
Abstract
Environmental exposures to factors such as toxicants or nutrition can have impacts on testis biology and male fertility. The ability of these factors to influence epigenetic mechanisms in early life exposures or from ancestral exposures will be reviewed. A growing number of examples suggest environmental epigenetics will be a critical factor to consider in male reproduction.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Environmental toxicants present in the environment, from either synthetic or natural origins, can influence physiological responses and developmental processes in organisms. Some of these compounds interfere with the action of endogenous hormones at several physiological levels and so are categorized as endocrine disruptors (Schug et al. 2011). Industrialization and the progressive accumulation of synthetic endocrine disruptors in the environment has altered the ecological balances in natural populations and affected human health (Balabanic et al. 2011). These compounds are present in cosmetics, food items and containers, packaging materials, toys, agrochemicals, and in practically every manufactured product with which humans have contact. These toxicants are often associated with increased incidence of reproductive disease (Balabanic et al. 2011; Caserta et al. 2011; Fowler et al. 2012). Research has demonstrated that exposure to environmental factors such as environmental contaminants, stress, or dietary compounds early during fetal and postnatal development have a significant impact on human health (Guillette and Iguchi 2012). Many common human diseases have seen a dramatic increase in incidence in the past decade. Exposure to environmental factors are estimated to account for 40 % of deaths worldwide (Pimentel et al. 2007), which includes the majority of cancers being linked to environmental exposures. Regional environmental influences are an important component in noninfectious disease incidence (Wallace 2010). For example, regional variations exist in cancer incidence worldwide (Forouzanfar et al. 2011; Parkin 2004). Differences in lifestyles, exposure to dietary compounds, or environmental toxicants are the primary factors involved. Regions of the world with high consumption of salt, processed meat, and N-nitroso compounds are associated with increased risk of gastric cancer (Tsugane and Sasazuki 2007). Other noninfectious diseases are correlated with exposure to environmental toxicants. For example, human populations that are highly exposed to arsenic tend to present increased susceptibility to develop liver, bladder, skin, and lung cancer (Anetor et al. 2007).
One of the disease states that has emerged as a result of exposure to environmental factors is the increasing incidence of abnormalities of the male reproductive system (Giwercman and Giwercman 2011). Recent epidemiological trends indicate changes in the incidence of several pathologies of the male reproductive tract in humans in recent decades such as decreases in sperm count and quality (Sharpe 2010) and increases in testicular cancer (Skakkebaek et al. 2007) or suggested increases in cryptorchidism or hypospadias (Main et al. 2010). These trends have led authors to group these male reproductive conditions into the complex disease trait of testicular dysgenesis syndrome, which has been associated with the environmental exposures that humans have been subjected to in recent decades (Giwercman and Giwercman 2011; Skakkebaek et al. 2001). A number of examples exist of a direct correlation between environmental toxicants and effects on male reproductive health. Accidental in utero exposures of humans to the synthetic organic pollutants polychlorinated biphenyls and polychlorinated dibenzofurans in Taiwan was reported to produce a marked effect in semen quality and motility (Guo et al. 2000, 2004). Cases of massive agroworker pesticide-induced sterilization have been observed in California (1970s) (Whorton et al. 1979) and in Costa Rica (early 1960s to 1984) (Thrupp 1991) due to exposure to nematicide 1,2-Dibromo-3-chloropropane (DBCP). Exposure to naturally available estrogenic compounds has also been associated with reduced fertility in male animals. For example, the identification of phytoestrogens as having estrogenic or reproductive effects in animals started with observations from farmers in New Zealand who found that ewes would become infertile after eating clover (Adams 1981, 1990). The same effect was further reported in cattle (Adams 1995). Understanding the basic developmental biology of the male reproductive tract (e.g., testis) and mechanisms of action of these environmental factors is reviewed below.
Epigenetic and Transgenerational Effects of Environmental Exposures
Epigenetics is defined as molecular factors around the DNA that regulate genome activity independent of DNA sequence and that are mitotically stable (Skinner 2011; Skinner et al. 2010). The factors involved include DNA methylation, histone modification, chromatin structure, and noncoding RNAs. Environmental epigenetics involves the ability of environmental factors to alter epigenetic marks that then alter genome activity and cellular function (Skinner et al. 2010). Since the vast majority of environmental factors cannot influence or alter DNA sequence, epigenetics provides an efficient mechanism to mediate environmental impacts on biology (Skinner 2011). Many research groups have documented the epigenetic actions of environmental exposures. Environmental factors can directly influence epigenetic marks that generate phenotypic variation that includes the induction of disease such as subfertility and imprinting disorders (Inbar-Feigenberg et al. 2013). Epigenetic tools will help identify etiological factors causing specific molecular pathologies (Ogino et al. 2013). For example, environmental effects such as trauma, stress, or disorganized attachment can induce epigenetic changes in the brain to cause long-term effects on the regulation of the genome function to promote psychopathology, such as schizophrenia (Gonzalez-Pardo and Perez Alvarez 2013). Studies have shown that early-life environment and epigenetics have an important role in a variety of diseases, such as cardiovascular disease (Sun et al. 2013), allergies (North and Ellis 2011), and asthma (Karmaus et al. 2013). Several environmental factors have been described as causing epigenetic effects, including hypoxia (Yuen et al. 2013), phytochemicals (Guerrero-Bosagna and Skinner 2012), organic environmental toxicants (Manikkam et al. 2012a), inorganic compounds (Cheng et al. 2012), and nanosized materials (Stoccoro et al. 2012).
A number of environmental exposures have been shown to produce transgenerational effects on disease and phenotypic variation (Anway et al. 2005; Skinner et al. 2010). Epigenetic transgenerational inheritance processes involve key features such as the action of environmental toxicants on gestating females during the period of fetal gonadal sex determination resulting in generational phenotypes (Skinner et al. 2010). Since the gestating female (F0 generation), fetus (F1 generation), and fetal germline (F2 generation) are directly exposed, phenotypes in these generations are due to multigenerational exposures. Interestingly, the occurrence of phenotypes for three generations or more, following the initial F0 generation exposure, constitutes an epigenetic transgenerational inheritance phenomenon (Skinner 2011; Skinner et al. 2010). The role of germline in transmitting epigenomes is essential for this phenomenon and is becoming well established in several different organisms (Arico et al. 2011; Carone et al. 2010; Dunn and Bale 2011; Guerrero-Bosagna et al. 2010; Morgan and Bale 2011). During the initiation of development of the germline, a major DNA methylation erasure occurs followed by a reestablishment of DNA methylation patterns (Lees-Murdock and Walsh 2008; Reik et al. 2001). DNA methylation erasure takes place during the migration of primordial germ cells to the genital ridge and gonad, and then remethylation is initiated during the first events of sex determination (Allegrucci et al. 2005; Durcova-Hills et al. 2006). This period in germ cell development and epigenetic programming represents a window of sensitivity to environmental factors, and when an altered epigenetic programming is induced, it can be perpetuated across generations (Anway et al. 2005; Skinner et al. 2010).
A number of different environmental toxicants have been shown to promote exposure-specific alterations in the F3 generation sperm epigenome (DNA methylation) (Manikkam et al. 2012a). These include dioxin (Bruner-Tran and Osteen 2011; Manikkam et al. 2012c), a mixture of plastic compounds [bisphenol A (BPA) and phthalates] (Manikkam et al. 2013), the pesticide methoxychlor (Anway et al. 2005), a mixture of pesticide and insecticide (permethrin and DEET) (Manikkam et al. 2012b), and a hydrocarbon mixture (JP8 jet fuel) (Tracey et al. 2013). In addition to environmental toxicants, nutritional compounds (Burdge et al. 2011; de Assis et al. 2012) and stress (Champagne 2008; Crews et al. 2012) can promote epigenetic transgenerational phenotypes.
Testis Development and Biology
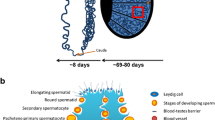
The process of gonadal development is essential for sex determination and the establishment of the germline. Cell lineages and cell populations are established during early embryonic development; they then influence adult gonadal function, endocrine responses, and fertility. Understanding the fetal basis of adult onset testis defects and infertility requires an elucidation of the molecular and cellular events during gonadal sex determination. The adult testis is a complex organ that is composed of seminiferous tubules enclosed by a surrounding interstitium. The seminiferous tubules are the site of spermatogenesis where germ cells develop into spermatozoa in close interaction with Sertoli cells. The Sertoli cell is an important testicular somatic cell that controls the germ cell environment by the secretion and transport of nutrients and regulatory factors (Fawcett 1975; Sertoli 1865). Tight junctional complexes between the Sertoli cells contribute to the maintenance of a blood–testis barrier (Setchell and Waites 1975) and create a unique environment within the tubule (Waites and Gladwell 1982). Surrounding the basal surface of the Sertoli cells is a layer of peritubular myoid cells that function to contract the tubule. The peritubular cells surround and form the exterior wall of the seminiferous tubule. The interstitial space around the seminiferous tubules contain another somatic cell type, the Leydig cell, which is responsible for testosterone production. Leydig cells have a major influence on spermatogenesis through the actions of testosterone on both the seminiferous tubule and the pituitary. Although the Leydig cell has numerous secretory products (Skinner 1991), testosterone is the most significant secretory product of the cells. Interaction of all three somatic cells, Sertoli, peritubular, and Leydig, is important for the regulation of normal spermatogenic function in the testis (Skinner 1991). The coordinated interactions of different testis cell populations are critical for the initial morphogenesis process through the adult stage of maintaining the process of spermatogenesis.
The process of fetal testis formation occurs late in fetal development (embryonic day 13, E13, in the rat). Initially this involves migration of primordial germ cells from the yolk sac to the hindgut and then from the hindgut to the genital ridge and gonad. After migration, germ cell differentiation in the gonad is dependent on gonadal sex determination and the induction of specific transcription factors (McLaren 1991; Takasaki et al. 2001). The gonad is bipotential after germ cell migration and can be distinguished morphologically from the adjoining mesonephros (E12 in rat) but cannot be identified as an ovary or a testis. A variety of genes such as Sry (sex determining region Y), Sox9 (SRY box 9), Sf1 (splicing factor 1), Dmrt1 (double sex and mab-3 related transcription factor), and Tcf21 (transcription factor 21) are involved in the transcriptional induction of Sertoli cell differentiation and testis development (Bhandari et al. 2012b; Clinton and Haines 2001; Drews 2000; Ikeda et al. 2001; McLaren 2000; Ostrer 2000; Parker et al. 2001; Raymond et al. 2000; Vaillant et al. 2001). Two morphological events occur early at embryonic day 13 (E13) during sex determination to alter the bipotential gonad. First, Sertoli cells, which are proposed to be the first cell in the testis to differentiate, aggregate around primordial germ cells (Jost et al. 1981; Magre and Jost 1980). Secondly, migration of mesenchymal cells occurs from the adjoining mesonephros and coelomic epithelium into the developing gonad to surround the Sertoli cell-germinal cell aggregates. It has been speculated that the migrating population of cells is preperitubular cells (Buehr et al. 1993; Merchant-Larios et al. 1993; Ricci et al. 1999). The mechanism for this migration signal is from the testis to promote cell migration (Clement et al. 2011) through observations that female mesonephros cells can also be stimulated to initiate cell migration after close interaction with a developing testis (McLaren 2000). In addition, using an organ culture system in which mesonephros and embryonic testis were separated by an embryonic ovary, mesonephros cells migrated through the ovary to the testis (Karl and Capel 1998). Another cell migration event required for cord formation involves endothelial cells from the coelomic epithelium to form the testis vasculature (Bott et al. 2008; Cool and Capel 2009; Cool et al. 2008). The cords develop neonatally into seminiferous cords and at the onset of puberty develop into seminiferous tubules.
Molecular Processes in Fetal Development
SRY is the testis-determining factor on the Y chromosome proposed by Jost that initiates the molecular events for Sertoli cell differentiation and male gonadal sex determination (McClelland et al. 2012; Parma and Radi 2012). The combined interactions between SRY and SOX9 are critical for male sex determination and precursor Sertoli cell differentiation (Kim and Capel 2006; Miyamoto et al. 2008; Ottolenghi et al. 2007). Upstream genes such as Wt1 (Wilms tumor 1) precede Sry (Gao et al. 2006; Kanai et al. 2005), but SRY initiates Sertoli cell differentiation, which subsequently involves an upregulation of SOX9 in Sertoli cells (Gao et al. 2006; Kidokoro et al. 2005; Sekido et al. 2004). SRY and SOX9 expression in Sertoli cells is associated with germ cell–Sertoli cell aggregation prior to cord formation (Moreno-Mendoza et al. 2003; Sekido et al. 2004). Abnormal SRY or SOX9 expression is associated with sex reversal and other disease states, including abnormal testis development (Barrionuevo et al. 2006; Bouma et al. 2005; Bullejos and Koopman 2005; Moreno-Mendoza et al. 2003; Nikolova and Vilain 2006). In regards to the regulation of the Sry promoter and inducing factors, very little is known outside the timing of the event in the genital ridge (Daneau et al. 2002; Hiramatsu et al. 2009; Nikolova and Vilain 2006). In regards to downstream genes to Sry, a large number of binding targets have recently been identified (Bhandari et al. 2012a). A downstream target of SRY is the basic helix loop factor TCF21 that promotes a secondary cascade of events associated with Sertoli cell differentiation (Bhandari et al. 2012b). Another downstream function of SRY/SOX9 is the production of prostaglandin D2 (Daneau et al. 2002; DiNapoli and Capel 2008; Malki et al. 2005; Wilhelm et al. 2005), but SOX9 appears to be the primary regulator of prostaglandin synthesis (Wallis et al. 2008; Wilhelm et al. 2007). Synergistic actions of SRY and SF1 have been shown on the Sox9 promoter (Sekido and Lovell-Badge 2008). Recent SRY downstream gene candidates have been suggested (Bhandari et al. 2012a; Bradford et al. 2009), such as the Cbln4 gene with no known function. Recently Wdr5 (WD repeat domain) has been shown to be a downstream target of SRY (Xu et al. 2012) and NTF3 (neurotropin 3) (Clement et al. 2011) and the bHLH factor TCF21 (Bhandari et al. 2011). Interestingly, NTF3 was previously shown to act as a Sertoli-cell-produced chemotactic factor to promote mesonephros cell migration into the developing testis to promote cord formation (Cupp et al. 2003). The induction of fetal testis cord function is an anticipated initial downstream function for SRY (Cupp et al. 2003), while TCF21 is proposed to be involved in the induction of Sertoli cell differentiation (Bhandari et al. 2011, 2012b).
Environmental Exposures and Fetal Testis Development
Early life exposures to nutritional alterations or environmental compounds have been shown to cause later life adult onset disease (Manikkam et al. 2012a; Skinner et al. 2010). The fetal basis of adult onset disease is now well established and one of the primary mechanisms involved is epigenetics (Skinner 2011). The fetal exposure to an environmental insult at a critical window of development for an organism can shift the epigenetic programming that is mitotically stable to then promote altered gene expression and adult onset disease (Skinner 2011; Skinner et al. 2010). The critical window of exposure for the testis and subsequent adult onset testis disease is the gonadal sex determination period. This is when the somatic cells fate, germline cell fate and initial differentiation develops. The later life adult onset disease associated with these fetal exposures are spermatogenic cell apoptosis and defects (Shukla et al. 2012), as well as male infertility (Anway et al. 2005, 2006). Fetal exposure to vinclozolin during male gonadal sex determination has been shown to promote later life testis spermatogenic cell defects (apoptosis) in 90 % of the males and in adult rats at 1 year of age a 30 % increase of male infertility (Anway et al. 2005, 2006). Vinclozolin is a commonly used agriculture fungicide which is an antiandrogenic endocrine disruptor. In addition to promoting adult onset testis disease in the F1 generation, the germline (sperm) epigenome becomes permanently programmed epigenetically to transmit the epigenetic alterations (epimutations) and disease phenotypes to subsequent generations (F1–F4) through epigenetic transgenerational inheritance of the disease phenotype (Anway et al. 2005, 2006; Skinner et al. 2010). Therefore, the in vivo exposure of a gestating female during the period of gonadal sex determination for the F1 generation fetus promotes adult onset testis disease in the F1 generation, as well as induces an epigenetic transgenerational inheritance of the testis disease phenotype to subsequent generations (Anway et al. 2005, 2006). Other authors have shown similar transgenerational effects on fertility after peritoneal exposure to bisphenol-A (Salian et al. 2009).
Epigenetic Alterations of Testis Cell Biology and Fertility
Epigenetic mechanisms are fundamental to ensuring normal gonadal development and spermatogenesis (Carrell 2012; Rajender et al. 2011; Skinner et al. 2010). One of the crucial processes that depends on epigenetic mechanisms is the exchange of histones for protamines, which results in the genome's becoming tightly compacted (heterochromatin) in the sperm and in inhibition of expression (Carrell 2012; Rajender et al. 2011). For this process to occur, hyperacetylation of histone H4 is needed (Sonnack et al. 2002). Recent experiments have shown that H4K12ac associates preferentially with regions near the transcription start site and in promoters that express transcripts stored in mature human sperm (Paradowska et al. 2012). Interestingly, decreased histone H4 acetylation in spermatids results in impaired spermatogenesis and decreased fertility (Sonnack et al. 2002). Additional histones such as H2AL1 and H2AL2 have also been described to mark pericentric regions in condensing spermatids and be involved in forming new nucleoprotein structures (Govin et al. 2007). Recently, several publications have highlighted the interaction between histone modifications and DNA methylation in several organism models (Du et al. 2012; Johnson et al. 2007; Ooi et al. 2007). This would also be the case for histone modifications during spermatogenesis. Observations suggest that the fertilized zygote inherits specific histones and histone-based chromatin organization from the sperm (Paradowska et al. 2012), but the potential random nature of this programming needs to be assessed. Histone binding and chromatin organization in the male germline would be a consequence of fine-scale base composition variation GC and CpG content (Vavouri and Lehner 2011). GC-rich regions in promoters would be prone to retain the nucleosomes and not exchange them for protamines, which happens in 4 % of the sperm genome (Vavouri and Lehner 2011). These regions with nucleosome retention would prevent reprogramming of DNA methylation after fertilization (Vavouri and Lehner 2011). Interestingly, it was recently shown that infertile men display abnormalities in both histone modifications (H3K4me and H3K27me) and DNA methylation at imprinted and developmental loci (Hammoud et al. 2011, 2010; Houshdaran et al. 2007) in the sperm DNA. Reduced histone methylation in the Brdt (bromo domain testis-specific) promoter is associated with reduced BRDT expression in subfertile men (Steilmann et al. 2010). Studies have also shown that sperm from men with fertility problems have altered DNA methylation patterns in imprinted genes (Boissonnas et al. 2010; Kobayashi et al. 2007; Marques et al. 2004, 2008, 2010), which would generate imprinting abnormalities in the offspring when this sperm is used in assisted reproductive technologies (Kobayashi et al. 2007; Marques et al. 2004). Adult exposure to butyl-paraben has been shown to produce DNA methylation changes in the sperm (Park et al. 2012). Prenatal exposure to ethanol has also been shown to induce decreased spermatogenesis and DNA methylation changes in imprinted genes (Stouder et al. 2011). It is postulated that the methylenetetrahydrofolate reductase (Mthfr) gene would have a central role in idiopathic male infertility. Some Mthfr-deficient strains of mice have alterations in sperm DNA methylation in a number of sites (Chan et al. 2010). Also, Mthfr DNA hypermethylation in sperm is associated with idiopathic male infertility in humans (Wu et al. 2010). In addition to the importance of DNA methylation changes in germline development in imprinted and developmental loci, the DNA methylation in repeat elements such as B1 SINEs has been proposed as having a role in transcriptional regulation of testis-specific genes (Ichiyanagi et al. 2011). Genes involved in the pathway of PIWI-associated small RNAs (piRNAs), such as Piwil2 (Piwi-like 2) and Tdrd1 (tudor domain containing 1), are hypermethylated in the testicular tissue of males with different forms of fertility problems (Heyn et al. 2012).
Epigenetic modifications have also been reported in the somatic cells controlling the process of spermatogenesis, such as Sertoli and Leydig cells. In Sertoli cells Rhox5 (reproductive homeobox 5) deletion produces subfertility, increases germ-cell apoptosis, and decreases sperm count and motility through two promoters repressed by DNA methylation (Shanker et al. 2008). Another interesting observation relates to epigenetic changes produced in the proximal promoter of the fatty acid amide hydrolase (Faah) gene (reduced DNA and histone H3 methylation) in response to estradiol in mouse Sertoli cell cultures (Grimaldi et al. 2012). Epigenetic changes have also been observed in Leydig cells after exposure to environmental contaminants. Changes in DNA methylation have been observed in mouse Leydig TM3 cell line cultures following exposure to either low or high doses of arsenic (Singh and DuMond 2007). Exposure of these cells to cadmium leads to reduced expression of DNA methyltransferase 1 (Singh et al. 2009). In utero exposure to di-(2-ethylhexyl)phthalate has been shown to produce postnatal alteration in demethylation in several nuclear receptor genes in Leydig cells, among them the estrogen receptor beta (ER-beta), Nr142 (thyroid receptor beta), peroxisome proliferator activated receptor alpha (PPAR-alpha), and mineralocorticoid receptor (MR) (Martinez-Arguelles et al. 2009). Interestingly, treatment of Leydig cells with luteinizing hormone causes cellular hypomethylation, suggesting that environmental exposures that alter DNA methylation in testicular cells may influence hormone actions (Reddy and Reddy 1990). Epigenetic modifications in somatic testicular tissues or germ cells that are associated with infertility or poor semen parameters are shown in Table 5.1.
Conclusions
Increasing concerns about the decrease in fertility in men have developed over the past few decades. An explanation for this trend is the exposure of the human population to toxicants derived from industrial products or processes. Many of these contaminating agents are capable of altering epigenetic programming in organisms. These alterations are generally produced during early developmental stages and generate diseases in adults. A number of reproductive and metabolic diseases have been shown to have an epigenetic and developmental component. Interestingly, these environmentally induced disease states can become transgenerationally transmitted. Strong evidence has accumulated in recent years showing that environmental toxicants alter developmental and epigenetic processes to promote abnormal spermatogenesis in men. Although the molecular mechanisms await full elucidation, there is no longer any doubt that an important component of the disruption of spermatogenic cell development is exposure to environmental toxicants. Therefore, future studies addressing fertility in humans should place special emphasis on the role of environmental epigenetics on testis development and spermatogenic-cell-associated disease.
References
Adams NR (1981) A changed responsiveness to oestrogen in ewes with clover disease. J Reprod Fertil Suppl 30:223–230
Adams NR (1990) Permanent infertility in ewes exposed to plant oestrogens. Aust Vet J 67: 197–201
Adams NR (1995) Detection of the effects of phytoestrogens on sheep and cattle. J Anim Sci 73:1509–1515
Allegrucci C, Thurston A, Lucas E et al (2005) Epigenetics and the germline. Reproduction 129:137–149
Anetor JI, Wanibuchi H, Fukushima S (2007) Arsenic exposure and its health effects and risk of cancer in developing countries: micronutrients as host defence. Asian Pac J Cancer Prev 8(1):13–23
Anway MD, Cupp AS, Uzumcu M et al (2005) Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308:1466–1469
Anway MD, Leathers C, Skinner MK (2006) Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology 147:5515–5523
Arico JK, Katz DJ, van der Vlag J et al (2011) Epigenetic patterns maintained in early Caenorhabditis elegans embryos can be established by gene activity in the parental germ cells. PLoS Genet 7:e1001391
Balabanic D, Rupnik M, Klemencic AK (2011) Negative impact of endocrine-disrupting compounds on human reproductive health. Reprod Fertil Dev 23:403–416
Barrionuevo F, Bagheri-Fam S, Klattig J et al (2006) Homozygous inactivation of Sox9 causes complete XY sex reversal in mice. Biol Reprod 74:195–201
Bhandari R, Sadler-Riggleman I, Clement TM et al (2011) Basic helix-loop-helix transcription factor TCF21 is a downstream target of the male sex determining gene SRY. PLoS One 6:e19935
Bhandari R, Haque Md M, Skinner M (2012a) Global genome analysis of the downstream binding targets of testis determining factor SRY AND SOX9. PLoS One 7:e43380
Bhandari RK, Schinke EN, Haque MM et al (2012b) SRY induced TCF21 genome-wide targets and cascade of bHLH factors during sertoli cell differentiation and male sex determination in rats. Biol Reprod 87:131
Boissonnas CC, Abdalaoui HE, Haelewyn V et al (2010) Specific epigenetic alterations of IGF2-H19 locus in spermatozoa from infertile men. Eur J Hum Genet 18:73–80
Bott RC, Clopton DT, Cupp AS (2008) A proposed role for VEGF isoforms in sex-specific vasculature development in the gonad. Reprod Domest Anim 43(Suppl 2):310–316
Bouma GJ, Albrecht KH, Washburn LL et al (2005) Gonadal sex reversal in mutant Dax1 XY mice: a failure to upregulate Sox9 in pre-Sertoli cells. Development 132:3045–3054
Bradford ST, Hiramatsu R, Maddugoda MP et al (2009) The cerebellin 4 precursor gene is a direct target of SRY and SOX9 in mice. Biol Reprod 80:1178–1188
Bruner-Tran KL, Osteen KG (2011) Developmental exposure to TCDD reduces fertility and negatively affects pregnancy outcomes across multiple generations. Reprod Toxicol 31:344–350
Buehr M, Gu S, McLaren A (1993) Mesonephric contribution to testis differentiation in the fetal mouse. Development 117:273–281
Bullejos M, Koopman P (2005) Delayed Sry and Sox9 expression in developing mouse gonads underlies B6-Y(DOM) sex reversal. Dev Biol 278:473–481
Burdge GC, Hoile SP, Uller T et al (2011) Progressive, transgenerational changes in offspring phenotype and epigenotype following nutritional transition. PLoS One 6:e28282
Carone BR, Fauquier L, Habib N et al (2010) Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143:1084–1096
Carrell DT (2012) Epigenetics of the male gamete. Fertil Steril 97:267–274
Caserta D, Mantovani A, Marci R et al (2011) Environment and women’s reproductive health. Hum Reprod Update 17:418–433
Champagne FA (2008) Epigenetic mechanisms and the transgenerational effects of maternal care. Front Neuroendocrinol 29:386–397
Chan D, Cushnie DW, Neaga OR et al (2010) Strain-specific defects in testicular development and sperm epigenetic patterns in 5,10-methylenetetrahydrofolate reductase-deficient mice. Endocrinology 151:3363–3373
Cheng TF, Choudhuri S, Muldoon-Jacobs K (2012) Epigenetic targets of some toxicologically relevant metals: a review of the literature. J Appl Toxicol 32:643–653
Clement TM, Bhandari RK, Sadler-Riggleman I et al (2011) Sry directly regulates the neurotrophin-3 promoter during male sex determination and testis development in rats. Biol Reprod 85:227–284
Clinton M, Haines LC (2001) An overview of factors influencing sex determination and gonadal development in birds. EXS 97–115
Cool J, Capel B (2009) Mixed signals: development of the testis. Semin Reprod Med 27:5–13
Cool J, Carmona FD, Szucsik J et al (2008) Peritubular myoid cells are not the migrating population required for testis cord formation in the XY gonad. Sex Dev 2:128–133
Crews D, Gillette R, Scarpino SV et al (2012) Epigenetic transgenerational inheritance of altered stress responses. Proc Natl Acad Sci U S A 109:9143–9148
Cupp AS, Uzumcu M, Skinner MK (2003) Chemotactic role of neurotropin 3 in the embryonic testis that facilitates male sex determination. Biol Reprod 68:2033–2037
Daneau I, Pilon N, Boyer A et al (2002) The porcine SRY promoter is transactivated within a male genital ridge environment. Genesis 33:170–180
de Assis S, Warri A, Cruz MI et al (2012) High-fat or ethinyl-oestradiol intake during pregnancy increases mammary cancer risk in several generations of offspring. Nat Commun 3:1053
DiNapoli L, Capel B (2008) SRY and the standoff in sex determination. Mol Endocrinol 22:1–9
Drews U (2000) Local mechanisms in sex specific morphogenesis. Cytogenet Cell Genet 91:72–80
Du J, Zhon X, Bernatavichute Y et al (2012) Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 151:167–180
Dunn GA, Bale TL (2011) Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 152:2228–2236
Durcova-Hills G, Hajkova P, Sullivan S et al (2006) Influence of sex chromosome constitution on the genomic imprinting of germ cells. Proc Natl Acad Sci U S A 103:11184–11188
Fawcett D (1975) The ultrastructure and functions of the Sertoli cell. In: Hamilton RGE (ed) Handbook of physiology. American Physiological Society, Washington, DC, pp 22–55
Forouzanfar MH, Foreman KJ, Delossantos AM et al (2011) Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet 378:1461–1484
Fowler PA, Bellingham M, Sinclair KD et al (2012) Impact of endocrine-disrupting compounds (EDCs) on female reproductive health. Mol Cell Endocrinol 355:231–239
Gao F, Maiti S, Alam N et al (2006) The Wilms tumor gene, Wt1, is required for Sox9 expression and maintenance of tubular architecture in the developing testis. Proc Natl Acad Sci U S A 103:11987–11992
Giwercman A, Giwercman YL (2011) Environmental factors and testicular function. Best Pract Res Clin Endocrinol Metab 25:391–402
Gonzalez-Pardo H, Perez Alvarez M (2013) Epigenetics and its implications for psychology. Psicothema 25:3–12
Govin J, Escoffier E, Rousseaux S et al (2007) Pericentric heterochromatin reprogramming by new histone variants during mouse spermiogenesis. J Cell Biol 176:283–294
Grimaldi P, Pucci M, Di Siena S et al (2012) The faah gene is the first direct target of estrogen in the testis: role of histone demethylase LSD1. Cell Mol Life Sci 69:4177–4190
Guerrero-Bosagna and Skinner (2012) Environmental epigenetics and phytoestrogen/phytochemical exposures. [epub ahead of print]. doi:10.1016/j.jsbmb.2012.12.011
Guerrero-Bosagna C, Settles M, Lucker B et al (2010) Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS One 5:e13100
Guillette LJ Jr, Iguchi T (2012) Ecology. Life in a contaminated world. Science 337:1614–1615
Guo YL, Hsu PC, Hsu CC et al (2000) Semen quality after prenatal exposure to polychlorinated biphenyls and dibenzofurans. Lancet 356:1240–1241
Guo YL, Lambert GH, Hsu CC et al (2004) Yucheng: health effects of prenatal exposure to polychlorinated biphenyls and dibenzofurans. Int Arch Occup Environ Health 77:153–158
Hammoud SS, Purwar J, Pflueger C et al (2010) Alterations in sperm DNA methylation patterns at imprinted loci in two classes of infertility. Fertil Steril 94:1728–1733
Hammoud SS, Nix DA, Hammoud AO et al (2011) Genome-wide analysis identifies changes in histone retention and epigenetic modifications at developmental and imprinted gene loci in the sperm of infertile men. Hum Reprod 26:2558–2569
Heyn H, Ferreira HJ, Bassas L et al (2012) Epigenetic disruption of the PIWI pathway in human spermatogenic disorders. PLoS One 7:e47892
Hiramatsu R, Matoba S, Kanai-Azuma M et al (2009) A critical time window of SRY action in gonadal sex determination in mice. Development 136:129–138
Houshdaran S, Cortessis VK, Siegmund K et al (2007) Widespread epigenetic abnormalities suggest a broad DNA methylation erasure defect in abnormal human sperm. PLoS One 2:e1289
Ichiyanagi K, Li Y, Watanabe T et al (2011) Locus- and domain-dependent control of DNA methylation at mouse B1 retrotransposons during male germ cell development. Genome Res 21:2058–2066
Ikeda Y, Takeda Y, Shikayama T et al (2001) Comparative localization of Dax-1 and Ad4BP/SF-1 during development of the hypothalamic-pituitary-gonadal axis suggests their closely related and distinct functions. Dev Dyn 220:363–376
Inbar-Feigenberg M, Choufani S, Butcher DT, Roifman M, Weksberg R. (2013) Basic concepts of epigenetics. Fertil Steril 1;99(3):607–615
Johnson LM, Bostick M, Zhang X et al (2007) The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr Biol 17:379–384
Jost A, Magre S, Agelopoulou R (1981) Early stages of testicular differentiation in the rat. Hum Genet 58:59–63
Kanai Y, Hiramatsu R, Matoba S et al (2005) From SRY to SOX9: mammalian testis differentiation. J Biochem 138:13–19
Karl J, Capel B (1998) Sertoli cells of the mouse testis originate from the coelomic epithelium. Dev Biol 203:323–333
Karmaus W, Ziyab AH, Everson T et al (2013) Epigenetic mechanisms and models in the origins of asthma. Curr Opin Allergy Clin Immunol 13:63–69
Khazamipour N, Noruzinia M, Fatehmanesh P et al (2009) MTHFR promoter hypermethylation in testicular biopsies of patients with non-obstructive azoospermia: the role of epigenetics in male infertility. Hum Reprod 24:2361–2364
Kidokoro T, Matoba S, Hiramatsu R et al (2005) Influence on spatiotemporal patterns of a male-specific Sox9 activation by ectopic Sry expression during early phases of testis differentiation in mice. Dev Biol 278:511–525
Kim Y, Capel B (2006) Balancing the bipotential gonad between alternative organ fates: a new perspective on an old problem. Dev Dyn 235:2292–2300
Kobayashi H, Sato A, Otsu E et al (2007) Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum Mol Genet 16:2542–2551
Lees-Murdock DJ, Walsh CP (2008) DNA methylation reprogramming in the germ line. Epigenetics 3:5–13
Magre S, Jost A (1980) The initial phases of testicular organogenesis in the rat. An electron microscopy study. Arch Anat Microsc Morphol Exp 69:297–318
Main KM, Skakkebaek NE, Virtanen HE et al (2010) Genital anomalies in boys and the environment. Best Pract Res Clin Endocrinol Metab 24:279–289
Malki S, Nef S, Notarnicola C et al (2005) Prostaglandin D2 induces nuclear import of the sex-determining factor SOX9 via its cAMP-PKA phosphorylation. EMBO J 24:1798–1809
Manikkam M, Guerrero-Bosagna C, Tracey R et al (2012a) Transgenerational actions of environmental compounds on reproductive disease and epigenetic biomarkers of ancestral exposures. PLoS One 7:e31901
Manikkam M, Tracey R, Guerrero-Bosagna C et al (2012b) Pesticide and insect repellent mixture (Permethrin and DEET) induces epigenetic transgenerational inheritance of disease and sperm epimutations. Reprod Toxicol 34:708–719
Manikkam M, Tracey R, Guerrero-Bosagna C et al (2012c) Dioxin (TCDD) induces epigenetic transgenerational inheritance of adult onset disease and sperm epimutations. PLoS One 7:e46249
Manikkam M, Tracey R, Guerrero-Bosagna C et al (2013) Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of adult-onset disease and sperm epimutations. PLoS One 8:e55387
Marques CJ, Carvalho F, Sousa M et al (2004) Genomic imprinting in disruptive spermatogenesis. Lancet 363:1700–1702
Marques CJ, Costa P, Vaz B et al (2008) Abnormal methylation of imprinted genes in human sperm is associated with oligozoospermia. Mol Hum Reprod 14:67–74
Marques CJ, Francisco T, Sousa S et al (2010) Methylation defects of imprinted genes in human testicular spermatozoa. Fertil Steril 94:585–594
Martinez-Arguelles DB, Culty M, Zirkin BR et al (2009) In utero exposure to di-(2-ethylhexyl) phthalate decreases mineralocorticoid receptor expression in the adult testis. Endocrinology 150:5575–5585
McClelland K, Bowles J, Koopman P (2012) Male sex determination: insights into molecular mechanisms. Asian J Androl 14:164–171
McLaren A (1991) Development of the mammalian gonad: the fate of the supporting cell lineage. Bioessays 13:151–156
McLaren A (2000) Germ and somatic cell lineages in the developing gonad. Mol Cell Endocrinol 163:3–9
Merchant-Larios H, Moreno-Mendoza N, Buehr M (1993) The role of the mesonephros in cell differentiation and morphogenesis of the mouse fetal testis. Int J Dev Biol 37:407–415
Miyamoto Y, Taniguchi H, Hamel F et al (2008) A GATA4/WT1 cooperation regulates transcription of genes required for mammalian sex determination and differentiation. BMC Mol Biol 9:44
Moreno-Mendoza N, Harley V, Merchant-Larios H (2003) Cell aggregation precedes the onset of Sox9-expressing preSertoli cells in the genital ridge of mouse. Cytogenet Genome Res 101:219–223
Morgan CP, Bale TL (2011) Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J Neurosci 31:11748–11755
Nikolova G, Vilain E (2006) Mechanisms of disease: transcription factors in sex determination–relevance to human disorders of sex development. Nat Clin Pract Endocrinol Metab 2:231–238
North ML, Ellis AK (2011) The role of epigenetics in the developmental origins of allergic disease. Ann Allergy Asthma Immunol 10:355–361, quiz 362
Ogino S, Lochhead P, Chan AT et al (2013) Molecular pathological epidemiology of epigenetics: emerging integrative science to analyze environment, host, and disease. Mod Pathol 26:465–484
Ooi SK, Qiu C, Bernstein E et al (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448:714–717
Ostrer H (2000) Sexual differentiation. Semin Reprod Med 18:41–49
Ottolenghi C, Uda M, Crisponi L et al (2007) Determination and stability of sex. Bioessays 29:15–25
Paradowska AS, Miller D, Spiess AN et al (2012) Genome wide identification of promoter binding sites for H4K12ac in human sperm and its relevance for early embryonic development. Epigenetics 7:1057–1070
Park CJ, Nah WH, Lee JE et al (2012) Butyl paraben-induced changes in DNA methylation in rat epididymal spermatozoa. Andrologia 44(Suppl 1):187–193
Parker KL, Schimmer BP, Schedl A (2001) Genes essential for early events in gonadal development. EXS 91:11–24
Parkin DM (2004) International variation. Oncogene 23:6329–6340
Parma P, Radi O (2012) Molecular mechanisms of sexual development. Sex Dev 6:7–17
Pimentel D, Cooperstein S, Randell H et al (2007) Ecology of increasing diseases: population growth and environmental degradation. Hum Ecol 35:653–668
Poplinski A, Tuttelmann F, Kanber D et al (2010) Idiopathic male infertility is strongly associated with aberrant methylation of MEST and IGF2/H19 ICR1. Int J Androl 33:642–649
Rajender S, Avery K, Agarwal A (2011) Epigenetics, spermatogenesis and male infertility. Mutat Res 727:62–71
Raymond CS, Murphy MW, O’Sullivan MG et al (2000) Dmrt1, a gene related to worm and fly sexual regulators, is required for mammalian testis differentiation. Genes Dev 14:2587–2595
Reddy PM, Reddy PR (1990) Differential regulation of DNA methylation in rat testis and its regulation by gonadotropic hormones. J Steroid Biochem 35:173–178
Reik W, Dean W, Walter J (2001) Epigenetic reprogramming in mammalian development. Science 293:1089–1093
Ricci G, Catizone A, Innocenzi A et al (1999) Hepatocyte growth factor (HGF) receptor expression and role of HGF during embryonic mouse testis development. Dev Biol 216:340–347
Salian S, Doshi T, Vanage G (2009) Perinatal exposure of rats to Bisphenol A affects the fertility of male offspring. Life Sci 85:742–752
Schug TT, Janesick A, Blumberg B et al (2011) Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol 127:204–215
Sekido R, Lovell-Badge R (2008) Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 453:930–934
Sekido R, Bar I, Narvaez V, Penny G et al (2004) SOX9 is up-regulated by the transient expression of SRY specifically in Sertoli cell precursors. Dev Biol 274:271–279
Sertoli E (1865) On the existence of special branched cells in hte seminiferous tubule of the human testes. Morgangni 7:31–39
Setchell BP, Waites GMH (1975) In: Hamilton D, Greep RO (eds) Handbook of physiology. American Physiological Society, Washington, DC, pp 143–172
Shanker S, Hu Z, Wilkinson MF (2008) Epigenetic regulation and downstream targets of the Rhox5 homeobox gene. Int J Androl 31:462–470
Sharpe RM (2010) Environmental/lifestyle effects on spermatogenesis. Philos Trans R Soc Lond B Biol Sci 365:1697–1712
Shukla KK, Mahdi AA, Rajender S (2012) Apoptosis, spermatogenesis and male infertility. Front Biosci Elite Ed 4:746–754
Singh KP, DuMond JW Jr (2007) Genetic and epigenetic changes induced by chronic low dose exposure to arsenic of mouse testicular Leydig cells. Int J Oncol 30:253–260
Singh KP, Kumari R, Pevey C et al (2009) Long duration exposure to cadmium leads to increased cell survival, decreased DNA repair capacity, and genomic instability in mouse testicular Leydig cells. Cancer Lett 279:84–92
Skakkebaek NE, Rajpert-De Meyts E, Main KM (2001) Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects. Hum Reprod 16:972–978
Skakkebaek NE, Rajpert-De Meyts E et al (2007) Testicular cancer trends as ‘whistle blowers’ of testicular developmental problems in populations. Int J Androl 30:198–204, discussion 204–195
Skinner MK (1991) Cell-cell interactions in the testis. Endocr Rev 12:45–77
Skinner MK (2011) Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics 6:838–842
Skinner MK, Manikkam M, Guerrero-Bosagna C (2010) Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab 21:214–222
Sonnack V, Failing K, Bergmann M et al (2002) Expression of hyperacetylated histone H4 during normal and impaired human spermatogenesis. Andrologia 34:384–390
Steilmann C, Cavalcanti MC, Bartkuhn M et al (2010) The interaction of modified histones with the bromodomain testis-specific (BRDT) gene and its mRNA level in sperm of fertile donors and subfertile men. Reproduction 140:435–443
Stoccoro A, Karlsson HL, Coppede F et al (2012) Epigenetic effects of nano-sized materials. Toxicology. Dec 10. [Epub ahead of print]. doi:10.1016/j.tox.2012.12.002
Stouder C, Somm E, Paoloni-Giacobino A (2011) Prenatal exposure to ethanol: a specific effect on the H19 gene in sperm. Reprod Toxicol 31:507–512
Sun C, Burgner DP, Ponsonby AL et al (2013) Effects of early-life environment and epigenetics on cardiovascular disease risk in children: highlighting the role of twin studies. Pediatr Res 73(4 Pt 2):523–530
Takasaki N, Rankin T, Dean J (2001) Normal gonadal development in mice lacking GPBOX, a homeobox protein expressed in germ cells at the onset of sexual dimorphism. Mol Cell Biol 21:8197–8202
Thrupp LA (1991) Sterilization of workers from pesticide exposure: the causes and consequences of DBCP-induced damage in Costa Rica and beyond. Int J Health Serv 21:731–757
Tracey R, Manikkam M, Guerrero-Bosagna C, Skinner MK (2013) Hydrocarbons (jet fuel JP-8) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. Reprod Toxicol 36:104–116
Tsugane S, Sasazuki S (2007) Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 10:75–83
Vaillant S, Magre S, Dorizzi M et al (2001) Expression of AMH, SF1, and SOX9 in gonads of genetic female chickens during sex reversal induced by an aromatase inhibitor. Dev Dyn 222: 228–237
Vavouri T, Lehner B (2011) Chromatin organization in sperm may be the major functional consequence of base composition variation in the human genome. PLoS Genet 7:e1002036
Waites GM, Gladwell RT (1982) Physiological significance of fluid secretion in the testis and blood-testis barrier. Physiol Rev 62:624–671
Wallace DC (2010) Bioenergetics and the epigenome: interface between the environment and genes in common diseases. Dev Disabil Res Rev 16:114–119
Wallis MC, Waters PD, Graves JA (2008) Sex determination in mammals–before and after the evolution of SRY. Cell Mol Life Sci 65:3182–3195
Whorton D, Milby TH, Krauss RM et al (1979) Testicular function in DBCP exposed pesticide workers. J Occup Med 21:161–166
Wilhelm D, Martinson F, Bradford S et al (2005) Sertoli cell differentiation is induced both cell-autonomously and through prostaglandin signaling during mammalian sex determination. Dev Biol 287:111–124
Wilhelm D, Hiramatsu R, Mizusaki H et al (2007) SOX9 regulates prostaglandin D synthase gene transcription in vivo to ensure testis development. J Biol Chem 282:10553–10560
Wu W, Shen O, Qin Y et al (2010) Idiopathic male infertility is strongly associated with aberrant promoter methylation of methylenetetrahydrofolate reductase (MTHFR). PLoS One 5:e13884
Xu Z, Gao X, He Y et al (2012) Synergistic effect of SRY and its direct target, WDR5, on Sox9 expression. PLoS One 7:e34327
Yuen RK, Chen B, Blair JD et al (2013) Hypoxia alters the epigenetic profile in cultured human placental trophoblasts. Epigenetics 8
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Guerrero-Bosagna, C., Skinner, M.K. (2014). Environmental Epigenetics and Effects on Male Fertility. In: Baldi, E., Muratori, M. (eds) Genetic Damage in Human Spermatozoa. Advances in Experimental Medicine and Biology, vol 791. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7783-9_5
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7783-9_5
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7782-2
Online ISBN: 978-1-4614-7783-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)