthase and nonribosomal synthetase genes in Fusarium. Int J Food Microbiol 155(3):128–136
Sa-Correia I, dos Santos SC, Teixeira MC, Cabrito TR, Mira NP (2009 Jan) Drug:H+ antiporters in chemical stress response in yeast. Trends Microbiol 17(1):22–31
Kretzschmar T, Burla B, Lee Y, Martinoia E, Nagy R (2011 Sep 7) Functions of ABC transporters in plants. Essays Biochem 50(1):145–160
Wendlandt S, Lozano C, Kadlec K, Gomez-Sanz E, Zarazaga M, Torres C et al (2013 Feb) The enterococcal ABC transporter gene lsa(E) confers combined resistance to lincosamides, pleuromutilins and streptogramin A antibiotics in methicillin-susceptible and methicillin-resistant Staphylococcus aureus. J Antimicrob Chemother 68(2):473–475
Ma SM, Li JW, Choi JW, Zhou H, Lee KK, Moorthie VA et al (2009 Oct 23) Complete reconstitution of a highly reducing iterative polyketide synthase. Science 326(5952):589–592
Rugbjerg P, Naesby M, Mortensen UH, Frandsen RJ (2013 Apr 4) Reconstruction of the biosynthetic pathway for the core fungal polyketide scaffold rubrofusarin in Saccharomyces cerevisiae. Microb Cell Fact 12(1):31
Lackner G, Misiek M, Braesel J, Hoffmeister D (2012 Dec) Genome mining reveals the evolutionary origin and biosynthetic potential of basidiomycete polyketide synthases. Fungal Genet Biol 49(12):996–1003
Proctor RH, Brown DW, Plattner RD, Desjardins AE (2003 Mar) Co-expression of 15 contiguous genes delineates a fumonisin biosynthetic gene cluster in Gibberella moniliformis. Fungal Genet Biol 38(2):237–249
Ro DK, Ouellet M, Paradise EM, Burd H, Eng D, Paddon CJ et al (2008) Induction of multiple pleiotropic drug resistance genes in yeast engineered to produce an increased level of anti-malarial drug precursor, artemisinic acid. BMC Biotechnol 8:83
Soukup AA, Chiang YM, Bok JW, Reyes-Dominguez Y, Oakley BR, Wang CC et al (2012 Oct) Overexpression of the Aspergillus nidulans histone 4 acetyltransferase EsaA increases activation of secondary metabolite production. Mol Microbiol 86(2):314–330
Christianson DW (2008 Apr) Unearthing the roots of the terpenome. Curr Opin Chem Biol 12(2):141–150
Davis EM, Croteau R (2000) Cyclization enzymes in the biosynthesis of monoterpenes, sesquiterpenes, and diterpenes. Biosynthesis: aromatic polyketides, isoprenoids, alkaloids. Springer, Berlin, p 53–95
Mitsuguchi H, Seshime Y, Fujii I, Shibuya M, Ebizuka Y, Kushiro T (2009 May 13) Biosynthesis of steroidal antibiotic fusidanes: functional analysis of oxidosqualene cyclase and subsequent tailoring enzymes from Aspergillus fumigatus. J Am Chem Soc 131(18):6402–6411
Kimura M, Kushiro T, Shibuya M, Ebizuka Y, Abe I (2010 Jan 1) Protostadienol synthase from Aspergillus fumigatus: functional conversion into lanosterol synthase. Biochem Biophys Res Commun 391(1):899–902
Cane DE, Kang I (2000 Apr 15) Aristolochene synthase: purification, molecular cloning, high-level expression in Escherichia coli, and characterization of the Aspergillus terreus cyclase. Arch Biochem Biophys 376(2):354–364
Proctor RH, Hohn TM (1993 Feb) Aristolochene synthase—isolation, characterization, and bacterial expression of a sesquiterpenoid biosynthetic gene (Ari1) from Penicillium roqueforti. J Biol Chem [Article] 268(6):4543–4548
Engels B, Heinig U, Grothe T, Stadler M, Jennewein S (2011 Mar 4) Cloning and characterization of an Armillaria gallica cDNA encoding protoilludene synthase, which catalyzes the first committed step in the synthesis of antimicrobial melleolides. J Biol Chem 286(9):6871–6878
McCormick SP, Alexander NJ, Harris LJ (2010 Jan) Clm1 of Fusarium graminearum encodes a longiborneol synthase required for culmorin production. Appl Environ Microbiol 76(1):136–141
Brock NL, Tudzynski B, Dickschat JS (2011 Oct 11) Biosynthesis of sesqui- and diterpenes by the gibberellin producer Fusarium fujikuroi. Chembiochem 12(17):2667–12
Quin MB, Flynn CM, Wawrzyn GT, Choudhary S, Schmidt-Dannert C (2013) Mushroom hunting using bioinformatics: application of a predictive framework facilitates the selective identification of sesquiterpene synthases in basidiomycota. Chembiochem 14(18):2480–91
Citron CA, Gleitzmann J, Laurenzano G, Pukall R, Dickschat JS (2012 Jan 23) Terpenoids are widespread in actinomycetes: a correlation of secondary metabolism and genome data. Chembiochem 13(2):202–214
Novak R (2011 Dec) Are pleuromutilin antibiotics finally fit for human use? Ann N Y Acad Sci 1241:71–81
Shen JW, Ruan Y, Ma BJ (2009 Jun) Diterpenoids of macromycetes. J Basic Microbiol 49(3):242–255
Toyomasu T (2008 May) Recent advances regarding diterpene cyclase genes in higher plants and fungi. Biosci Biotechnol Biochem 72(5):1168–1175
Troncoso C, Gonzalez X, Bomke C, Tudzynski B, Gong F, Hedden P et al (2010 Aug) Gibberellin biosynthesis and gibberellin oxidase activities in Fusarium sacchari, Fusarium konzum and Fusarium subglutinans strains. Phytochemistry 71(11–12):1322–1331
Bomke C, Tudzynski B (2009 Oct-Nov) Diversity, regulation, and evolution of the gibberellin biosynthetic pathway in fungi compared to plants and bacteria. Phytochemistry 70(15–16):1876–1893
Bromann K, Toivari M, Viljanen K, Vuoristo A, Ruohonen L, Nakari-Setala T (2012) Identification and characterization of a novel diterpene gene cluster in Aspergillus nidulans. PLoS ONE 7(4):e35450
Noike M, Liu C, Ono Y, Hamano Y, Toyomasu T, Sassa T et al (2012 Mar 5) An enzyme catalyzing O-prenylation of the glucose moiety of fusicoccin A, a diterpene glucoside produced by the fungus Phomopsis amygdali. Chembiochem 13(4):566–573
Noike M, Ono Y, Araki Y, Tanio R, Higuchi Y, Nitta H et al (2012) Molecular breeding of a fungus producing a precursor diterpene suitable for semi-synthesis by dissection of the biosynthetic machinery. PLoS ONE 7(8):e42090
Ono Y, Minami A, Noike M, Higuchi Y, Toyomasu T, Sassa T et al (2011 Mar 2) Dioxygenases, key enzymes to determine the aglycon structures of fusicoccin and brassicicene, diterpene compounds produced by fungi. J Am Chem Soc 133(8):2548–2555
Toyomasu T, Kaneko A, Tokiwano T, Kanno Y, Kanno Y, Niida R et al (2009 Feb 20) Biosynthetic gene-based secondary metabolite screening: a new diterpene, methyl phomopsenonate, from the fungus Phomopsis amygdali. J Org Chem 74(4):1541–1548
Toyomasu T, Niida R, Kenmoku H, Kanno Y, Miura S, Nakano C et al (2008 Apr) Identification of diterpene biosynthetic gene clusters and functional analysis of labdane-related diterpene cyclases in Phomopsis amygdali. Biosci Biotechnol Biochem 72(4):1038–1047
Fujii R, Minami A, Tsukagoshi T, Sato N, Sahara T, Ohgiya S et al (2011) Total biosynthesis of diterpene aphidicolin, a specific inhibitor of DNA polymerase alpha: heterologous expression of four biosynthetic genes in Aspergillus oryzae. Biosci Biotechnol Biochem 75(9):1813–1817
Chiba R, Minami A, Gomi K, Oikawa H (2013 Feb 1) Identification of ophiobolin F synthase by a genome mining approach: a sesterterpene synthase from Aspergillus clavatus. Org Lett 15(3):594–597
Itoh T, Tokunaga K, Matsuda Y, Fujii I, Abe I, Ebizuka Y et al (2010 Oct) Reconstitution of a fungal meroterpenoid biosynthesis reveals the involvement of a novel family of terpene cyclases. Nat Chem 2(10):858–864
Guo CJ, Knox BP, Chiang YM, Lo HC, Sanchez JF, Lee KH et al (2012 Nov 16) Molecular genetic characterization of a cluster in A. terreus for biosynthesis of the meroterpenoid terretonin. Org Lett 14(22):5684–5687
Matsuda Y, Awakawa T, Itoh T, Wakimoto T, Kushiro T, Fujii I et al (2012 Aug 13) Terretonin biosynthesis requires methylation as essential step for cyclization. Chembiochem 13(12):1738–1741
Itoh T, Tokunaga K, Radhakrishnan EK, Fujii I, Abe I, Ebizuka Y et al (2012 May 29) Identification of a key prenyltransferase involved in biosynthesis of the most abundant fungal meroterpenoids derived from 3,5-dimethylorsellinic acid. Chembiochem 13(8):1132–1135
Lin HC, Chooi YH, Dhingra S, Xu W, Calvo AM, Tang Y (2013 Mar 27) The fumagillin biosynthetic gene cluster in Aspergillus fumigatus encodes a cryptic terpene cyclase involved in the formation of beta-trans-bergamotene. J Am Chem Soc 135(12):4616–4619
Lo HC, Entwistle R, Guo CJ, Ahuja M, Szewczyk E, Hung JH et al (2012 Mar 14) Two separate gene clusters encode the biosynthetic pathway for the meroterpenoids austinol and dehydroaustinol in Aspergillus nidulans. J Am Chem Soc 134(10):4709–4720
Tagami K, Liu C, Minami A, Noike M, Isaka T, Fueki S et al (2013 Jan 30) Reconstitution of biosynthetic machinery for indole-diterpene paxilline in Aspergillus oryzae. J Am Chem Soc 135(4):1260–1263
Saikia S, Nicholson MJ, Young C, Parker EJ, Scott B (2008 Feb) The genetic basis for indole-diterpene chemical diversity in filamentous fungi. Mycol Res 112(Pt 2):184–199
Young C, McMillan L, Telfer E, Scott B (2001 Feb) Molecular cloning and genetic analysis of an indole-diterpene gene cluster from Penicillium paxilli. Mol Microbiol 39(3):754–764
Young CA, Felitti S, Shields K, Spangenberg G, Johnson RD, Bryan GT et al (2006 Oct) A complex gene cluster for indole-diterpene biosynthesis in the grass endophyte Neotyphodium lolii. Fungal Gen Biol 43(10):679–693
Young CA, Tapper BA, May K, Moon CD, Schardl CL, Scott B (2009 Apr) Indole-diterpene biosynthetic capability of Epichloë endophytes as predicted by ltm gene analysis. Appl Environ Microbiol 75(7):2200–2211
Teixeira MC, Godinho CP, Cabrito TR, Mira NP, Sa-Correia I (2012) Increased expression of the yeast multidrug resistance ABC transporter Pdr18 leads to increased ethanol tolerance and ethanol production in high gravity alcoholic fermentation. Microb Cell Fact 11:98
Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D et al (2013 Apr 25) High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496(7446):528–532
Chanda A, Roze LV, Linz JE (2010 Nov) A possible role for exocytosis in aflatoxin export in Aspergillus parasiticus. Eukaryot Cell 9(11):1724–1727
Chanda A, Roze LV, Kang S, Artymovich KA, Hicks GR, Raikhel NV et al (2009 Nov 17) A key role for vesicles in fungal secondary metabolism. Proc Natl Acad Sci USA 106(46):19533–19538
Menke J, Weber J, Broz K, Kistler HC (2013) Cellular development associated with induced mycotoxin synthesis in the filamentous fungus Fusarium graminearum. PLoS ONE 8(5):e63077
Menke J, Dong Y, Kistler HC (2012 Nov) Fusarium graminearum Tri12p influences virulence to wheat and trichothecene accumulation. Mol Plant Microbe Interact 25(11):1408–1418
Bai J, Swartz DJ, Protasevich, II, Brouillette CG, Harrell PM, Hildebrandt E et al (2011) A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PLoS ONE 6(8):e22577
Bernier SG, Lazarus DD, Clark E, Doyle B, Labenski MT, Thompson CD et al (2004 Jul 20) A methionine aminopeptidase-2 inhibitor, PPI-2458, for the treatment of rheumatoid arthritis. Proc Natl Acad Sci USA 101(29):10768–10773
Schobert R, Knauer S, Seibt S, Biersack B (2011) Anticancer active illudins: recent developments of a potent alkylating compound class. Curr Med Chem 18(6):790–807
Seong KY, Pasquali M, Zhou X, Song J, Hilburn K, McCormick S et al (2009 Apr) Global gene regulation by Fusarium transcription factors Tri6 and Tri10 reveals adaptations for toxin biosynthesis. Mol Microbiol 72(2):354–367
Acknowledgements
Research on terpenoid biosynthesis in C.S-D’s laboratory is supported by the National Institute of Health Grant GM080299 (to C.S-D.). G.T.W. and S.E.B. were supported by the predoctoral National Institute of Health traineeship: GM08700 (G.T.W.) and GM008347 (S.E.B.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Wawrzyn, G., Held, M., Bloch, S., Schmidt-Dannert, C. (2015). Genome Mining for Fungal Secondary Metabolic Gene Clusters. In: Zeilinger, S., Martín, JF., García-Estrada, C. (eds) Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites, Volume 2. Fungal Biology. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2531-5_4
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2531-5_4
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2530-8
Online ISBN: 978-1-4939-2531-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)