
Abstract
The discovery of central sleep apnea (CSA) at high altitude is usually attributed to Angelo Mosso who published in 1898. It can occur in susceptible individuals at altitude above 2000 m, but at very high altitude, say above 5000 m, it will occur in most subjects. Severity is correlated with ventilatory responsiveness, particularly to hypoxia. Theoretically, it should spontaneously improve with time and acclimatization. Although the time course of resolution is not well described, it appears to persist for more than a month at 5000 m.
It occurs due to the interaction of hypocapnia with stages 1 and 2 NREM sleep, in the presence of increased loop-gain. The hypocapnia is secondary to hypoxic ventilatory drive. With acclimatization, one might expect that the increase in PaO2 and cerebral blood flow (CBF) would mitigate the CSA. However, over time, both the hypoxic and hypercapnic ventilatory responses increase, causing an increase in loop gain which is a counteracting force.
The severity of the CSA can be reduced by descent, supplemental oxygen therapy, oral or intravenous acetazolamide. Recent studies suggest that acute further increases in cerebral blood flow will substantially, but temporarily, reduce central sleep apnea, without altering acid based balance. Very recently, bi-level noninvasive ventilation has also been shown to help (mechanism unknown). Sleep quality can be improved independent of the presence of CSA by the use of benzodiazepine sedation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Central sleep apnea (CSA) at high altitude typically consists of 2–4 breaths, separated by an apnea from the next burst of 2–4 breaths, which in appearance closely resembles the periodic breathing of the premature infant [34]. It is different from the typical waxing and waning of tidal volume that one sees in the periodic breathing of heart failure [6], or the somewhat chaotic or irregular appearance of apneas associated with opiate use [29]. The description of CSA at high altitude is usually attributed to Angelo Mosso who published his description in 1898 and included an illustration of the periodic breathing recorded on his brother [21]. Typically it occurs at altitudes above 2000 m of varying severity, depending on characteristics of the individuals, but above 5000 m altitude it occurs in most people [4, 33]. The bursts of breathing (hyperpneas) are associated with arousal from sleep and sometimes full wakefulness, which causes tiredness during the day and cognitive impairment [1], similar to that seen from other causes of sleep disruption. The severity of CSA has been correlated with ventilatory responsiveness, particularly to hypoxia [17, 18, 32]. Intuitively, one might expect it to improve with acclimatization; however, the time course of resolution is not well described. Our experiments, of up to 2 weeks duration at 5000 m, have shown worsening of the CSA with acclimatization [3, 4]. Salvaggio et al. [24], over a period of 1 month at the same altitude, but in only five subjects, showed no diminution in the severity of CSA over that period.
2 Mechanisms
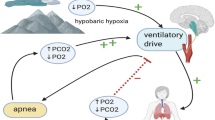
Although severity of CSA has been traditionally linked to hypoxic ventilatory responsiveness, there are other concepts that are also probably important to our understanding of the mechanisms of OSA at high altitude: The concept that CSA is caused by a disproportionate elevation of either hypoxic or hypercapnic ventilation response compared to the other is a relatively new and plausible theory [28]. The engineering concept of “loop gain” has been around since the 1980s in the respiratory control literature as a key cause of CSA [16]. More recently, alterations of cerebral blood flow have been proposed as a potential key factor in CSA [35] (see Fig. 19.1). Figure 19.1 shows a very tight correlation between the degree of fall in cerebral blood flow (CBF) at sleep onset and the subsequent degree of CSA during sleep. This suggests that a fall in CBF during sleep promotes CSA.

(a) The relationship between change in CBF at sleep onset and subsequent CSA during sleep. (b) Summary of known effects of acclimatization at 5050 m relevant to central sleep apnea. (c) A summary of our speculation as to the mechanisms of worsening CSA during 2 weeks acclimatization
CSA occurs in light sleep, typically Stages 1 and 2 of non-rapid eye movement (NREM ) sleep, when the patient has crossed the “apnea threshold” [7], which often means in practice that their arterial PCO2, [and hence brain PCO2, (PbCO2)] has fallen a few millimeters lower than the resting PbCO2 when they go into light sleep. The acute event is often triggered by a sigh or arousal from sleep, which causes a sudden drop in PaCO2. The baseline hypocapnia at altitude is secondary to hypoxic ventilatory drive [17].
For the initial apnea to develop into sustained periodic breathing or CSA, there must be an increase in the “loop gain” of the feedback control system above 1 [15]. The calculation of absolute loop gain is problematic, although Edwards et al. [11] believe they have discovered a mathematical formula that is applicable to the periodic breathing of newborns and premature animals, and possibly to the clinical context of high altitude CSA. Commonly one uses a surrogate for absolute loop gain, which can be derived from the polysomnogram and compared from subject to subject and from time period to time period. It is the relationship between the lengths of the hyperpneas and apneas. Put simply, “loop gain” is the response of a feedback control system divided by the stimulus [30]. Different groups have regarded the hyperpneic phase of CSA as either the stimulus [11] or the response [30], and vice versa for the apnea. In practice, it does not matter: Let us assume that hyperpnea is the stimulus, in which case, if a subject has a long apnea after a short hyperpnea , then by definition the response, (the length of the apnea), over the stimulus, (the length of the hyperpnea), is large, whereas if the hyperpnea was long and the apnea short, then loop gain must be much smaller in that situation. We have previously shown an increase in loop gain with acclimatization at 5000 m [3].
It is generally accepted that the severity of CSA at high altitude is strongly correlated with the ventilatory response to hypoxia. Kellogg [17] was one of the first to show a correlation between the slope of the ventilatory response to hypoxia and the severity of CSA. Supporting that view, Hackett and Roach have shown that almitrine, (a stimulus to hypoxic ventilatory response ), will increase CSA in normal volunteers, whereas acetazolamide, (which among other actions acutely inactivates carotid chemoreceptors), would suppress CSA [12]. But that may be only part of the explanation. Studies of over 2 weeks duration in 2005 at 3840 m and in 2008 at 5050 m, have found an approximate doubling of both the hypoxic and hypercapnic ventilatory responses in normal volunteers in the transition between low altitude and high altitude [2]. They confirm a similar observation by White et al. [31]. Topor and Remmers have shown, in a computer model, that unstable breathing, due to high loop gain, is more likely to occur at high altitude if there is a disproportion between the hypoxic and hypercapnic ventilatory responses [28]. So a high hypercapnic response coupled with a low hypoxic ventilatory response, could also cause CSA.
It is somewhat surprising that CSA would increase in severity over 2 weeks of acclimatization at 5000 m, and yet not begin to improve by 4 weeks at the same altitude, because by then arterial blood gas values have started to return towards sea level values.
Upon arrival at high altitude, normal subjects will already have established hypocapnia, secondary to increased minute ventilation, due to the hypoxic stimulus to breathe. However, their PaCO2 will not have reached its optimal and lowest level initially because of “hypocapnic braking” [20], which is an effect of the acute respiratory alkalosis affecting the central chemoreceptors inhibiting ventilation. Over time, renal excretion of bicarbonate starts to restore the arterial (and presumably brain) pH from alkaline towards neutral values [8]. This reduces the braking effects of the alkalosis and allows minute ventilation to increase further, with a further fall in PaCO2 and (through the alveolar gas equation) a rise in PaO2.
Initially, the sympathetic system is activated, so that cardiac output and mean arterial pressures (and CBF) are higher than at sea level [13]. Over a period of 2 weeks at 5000 m, cerebral blood flow returns to, or close to, sea level values [19], although sympathetic activation remains high [13]. Ventilatory responses to hypoxia and hypercapnia will approximately double for a group of subjects over that 2 week period [2]. Many of these changes generate counteracting forces; the fall in PaCO2 and presumably PbCO2, could be expected to increase the propensity to CSA, however the increase in PaO2 could tend to counteract that. The initial high cerebral blood flow could be expected to wash out CO2 from around the brain stem central chemoreceptors and so initially reduce the ventilatory response to CO2 and hence perhaps reduce periodic breathing. Over time, as cerebral blood flow returns to sea level values [19], PbCO2 may rise despite a lower arterial PaCO2, although that is a speculation.
Experiments designed to tease out the relative importance of changes in PaCO2, ventilatory responses and cerebral blood flow, have not so far clarified this complex issue. Intravenous acetazolamide has been shown to increase cerebral blood flow by approximately 30 % at high altitude and this has been associated with a significant fall in central apnea-hypopnea index (AHI) [3]. Oral indomethacin has been shown to reduce cerebral blood flow by approximately 30 %, but this has been associated with an insignificant increase in central AHI [3]. There was no significant difference in ventilatory responses to hypercapnia between the two post drug conditions and yet there was a strong negative correlation between change in CBF and change in CSA severity. The issue, however, is clouded by the effect of the acetazolamide on arterial PCO2, which caused an acute rise of 3 mmHg. An acute rise of that size could be expected to inhibit CSA by itself.
3 Treatments
Since there is a strong correlation between absolute altitude and severity of CSA [4, 33] the obvious treatment would be to reverse that process and descend. If that were not feasible, or desirable, then Lahiri has shown elegantly the curative effects of supplemental oxygen therapy on a subject with sustained CSA at 5300 m [18] (see Fig. 19.2). We have shown similar transient benefits in the artificial situation of normobaric hypoxia, created using a nitrogen tent, in which a patient with established obstructive sleep apnea (OSA) could be converted, after several hours’ exposure, to a simulated altitude of 2750 m, (approximately 15 % oxygen environment), to sustained CSA [5]. Introduction of supplemental oxygen into the subject’s face mask quickly terminated the CSA and allowed the underlying OSA to reemerge.

The effect of oxygen breathing upon periodic breathing (above) and arterial oxygen saturation (below) during sleep at 17,700 ft (5400 m). Periodic breathing is replaced by shallow, continuous breathing as arterial oxygen saturation is increased. From Lahiri et al. [18]
Oral acetazolamide has been shown by a number of authors to effectively suppress CSA at high altitude [12, 25, 27]. This has been attributed to the development of a metabolic acidosis, rather than the effect that one sees with rapid intravenous infusion of acetazolamide. In the acute intravenous administration situation, acid based balance does not change, nor ventilatory responses, but cerebral blood flow increases [35] due to paralysis of vasoconstriction in the cerebral arteries and there is a step up of PaCO2 [3], presumably due to paralysis of carbonic anhydrase in the subject’s red cells [26]. Regular oral administration, on the other hand, causes metabolic acidosis, which moves the subjects away from their apnea threshold and has a similar effect to adding CO2 to the subject’s breathing mix.
A very different treatment (bi-level ventilation) has recently been shown in a pilot study to halve the severity of CSA in seven volunteers at 3800 m at White Mountain. The author [14] unfortunately did not collect arterial blood gases, nor measure ventilatory responses or cerebral blood flow, so the mechanism of that effect is uncertain. One could speculate that the ventilation further reduced PaCO2 and raised PaO2, however, one would expect the further fall in CO2, would favor CSA. Noninvasive positive pressure ventilation (NIPPV), like continuous positive airway pressure (CPAP), raises functional residual capacity (FRC), which would increase oxygen stores and hence lower loop gain. That may be the main mechanism with NIPPV because Edwards et al. have shown a reduction in loop gain in premature lambs by the application of CPAP, with resolution of CSA [10]. In the context of chronic severe heart failure with CSA, CPAP has been shown to have a sympatholytic effect [22], which may also have been a factor.
Separate from treatments that affect the severity of CSA, other treatments for the sleep disturbances associated with the CSA, have been used with varying results (see Table 19.1). Dubowitz [9] and Nickol et al. [23] have used temazepam at 5400 m. Both have shown a subjective improvement in sleep quality, but with varying effects on saturation and CSA severity. Dubowitz, in a group of 11 subjects, showed no change in mean arterial saturation, but appeared to show a reduction in “desaturation events”, probably indicating a reduction in CSA severity linked to arousal from sleep, although no measurements of sleep state were recorded [9].
Nickol et al. [23], on the other hand, showed a modest but significant reduction in CSA index, from 16/h to 9/h, in a group of 33 healthy volunteers. There was a small reduction in mean saturation from 78 to 76 %. They claim to have found a reduction in acute mountain sickness scores.
New non-benzodiazepine sedative hypnotics have also been studied at high altitude [1]. Sleep quality was improved, but no direct data were provided about effects on CSA, although there was no change in oxygen desaturatio n index (see Table 19.1).
References
Beaumont M, Batejat D, Pierard C, Van Beers P, Phillippe M, Leger D, Savourey G, Jouanin JC. Zaleplon and zolpiden objectively alleviate sleep disturbances in mountaineers at a 3,613 meter altitude. Sleep. 2007;30(11):1527–33.
Burgess K, Burgess K, Subedi P, Ainslie P, Topor Z, Whitelaw W. Prediction of periodic breathing at altitude. Adv Exp Med Biol. 2008;605:442–6.
Burgess KR, Dawson A, Shepherd K, Swart M, Thomas KN, Fan JL, Lucas RAI, Lucas SJE, Cotter JD, Peebles KC, Basnyat R, Ainslie P (2009) Separate effects of acclimatisation and cerebral blood flow on central sleep apnea at high altitude. Abstract. International Hypoxia Symposium, Lake Louise, Canada
Burgess KR, Johnson PL, Edwards N. Central and obstructive sleep apnoea during ascent to high altitude. Respirology. 2004;9:222–9.
Burgess KR, Saye P, Cheong M, Lee CS, Worthington JM. Upper airway resistance in OSA patients during exposure to simulated high altitude. Am J Respir Crit Care Med. 2008;177:A595.
Cheyne J. A case of apoplexy in which the fleshy part of the heart was converted to fat. In: Dublin Hospital Records, vol. 2. Cambridge, MA: Harvard University Press; 1818. p. 216–23.
Dempsey JA. Crossing the apnoeic threshold: causes and consequences. Exp Physiol. 2005;90(1):13.
Dempsey JA, Forster HV, De Pico GA. Ventilatory acclimatisation to moderate hypoxemia in man. The role of spin fluid (H+). J Clin Invest. 1974;53(4):1091–100.
Dubowitz G. Effect of temazepam on oxygen saturation and sleep quality at high altitude: randomised placebo controlled crossover trial. Br Med J. 1998;316(7131):587–9.
Edwards BA, Sands SA, Feeney C, Skuza EM, Brodecky V, Wilkinson MH, Berger PJ. Continuous positive airway pressure reduces loop gain and resolves periodic central apnoeas in the lamb. Respir Physiol Neurobiol. 2009;168:239–49.
Edwards BA, Sands SA, Skuza EM, Stocks EM, Brodecky V, Wilkinson M, Berger P. Increased peripheral chemosensitivity via dopaminergic manipulation promotes respiratory instability in lambs. Respir Physiol Neurobiol. 2008;164:419–28.
Hackett PH, Roach RC, Harrison GL, Schoene RB, Mills Jr WJ. Respiratory stimulants and sleep periodic breathing at high altitude. Almitrine versus acetazolamide. Am Rev Respir Dis. 1987;135(4):896–8.
Hansen J, Sander M. Sympathetic neural overactivity in healthy humans after prolonged exposure to hypobaric hypoxia. J Physiol. 2003;546(3):921–9.
Johnson P, Popa DA, Prisk GK, Edwards N, Sullivan CE. Non-invasive positive pressure ventilation during sleep at 3800 m: relationship to acute mountain sickness and sleeping oxyhaemoglobin saturation. Respirology. 2010;15(2):191–3.
Khoo MCK, Anholm JD, Ko SW, Downey III R, Powles AC, Sutton JR, Houston CS. Dynamics of periodic breathing and arousal during sleep at extreme altitude. Respir Physiol. 1996;103:33–43.
Khoo MCK, Kronauer RE, Strohl KP, Slutsky AS. Factors inducing periodic breathing in humans: a general model. J Appl Physiol. 1982;53(3):644–59.
Kellogg R. Altitude acclimatisation. A historical introduction emphasizing the regulation of breathing. Physiologist. 1968;11:37–57.
Lahiri S, Maret K, Sherpa MG. Dependence of high altitude sleep apnea on ventilatory sensitivity to hypoxia. Respir Physiol. 1983;52:281–301.
Lucas SJ, Burgess KR, Thomas KN, Donnelly J, Peebles KC, Lucas RA, Fan JL, Cotter JD, Basnyat R, Ainslie PN. Alterations in cerebral blood flow & cerebrovascular reactivity during 14 days at 5050. J Physiol. 2011;589:741–53.
Moore LG, Huang SY, McCullough RE, Sampson JB, Maher JT, Weil JV, Grover RF, Alexander JK, Reeves JT. Variable inhibition by falling CO2 of hypoxic ventilatory response in humans. J Appl Physiol. 1984;56:207–10.
Mosso A. A life of man on the high alps. London: Unwin; 1898.
Naughton MT, Bernard DC, Lui PP, Rutherford R, Rankin F, Bradley TD. Effects of nasal CPAP therapy on sympathetic activity in patients with hart failure and central sleep apnea. Am J Respir Crit Care Med. 1995;152:473–9.
Nickol AH, Leverment J, Richards P, Seal P, Harris GA, Cleland J, Dubowitz G, Collier DJ, Milledge J, Stradling JR, Morrell MJ. Temazepam at high altitude reduces periodic breathing without impairing next-day performance: a randomized cross-over double-blind study. J Sleep Res. 2006;15(4):445–54.
Salvaggio A, Insalaco G, Marrone O, Romano S, Braghiroli A, Lanfranchi P, Patruno V, Donner CF, Bonsignore G. Effects of high-altitude periodic breathing on sleep and arterial oxyhaemoglobin saturation. Eur Respir J. 1998;12:408–13.
Sutton JR, Gray GW, Houston CS, Powles AC. Effects of duration at altitude and acetazolamide on ventilation and oxygenation during sleep. Sleep. 1980;3(3/4):455–64.
Swenson ER. Respiratory and renal roles of carbonic anhydrase in gas exchange and acid base regulation. In: Chegwidden WR, Carter ND, Edwards YH, editors. The carbonic anhydrases: new horizons. London: Birkhauser Press; 1999.
Swenson ER, Leatham KL, Roach RC, Schoene RB, Mills Jr WJ, Hackett PH. Renal carbonic anhydrase inhibition reduces high altitude sleep periodic breathing. Respir Physiol. 1991;86(3):333–43.
Topor ZL, Vasilakos K, Remmers JE. Stability analysis of the respiratory control system during sleep. Adv Exp Med Biol. 2004;551:203–9.
Wang D, Teichtahl H. Opiods, sleep architecture and sleep disordered breathing. Sleep Med Rev. 2007;11(1):35–46.
White DP, Gleeson K, Pichett CK, Douglas NJ, Findley LJ, Weil JV. Central sleep apnoea. Improvement with acetazolamide therapy. Arch Intern Med. 1982;142:1816–9.
White DP, Gleeson K, Pickett CK, Rannels AM, Cymerman A, Weil JV. Altitude acclimatisation: influence on periodic breathing and chemoresponsiveness during sleep. J Appl Physiol. 1987;63:401–12.
Weil JV, Byrne-Quinn E, Sodal IE, Friesen WO, Underhill B, Filley GF, Grover RF. Hypoxic ventilatory drive in normal man. J Clin Invest. 1970;49:1061–72.
West J, Peters Jr RM, Aksnes G, Maret KH, Milledge JS, Schoene RB. Nocturnal periodic breathing at altitudes of 6300 and 8050m. J Appl Physiol. 1986;61:280–7.
Wilkinson MH, Berger PJ, Blanch N, Brodecky V, Jones C. Source of respiratory drive during periodic breathing in lambs. Respir Physiol. 1996;104:115–26.
Xie A, Skatrud JB, Morgan B, Chenuel B, Khayat R, Reichmuth K, Lin J, Dempsey JA. Influence of cerebrovascular function on the hypercapnic ventilatory response in healthy humans. J Physiol. 2006;577:319–29.
Acknowledgements
We would like to thank Ms Sue Coulson for preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this chapter
Cite this chapter
Burgess, K.R., Ainslie, P.N. (2016). Central Sleep Apnea at High Altitude. In: Roach, R., Hackett, P., Wagner, P. (eds) Hypoxia. Advances in Experimental Medicine and Biology, vol 903. Springer, Boston, MA. https://doi.org/10.1007/978-1-4899-7678-9_19
Download citation
DOI: https://doi.org/10.1007/978-1-4899-7678-9_19
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4899-7676-5
Online ISBN: 978-1-4899-7678-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)